2.7.1
生物薬剤学試験及び関連する分析法の目次
2.7.1 生物薬剤学試験及び関連する分析法の目次 ... 1 2.7.1.1 背景及び概観 ... 2 2.7.1.1.1 要旨 ... 2 2.7.1.1.2 製剤開発の経緯 ... 3 2.7.1.1.3 溶出特性 ... 7 2.7.1.1.4 生体試料分析法 ... 8 2.7.1.1.5 薬物動態検討用採血及び薬物動態の評価 ... 9 2.7.1.1.5.1 多数点採血による薬物動態評価 ... 9 2.7.1.1.5.2 少数点採血による薬物動態評価 ... 10 2.7.1.1.6 生物薬剤学試験の概観 ... 11 2.7.1.1.6.1 溶解度及び透過性 ... 11 2.7.1.1.6.2 バイオアベイラビリティ ... 11 2.7.1.1.6.2.1 相対的バイオアベイラビリティ:普通錠及び 錠 と経口液剤との比較 ... 12 2.7.1.1.6.2.2 錠のバイオアベイラビリティ:申請製剤(40 ㎎錠 4 錠)と自社標準フィルムコーティング 錠 (100 ㎎錠 1 錠+20 ㎎錠 3 錠)との比較 ... 13 2.7.1.1.6.2.3 絶対的バイオアベイラビリティ ... 14 2.7.1.1.6.2.4 マスバランス試験 ... 14 2.7.1.1.6.3 食事の影響 ... 14 2.7.1.2 個々の試験結果の要約 ... 16 2.7.1.2.1 試験 11650 3 週間投与/1 週間休薬 用量漸増試験にお ける相対的バイオアベイラビリティの検討 ... 16 2.7.1.2.2 試験 12437 バイオアベイラビリティ試験... 20 2.7.1.2.3 試験 14656 食事の影響試験 ... 22 2.7.1.3 全試験を通しての結果の比較と解析 ... 27 2.7.1.4 付録 ... 29 参考文献 ... 292.7.1
生物薬剤学試験及び関連する分析法
2.7.1.1
背景及び概観
本項では、レゴラフェニブ開発中に検討した生物薬剤学試験に関する概要について2.7.1.1.1 に示した。各臨床試験で用いられた製剤及び溶出特性については2.7.1.1.2及び2.7.1.1.3に記載 した。また、ヒト生体試料中のレゴラフェニブ及び代謝物の定量に用いた分析法、並びに薬物動 態の評価方法については2.7.1.1.4及び2.7.1.1.5に記載した。2.7.1.1.6にはレゴラフェニブの 生物薬剤学な分類及び生物薬剤学試験に関する概要を示した。 個々の生物薬剤学試験の要約及び薬物動態に関する成績は2.7.1.2に記載した。また、全試験 をとおしての結果と解析を2.7.1.3に要約した。2.7.1.1.1
要旨
背景 経口バイオアベイラビリティが高いレゴラフェニブの即放性製剤を提供することを目的として、 製剤の開発を行った。 の原薬(レゴラフェニブ水和物)は水性溶媒にはほとんど溶けないため、高い経口バイ オアベイラビリティを目的に、 ( )錠、すなわち ではなく のレゴラ フェニブを含有する錠剤を選択した(2.3.P.2.1.1 参照)。 することにより、in vivo
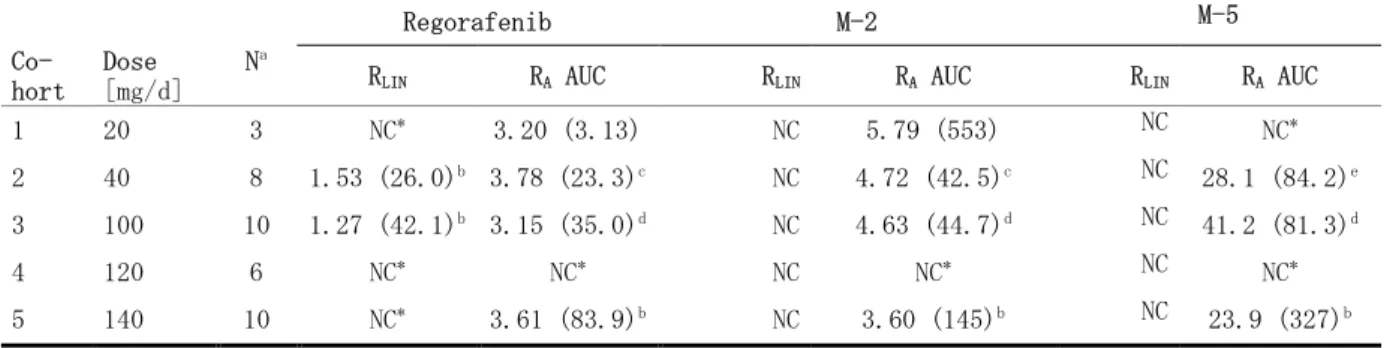
での溶出性が改善し、レゴラフェニブの経口バイオアベイラビリティが最適化された。 製剤 レゴラフェニブ 20mg、40mg 及び 100mg を含有する即放性の 錠(以下、 錠)を開 発した。レゴラフェニブ 20mg、40mg 及び 100mg は、レゴラフェニブ水和物として、それぞれ、 20.75mg、41.49mg 及び 103.73mg に相当する。レゴラフェニブ水和物の水和水は製造中に除去さ れ、 のレゴラフェニブ(無水物)となる。 申請製剤 40mg 錠(市販のフィルムコーティング剤オパドライ を使用)以外に、自社 標準フィルムコーティングを施した 20mg 及び 100mg 錠、並びにオパドライ でフィルム コーティングを施した 20mg 錠を開発し、臨床試験に使用した。すべての 錠の素錠 の組成及び製造工程の原理は、開発過程をとおして変更しなかった。開発中に行った主な変更は、 フィルムコーティングの変更(自社標準フィルムコーティングからオパドライ を用いたフィル ムコーティングに変更)であった(2.3.P.2.2 及び2.7.1.1.2参照)。 また、 錠開発前には、 のレゴラフェニブ 20mg を含有する通常の製造方法による 即放錠(以下、普通錠)及びレゴラフェニブの 2%(w/v)経口液剤も製剤化した。レゴラフェ ニブで実施した最初の第Ⅰ相臨床試験(試験 11650)には、普通錠、 錠及び経口液剤を 使用した。また、マスバランス試験(試験 12436)は、経口液剤を用いて実施された。 各試験で使用した製剤の概観を表 2.7.1.1-1に示した。一部の初期臨床試験は、自社標準フィ ルムコーティングを施した旧 錠を用いて実施されたが、第Ⅲ相臨床試験(試験 14387)を 含めた、ほとんどの第Ⅰ相及び第Ⅱ相臨床試験において、申請製剤である 40mg 錠を使用 した(表 2.7.1.1-1)。溶出特性
申請製剤の溶出性の規格として、 で一般的に定められている規格に準じた % ( 分)を適用した。申請製剤は、この規格を満たしていた(2.3.P.5.4 及び2.7.1.1.3参照)。
バイオアベイラビリティ
バイオアベイラビリティに関して、レゴラフェニブは 溶解性/ 透過性の化合物であり、す なわち Biopharmaceutical Classification System(BCS)基準 1)による Class に分類される薬
物である。原薬は、水及び pH ~ の水性溶媒にほとんど溶けない。また、バリデートされた Caco-2 アッセイ系を用いた
in vitro
試験において、レゴラフェニブは、BCS 基準による 透過 性に分類されることが示された(2.7.1.1.6.1参照)。 原薬が水性溶媒にほとんど溶けないため、静脈注射剤を開発することができず、絶対的バイオ アベイラビリティ試験は実施していない。ただし、[14C]レゴラフェニブを用いたマスバランス 試験(試験 12436)の結果からは、レゴラフェニブを経口投与した時のバイオアベイラビリティ は高いことが示唆された(2.7.1.1.6.2.4及び 2.7.2.2.5.2.1 参照)。 生物薬剤学試験 生物薬剤学試験 3 試験(表 2.7.1.1-2)が、国外にて実施されている。これら試験におけるバ イオアベイラビリティの検討は、バリデートされた生体試料分析法(2.7.1.1.4参照)及び適切 な薬物動態解析方法(2.7.1.1.5参照)によった。 固形がん患者を対象とした用量漸増試験(試験 11650)において普通錠及び 錠の経口液 剤に対する相対的バイオアベイラビリティを検討した。 のレゴラフェニブを含有する 20mg 普 通 錠 の 経 口 液 剤 に 対 す る 相 対 的 バ イ オ ア ベ イ ラ ビ リ テ ィ は 低 く ( 2.7.1.1.6.2 、 2.7.1.2.1参照)、普通錠の開発は中止することとした。一方、 錠の経口液剤に対する相 対的バイオアベイラビリティは良好であり(試験 11650、自社標準フィルムコーティングを使 用)、 錠は、製剤の開発目的に合致した即放錠であることが示されたため、以降の開発に は 錠が選択された。 錠に対して開発中に行った主な変更は、フィルムコーティングの変更であった。また、 第Ⅲ相臨床試験(試験 14387)の開始前に、錠剤の含量を 20mg 又は 100mg から 40mg に変更した。 これらの変更がバイオアベイラビリティに及ぼす影響について、試験 12437 で評価した。両 錠は生物学的に同等であることが示された(2.7.1.1.6.2、2.7.1.2.2参照)。そこで、この 錠 40mg 錠を第Ⅲ相臨床試験(試験 14387)に使用することとした。その後、製剤に変更は なく、申請に至った。 今回の申請に際して、本剤のバイオアベイラビリティに及ぼす食事の影響を検討するために、 申請製剤を用いて食事の影響試験(試験 14656)を実施した(2.7.1.1.6.3、2.7.1.2.3参照)。 上述のような生物薬剤学試験の結果、申請製剤である 錠 40 ㎎錠はバイオアベイラビリ ティの観点から優れた製剤であることが示されている。2.7.1.1.2
製剤開発の経緯
抗がん剤としてのレゴラフェニブの開発は 20 年に開始した。20 年に開始した国外第Ⅰ相 臨床試験には経口液剤、続いて、普通錠及び 錠を用いた。経口バイオアベイラビリティが高いレゴラフェニブの即放性製剤を供給することを目的として、 製剤の開発を行った。申請製剤には、 錠、すなわち ではなく のレゴラフェニ ブを含有する錠剤を選択した。 することにより、
in vivo
