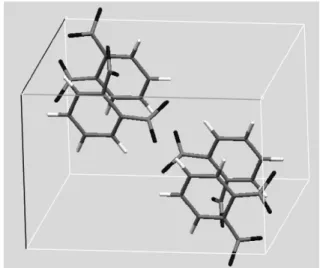
1.はじめに 最近になって構造が比較的複雑な有機物, 有機金属錯 体でも粉末回折データから未知構造が決定されるように なってきた. この背景には 1990 年代より欧米を中心に勢 力的に進められた粉末未知構造決定法の開発と, 放射光 を始めとした光源・光学系, 測定装置の進歩, 安価で高速 な計算を可能とするコンピュータの普及がある. 本稿で は粉末回折からの有機物質の構造決定において中心的な 役割を果たしている構造決定法,“実空間法”1) について実 例を交えながら紹介する. 2.粉末 X 線回折パターンと結晶構造 実空間法は, 結晶構造があれば粉末回折パターンが計 算できることに基づいている. これを理解するために, 最 初に結晶構造からの粉末回折パターンの計算について述 べる. 結晶構造として, o-ジニトロベンゼンを使用する. 本稿では例としてこの物質を一貫して用いる. 図 1 に結 晶構造を示した. 結晶系は単斜晶, 空間群は P21/aであり, 格子定数は a = 7.258[Å], b= 12.866[Å], c= 7.915[Å], β = 113.08[°] である. この格子の情報から, Bragg の式 2dhklsinθhkl=λ よ り回折線のピーク位置を求めることができる. ここで dhkl はミラー指数 hkl をもつ回折線の面間隔, λ は波長, θhklは 回折角である. 単斜晶の場合, 1/dhkl= 1/sin2β(h2/a2+ k2/b2+ l2/c2− 2hlcosβ/ac)から d hklを計算する. 波長を 1.0 [Å]として dhklから計算した回折ピーク位置を図 2a に示 す. 横軸は 2θ である. 空間群 P21/aの反射の出現則から h0l: h = 2n, 0k0 : k = 2n, h00 : h = 2n 以外の反射は現 れないため, 例えば 100, − 101, 030, 101, − 102 反射は 消滅する. 2θ = 5 ∼ 20 °の領域の反射数は合計 31 本とな る. 各回折線の計算積分強度 Ihklは Ihkl=|Fhkl|2LPmhklで表 される. ここで Fhklは結晶構造因子, LP はローレンツ偏 光因子, mhklは多重度である. Fhklは単位格子に含まれる全 原子数を N, n 番目の原子の分極座標(xn, yn, zn)と原子散 乱因子 fn, 熱振動の項 Tnを用いて以下の形に書かれる. (1) |Fhkl|2= FhklFhkl*である. ローレンツ偏光因子は, 試料と検 出器間の距離を一定とし定数を取り除くと LP =(1 + cos22θ)/(sinθsin2θ)となる. m hklは単斜晶の場合 mhkl= 4, mh0l= 2, m0k0= 2 となる. 例えば, 321 反射の場合, 3-21, 32-1, 3-2-1の 3 本の反射が同じ強度, 同じ回折線位置で Fhkl fn i hxn kyn lzn Tn n N =
∑
exp 2{
π(
+ +)
}
240実空間法による構造決定
名古屋大学工学研究科西堀英治
Eiji NISHIBORI: Structure Determination from Powder Diffraction
by Direct-Space Strategy
The crystal structure determination from powder diffraction(SDPD) has attracted wide interests for its huge potential to accelerate a design, synthesis, and characterization of the materials in the fields of nano-and bio- technologies. One of the most important progresses of the SDPD is the development of the direct-space strategy. In this report, SDPD by direct-space strategy has been described. Fundamental aspects of the strategy have been demonstrated with some examples. Global optimization methods within direct-space strategy such as Grid Search, Simulated Annealing, and Genetic Algorithm have been described including detailed methodologies.
有機粉末構造解析をはじめよう!(4)
日本結晶学会誌 53,240-248(2011)
図 1 o-ジニトロベンゼンの結晶構造.(Crystal Structure of o-dinitrobenzene.)