での溶出性が改善し、レゴラ フェニブの最適な経口バイオアベイラビリティが得られる。 申請製剤である 40mg 錠に加えて、含量 20mg 及び 100mg の 錠を開発し、臨床試験 に使用した。臨床試験で使用した含量の異なる錠剤(20mg、40mg 及び 100mg)は、最適な溶出特 性を有する同じ 造粒末を用いて、同一の製造工程で製造した。錠剤の重量及び大きさを調 整し、含量の異なる錠剤を製造した。盲検化を容易にするため、臨床試験で使用する錠剤にはす べて刻印を行わなかった。 申請製剤である 40mg 錠は、長径 16mm、短径 7mm、重量 472mg の楕円形のフィルムコー ティング錠である。本品は、着色による識別性を高め、錠剤取扱い時の飛散性を軽減し、嚥下し やすくするために、淡赤色の非機能性フィルムコーティングを施している。また、臨床試験にお ける盲検化のためにもフィルムコーティングを行った。 錠の開発中は、フィルムコーティングに関して軽微な変更は加えたものの、組成及び製 造工程の原理は変更していない。水系コーティング工程における温度負荷を最小限にするため、 最初の錠剤で使用した自社標準フィルムコーティングを、市販のポリビニルアルコールベース (オパドライ )のフィルムコーティングに変更した。 臨床試験で使用した申請製剤であるオパドライ フィルムコーティングの 40mg 錠(開 発番号 142)の組成に関する詳細は、2.3.P.2.2 に示した。第Ⅲ相臨床試験(試験 14387)は、 申請製剤を使用して実施した(表 2.7.1.1-1)。また、最初の国外第Ⅰ相臨床試験(試験 11650)及びマスバランス試験(試験 12436、経口液剤のみ)で使用した普通錠及び経口液剤の 組成に関する詳細は、2.3.P.2.2 に記載した。表 2.7.1.1-1 臨床試験に使用した製剤の一覧表
Sorted by protocol number
Drug products used a
Location of study report (study
number) Abbreviated title Ora
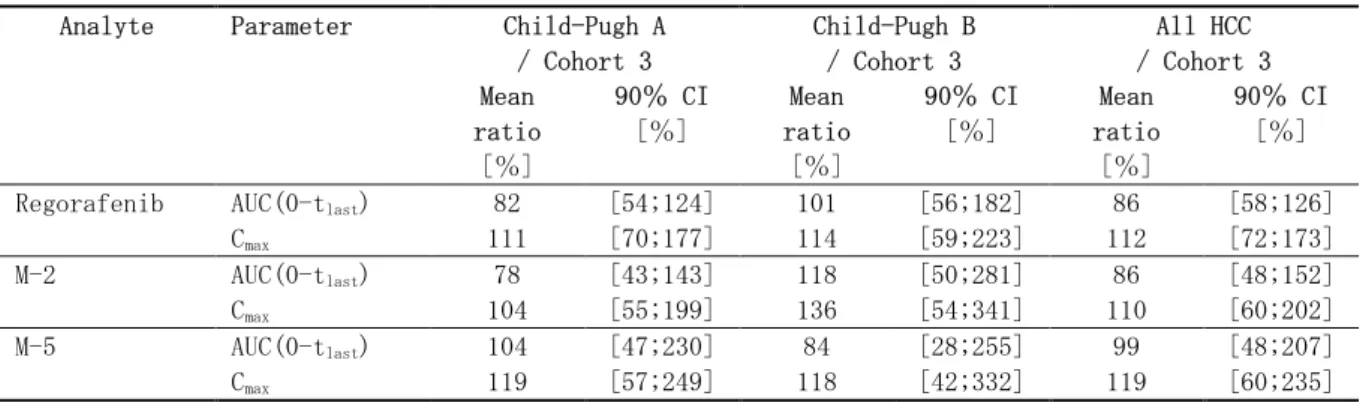
l s ol u ti o n b 2 % (w / v) C o n ve n ti o na l t a bl e t c 2 0 m g ( #0 2 0) t a b le t 2 0 mg (# 0 21 ) t a b le t 1 0 0m g ( # 10 1 ) t a b le t 2 0 mg (# 1 22 ) d t a b l e t 4 0 mg ( #1 4 2 ) d 5.3.3.2.1, PH-36733 (11650)
Safety, tolerability, maximum tolerated dose, PK, relative BA of
tablet vs. oral solution and biomarker status of regorafenib x x x x 5.3.3.2.3, PH-36984, 5.3.3.2.2, PH-36742 (11651)
Safety, tolerability, maximum tolerated dose, PK, and biomarker status of regorafenib
x x
5.3.3.2.6,
PH-36735
(11656)
Safety, PK, and PD of regorafenib in comb-ination with "mFOLFOX6" or "FOLFIRI" in patients with metastatic colorectal cancer (CRC) x x x 5.3.5.2.2, A46572 5.3.5.2.3, A55873 (11726)
Phase II uncontrolled study of
regorafenib in patients with renal cell cancer (RCC)
x x
5.3.3.4.4,
PH-36865
(12434)
Effect of regorafenib on probe
substrates of CYP 2C9, 2C19, 3A4 and 2C8
x
5.3.3.4.1,
PH-36717
(12435)
Effect of ketoconazole on the PK of regorafenib
x
5.3.3.1.1, PH-36734 (12436)
Metabolism, excretion pattern, mass balance, safety, tolerability and PK of [14C] regorafenib
x
5.3.1.2.1, PH-36595 (12437)
Relative bioavailability of regorafenib (1 x 100mg + 3 × 20mg vs. 4 × 40mg)
x x x
5.3.3.2.4, A51164 (13172)
PK and safety of regorafenib in Japanese patients with advanced solid tumors
x x x x
5.3.5.1.1, A53306 (14387)e
Study in patients with metastatic colorectal cancer (mCRC) treated with regorafenib or placebo after failure of standard therapy
表 2.7.1.1-1 臨床試験に使用した製剤の一覧表(続き)
Sorted by protocol number
Drug products used 1
Location of study report (study
number) Abbreviated title Ora
l s ol u ti o n b 2 % (w / v) C o n ve n ti o na l t a bl e t c 2 0 m g ( #0 2 0) t a b le t 2 0 mg (# 0 21 ) t a b le t 1 0 0m g ( # 10 1 ) t a b le t 2 0 mg (# 1 22 ) d t a b l e t 4 0 mg ( #1 4 2 ) d 5.3.5.2.1, A51601 (14596)
Safety in patients with hepatocellular carcinoma (HCC)
x
5.3.1.1.1, PH-36525 (14656)
The effect of food on the PK of regorafenib
(high-fat breakfast vs. low-fat breakfast vs. fasting state)
x
5.3.4.2.2, PH-36866 (14814)
Cardiovascular safety, tolerability, PK and antitumor activity of regorafenib
x
5.3.3.2.5, A51600 (14996)
PK and safety,tolerability and efficacy of regorafenib in Chinese patients
x
5.3.3.4.2, PH-36716 (15524)
Effect of rifampin on the PK of regorafenib
x
5.3.5.4.2
(11728)f
First line treatment of metastatic colorectal cancer with "mFOLFOX6" in combination with regorafenib
x
5.3.5.4.1
(14458)f
Safety, efficacy, and pharmacokinetics of regorafenib combined with pemetrexed and cisplatin in patients with
nonsquamous non-small cell lung cancer
x x
5.3.5.4.3
(14874)e,f
Regorafenib as a 3rd-line or greater treatment for gastrointestinal stromal tumors (GIST) (GRID)
x
a All tablets were immediate release tablets
b 2% (w/v) solution of regorafenib in a macrogol based vehicle
c IR tablets comprising the drug in , micronized form
d Opadry® coating
e Phase III study; all other studies were Phase I or II studies
f Ongoing study (as of 31 Dec 20 ). (Only study protocols are provided for these studies in
Module 5.)
PK pharmacokinetics PD pharmacodynamics
2.7.1.1.3
溶出特性
臨床試験用製剤のin vitro
溶出試験は、すべて、標準的な溶出試験法を用いて実施した(パ ドル法、900mL、試験液:酢酸緩衝液 pH4.5+0.1%ラウリル硫酸ナトリウム、37℃、75rpm)。 レゴラフェニブの定量は、特異性、直線性、真度、及び精度に関してバリデートされた紫外分光 法( nm)により行った。試験法の詳細は 2.3.P.5.2 に記載した。 申請製剤の溶出規格として、 で一般的に定められている規格に準じた %( 分)を適用した。 申請製剤はこの規格を満たしていた(2.3.P.5.4 参照)。図 2.7.1.1-1にバイオアベイラビリ ティ試験(試験 12437)で使用した製剤、すなわち申請製剤である 40mg 錠並びに自社標 準フィルムコーティングを施した 100mg 及び 20mg 錠の溶出特性(平均溶出率)を示す。 溶出特性の類似性(特に 20mg 錠と 40mg 錠)から、フィルムコーティングの変更は本剤の溶出特 性に大きな影響を及ぼさないことが示された。Mean % dissolved in acetate buffer pH 4.5 + 0.1% sodium dodecyl sulfate
図 2.7.1.1-1 試験 12437 で使用した錠剤の溶出特性
2.7.1.1.4
生体試料分析法
臨床開発段階において、高速液体クロマトグラフィー/タンデム質量分析(LC-MS/MS)法を用 いて、血漿試料中の未変化体(レゴラフェニブ:BAY 73-4506)並びにその代謝物 M-2(BAY 75-7495)、M-3(BAY 81-8753)、M-4(BAY 75-1098)及び M-5(BAY 81-8752)の同時定量法を開 発し、分析法バリデーションを実施した上で、生体試料分析に供した。尿試料中のレゴラフェニ ブ及び代謝物 M-2 は、バリデートされた LC-MS/MS 法を用いて測定し、レゴラフェニブ及び M-2 のグルクロン酸抱合体である M-7(BAY 86-6651)及び M-8(BAY 86-6652)は、酵素による加水 分解処理後にレゴラフェニブ及び M-2 として間接的に測定した。血漿試料は、たん白質除去後、 逆相クロマトグラフィーにより分離した検体を質量分析計で定量した。尿試料は、希釈後、血漿 試料と同様に逆相クロマトグラフィーにより分離した検体を質量分析計で定量した。
分析法バリデーション及び試料分析は、関連する FDA ガイダンス2)に従って実施した。 臨床試験で使用した生体試料分析法の詳細並びにバリデーション及び試料の安定性に関する結 果の要約を、5.3.1.4.1 A59117 にバリデーション報告書として添付した。 臨床開発段階においては、最新の要件を満たすため、既存の定量法と異なるバージョンを用い ることがあった。各試験で実際に使用した定量方法及びその特性については、対応する試験総括 報告書に添付した。 レゴラフェニブ及び代謝物 M-2、M-3、M-4 及び M-5 の血漿中濃度の定量下限(LLOQ)は、 0.002~0.005mg/L であった。また検量線の上限は、すべての分析対象において 2mg/L であった。 尿中濃度定量法の LLOQ は、レゴラフェニブ及び M-2 の場合がおよそ 0.010mg/L であった。間 接的に測定する必要がある M-7 及び M-8 の LLOQ は、換算を要するため、およそ 0.014mg/L で あった。検量線範囲の上限は、レゴラフェニブ及び M-2 が、およそ 5mg/L、M-7 及び M-8 が、お よそ 7mg/L であった。 低、中及び高濃度の精度管理用(QC)試料に関して、すべての分析対象の精度は 15%以内、 真度は 85~115%以内であった。また LLOQ に相当する濃度の QC 試料については、精度は 20%以 内、真度は 80~120%以内であり、いずれも分析法バリデーションのガイダンス 2)の要件を満た していた。 さらに、予想される検体取扱い条件下(血漿試料:37℃で 3 時間保存、-15℃で 18 ヵ月間保存、 な ど ) で 安 定 性 を 検 討 し た 結 果 、 す べ て の 分 析 対 象 は こ れ ら の 条 件 下 で 安 定 で あ っ た (5.3.1.4.1 A59117 参照)。 使用した生体試料分析法は、検量線用標準曲線及び精度管理用 QC 試料測定の結果から定量実 施期間を通じて信頼し得るものであることが示された。
2.7.1.1.5
薬物動態検討用採血及び薬物動態の評価
2.7.1.1.5.1
多数点採血による薬物動態評価
生物薬剤学的検討を行った 3 つの臨床薬理試験(試験 11650、12437 及び 14656)では、多数 点採血によりデータを収集することを計画した。詳細は、表 2.7.1.1-2、並びに 2.7.2.2 の個々 の試験結果の要約に記載した。各試験において、治験実施計画書に規定されている検体採取日に レゴラフェニブ及び主代謝物の血漿中濃度の測定用に静脈血を採取した。標準的な採血スケ ジュールとして、投与前に 1 点採血後、投与後 12 時間目までは比較的短い採血間隔により、翌 日以降はやや長い採血間隔により多数点の採血を行った。すなわち、申請製剤のバイオアベイラ ビリティを評価した試験 12437 では、投与前、投与後 1、2、3、4、6、8、12、16 及び 24 時間目、 並びに投与後 2、3、4、6 及び 7 日目に採血した(6、7 日目は 2 期のみ、2.7.6.2 参照)。後述 する食事の影響試験(試験 14656)及び 3 週間投与/1 週間休薬 用量漸増試験(試験 11650: 相対的バイオアベイラビリティを検討したパート)においても同様のスケジュールで採血した。 薬物動態学的パラメータは、それぞれの治験実施計画書に従い、以下のパラメータなどを算出 した。パラメータはレゴラフェニブ及び代謝物の血漿中濃度、並びに時間データを基に、 WinNonlin ソフトウェア Version 4.1(Pharsight Corporation, Mountain View/CA、米国)を 用い、ノンコンパートメント法により算出した。単回又は初回投与後の薬物動態学的パラメータ AUC 投与後 0~無限大時間までの血漿中濃度-時間曲線下面積 AUC(0-tlast) 投与後 0 時間から最終データ時点(t)までの血漿中濃度-時間曲線下面積 Cmax 最高血漿中濃度 tmax 最高血漿中濃度到達時間 t1/2 消失半減期 CL/F 経口投与時の見かけの全身クリアランス(経口クリアランス) Vz/F 見かけの分布容積 定常状態における薬物動態学的パラメータ AUC,ss, 定常状態における投与間隔での AUC Cmax,ss 定常状態における Cmax tmax,ss 定常状態における tmax CLss/F 定常状態におけるCL/F Vz,ss/F 定常状態におけるVz/F RA 蓄積率:
RLIN linearity factor:
さらに、必要に応じて投与量で補正したパラメータ(Cmax/D 及び AUC/D)並びに体重(kg)あ
たりの投与量で補正したパラメータ(Cmax,norm及び AUCnorm)を算出した。
血漿中濃度-時間曲線下面積(AUC)の算出には、台形法を用いた。消失半減期(t1/2)は、終 末相の血漿中濃度を対数変換後に線形最小二乗法により回帰させ推定した。 薬物動態学的パラメータ(tmaxを除く)は幾何平均値並びに幾何変動係数(%)又は範囲で示 し、tmaxは中央値及び範囲で示した。また、血漿中薬物濃度(2/3 以上において LLOQ を上回って いた場合に算出)は幾何平均値で示した。
2.7.1.1.5.2
少数点採血による薬物動態評価
少数点採血による薬物動態評価又は母集団薬物動態解析は、第Ⅲ相臨床試験(試験 14387)の 1 試験でのみ実施した。現在、解析中であり、別途報告書を作成する予定である。2.7.1.1.6
生物薬剤学試験の概観
2.7.1.1.6.1
溶解度及び透過性
原薬は、水性溶媒にほとんど溶けない(0.1mol/L HCl、水、及び 0.1mol/L NaOH に 25℃で mg/L 以下)(2.3.S.1.3.6 参照)。溶出性を向上し、経口バイオアベイラビリティを改善す るために、レゴラフェニブ製剤は 錠として製造することとした。溶出試験液に接すると、 錠剤が崩壊し、 の原薬の溶解性から予想されるよりもはるかに高濃度のレゴラフェニブを 含む を形成しながら、 が溶解する。 レゴラフェニブの
in vitro
における透過性は、バリデートされた Caco-2 アッセイ系を用いて 検 討 し た 。 試 験 結 果 か ら 、 レ ゴ ラ フ ェ ニ ブ は BCS 基 準 に お け る 透 過 性 に 分 類 さ れ た (3.2.P.2.1.1.1 参照)。 以上より、レゴラフェニブは、BCS 基準による Class の薬物( 溶解性/ 透過性の化合 物)である1)。2.7.1.1.6.2
バイオアベイラビリティ
生物薬剤学試験の一覧を表 2.7.1.1-2に示した。最初に実施した試験 11650 では、普通錠及び 錠(自社標準フィルムコーティングの 20mg 及び 100mg 錠)の経口液剤に対する相 対的バイオアベイラビリティを検討した。試験 12437 では、申請製剤である 40mg 錠のバ イオアベイラビリティを、初期の臨床試験で使用した 錠(自社標準フィルムコーティング の 20mg 及び 100mg 錠)と比較した。試験 14656 では、申請製剤である 40mg 錠のバイオア ベイラビリティに及ぼす食事の影響を検討した。絶対的バイオアベイラビリティ試験は実施しな かった。表 2.7.1.1-2 生物薬剤学試験の一覧 Location of study report (study number)
Full title Biopharmaceutically relevant
comparison Clinical part (year) Bioavailability studies 5.3.3.2.1, PH-36733 (11650)
Open label, Phase I study to determine the safety,
tolerability, maximum tolerated dose, pharmacokinetics, relative
BA of tablet and
liquid solution and biomarker status of BAY 73-4506 in patients with advanced malignancies
60mg: 20mg conventional tablets (#020) or 20mg tablets (#021) vs. oral solution (#201) 100mg: 100mg tablet (#101) vs. oral solution (#201) 2005 – 2009* 5.3.1.2.1, PH-36595 (12437)
A two-way, single dose,
randomized, open label, Phase I study to evaluate the relative bioavailability of 160mg of BAY 73-4506 administered as 1 × 100mg + 3 × 20mg tablets and 4 × 40mg tablets. 160mg: 40mg tablets with Opadry® coating (#142) vs. 100mg (#101) + 20mg (#021) tablets with standard in-house coating
2009
Food-drug interaction studies 5.3.1.1.1,
PH-36525 (14656)
A Phase I, randomized, open label, 3-way cross-over study to determine the effect of a high-fat breakfast, a low-high-fat breakfast and fasting state on the pharmacokinetics of a single oral dose of 160mg regorafenib (BAY 73-4506) in healthy volunteers
160mg:
(40mg tablets
with Opadry® coating (#142))
- with high-fat breakfast vs. - with low-fat breakfast vs. - fasting