重なり合うため mhkl= 4 となる. o-ジニトロベンゼンの結 晶構造から計算した Ihklを図 2b に棒グラフとして示した. なお, T はすべての原子に同一の値を与えている. 現実の粉末回折データは図 2b のような棒グラフの形で は観測されない. 試料と光源のサイズが有限であり試料 粒のサイズも有限である効果のため, 各 Ihklが広がりをも ったプロファイルとして観測される. 粉末回折のデータ 解析ではこのプロファイルをプロファイル関数 P(2θ)を 用いて表す. 代表的な関数として, ガウス関数とローレン ツ関数を足し合わせた pesudo-Voigt 関数やローレンツ関 数の指数部を変化させた Pearson Ⅶ関数などが用いられ る. pesudo-Voigt 関数は次式で書かれる. (2) ここでη はローレンツ関数の比率, Hhklは回折線の反値幅 である. PearsonVII 関数は次式で書かれる. (3) ここで m は精密化可能な指数部を表す. pesudo-Voigt関数 P(2θ)を図 2c に示す. これらの関数 は, 一般に 2θhklを中心として低角, 高角側にそれぞれ 2θ 数度の範囲で定義される. また, 積分強度に相当する関数 の内部の面積が 1 になるよう規格化されている. よって, P(2θ)に Ihklをかけ合わせることで図 2d に示す粉末回折 パターンを計算できる. 必要となるのはプロファイルの幅 Hhklとピーク形状を決めるη や m の値である. 以上のよ うに, 粉末回折データを計算するには, 格子定数, 空間群, 単位格子内の原子の配置とプロファイル関数のパラメー タが必要であることがわかる. 現実には, データはバック グラウンドを含むためモデル化(関数化)されたバックグ ラウンドも必要になる. 粉末回折パターンは反射の重なり合いがあるため Ihklの 抽出が難しいことが知られている. このことを計算した回 折パターンを使って見てみる. o-ジニトロベンゼンの粉 末回折パターンの拡大図を図 3 に示す. 図 3a に点で示し た観測データで 1 本に見えるこの反射は下に示す 200 と − 122 反射が 6/1000 °の間隔で重なり合っている. 200 と− 122 のそれぞれの観測強度を求めることは非常に困 難でありほぼ不可能なことが容易にわかる. また, 図 3b の領域には 022, − 221, − 202 の 3 本の反射が重なり合 っている. − 221 と− 202 の間隔は 5/1000 °であり, この 場合も各反射の強度を求めるのは困難である. 一般に NA 個の原子の座標を単結晶構造解析で用いられる直接法な どで決定するには各原子で決めるべき x, y, z の 3 変数の 10倍である 30NA本の Ihklが必要とされる. o-ジニトロベ P m m H H m hkl m i hkl hkl m 2 2 2 1 0 5 1 4 2 1 2 2 1 1 2 2 θ π θ θ
( )
=(
−)
−(
)
+(
−)
−(
)
⎡ ⎣ ⎢ ⎢ ⎤ ⎦ ⎥ ⎥ − / / . P H H H H hkl i hkl hkl hkl i hkl hkl 2 2 1 4 2 2 1 4 2 4 2 2 2 2 2 2 2 θ η π θ θ η π θ θ( )
= ⎡ +(
−)
⎣ ⎢ ⎢ ⎤ ⎦ ⎥ ⎥ + −( )
⎛−(
−)
⎝ ⎜ ⎞ ⎠ ⎟ ln exp ln 241 図 2 結晶構造から計算される(a)粉末回折ピーク位置と(b)粉末回折積分反射強度,(c)pesudo-Voigt 関数(左)とPearsonⅦ関数(右)(d)計算された粉末回折パターン.(, (a)peak positions of powder peaks,(b)bar chart of integrated bragg intensity,(c)pseudo-Voigt Function,(d)calculated powder diffraction patterns.)
ンゼンの水素を除いた独立原子の数は C6N2O4で 12 個で
あるから 30NAは 360 となる. 360 本は 2θ > 48 °(d > 1.26
Å)で達成される. Christensen, Lehmann, Nielsen2) によっ
て導出された d 領域における反射数 NBの大雑把な見積も り NB ≈ 4πVc/d3NAを用いると d≈ 1.26 Å の領域における 反射数は, d ≈ 2.9 Å の反射数の 10 ∼ 20 倍に達する. こ のため, 2.9 Å > d > 1.26 Å の Ihklの見積もりは絶望的と なる. 以上のように有機物質の場合, 簡単な分子でも各反 射の強度を抽出し直接法などで構造を決定することは非 常に困難となる. 3.実空間法 実空間法は, 実験データと独立に実空間で構造モデル を作製し, 構造モデルから計算された粉末回折パターン と実験データを比較することでモデルの良否を判断し構 造を求めていく手法である. モデル作製に Ihklは必要なく, モデルから計算されるパターンには反射の重なり合いも含 まれるため Ihkl抽出の問題を克服している. この方法は古
くから粉末構造解析で行われてきた Trial and Error を組
織的に行うことに相当する. コンピュータを用いたグリッ ト探索や Simulated Annealing(SA),3) Genetic Algorithm
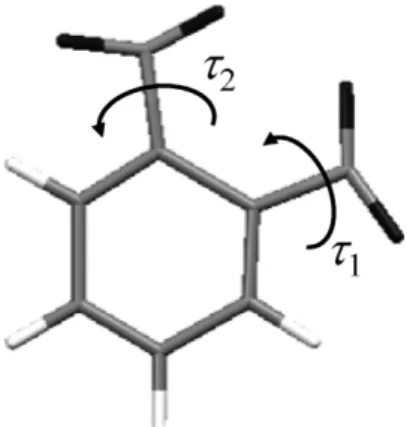