2009 -2010
* cut off date for study report
2.7.1.1.6.2.1
相対的バイオアベイラビリティ:普通錠及び
錠と経口液剤との比
較
試験 11650(2.7.1.2.1参照)では、進行性固形がん患者を対象として、20mg 普通錠及び自社 標準フィルムコーティングを用いた 20mg 及び 100mg 錠のレゴラフェニブの経口液剤に対 する相対的バイオアベイラビリティ(Frel:錠剤/経口液剤)を検討した。 20mg 普通錠は、経口液剤と比べて未変化体の相対的バイオアベイラビリティは 10%未満で あった。一方、20mg 及び 100mg 錠は、経口液剤と比べて未変化体の相対的バイオアベイラビリティがそれぞれ約 70%及び 83%であった(表 2.7.1.1-3)。この結果に基づき、普通錠 の開発を中止し、以降の臨床開発には 錠が選択された。
表 2.7.1.1-3 各種錠剤の経口液剤に対するレゴラフェニブの相対的バイオアベイラビリティ
Analyte Parameter N Test * Reference * (oral sol.) Point estimate [%] (Test/Ref.) 2-sided 90% confidence interval [%] 20mg conventional tablets (60mg) vs. oral solution (60mg)
Parent AUC(0- tlast) [mg∙h/L] 6 2.16 (37.7) 26.5 (23.0) 8.15 [6.52 ; 10.2] Cmax [mg/L] 6 0.0847 (63.7) 1.78 (36.1) 4.75 [3.35 ; 6.73]
20mg tablets (60mg) vs. oral solution (60mg)
AUC(0- tlast) [mg∙h/L] 6 18.4 (33.2) 26.5 (23.0) 69.5 [55.6 ; 86.9] Cmax [mg/L] 6 1.05 (85.0) 1.78 (36.1) 59.0 [41.6 ; 83.5]
100mg tablet (100mg) vs. oral solution (100mg)
Parent AUC(0- tlast) [mg∙h/L] 7 33.8 (35.3) 40.6 (27.0) 83.2 [74.1 ; 93.4] Cmax [mg/L] 7 1.82 (63.9) 3.31 (40.4) 54.9 [41.0 ; 73.5] Source: Study report 5.3.3.2.1, PH-36733, Table 9-3, 9-6, 9-7, and 9-10
* Geometric means (%CV)
Study population: men and women with advanced malignancies 60mg group: 4 men and 2 women (54-68 years of age)
who received three single doses of 60mg regorafenib:
oral solution / 20mg conventional tablets / 20mg tablets
100mg group: 5 men and 2 women (35-70 years of age)
who received two single doses of 100mg regorafenib:
oral solution / 100mg tablet
2.7.1.1.6.2.2
錠のバイオアベイラビリティ:申請製剤(40 ㎎錠 4 錠)と自社
標準フィルムコーティング
錠(100 ㎎錠 1 錠+20 ㎎錠 3 錠)との
比較
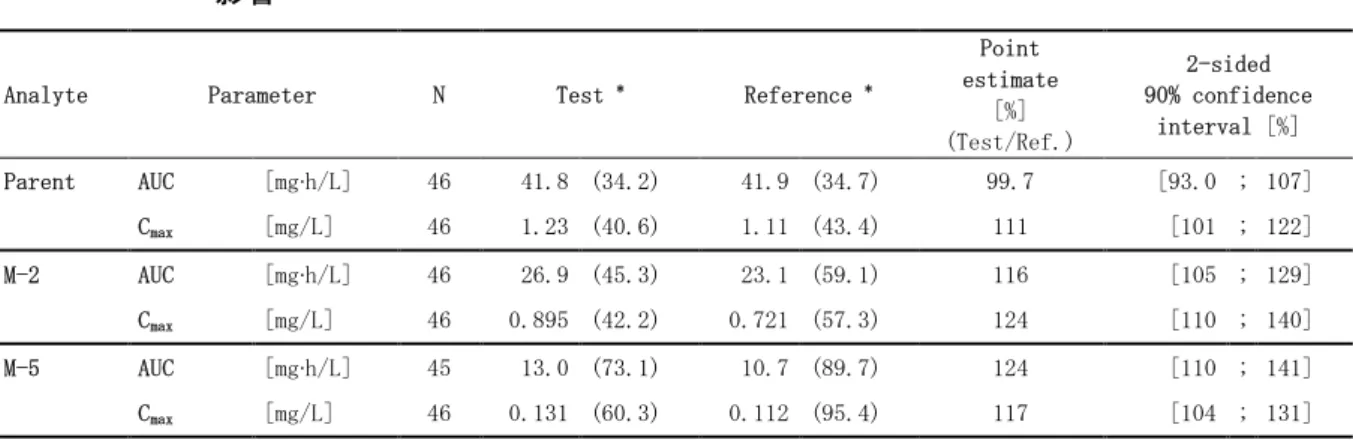
2.7.1.1.2に述べたように、レゴラフェニブの臨床試験で使用した各種 錠の組成は同一 のものであるが、フィルムコーティングについては、自社標準フィルムコーティングからオパド ライ フィルムコーティングに変更された。 初期の臨床試験相で行われたフィルムコーティングの変更、並びに 20mg 錠及び 100mg 錠から 40mg 錠への含量が異なる錠剤への変更がバイオアベイラビリティに及ぼす影響について検討す る目的で、健康男性被験者を対象として 錠のバイオアベイラビリティを評価する試験(試 験 12437)を実施した。推奨用量 160mg 投与時におけるレゴラフェニブの曝露量を評価項目とし て、申請製剤である 40mg 錠 4 錠投与時と、自社標準フィルムコーティングの 100mg 錠 1 錠+20mg 錠 3 錠投与時とで比較したところ、レゴラフェニブの AUC 及び Cmaxの比 (40mg 錠 4 錠/100mg 錠 1 錠+20mg 錠 3 錠)の 90%信頼区間(CI)が生物学的同等性の判定基 準の範囲内(80~125%)にあり、両剤は生物学的に同等であることが示された。本試験の結果 に基づき、第Ⅲ相臨床試験(試験 14387)には、 錠 40 ㎎錠 4 錠(160mg)を使用することと した。表 2.7.1.1-4 錠のバイオアベイラビリティ評価:フィルムコーティング及び含量変更の 影響
Analyte Parameter N Test * Reference *
Point estimate [%] (Test/Ref.) 2-sided 90% confidence interval [%] Parent AUC [mg∙h/L] 46 41.8 (34.2) 41.9 (34.7) 99.7 [93.0 ; 107] Cmax [mg/L] 46 1.23 (40.6) 1.11 (43.4) 111 [101 ; 122] M-2 AUC [mg∙h/L] 46 26.9 (45.3) 23.1 (59.1) 116 [105 ; 129] Cmax [mg/L] 46 0.895 (42.2) 0.721 (57.3) 124 [110 ; 140] M-5 AUC [mg∙h/L] 45 13.0 (73.1) 10.7 (89.7) 124 [110 ; 141] Cmax [mg/L] 46 0.131 (60.3) 0.112 (95.4) 117 [104 ; 131] Source: Study report 5.3.1.2.1, PH-36595, Table 9-1,9-2
* Geometric means followed by geometric coefficients of variation [%] in brackets Study population: healthy men (18 – 45 years of age)
Test: 160mg regorafenib as 40mg tablets (#142)
Reference: 160mg regorafenib as 1 x 100mg (#101) + 3 x 20mg tablets (#021)
2.7.1.1.6.2.3
絶対的バイオアベイラビリティ
前述のように、原薬が水性溶媒にほとんど溶けないため、レゴラフェニブの静脈注射剤を開発 することができなかった。したがって、レゴラフェニブの絶対的バイオアベイラビリティ試験は 実施していない。2.7.1.1.6.2.4
マスバランス試験
マスバランス試験(試験 12436)において、レゴラフェニブは経口投与後速やかに吸収され、 腸肝循環を受けた後、主として胆汁/糞を介して排泄されることが示された。 マスバランス試験において、健康男性被験者 4 例に[14C]レゴラフェニブ 120mg を経口液剤とし て単回投与した。投与された放射能の約 90%が、採取期間終了時までに回収され、そのうち約 71%(未変化体 47%、代謝物 24%)が糞中に、約 19%が尿中に排泄された(2.7.2.2.5.2.1 参 照)。このデータから、直接、本剤のバイオアベイラビリティを推定することはできないが、本 薬経口投与時のバイオアベイラビリティは高いことが示唆された。なお、投与後 48 時間目まで の糞中排泄率が投与量の約 23%のみであり、全被験者が少なくとも 1 回の排便を 48 時間までに 済ませていることから、薬物の吸収不良が原因で、糞中への排泄が高くなったのではないと考え られる(2.7.2.2.5.2.1 参照)。2.7.1.1.6.3
食事の影響
申請製剤のバイオアベイラビリティに及ぼす食事の影響を、健康男性被験者を対象とした 3 期 クロスオーバー試験(試験 14656)により検討した。本試験では、空腹時、高脂肪食摂取後、及 び低脂肪食摂取後にレゴラフェニブ 160mg(申請製剤 40mg 錠 4 錠)を投与した。未変化体の AUC 及び Cmaxは、低脂肪及び高脂肪の朝食摂取後で同様であり、AUC 及び Cmaxの比
(高脂肪食/低脂肪食)の 90%CI も、生物学的同等性の判定基準の範囲(80~125%)内であっ た。空腹時投与時の AUC 及び Cmaxを食後(低脂肪又は高脂肪の朝食)と比較したとき、食後投与
総曝露量を考えると、低脂肪食摂取後にレゴラフェニブを投与した場合に、曝露量が最も高い ことが示唆された(2.7.1.2.3参照)。
国際共同第Ⅲ相臨床試験等、がん患者を対象とした臨床試験の試験計画書において、本剤は軽 食又は低脂肪の朝食摂取後に投与するように規定されている。
表 2.7.1.1-5 申請製剤のバイオアベイラビリティに及ぼす食事の影響
Analyte Parameter N Test * Reference *
Point estimate [%] (Test/Ref.) 2-sided 90% confidence interval [%] Low fat vs. fasted
Parent AUC [mg∙h/L] 24 61.8 (31.4) 45.4 (36.9) 136 [123 ; 150
]
Cmax [mg/L] 24 1.93 (28.0) 1.25 (36.9) 154 [138 ; 173]
High fat vs. fasted
AUC [mg∙h/L] 24 67.3 (35.6) 45.4 (36.9) 148 [134 ; 164
]
Cmax [mg/L] 24 2.16 (31.8) 1.25 (36.9) 173 [154 ; 193
]
High fat vs. low fat
AUC [mg∙h/L] 24 67.3 (35.6) 61.8 (31.4) 109 [98.6 ; 120
]
Cmax [mg/L] 24 2.16 (31.8) 1.93 (28.0) 112 [100 ; 125
]
Source: Source: Study report 5.3.1.1.1, PH-36525, Table 9-1,9-2* Geometric means followed by geometric coefficients of variation [%] in brackets
Study population: healthy men (18 – 45 years of age)
Fasted: 4 x 40mg regorafenib (#142) administered after overnight fasting
Low fat: 4 x 40mg regorafenib (#142) administered immediately after a low fat breakfast High fat: 4 x 40mg regorafenib (#142) administered immediately after a high fat breakfast
2.7.1.2
個々の試験結果の要約
本項では、生物薬剤学の観点から重要と考えられる試験結果について記述した。なお、これら の試験で得られた安全性成績は 2.7.4 に要約し、有効性成績は 2.7.3 に、薬力学的成績及び薬 物動態学的成績は 2.7.2 に示した。 レゴラフェニブ及び代謝物の血漿中濃度は、バリデートされた LC-MS/MS 法により測定した (2.7.1.1.4参照)。薬物動態学的パラメータは WinNonlin を用いて算出した(2.7.1.1.5.1参 照)。薬物動態に関する解析として記述統計量を算出した。探索的な検討の場合、統計的な被験 者数の算出は行っていないが、バイオアベイラビリティ試験(試験 12437)は、未変化体の AUC 及び Cmaxを主要評価項目として、製剤間の生物学的同等性を証明するのに必要な被験者数に設定 した。2.7.1.2.1
試験 11650
3 週間投与/1 週間休薬 用量漸増試験における相対的バイオ