(GA)4) などの大域的探索法と組み合わされて用いられる ことで粉末構造決定の中心的手法になってきた. この方法 は特に有機物などの分子性の物質で威力を発揮している. 単位格子と空間群が既知で, 単位格子に含まれる原子の 種類と個数 N がわかっている場合, 結晶構造の決定は, 単 位格子の非対称単位における N 個の独立した原子の座標 (x, y, z)を決定することに等しい. 決定するパラメータ数 は 3N 個となる. 例えば, 非対称単位に 2 個の原子が入っ ている場合を考えてみよう. この物質の粉末回折実験デー タが得られており, 格子定数と空間群は既知とする. 2 個 の原子の位置を実空間法でグリッド探索する. 非対称単位 を x 方向, y 方向, z 方向にそれぞれ 10 分割し, グリット点 に原子を配置して計算回折パターンを計算し, 実験データ と比較する. 正しい位置に原子が配置されれば, 実験値に 近い計算回折パターンが得られる. 3 方向に 10 分割のグ リット点に原子を 1 個配置する場合の数は 103であるか ら, 原子 2 個の場合(103)2= 106通りの中から解を探すこ とになる. この方法は単純で, 適切に分割したグリットが 使用されれば大域的最小値が見つかることを保証する利 点をもつ. 有機分子 o-ジニトロベンゼンの原子配列をグリット探 索で原子配置を探す場合を考える. 問題を簡単にするため に, 水素原子を無視して残った C, N, O の 12 原子で考え る. 先ほどと同じ 10 分割で原子を同一原子の区別など何 も考慮に入れず配置することを考えると変数の数は各原 子当たり x, y, z の 3 で 12 × 3 = 36 個となるため 1036の 場合の数があることになる. 例えば 1 秒で 1 万の構造がデ ータに合うか確認できるとしてもすべてを試すには 1032 秒= 3.2 × 1024年が必要となり実用的には探索不可能と なる(図 4). 結合した原子の集団として分子を用いることができる. o-ジニトロベンゼンの場合, 結合の長さと結合角が十分 西堀英治 242 図 3 計算回折パターンにおける反射の重なりあいの様 子.(Peak overlaps in calculated powder pattern.)
図 4 o-ジニトロベンゼン分子の分子構造とねじれ角. ( Molecular Structure and torsion angles of
な精度で推測できる. わからないのは図 3 にあるように NO2の 2 つのねじれ角τ1, τ2だけである. 単位格子の非対 称単位における o-ジニトロベンゼン分子の位置と方向は, x, y, z座標と 3 つのオイラー角θ, φ, ψ および τ1とτ2によ って表現できる. これらを用いれば, 8 個の独立した変数 のみで試行結晶構造が表せる. 解は, 36 次元の原子座標 空間の 8 変数によるサブ空間内に制限される. 12 個の原 子の問題の解はグリッドを 10 分割した場合は 108のグリ ッド検索に減らされる. ここまで減少すれば, 分子の構造 の使用により実空間法で結晶構造が解ける可能性が生ま れる. このように有機物など分子性の物質では, 分子を単 位とした構造決定が可能となる. このことが, 有機物の構 造決定に実空間法が威力を発揮している理由である. では, 8 変数の問題を現実的に取り扱ってみよう. 非対 称単位中の正しい o-ジニトロベンゼン分子の位置を 0.5 Å 以下の範囲で求めようとすれば, およそ a = 7, b = 13/4, c= 8[Å]の非対称単位を 14 × 13 × 16 のグリットに分 割し探索する必要がある. 向きとねじれ角についても正 解± 10 °の解を要求し位置のグリットに加えると, NO2は 180°回れば同じ形になるため 180 °の範囲制限をつけた としても 14 × 13 × 16 × 36 × 36 × 18 × 18 × 18 = 2.2 × 1010で, 1 秒間に 10,000 個の構造を評価したとしても約 1 カ月の時間がかかる. Rietveld 解析5) の経験のある人な ら, 適当に分子を置いてみて Rietveld 解析で精密化すれば よいと思うかもしれない. 最小二乗法を基本原理とした方 法は, 近傍の最小値を探すことしかできないため上述した 構造だけから構造を決定するには通常至らない. オイラー角θ, ψ の 2 変数のグリット探索における実験 データと計算値の一致度 Rwpの分布を図 5 に示す. Rwpは Rietveld解析など粉末構造解析で用いられる評価関数で あり以下の式で書かれる. (4) ここで yobs(2θi)はデータ点 i における観測値, ycal(2θi) は計算値, wiは重みである. わかりやすくなるよう, デー タとの一致度が高い Rwpの小さい値の領域を上にして示 した. たった 2 変数の場合でも矢印で示した部分に高さの 異なるピークが複数存在している. 変数が増えるほど, こ の分布は複雑化し, 局所解を多数示す多数のピークをもつ 分布となる. Rietveld 解析では, 近傍のピークに解を落ち 込ませることはできるが, パラメータ空間から最も一致度 の高い分布を探すことはできない. このような局所解を多 数もつ解空間の中からの解の探索を大域的最適化と呼ぶ. 大域的最適化の問題は至るところに存在し, 問題に取り 組むための多くの方法が開発されている. 例えば, 多数の 素子を必要とする回路設計やタンパク質など巨大分子の 配座探索などが挙げられる. これらの問題では, ヒューリ R w y y w y i i i i i wp obs cal obs =
(
( )
−( )
)
( )
(
)
⎧ ⎨ ⎪ ⎩⎪ ⎫ ⎬ ⎪ ⎭⎪∑
∑
2 2 2 2 2 θ θ θ 243 図 5 オイラー角θ, ψ の 2 変数のグリット探索における実験データと計算値の一致度 Rwp(Hyper surface of R. wp forスティックアルゴリズムと呼ばれる厳密に最適な解を求 めることを放棄して実用的な計算時間で解を求める SA や GA などが広く用いられている. 実空間法による粉末 回折データの構造決定は一種の大域的探索とみなせるた め SA, GA が用いられるわけである. 3.1 実空間法の前準備 実空間法を行うのに必要な情報は, 格子定数, 空間群 (候補複数でも可), 単位格子の非対称単位に含まれる原子 もしくは分子の数, 分子構造と, 後述する Pawley6) 法や LeBail7) 法を用いて得られた Ihklもしくは, Pawley 法や LeBail法の解析結果のプロファイルパラメータと粉末回 折データである. 格子定数と空間群(の候補)は前号の記 事に基づき求められているとする. 単位格子の非対称単位 に含まれる原子もしくは分子の数は, 非対称単位の体積と 分子(原子)の体積から求めることになる. 分子の体積は, 大雑把な見積もりとして C, N, O を 15 Å3, Cl, Sを 25 Å3, H 5 Å3とし計算する方法がある.8) o-ジニトロベンゼンの 場合, 非対称単位の体積は 169.5 Å3, C6H4(NO2)は 15 × 6+ 4 × 5 + 2 ×(15 × 3)= 200 Å3となり非対称単位に 1分子であることが推定できる. 空間群の候補が複数ある 場合, 分子数の情報から空間群を絞り込める場合もある. ほかには, ケンブリッジ結晶構造データベース(CCDC) から類似の分子の体積を調べその値から見積もるなどの 方法がある. 実空間法では直交座標系で記述された分子モデルが必 要となる. 多くのソフトウェアでは mol, mol2 形式や pdb 形式のファイルの入力を求めることが多い. 一般的なモデ ル作成法は, Symyx(ISIS)Draw などで分子の構造式を書 き, DS Visualizer などで三次元の座標を含むファイルに 変換する方法である. Chem Draw, Chem3D や MOE など の統合計算科学ソフトウェアでも同様の分子モデル作製 を行うことができる. 作製したモデルを分子力場計算や量 子化学計算で最適化して用いることもできる. この最適化 したモデルの作成はできればやってみることをお勧めす る. 最適化したモデルが役に立つと言っているわけではな く, 作製しただけのモデルと最適化したモデルの両方を使 って解析を行ってみることで解が求まる可能性が高まる ためである. 分子内の結合長のわずかな変化の積み重ねに より分子全体の長さが Å オーダーで変わることもあるた め, 複数のモデルを作っておくことをお勧めする. その他 の方法として, CCDC などからすでに構造が解かれた類似 構造を基に分子モデルを作製する方法がある. 筆者の経験 では, 構造を解かれた類似構造を変形して用いる方法が成 功に結び付くことも多いと感じている. 次に行うプロセスは, Pawley 法や LeBail 法による回折 パターンのフィッティングである. どちらも回折ピーク位 置 2θhklにプロファイル関数を仮定し, 格子定数, 半値幅 などのパラメータを精密化し, 計算プロファイルをデータ に一致させる. Pawley 法では, 各反射の強度 Ihklをパラメ ータとして精密化に含める. 一方, LeBail 法では Ihklは観 測データからプロファイル関数を用いて Ihklを見積もるた めパラメータには含まれない. このため, 一般的に Pawley 法は計算量が多いが LeBail 法と比較して精度が高い Ihklを 得やすいと考えられている. ただし計算時間や収束の容易 さについては LeBail 法が優れている. これらのプロセス では, 図 3 で示したようなほぼ完全に重なった反射は等し い強度に分配されることが多い. 以上の情報が揃えば実空間法による構造決定が可能と なる. 実空間法における解の探索に用いるデータと評価関数 には大きく分けて以下の 2 種類が存在する. 1 つは Pawley 法や LeBail 法により抽出された Ihklから LP 因子, 多重度 の効果を除いた|Fhkl|2を求め, |Fhkl|2とモデルから計算さ れた|Fhklcal|2との間でχ2などの評価関数を用いる方法であ る. 抽出した強度を用いているため, 反射の重なり合いの 問題を解決しているように見えないが, この点については ソフトウェアごとにさまざまな工夫がこらされている. 例 えばプログラム DASH9) では強度抽出に用いる Pawley 法 の共分散行列 Vhkをχ2の数式に含めることで反射の重な りあいを考慮したχ2を求めている. これについては, David10) による詳しい解説がある. もう 1 つは, 粉末回折 データそのものを用いて, モデルから計算した計算回折パ ターンとの一致度を Rietveld 法における重み付き信頼度 因子 Rwpを用いて評価する方法である. この方法は, 強度 抽出を必要としないため反射の重なり合いの問題は本質 的に回避できる. ただし, χ2の計算と比較して計算量が多 い. どちらを使用するかについての優劣をつけるのは難し い. 