アベイラビリティの検討
試験 11650(5.3.3.2.1 PH-36733):進行性固形がん患者を対象としたレゴラフェニブの安全性、 忍容性、最大耐用量、薬物動態及びバイオマーカーを検討した非盲検、第 I 相臨床試験 本試験は、進行性固形がん患者を対象にレゴラフェニブを単回及び反復投与した際の薬物動態、 薬理学的作用、安全性及び忍容性を検討することを目的とした試験であり、試験目的の 1 つとし て、普通錠及び 錠におけるレゴラフェニブの相対的バイオアベイラビリティを経口液剤と 比較検討した。本試験は第Ⅰ相用量漸増試験であり、20 年~20 年にドイツで実施された。治験総括報告 書は、20 年 月 日をデータベースカットオフ日として作成された。 ここでは、試験方法、被験者背景、並びに本試験で得られた薬物動態の成績の順に記載し、相 対的バイオアベイラビリティに関する成績(普通錠と経口液剤との比較、 錠と経口液剤と の比較)について述べる。 本試験における相対的バイオアベイラビリティの検討は、コホート 4 及び 6 の反復投与サイク ルの前に実施し、各コホートの被験者に対して、レゴラフェニブ 60mg(コホート 4)及び 100mg (コホート 6)を異なる製剤にて単回投与した。すなわち、コホート 4 の 60mg の経口液剤投与 後、同じ被験者に対して、 錠、普通錠の順に、それぞれ 20mg 錠 3 錠を投与した。また、 コホート 6 終了後の被験者に対しては、レゴラフェニブ 100mg を経口液剤及び 錠にて、そ れぞれ単回投与した。レゴラフェニブの投与は、8 時間以上の絶食後に行い、各投与間には 1 週 間の休薬期間を設けた。 レゴラフェニブ及び主代謝物 M-2 及び M-5 の血漿中濃度測定用の血液検体は、投与後 48 時間 まで採取した。 60mg 投与群(コホート 4)は被験者 6 例(男性 4 例、女性 2 例)からなり、平均年齢 61.7 歳 (範囲:54~68 歳)、平均体重 84.1kg(範囲:61.9~105kg)、平均 BMI 27.3kg/m2(範囲: 21.4~34.9kg/m2)であった。また、100mg 投与群(コホート 6)は被験者 7 例(男性 5 例、女性 2 例)からなり、平均年齢 60.0 歳(範囲:35~70 歳)、平均体重 81.0kg(範囲:55.5~ 98.7kg)、平均 BMI 27.2kg/m2(範囲:18.5~36.9kg/m2)であった。 レゴラフェニブ 60mg を普通錠 20mg 錠 3 錠、 錠 20mg 3 錠、及び経口液剤にて単回経口 投与した際の未変化体並びに代謝物 M-2 及び M-5 の血漿中薬物動態学的パラメータを表 2.7.1.2-1に要約した。レゴラフェニブ 100mg を 錠 100 ㎎錠1錠及び経口液剤にて単回経 口投与した際の薬物動態学的パラメータを表 2.7.1.2-3に、統計解析の結果をそれぞれ表 2.7.1.2-2及び表 2.7.1.2-4に示した。 薬物動態評価において、相対的バイオアベイラビリティ(Frel:錠剤/経口液剤)は、AUC の外 挿に必要な t1/2を、全被験者で推定することができなかったため、AUC(0-tlast)に基づき算出した。 また、レゴラフェニブの消失は緩徐であったため、投与前に定量下限(LLOQ)を若干上回る血漿 中レゴラフェニブ濃度が検出された。しかしながら、この濃度はわずか(各投与期間の平均最高 濃度の 3.5%以下)であったことから、相対的バイオアベイラビリティの評価に大きな影響を及 ぼすものではないと考えられた。 レゴラフェニブ 60mg 投与時において、普通錠の経口液剤に対する未変化体の相対的バイオア ベイラビリティは、AUC と Cmaxのいずれで評価しても 10%未満であった。一方、 錠の経口 液剤に対する未変化体の相対的バイオアベイラビリティは、AUC で約 70%、Cmaxで約 60%であっ た。また、100mg 錠投与時における未変化体の経口液剤に対する相対的バイオアベイラビ リティは、AUC で約 83%、Cmaxで約 55%であった。 なお、レゴラフェニブの主代謝物 M-2 及び M-5 は、含量が異なる 錠(20mg 及び 100mg) 投与時には、ともに比較的低い血漿中濃度が観察されたが、普通錠投与時には、いずれの代謝物 の血漿中濃度も LLOQ 以下であった。 以上、がん患者を対象とした用量漸増試験(試験 11650)において普通錠及び 錠の経口 液剤に対する相対的バイオアベイラビリティを検討した。 のレゴラフェニブを含有する 20mg 普通錠の経口液剤に対する相対的バイオアベイラビリティは低く、普通錠の開発は中止す ることとした。一方、 錠(自社標準フィルムコーティングを使用)の経口液剤に対する相
対的バイオアベイラビリティは良好であり、 錠は、製剤の開発目的に合致した即放錠であ ることが示された。本試験の結果をもとに、普通錠の開発は行わず、 錠で臨床開発を行う との結論に至った。 表 2.7.1.2-1 がん患者にレゴラフェニブ 60mg を各種製剤にて単回経口投与(空腹時)した際 の血漿中レゴラフェニブ並びに主代謝物 M-2 及び M-5 の薬物動態学的パラメー タ[幾何平均値(%CV)、N = 6]
Analyte Treatment AUC(0- tlast)
[mg*h/L] Cmax [mg/L] tmax a [h] t½ [h] Parent Conv. tablets 2.16 (37.7) 0.0847 (63.7) 3.5 (3-71) n.d.b
tablets 18.4 (33.2) 1.05 (85.0) 3.0 (2-4) n.d.b
Oral solution 26.5 (23.0) 1.78 (36.1) 1.0 (1-2) n.d.b
M-2 Conv. tablets M-2 plasma concentrations were < LLOQ
tablets 6.09 (54.0) 0.370 (89.7) 3.0 (2-4) 24.0 (33.5)c
Oral solution 14.5 (37.9) 0.933 (42.0) 2.0 (1-3) 23.6 (30.8)d
M-5 Conv. tablet M-5 plasma concentrations were < LLOQ tablets 0.883 (140) 0.0192 (137) 30.0 (8-72) n.d.b
Oral solution 4.23 (97.4) 0.107 (105) 47.0 (24-72) n.d.b Source: Study report 5.3.3.2.1, PH-36733, Table 9-3,9-4,9-5
a median (minimum – maximum)
b insufficient data points during the terminal phase c N = 5
d N = 3
n.d. = not determined
LLOQ = lower limit of quantitation
Conv. tablets = three 20mg conventional tablets
tablets = three 20mg tablets
Oral solution = oral solution containing 60mg regorafenib
表 2.7.1.2-2 レゴラフェニブ 60mg 投与時の普通錠及び 錠の経口液剤に対する相対的バ イオアベイラビリティに関する解析結果
Analyte Comparison Parameter N
Point estimate [%] (Ratio) 2-sided 90% confidence interval [%] Parent Conv. tablets/solution AUC(0- tlast) 6 8.15 [6.52 ; 10.2]
Cmax 6 4.75 [3.35 ; 6.73]
tablets/solution AUC(0- tlast) 6 69.5 [55.6 ; 86.9]
Cmax 6 59.0 [41.6 ; 83.5]
M-2 Conv. tablets/solution M-2 plasma concentrations were < LLOQ after administration of conventional tablets tablets/solution AUC(0- tlast) 6 41.9 [33.5 ; 52.5]
Cmax 6 39.7 [27.2 ; 57.9]
M-5 Conv. tablets/solution M-5 plasma concentrations were < LLOQ after administration of conventional tablets tablets/solution AUC(0- tlast) 6 20.9 [15.2 ; 28.8]
Source: Study report 5.3.3.2.1, PH-36733, Table 9-6 LLOQ = lower limit of quantitation
Conv. tablet = three 20mg conventional tablets
tablets = three 20mg tablets
Solution = oral solution containing 60mg regorafenib All medications were administered under fasting conditions.
表 2.7.1.2-3 がん患者にレゴラフェニブ 100mg を 錠にて単回経口投与(空腹時)した際 の血漿中レゴラフェニブ並びに主代謝物 M-2 及び M-5 の薬物動態学的パラメータ [幾何平均値(%CV)、N = 7]
Analyte Treatment AUC(0-tlast)
[mg*h/L] Cmax [mg/L] tmax a [h] t½ [h] Parent tablet 33.8 (35.3) 1.82 (63.9) 4.08 (2-6) 22.6 (76.4)b Oral solution 40.6 (27.0) 3.31 (40.4) 1.05 (0.6-4) 21.2 (59.2)b M-2 tablet 9.31 (62.1) 0.498 (95.1) 4.17 (2-12) 17.7 (47.0)b Oral solution 23.5 (33.5) 1.56 (67.7) 2.00 (1-4) 18.7 (35.8)c M-5 tablet 1.00 (112) 0.0282 (130) 47.5 (22-71) n.d.d Oral solution 6.00 (99.1) 0.134 (109) 11.9 (3-70) n.d.d Source: Study report 5.3.3.2.1, PH-36733, Table 9.7, 9-8, 9-9
a median (minimum – maximum) b N = 4
c N = 6
d insufficient data points during the terminal phase
n.d. = not determined
tablet = one 100mg tablet
Oral solution = oral solution containing 100mg regorafenib
表 2.7.1.2-4 レゴラフェニブ 100mg 投与時の 錠の経口液剤に対する相対的バイオアベイ ラビリティに関する解析結果
Analyte Comparison Parameter N
Point estimate [%] (Ratio) 2-sided 90% confidence interval [%] Parent tablet/solution AUC(0- tlast) 7 83.2 [74.1 ; 93.4]
Cmax 7 54.9 [41.0 ; 73.5]
M-2 tablet/solution AUC(0- tlast) 7 39.6 [27.3 ; 57.5]
Cmax 7 32.0 [18.9 ; 54.2]
M-5 tablet/solution AUC(0- tlast) 7 16.7 [8.74 ; 31.9]
Cmax 7 21.1 [13.1 ; 33.9]
Source: Study report 5.3.3.2.1, PH-36733, Table 9-10
tablet = one 100mg tablet
Solution = oral solution containing 100mg regorafenib All medications were administered under fasting conditions.