明らかに対象とする物質に依存するためである. 一般 にはプロファイル P(2θ)は規格化した各反射の値を一度 計算すればよいだけであり, 各サイクルでの計算量は |Fhklcal|2の計算と比較して小さい. 原子数が 100 を超えて増 え|Fhklcal|2が増加するとプロファイル計算の部分はほぼ無 視できる量になる. この場合には両者の計算速度の差はほ とんど見られなくなる. また, Vhkを用いたχ2の計算は, Ihkl を精密化する Pawley 法で求める必要があるため, この部 分で収束に時間がかかる場合がある. 本稿ではこの 2 つの 手法の違いについては述べず, 両方の手法の共通部分を述 べていくこととする. 3.2 実空間法による構造決定 以下では, 実空間法において, x, y, z, θ, φ, ψ などの構 造を表すパラメータのセットを表すベクトルを P で表す. o-ジニトロベンゼンでは, 分子は, 2 つのニトロ基と六員 環からなる. 非対称ユニット中の o-ジニトロベンゼン分 子では, 結晶構造は 8 の自由度, 分子の中心位置(x, y, z) と分子の方向(θ, φ, ψ), 2 つのねじれ角(τ1, τ2)で定義さ れる. P は 8 つの変数からなるベクトル P ={x, y, z, θ, φ, 西堀英治 244
ψ, τ1, τ2}で定義される.
3.2.1 Simulated annealing
Simulated annealing(SA)は, 粉末回折データからの構 造決定に最も使われている最適化法である. SA の最適化 手法としての利点は, コンピュータでプログラムを作製す ることが簡単で, 使用の際のコントロールパラメータが少 ない点にある. このため, アルゴリズムに精通していなく ても使用しやすい. SAの原理と最小化における特徴は,(融体)液体から冷 却し固体を作るプロセスとの類似性から理解できる. 液体 から冷却され作製される固体相が結晶かアモルファスの どちらかになると仮定する. 融点より上の温度で, 原子は ランダムに動き回っており集団の総エネルギーは高い. こ の系のエネルギー最小は結晶固体であるとする. 液体が固 体になるのに 2 つの極端な経路がある. ゆっくり冷やすか 早く冷やす(クエンチする)である. クエンチでは, ランダ ムな原子配置が凍結され, 結晶状態よりエネルギーの高い ガラス状態になる. 温度を少しずつ減少させる冷却がアニ ーリングである. 原子の動きが徐々に小さくなり, 原子集 団はエネルギー空間を十分に探索し, 最もエネルギー的に 安定な結晶に向かう. この熱力学的状態を粉末回折データ からの結晶構造決定に当てはめるには, 液体状態の原子と 構造パラメータを入れ替え, エネルギーを評価関数と入れ 替える. SA では, 局所解を避けて大域的最適解を探すた めに評価関数が悪化する方向へのパラメータの変更も許 す必要がある. 評価関数が悪化する方向へのパラメータの 変更の頻度は最適化が大域解に向かうために減衰関数を 導入して少しずつ減らす必要がある. これは, アニーリン グの減少因子として働く温度に類似している. SAの結晶構造決定におけるパラメータ操作を以下に示 す. 評価関数はχ2(P)とする. 新たなパラメータ P iは か であるとき新しいパラメータとして受け入れられる. こ こで Pi-1は前のサイクルで受け入れられたパラメータ, ∆χ2curは現在のχ2の境界値, R は 0 ∼ 1 の乱数である. 最適 化が続く場合, P に含まれるパラメータを j でラベルする と Piの中の j 番目のパラメータ Pijは Pi-1jの値を通して Pi-1 のベクトルからモンテカルロ的な方法で計算される. ここで∆pjは最初に定義した最大移動量であり rjは− 1 ∼ 1の乱数である. piが受け入れられると Pi-1は Piになり手 順が繰り返される. “温度下降”法のさまざまなタイプの説明は Press らに よって与えられている. ここでは一例を示すにとどめる. この方法は粉末構造決定に使用された方法の 1 つである. 下降速度を調整するために f1, f2の 0 ∼ 1 の範囲で定義さ れる変数をあらかじめ与える. ∆χ2 curが与えられたとき, SAは, その時点までに拒否もしくは受け入れられた Pjベ クトルの総数が事前に与えた値 Ntotに到達するか, 受け入 れられた移動の数が事前に与える f1を使って f1Ntotより大 きくなるまで繰り返される. 上述した条件に到達したと き, ∆χ2curは事前に決めた値 f2を使って(1 − f2)∆χ2curにリ セットされ, その後また全体のプロセスが続けられる. 最 適化は∆χ2curの現在値の下降方向の移動がなくなると終 了する. 図 6 に SA での解探索の様子を模式的に示す. 温度が高 いうちは, パラメータは山から山へと変化しながら広い領 域を探索する. 温度の減少に伴い, 1 つの山の中を駆けあ がるように探索が進められる. 最終的に山の頂点のパラメ ータが得られ. 解探索は終了となる. SAを改良した方法には数多くのバリエーションがみら れる. 単純な改良としては指数関数的な温度減少を用いて 摂動を可能にすることがある. また, Parallel tempering を 用いた方法や, 並列で SA を行う multiple SA なども SA の改良型としてみることができる. これらの方法も実際の 構造決定で用いられている. 3.2.2 Genetic algorithms
Genetic algorithms(GA)は Darwin の進化論に基づく大
Pij=Pi−1j+ ×rj ∆pj exp
(
−(
χ2( )
Pi −χ2( )
Pi−1)
∆χ2cur)
>R χ2 χ2 1 Pi Pi( )
<( )
− 245 図 6 SA における解探索の様子.(Schematic Feature of SA process.)域的最適化の方法である. GA は交叉, 突然変異, 自然淘汰 などの進化論的操作を含んでいる. 典型的な GA のコント ロールパラメータは, 集団の大きさ, 交叉の割合, 交叉の タイプ(single-point, multi-point, simple, arithmetic, heuris-tic), 突然変異の割合, 突然変異のタイプ(single-point, mul-ti-point, uniform, non-uniform, each prossessing an indi-vidual rate), 生き残りの選択法則などである. 粉末構造決 定における GA の利用は K. D. M. Harris のグループによ って行われ,11) その後, Harris のグループ自身による改良 も含め手法の改良が進められてきた. ここでは K. D. M. Harrisらが 1998 年に報告した最も基本的と考えられる GA による構造決定法を o-ジニトロベンゼンを例に示す. GAの初期集団 G0(0 番目の世代)は Np 個のランダムに 作られた試行構造からなる. GA の間, 集団は交叉, 突然変 異, 自然淘汰の操作により, 世代 j の集団 Gjから作られた 世代 j + 1 の集団 Gj+1の生成により進化する. GA の Gjか らの Gj+1の生成のフローチャートを図 7 に示す. すべての 世代で集団の構造の数(Np)と交叉操作の数 Nm と突然変 異操作の数 Nx は一定である. GA では, 評価関数から各 世代で各固体に適応度を定義する. 適応度は, 各世代の集 団の個体の評価関数の最大値と最小値を用いて表される. このため, 評価関数は同じ値でも適応度は世代ごとに変 化する. 交叉における親の選択は, その適応度に依存する. 高い 適応度をもつ構造が選択される可能性が高い. 実際の選 択では, 適応度 Fs の構造が集団からランダムに選ばれ, 乱 数 R(0 ≦ R ≦ 1)が生成される. ランダムに選ばれた構造 は, Fs > R なら交叉に参加する. この選択は 2 つ目の構 造が見つかるまで続けられる. これが Nm 回繰り返される. 同じ構造が交叉の親として何度か選択される可能性があ る. この方法は‘ルーレット選択’の改良型である. 2つの選ばれた親から子孫を生成する. 8 つのパラメー タにより定義される o-ジニトロベンゼンでは各固体の 8つのパラメータは 4 つのグループ{x, y, z|θ, φ, ψ|τ1|τ2} からなる. 2 つの選ばれた親の交叉のために, 4 つのグループ は 2 つのグループの 2 つの組に分割される. その組み合わ せは(i){ x, y, z|θ, φ, ψ}と{τ1|τ2}(ii){ x, y, z|τ1}と{θ, φ, ψ|τ2}(iii){x, y, z|τ2}と{θ, φ, ψ|τ1}の 3 種類である. 交叉で は, 4 つのグループの分割の方法のうちの 1 つが同じ確率 で選択され, 最初の親からの 2 つのグループの最初の組を 選び, 2 つ目の親からの 2 つのグループの 2 つ目の組を選ぶ ことで, 2 つの子孫が生み出される. したがって, 親{xa, ya, za|θa, φa, ψa|τ1a|τ2a}と{xb, yb, zb|θb, φb, ψb|τ1b|τ2b}の交叉は 同じ確率で,(i){xa, ya, za|θa, φa, ψa|τ1b|τ2b}と{xb, yb, zb|θb, φb, ψb|τ1a|τ2a},(ii){ xa, ya, za|θb, φb, ψb|τ1a|τ2b}と{ xb, yb, zb|θa, φa, ψa|τ1b|τ2a(iii)} {xa, ya, za|θb, φb, ψb|τ1b|τ2a}と{xb, yb, zb|θa, φa, ψa|τ1a|τ2b}の 3 つの組の子孫のうちの 1 つになる. 交叉の数は Nm である. 交叉は 2 つの子孫を生成するた め, 生み出される子孫の数は 2Nm となる. これは Np + 2Nmの構造からなる中間集団(Ij+1)を作り出す. すなわち, Gj世代に生まれた Np の構造と交叉による 2Nm の子孫で ある. Nm の交叉の後, すべての子孫に対する評価関数 が計算され, 最小値と最大値が決定される. この最大値 (Rmax)と最小値(Rmin)を用いて, 中間集団の全固体の適 応度を計算する. 2 つかそれ以上の構造がまったく同じと き, これらのうち 1 つを除いて全部が中間集団から除かれ る. 前の生成から運ばれた Np の各構造は, 同じ Rwpの値を もつ. その適応度の値は, 適応度が Rminと Rmaxの現在の値 に依存するために変化する. 中間集団での構造は適応度に よりランク付けされる. 各世代で, 突然変異の構造が作られる. 突然変異では, Nx個の親構造が中間集団からランダムに選ばれる. 