2.7.1.2.2
試験 12437
バイオアベイラビリティ試験
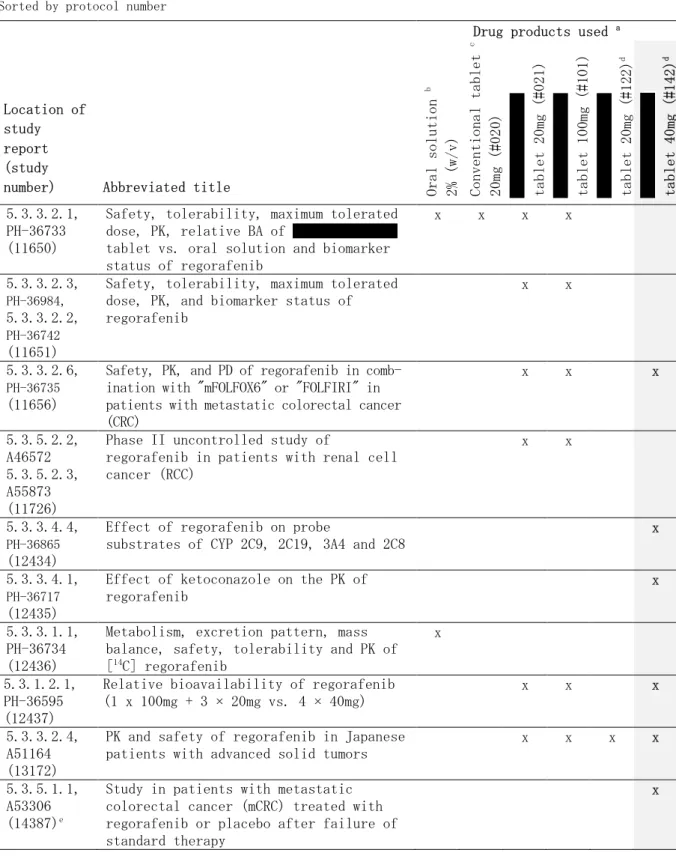
試験 12437(5.3.1.2.1 PH-36595):レゴラフェニブ 40mg 錠 4 錠と 100mg 錠 1 錠+20mg 錠 3 錠 による 160mg 投与時のバイオアベイラビリティの評価を目的とした、非盲検、無作為化、2 期ク ロスオーバー法による第 I 相臨床試験 本試験の主目的は、レゴラフェニブ 40mg 錠 4 錠(試験製剤:オパドライ フィルムコーティ ングの 錠/申請製剤)と 100mg 錠 1 錠+20mg 錠 3 錠(標準製剤:自社標準フィルムコー ティングの 錠/旧 錠)による 160mg 投与時の相対的バイオアベイラビリティを評価 することであった。 本試験は、健康男性被験者を対象とした、単施設、非盲検、無作為化、2 期クロスオーバー試 験であり、20 年に米国にて実施された。各被験者に対して、レゴラフェニブとして 160mg を、 クロスオーバー法により、40mg 錠 4 錠及び 100mg 錠 1 錠+20mg 錠 3 錠にて、それぞれ単回投与 した。各投与期の間には 7 日間の休薬期間を設けた。レゴラフェニブ及び主代謝物 M-2 及び M-5 の血漿中濃度測定用の血液検体は投与後 168 時間まで採取した。 48 例を安全性解析対象とし、うち 46 例を薬物動態解析対象とした。被験者の平均年齢は 34.0 歳(範囲:18~45 歳)、平均体重は 81.0kg(範囲:54.0~110kg)、平均 BMI は 25.7kg/m2(範 囲:18.5~31.8kg/m2)であった。レゴラフェニブ及び M-2 の平均血漿中濃度推移は、両製剤で 類似していた。M-5 の平均血漿中濃度には、製剤間で若干の違いがみられたものの、全体的には 類似した血漿中濃度推移がみられた(図 2.7.1.2-1)。 レゴラフェニブ並びに代謝物 M-2 及び M-5 の薬物動態学的パラメータを表 2.7.1.2-5に、バイ オアベイラビリティに関する解析(生物学的同等性)結果を、表 2.7.1.2-6にそれぞれ示した。 レゴラフェニブの AUC 及び Cmaxは、申請製剤と自社標準フィルムコーティングによる 錠で同様であり、AUC 及び Cmaxの比(40mg 錠 4 錠/100mg 錠 1 錠+20mg 錠 3 錠)の 90%CI は、生
物学的同等性の判定基準の範囲(80~125%)内であった。なお、代謝物については、AUC 及び Cmaxの比の 90%CI が生物学的同等性の判定基準の上限をわずかに逸脱したが、臨床的に意味のあ る差ではないと考えられた。 以上、申請製剤 40mg 錠 4 錠と自社標準フィルムコーティングによる 錠 100mg 錠 1 錠+ 20mg 錠 3 錠によるレゴラフェニブ 160 ㎎投与時の AUC 及び Cmaxから、両製剤は生物学的に同等で あることが示された。
Time (hr) 0 24 48 72 96 120 144 168 192 B A Y 7 3 -4 50 6 co n ce nt ra tio n (m cg /L ) 0 200 400 600 800 1000 1200 1400 4 x 40 mg 1 x 100 mg + 3 x 20 mg Time (hr) 0 20 40 60 80 100 120 140 160 180 B A Y 7 5 -7 4 95 c on ce n tr at io n (mc g/ L) 0 200 400 600 800 1000 4 x 40 mg 1 x 100 mg + 3 x 20 mg Time (hr) 0 20 40 60 80 100 120 140 160 180 BAY 81 -8 7 52 c on ce n tr a tio n ( m cg /L ) 0 20 40 60 80 100 120 4 x 40 mg 1 x 100 mg + 3 x 20 mg
Source: Study report 5.3.1.2.1, PH-36595, Figure 9-1, 9-2, 9-3 Geometric means BAY 73-4506 = regorafenib BAY 75-7495 = metabolite M-2 BAY 81-8752 = metabolite M-5 図 2.7.1.2-1 申請製剤(40mg 錠 4 錠)と自社標準フィルムコーティング 錠(100mg 錠 1 錠+20mg 錠 3 錠)のバイオアベイラビリティ試験において健康男性被験者を対 象にレゴラフェニブ 160mg を単回経口投与(空腹時)した際のレゴラフェニブ並 びに主代謝物 M-2 及び M-5 の平均血漿中濃度推移[幾何平均値、N=24]
表 2.7.1.2-5 申請製剤(40mg 錠 4 錠)と自社標準フィルムコーティング 錠(100mg 錠 1 錠+20mg 錠 3 錠)のバイオアベイラビリティ試験において健康男性被験者を対 象にレゴラフェニブ 160mg を単回経口投与(空腹時)した際のレゴラフェニブ並 びに主代謝物 M-2 及び M-5 の薬物動態学的パラメータ[幾何平均値(%CV)、N = 46]
Analyte Treatment AUC
[mg*h/L] Cmax [mg/L] tmax a [h] t½ [h] Parent Test 41.8 (34.2) 1.23 (40.6) 4 (2-16) 31.1 (35.2) Reference 41.9 (34.7) 1.11 (43.4) 4 (2-16) 30.1 (36.3) M-2 Test 26.9 (45.3) 0.895 (42.2) 4 (2-16) 25.1 (25.4) Reference 23.1 (59.1) 0.721 (57.3) 4 (2-16) 24.9 (27.9) M-5 Test 13.0 (73.1) 0.131 (60.3) 36 (12-72) 51.8 (59.0) Reference 10.7 (89.7) 0.112 (95.4) 24 (8-96) 50.6 (51.0)
Source: Study report 5.3.1.2.1, PH-36595, Table 9-1 and Table 14.4/2
a median (minimum – maximum)
Test: 160mg regorafenib as 40mg tablets (#142)
Reference: 160mg regorafenib as 100mg (#101) + 20mg tablets (#021)
表 2.7.1.2-6 申請製剤(40mg 錠 4 錠)と自社標準フィルムコーティング 錠(100mg 錠 1 錠+20mg 錠 3 錠)のバイオアベイラビリティに関する解析結果 Analyte Parameter N CV [%] Point estimate [%] (Test/Reference) 2-sided 90% confidence interval [%] Parent AUC 46 20.2 99.7 [93.0 ; 107] Cmax 46 28.3 111 [101 ; 122] M-2 AUC 46 30.3 116 [105 ; 129] Cmax 46 35.2 124 [110 ; 140] M-5 AUC 45 36.4 124 [110 ; 141] Cmax 46 33.3 117 [104 ; 131]
Source: Study report 5.3.1.2.1, PH-36595, Table 9-2
Test: 160mg regorafenib as 40mg tablets (#142)
Reference: 160mg regorafenib as 100mg (#101) + 20mg tablets (#021)
2.7.1.2.3
試験 14656
食事の影響試験
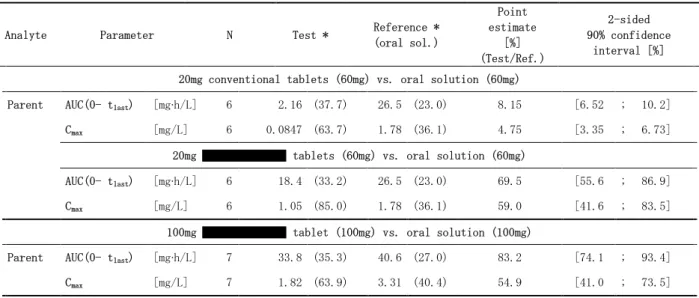
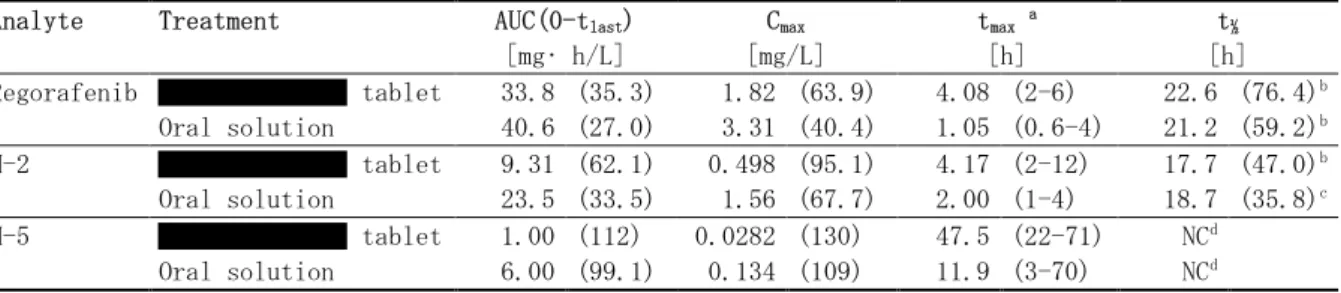
試験 14656(5.3.1.1.1 PH-36525):健康男性被験者を対象とした、レゴラフェニブ 160mg 単回 経口投与時の薬物動態に及ぼす低脂肪食及び高脂肪食及び空腹時の影響を検討する、非盲検、無 作為化、3 期クロスオーバー、第 I 相臨床試験 本試験は、申請製剤を用いて、健康男性被験者を対象にレゴラフェニブの薬物動態に及ぼす食 事の影響を検討した試験である。主目的は、レゴラフェニブの薬物動態に及ぼす食事(低脂肪食 及び高脂肪食)の影響を評価することであり、副次目的は、同時に主代謝物の薬物動態に及ぼす 食事の影響を評価することであった。本試験は、単施設、非盲検、無作為化 3 期クロスオーバー試験であり、20 年~20 年に米 国で実施された。レゴラフェニブの投与は、申請製剤を用いて、(a)一晩絶食後にレゴラフェニ ブ 160mg(40mg 錠×4)投与、(b) 低脂肪の朝食摂取直後にレゴラフェニブ 160mg(40mg 錠×4) 投与、(c) 高脂肪の朝食摂取直後にレゴラフェニブ 160mg(40mg 錠×4)投与を各投与期に行っ た。投与後 4 時間は絶食とし、各投与期の間には 14 日間の休薬期間を設けた。血漿中レゴラ フェニブ及び主代謝物(M-2 及び M-5)測定用の血液検体は、投与後 336 時間にわたり採取した。 本試験のデザイン及び食事の内容は、関連する FDA ガイダンス3),4)に従った。 24 例を無作為割り付けし、全例を薬物動態解析対象とした。被験者の平均年齢は 32.5 歳(範 囲:18~45 歳)、平均 BMI 26.4±3.5kg/m2であった。 レゴラフェニブ並びに代謝物 M-2 及び M-5 の平均血漿中濃度推移を図 2.7.1.2-2に示した。ま た、薬物動態学的パラメータを表 2.7.1.2-7に、バイオアベイラビリティに関する解析結果を表 2.7.1.2-8に示した。
未変化体の AUC 及び Cmaxは、低脂肪食及び高脂肪食摂取後で同様であった。AUC 及び Cmaxの比
(高脂肪食/低脂肪食)の 90%CI は、生物学的同等性の判定基準の範囲(80~125%)内であっ た。空腹時投与時の AUC 及び Cmaxを食後投与時(低脂肪又は高脂肪食摂取後)と比較したとき、
食後投与時の AUC 及び Cmaxは空腹時投与時に比べ増加した(AUC の点推定値:136%、90%CI:
123~150%)。 代謝物 M-2 及び M-5 の AUC 及び Cmaxについては、低脂肪食摂取後投与時において空腹時投与時 より高かったが、高脂肪食摂取後投与時では空腹時投与時より低かった。 以上の成績から、低脂肪食摂取後にレゴラフェニブを投与した場合、空腹時又は高脂肪食摂取 後投与に比べて、未変化体並びに代謝物 M-2 及び M-5 の曝露量が 20~40%増加し、最も高い曝 露量が得られた。