突然 変異を作るのに使った親構造は中間集団に残される. 突然 変異で生まれた構造が交叉に参加することは, 多様性の確 保のために重要である. 突然変異で生まれた新たな情報を 交叉を通して集団に渡すことができるためである. o-ジニトロベンゼンでは, 突然変異は 4 つの組の 2 つを ランダムに選び出し, 選ばれた組の中の 1 つのパラメータ に新しい乱数に入れ替える.(x, y, z)の組か(θ, φ, ψ)の 組が選ばれると組の中の 3 つのパラメータのうちランダ ムに選ばれた 1 つに新しい値が与えられる. Nx の突然変異の構造と中間集団 Ij+1から Np − Nx 個の 最も適応した構造を取り出すことで,( j + 1)番目の世代 (Gj+1)の集団が作られる. 突然変異の Rwpの値も計算され, Rminと Rmaxの新しい値が計算され, 新しい集団の構造の適 応度が決定される. 交叉, 突然変異, 自然淘汰を必要とす 西堀英治 246
るサイクルは, 指定した世代の数(Ng)までか予定された 終了基準まで繰り返される. 中間集団 Ij+1は 2Nm 個の新しい子の構造をもつ前の世 代 Gjのすべての構造を含むため, 集団 Gj+1における最良 の Rwpは, 集団 Gjにおける最良の Rwpの値より低いか同じ であることが保障される. 集団の大きさ(Np)は世代を経 ても一定のままである. 図 8 に GA での解探索の様子を模式的に示す. 初期状 態で多数の個体が存在する. これらの中から交叉の親が 適応度に応じて選ばれる. この親の組み合わせにより新 しい個体が生成される. この様子を図に示した. 最終的に は, 山の周りに個体が集まるか, いくつかの山に分散した 形で各個体が分布する. GAの方法のバリエーションは非常に多く, 構造決定だ けでもさまざまなタイプがみられる. ある GA をほかの GAのコントロールパラメータの最適化に使う方法まで 存在する. また Darwin の進化論に, 各固体がその一生の 間に適応度を向上する Lamarckian 進化論を組み合わせた 方法もある. 3.3 解けない場合の対策 以上, SA, GA を用いた実空間法に粉末未知構造決定に ついて述べてきた. 実際に行う際に留意しておくべきこ とや, うまくいかない場合の対策について筆者の経験の範 囲で述べる. SA, GA の解析は一度行ったら終わりではな く, 同じ条件でも複数回繰り返して行う必要がある. これ らの手法は, 前述したとおり, 厳密に最適な解を求めるこ とを放棄しているため, 安定して同じ解が得られるか確認 することは重要である. 当然, 解が得られたといっても正 しい保証はないため, 続く記事にあるように Rietveld 解 析まで行ってみて確認する必要がある. SA や GA の解析 の際のパラメータに関しては, Default 値の使用でほとん どの場合問題ない. 1990 年代から 2000 年代の初めごろに はこれらを調整したりする研究もいくつか見られたが, そ の頃から見て計算力は 10 倍以上に達しているため, 繰り 返してやれば解は得られるだろう. 複数回の試行を繰り返しても, 解が得られたとは思えな い状況になることもある. そのような場合の対策は, その 状態での解の出方によって異なる. 以下の 3 つの場合につ いて述べることとする. ①一致度の良い解がまったく出 ず, 複数試行しても出てくる解の構造もばらばらな場合, ②一致度は低いが, 出てくる解の構造は複数試行してどれ も似ている場合③複数試行で一致度が高く, よく似た解が 多数出てくるがそれらの構造の細部が異なる場合. ①の状況になる場合は, 解析の初期段階である指数付け や分子数などが間違っていることが多い. 格子定数と空間 群の取り直しからやり直すことをお勧めする. 粉末回折の 場合, 弱い強度を観測することが難しいため, 格子定数, 空間群を誤りやすい. もし, 格子が小さくて分子がうまく 入らないなどの場合は軸を 2 倍にして取り直してみる, 格 子が大きすぎて分子数が多くなりすぎるなら対称性の高 い Cell を探してみる, などこの部分の見直しでうまくい くことも多い. また, 分子数を見積もると 1.5 分子などあ り得ない数になるときも格子の取り直しを検討するとよ い. その他, 考えられることとしては, 結晶化(粉末化, 個 体化)の際に結晶化に用いた溶媒が混入し単位胞に含まれ る原子数が異なっている場合, 結晶多形が混じっている場 合などが考えられる. このように, 状況として考えられる ことは多数あるが, データだけから決めることは難しい. このような場合には, 温度を変化させてデータを測定して みる, 結晶化条件を変えた複数の試料でパターンを測定し て比較する, など実験に立ち返ったほうがうまくいくこと が多い. 特に結晶多形は熱力学的性質が違う場合があり, 温度変化をするだけで見分けられることもある. ②の場合に考えられるのは, 粉末回折データに選択配向 の影響が存在する. 結晶化の際の溶媒などの存在を無視し ている. などである. 前出と同じであるが, この場合には おおまかなフレームワークは決定できていると思われる ので, Rietveld 解析に進んでそちらで, モデルをチェック しモデルを再構築することができると思われる. ③の場合は, 実空間法としては解が出ている状況と言っ 247 図 8 GA における解探索の様子.(Schematic Feature of SA Analysis.)