Source: Study report 5.3.1.1.1, PH-36525, Figure 9-1, 9-2, 9-3 Geometric means BAY 73-4506 = regorafenib BAY 75-7495 = metabolite M-2 BAY 81-8752 = metabolite M-5 図 2.7.1.2-2 申請製剤の食事の影響試験において健康男性被験者を対象に空腹時、低脂肪及び 高脂肪の朝食摂取後にレゴラフェニブ 160mg を単回経口投与した際のレゴラフェ ニブ並びに代謝物 M-2 及び M-5 の平均血漿中濃度推移[幾何平均値、N=24]
表 2.7.1.2-7 申請製剤の食事の影響試験において健康男性被験者を対象に空腹時、低脂肪及び 高脂肪の朝食摂取後にレゴラフェニブ 160mg を単回経口投与したときの血漿中の レゴラフェニブ並びに主代謝物 M-2 及び M-5 の薬物動態学的パラメータ[幾何平 均値(%CV)、N = 24 ]
Analyte Treatment AUC
[mg*h/L] Cmax [mg/L] tmax a [h] t½ [h] Parent Fasted 45.4 (36.9) 1.25 (36.9) 4 (2-24) 37.9 (28.7) Low fat 61.8 (31.4) 1.93 (28.0) 4 (2-16) 35.0 (20.9) High fat 67.3 (35.6) 2.16 (31.8) 6 (3-6) 35.0 (21.7) M-2 Fasted 27.4 (52.8) 0.895 (45.7) 4 (2-24) 28.1 (21.6) Low fat 38.3 (37.2) 1.16 (34.6) 6 (3-16) 26.2 (21.5) High fat 21.9 (70.2) 0.647 (66.3) 6 (3-12) 27.5 (23.0) M-5 Fasted 12.8 (68.6) 0.125 (64.0) 24 (4-48) 64.1 (28.0) Low fat 15.7 (41.5) 0.139 (41.0) 48 (12-96) 56.8 (17.3) High fat 6.22 (71.6) 0.0508 (78.2) 48 (12-96) 65.5 (36.6)
Source: Study report 5.3.1.1.1, PH-36525, Table 9-1, 14.4/2
a median (minimum – maximum)
Fasted: 4 × 40mg regorafenib (#142) administered after overnight fasting
Low fat: 4 × 40mg regorafenib (#142) administered immediately after a low fat breakfast High fat: 4 × 40mg regorafenib (#142) administered immediately after a high fat breakfast
表 2.7.1.2-8 申請製剤の食事の影響試験におけるバイオアベイラビリティに関する解析結果
Analyte Comparison Parameter N
Point estimate [%] (Ratio) 2-sided 90% confidence interval [%]
Parent Low fat / fasted AUC 24 136 [123 ; 150
]
Cmax 24 154 [138 ; 173
]
High fat / fasted AUC 24 148 [134 ; 164
]
Cmax 24 173 [154 ; 193
]
High fat / low fat AUC 24 109 [98.6 ; 120
]
Cmax 24 112 [100 ; 125
]
M-2 Low fat / fasted AUC 24 140 [115 ; 169
]
Cmax 24 130 [106 ; 159
]
High fat / fasted AUC 24 80.0 [66.1 ; 96.8
]
Cmax 24 72.3 [59.1 ; 88.4
]
High fat / low fat AUC 24 57.3 [47.3 ; 69.4
]
Cmax 24 55.5 [45.4 ; 67.9
]
M-5 Low fat / fasted AUC 24 123 [101 ; 149
]
Cmax 24 112 [89.1 ; 140
]
High fat / fasted AUC 24 48.7 [40.0 ; 59.3
]
Cmax 24 40.7 [32.5 ; 51.1
]
High fat / low fat AUC 24 39.7 [32.6 ; 48.3
]
Cmax 24 36.5 [29.1 ; 45.7
]
Source: Study report 5.3.1.1.1, PH-36525, Table 9-2
Fasted: 4 × 40mg regorafenib (#142) administered after overnight fasting
Low fat: 4 × 40mg regorafenib (#142) administered immediately after a low fat breakfast High fat: 4 × 40mg regorafenib (#142) administered immediately after a high fat breakfast
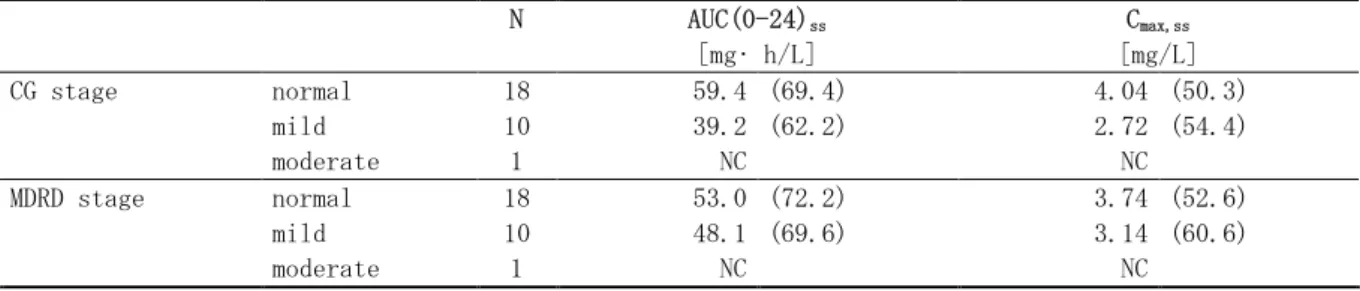
なお、低脂肪食摂取後投与により、未変化体及び主代謝物 M-2 及び M-5 の曝露量は、空腹時投 与に比べて増加したが、高脂肪食摂取後の食事の影響は未変化体と代謝物とで異なる可能性が示 唆された。すなわち、高脂肪食摂取後投与時において、未変化体は低脂肪食摂取後と同様に曝露 量の増加が認められたのに対して、代謝物の曝露量は低下していた。本剤のバイオアベイラビリ ティに及ぼす食事の影響の機序としては、食事の摂取により難溶性のレゴラフェニブの溶解性が 増したことにより、消化管内での未変化体の吸収が高まり、曝露量が増加したことが考えられる。 一方、代謝物 M-2 及び M-5 の曝露量が高脂肪食摂取後においてのみ低下した理由は不明である。 しかしながら、本薬の代謝、排泄経路から、低脂肪食摂取後と高脂肪食摂取後で胆汁への排泄及 び腸肝循環の過程において何らかの差異があったことが推察される。すなわち、M-5 の曝露量の 低下は M-2 の吸収量の低下と直接的に関連すると考えられることから、食事によって小腸上皮細 胞に存在している CYP3A の代謝能が何らかの理由により低下あるいは飽和しており、M-2 の生成 量が減少した可能性や、あるいは高脂肪食の存在により消化管での腸内細菌の活動が抑えられた ことで、M-2 のグルクロン酸抱合体の開裂が阻害された結果、消化管からの M-2 の吸収量が減少 したなど、主代謝物の吸収阻害が生じたことも否定し得えない。いずれも推論であり、その機序 は不明であるが、未変化体が循環血中における主要な薬理活性成分と考えられることから、食事 の影響によって代謝物の曝露量が変化したとしても、それらの変動が直接的に安全性及び有効性 に及ぼす影響は比較的小さいと考えられる。
2.7.1.3
全試験を通しての結果の比較と解析
レゴラフェニブ錠の製剤開発において、 のレゴラフェニブを含有する 錠と のレゴラフェニブを含有する普通錠を検討した。開発初期において、進行性固形がん患者を対象 に、 錠、普通錠の経口液剤に対する相対的バイオアベイラビリティを検討したところ、 錠のバイオアベイラビリティは、普通錠と比べて明らかに高かった(2.7.1.2.1参照)。本 試験の結果をもとに、普通錠の開発は行わず、 錠で臨床開発を行うとの結論に至った。 臨床試験に使用した 錠の組成に変更はないが、フィルムコーティングの軽微な変更を 行った。また、第Ⅲ相臨床試験の開始前に、錠剤の含量を 20mg 又は 100mg から 40mg に変更した ため、これらの変更がバイオアベイラビリティに及ぼす影響について検討することを目的として、 健康男性を対象に新旧 錠のバイオアベイラビリティ試験(試験 12437:2.7.1.2.2参照) を実施した。その結果、推奨用量 160mg 投与時の未変化体の曝露量は、申請製剤 40mg 錠 4 錠と 自社標準フィルムコーティングの 100mg 錠 1 錠+20mg 錠 3 錠投与時とで生物学的に同等である ことが示された。この結果は、各製剤のin vitro
における溶出プロファイルの類似性からも支 持されるものであった(図 2.7.1.1-1)。これらの成績より、第Ⅲ相臨床試験(試験 14387)は、 錠 40 ㎎錠 4 錠(160mg)を使用して実施することとした。 本剤のバイオアベイラビリティに及ぼす食事の影響を検討するため、健康男性を対象とした食 事の影響試験(試験 14656)を、申請製剤を用いて実施した。未変化体並びに主代謝物の血漿中 濃度は、低脂肪食摂取後投与時に最も高く、曝露量は空腹時投与と比較して 20~40%増加した。 また、本試験において得られた申請製剤 40mg 錠 4 錠投与時の薬物動態の成績は、先に実施した バイオアベイラビリティ試験(試験 12437)の結果とよく一致していた(表 2.7.1.3-1)。 健康男性を対象とした生物薬剤学的試験 2 試験(試験 12437、試験 14656)の成績を基に、未 変化体の AUC の個体間変動を検討したところ、AUC の変動係数(CV)は、それぞれ 34%及び 37%であった。健康男性を対象として食事の影響を検討した試験(試験 14656)で得られた CV は、進行性固形がん患者を対象とした試験(試験 11650)における CV と同様であり、経口液剤 投与時においても近似した CV がみられている(表 2.7.1.2-1及び表 2.7.1.2-3)。さらに、 錠の相対的バイオアベイラビリティが高いことを勘案すると、レゴラフェニブの薬物動態学 的パラメータに認められた比較的大きな個体間変動は、申請製剤に起因するものではないと考え られた。 以上、バリデートされた生体試料分析法及び適切な薬物動態評価方法を用いて、国外で実施さ れた生物薬剤学試験 3 試験の結果より、申請製剤である 錠 40 ㎎錠はバイオアベイラビリ ティの観点から優れた製剤であることが示された。また、食事の影響の検討により、本剤は低脂 肪食摂取後に投与することにより、レゴラフェニブ及び活性代謝物の最大の曝露量が得られるこ とを確認した。表 2.7.1.3-1 申請製剤の生物薬剤学試験間の比較:健康男性被験者を対象にレゴラフェニブ 160mg を空腹時に単回経口投与した際の血漿中レゴラフェニブ並びに代謝物 M-2 及び M-5 の薬物動態学的パラメータ[幾何平均値(%CV)] Analyte Study no. AUC [mg*h/L] Cmax [mg/L] tmax * [h] Parent 12437 41.8 (34.2) 1.23 (40.6) 4 (2-16) 14656 45.4 (36.9) 1.25 (36.9) 4 (2-24) M-2 12437 26.9 (45.3) 0.895 (42.2) 4 (2-16) 14656 27.4 (52.8) 0.895 (45.7) 4 (2-24) M-5 12437 13.0 (73.1) 0.131 (60.3) 36 (12-72) 14656 12.8 (68.6) 0.125 (64.0) 24 (4-48)
Source: Study reports 5.3.1.2.1, PH-36595, 5.3.1.1.1, PH-36525
* median (minimum – maximum)
Study 12437: N = 46 healthy men aged 18 – 45 years; body weight: 54.0 – 110kg Study 14656: N = 24 healthy men aged 18 – 45 years; body weight: 60.0 – 102kg
2.7.1.4
付録
該当なし
参考文献
(1) U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System 2000(5.4.26 参照)
(2) U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) & Center for Veterinary Medicine (CVM), Guidance for Industry - Bioanalytical Method Validation 2001(5.4.27 参照)
(3) U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Guidance for Industry: Food-effect
Bioavailability and Fed Bioequivalence Studies 2002(5.4.28 参照)
(4) U.S. Department of Health and Human Services, Food and Drug Administration, Center for Food Safety and Applied Nutrition, Office of Nutrition, Labeling and Dietary Supplements,Guidance for Industry: A Food Labeling Guide 2008(5.4.29 参照)
2.7.2 臨床薬理試験の目次
2.7.2.1 背景及び概観 ... 4 2.7.2.1.1 緒言 ... 4 2.7.2.1.2 要旨 ... 4 2.7.2.1.2.1 背景 ... 4 2.7.2.2 個々の試験結果の要約 ... 11 2.7.2.2.1 緒言 ... 11 2.7.2.2.2 がん患者を対象とした第Ⅰ相臨床試験 ... 11 2.7.2.2.2.1 試験 11650 3 週間投与/1 週間休薬 用量漸増試験... 11 2.7.2.2.2.2 試験 11651 連日経口投与 用量漸増試験... 24 2.7.2.2.2.3 試験 13172 日本人患者を対象とした試験... 31 2.7.2.2.2.4 試験 14996 中国人患者を対象とした試験... 37 2.7.2.2.3 薬物相互作用試験 ... 39 2.7.2.2.3.1 試験 11656 mFOLFOX6 又は FOLFIRI との薬物相互作用試 験 ... 39 2.7.2.2.3.2 試験 12434 CYP プローブ基質との薬物相互作用試験 ... 45 2.7.2.2.4 がん患者を対象とした特別な試験 ... 55 2.7.2.2.4.1 試験 14814 QT/QTc への影響を検討した試験 ... 55 2.7.2.2.5 健康被験者を対象とした第Ⅰ相臨床試験 ... 61 2.7.2.2.5.1 生物薬剤学的試験 ... 61 2.7.2.2.5.1.1 試験 14656 食事の影響試験... 61 2.7.2.2.5.1.2 試験 12437 バイオアベイラビリティ試験... 65 2.7.2.2.5.2 健康被験者を対象とした代謝試験 ... 68 2.7.2.2.5.2.1 試験 12436 [14C]マスバランス試験 ... 68 2.7.2.2.5.2.2 試験 12435 ケトコナゾールとの薬物相互作用試験 ... 72 2.7.2.2.5.2.3 試験 15524 リファンピシンとの薬物相互作用試験 ... 77 2.7.2.2.6 がん患者を対象とした第Ⅱ/Ⅲ相臨床試験 ... 81 2.7.2.2.6.1 試験 14596 韓国人 HCC 患者及び白人 HCC 患者の薬物動 態 ... 81 2.7.2.2.6.2 試験 11726 RCC 患者の薬物動態 ... 84 2.7.2.2.7 がん患者を対象とした薬物動態/薬力学評価を含む第Ⅲ相 臨床試験(試験 14387) ... 85 2.7.2.2.7.1 試験方法/対象集団 ... 86 2.7.2.2.7.2 母集団薬物動態解析(試験 14387、試験 11650) ... 86 2.7.2.2.7.3 曝露量-有効性関係の解析(試験 14387)... 892.7.2.2.7.4 曝露量-安全性関係の解析(試験 14387)... 90 2.7.2.2.7.5 遺伝子バイオマーカーの解析(試験 14387)... 91 2.7.2.2.7.6 非遺伝子バイオマーカーの解析(試験 14387)... 92 2.7.2.3 全試験を通しての結果の比較及び解析 ... 92 2.7.2.3.1 全試験を通しての薬物動態及び代謝成績の要約 ... 92 2.7.2.3.1.1 吸収速度と吸収率 ... 92 2.7.2.3.1.2 分布 ...102 2.7.2.3.1.3 代謝 ...103 2.7.2.3.1.4 排泄 ...106 2.7.2.3.1.5 薬物動態の時間依存性 ...108 2.7.2.3.1.6 健康被験者とがん患者の薬物動態成績の比較 ...111 2.7.2.3.2 内因性因子の影響 ...112 2.7.2.3.2.1 年齢 ...112 2.7.2.3.2.2 性差 ...114 2.7.2.3.2.3 体重 ...115 2.7.2.3.2.4 腎機能 ...117 2.7.2.3.2.5 肝機能 ...121 2.7.2.3.2.6 薬物動態の民族差比較 ...127 2.7.2.3.3 外因性因子の影響 ...133 2.7.2.3.3.1
In vitro
相互作用データ...134 2.7.2.3.3.2In vivo
相互作用データ...137 2.7.2.3.3.2.1 ケトコナゾールの影響 ...137 2.7.2.3.3.2.2 リファンピシンの影響 ...138 2.7.2.3.3.2.3 CYP プローブ基質に及ぼすレゴラフェニブの影響 ...139 2.7.2.3.3.2.4 mFOLFOX6 又は FOLFIRI との薬物相互作用...142 2.7.2.3.4 バイオマーカー ...144 2.7.2.3.4.1 分子バイオマーカー ...144 2.7.2.3.4.2 血漿中バイオマーカー ...148 2.7.2.3.4.2.1 レゴラフェニブによる血漿中バイオマーカー値の変化 ...148 2.7.2.3.4.2.2 血漿中たん白濃度とレゴラフェニブの反応による臨床 的有効性の相関性の評価 ...151 2.7.2.3.4.2.3 予後的価値の可能性がある血漿中たん白の評価 ...152 2.7.2.3.4.3 DCE-MRI ...152 2.7.2.3.5 用量/曝露量と反応との関係 ...153 2.7.2.3.5.1 定常状態におけるレゴラフェニブの濃度と細胞性 IC50の 関係 ...153 2.7.2.3.5.2 用量設定 ...1532.7.2.3.5.3 用量と効果との関係 ...156 2.7.2.3.5.4 用量と毒性との関係 ...156 2.7.2.3.5.5 曝露量と効果との関係 ...161 2.7.2.3.5.6 Cmax及び AUC と毒性との関係 ...167 2.7.2.3.6 心血管安全性 ...170 2.7.2.4 付録 ...170 参考文献 ...170
![表 2.7.1.2-6 申請製剤(40mg 錠 4 錠)と自社標準フィルムコーティング 錠(100mg 錠 1 錠+20mg 錠 3 錠)のバイオアベイラビリティに関する解析結果 Analyte Parameter N CV [%] Point estimate[%] (Test/Reference) 2-sided 90% confidence interval [%] Parent AUC 46 20.2 99.7 [93.0 ; 107] C max 46 28.3 111 [101 ; 122]](https://thumb-ap.123doks.com/thumbv2/123deta/6486080.657016/22.892.118.789.602.821/フィルムコーティング錠バイオアベイラビリティに関するParentC.webp)
![表 2.7.1.2-7 申請製剤の食事の影響試験において健康男性被験者を対象に空腹時、低脂肪及び 高脂肪の朝食摂取後にレゴラフェニブ 160mg を単回経口投与したときの血漿中の レゴラフェニブ並びに主代謝物 M-2 及び M-5 の薬物動態学的パラメータ[幾何平 均値(%CV)、N = 24 ]](https://thumb-ap.123doks.com/thumbv2/123deta/6486080.657016/25.892.103.789.242.491/空腹時低及びレゴラフェニブ血漿レゴラフェニブ並びパラメータ.webp)
![表 2.7.2.2-10 腎機能で層別したレゴラフェニブ 160mg 1 日 1 回反復経口投与後の血漿中 M-5 の薬物動態学的パラメータ(試験 11650) Geometric means (%CV) N AUC(0-24) ss [mg∙ h/L] C max,ss [mg/L] CG stage normal 18 69.1 (129) 4.11 (138) mild 10 49.5 (175) 3.21 (149) moderate 1 NC NC MDRD stage normal 18 55.](https://thumb-ap.123doks.com/thumbv2/123deta/6486080.657016/51.892.106.785.595.742/腎機能レゴラフェニブ回反与後血漿薬物動態学的パラメータGeometric∙.webp)