てよい. 細部の構造を表すパラメータのみ異なった解が 多数出る場合, 実空間法で探しきるのは難しいと思われ る. 解決策としては, 実空間法の解析に用いる分解能範囲 を広げて高角のデータまで使うようにする方法が考えら れる. むしろ, Rietveld 解析に進んで, 各モデルの違いを精 密化してパターンの変化を調べる, もしくは, マキシマム エントロピー法(MEM)やフーリエ合成で電子密度を求 めてモデルを作るなどの方法をとったほうが早い場合が 多い. この場合ほとんどの構造は正しいと考えられるた め, 電子密度からモデルを作る方法も粉末回折における 反射の重なり合いの問題があっても十分機能すると思わ れる. 4.おわりに 本稿では粉末回折からの有機物質の構造決定において 中心的な役割を果たしている構造決定法,“実空間法”に ついて述べた. いくつかのソフトウェアが販売され研究 例も増えてきているため, 50 原子以下, 自由度が 15 以下 程度であれば, 適切に格子と空間群を選ぶことができれ ば比較的簡単に構造が得られるようになってきている. 実 空間法の部分については, 手法論の開発も進み, 上述した 程度のサイズの解析であればブラックボックスとして使 える状態だと思われる. ソフトウェアとしても, 先に紹介 した DASH のほかに, Materials Studio Refilex Plus,12)
Crystal Profiler13) が市販ソフトウェアとして利用できる. フリーソフトウェアとしても FOX14) などがある. それよ りも, データ測定や指数付け, Pawly 法の解析や Rietveld 解析など粉末回折で一般に用いられるほかの実験・解析 部分のほうが技術や経験を要する場合が多い. これらに 関しては, 入門講座のほかの記事が役立つだろう. 文 献
1)K. D. M. Harris and M. Tremayne: Chem. Mater. 8, 2554 (1996). 2)A. N. Christensen, M. S. Lehmann and M. Nielsen: Aust. J.
Phys. 38, 497 (1985).
3)S. Kirkpatrick, C. D. Gelatt and M. P. Vecchi: Science 220, 671 (1983).
4)J. H. Holland: Adaptation in Natural and Artificial Systems, University
of Michigan Press (1975).
5)H. M. Rietveld: J. Appl. Cryst. 2, 65 (1969). 6)G. S. Pawley: J. Appl. Cryst. 14, 357 (1981).
7)A. Le Bail, H. Duroy and J. L. Fourquet: Mater. Res. Bull. 23, 447 (1988).
8)例えば http://www.ccdc.cam.ac.uk/support/documentation/dash/ dash_32.pdf
9)K. Shankland, et al.: J. Appl. Cryst. 35, 443 (2002). 10)W. I. F. David: J. Appl. Cryst. 37, 621 (2004).
11)K. D. M. Harris, R. L. Johnston and B. M. Kariuki: Acta. Cryst. A54, 632 (1998). 12)http://accelrys.co.jp/products/materials-studio/analytical-and-crystallization-software.html 13)http://www.rsi.co.jp/kagaku/cs/crystal_profiler/index.html 14)http://vincefn.net/Fox/ プロフィール 西堀英治 Eiji NISHIBORI 名古屋大学工学研究科応用物理学分野
Department of Applied Physics, Nagoya University 〒 464-8603 名古屋市千種区不老町 1
Furo-cho, Chikusa, Nagoya 464-8603, JAPAN e-mail: [email protected] 最終学歴:名古屋大学工学研究科博士課程前期課 程修了 専門分野:回折物理学 現在の研究テーマ:結晶学と計算科学の融合 西堀英治 248