メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 32 -
2.7.3 臨床的有効性の概要
2.7.3.1 背景及び概観
国内臨床試験は健康女性を対象とした第
I
相臨床試験1
試験[Study 107],閉経後健康女性を 対象とした第I
相臨床試験2
試験[Study 108,Study 109],更年期障害又は卵巣欠落症患者を対 象とした第II
相臨床試験2
試験[Study 203,Study 205]及び第III
相臨床試験1
試験[Study305]の 6
試験を実施し,健康女性12
例,閉経後健康女性56
例,更年期障害又は卵巣欠落症患 者1,000
例が試験に組み込まれた.国内で実施した臨床試験6
試験のうち,第I
相臨床試験を除 く第II
相臨床試験2
試験及び第III
相臨床試験1
試験の3
試験の結果に基づいて,更年期障害又 は卵巣欠落症患者に対するRPR106522
製剤の有効性を評価した.海外臨床試験データは,国内での臨床試験データを補完することを目的として,欧米の承認申 請時に評価対象とした臨床試験のうち,子宮内膜増殖症の発現抑制を検討した主要な臨床試験
[Study 202(参考資料)]の成績を参照することとした.国内臨床試験では,海外臨床試験と同 様に,3種類の
RPR106522
製剤(9 cm2製剤,16 cm2製剤,26 cm2製剤)を用いて検討を行った.E
2を単独で含有する市販の経皮投与製剤(フェミエスト®4.33 mg
及び)の臨床試験成績5-8)及び文献9-11)などの情報において,更年期障害及び卵巣欠落症の改 善に必要とされる血清中
E
2濃度は約30~70 pg/mL
であることが示されている.RPR106522製剤 は,国内第I
相臨床試験[Study 108,Study 109]成績から血清中E
2濃度が約50~70 pg/mL
を3
~4日間維持することが確認されている.また,血清中
NET
濃度はNETA
用量に依存した用量 線形性が見られることが確認されている.したがって,RPR106522製剤の国内臨床開発における 有効性面での評価として以下の2
点に着眼し,第II
相及び第III
相臨床試験を実施した.1.
更年期障害に対する効果:配合されたNETA
の用量に関わりなく,血管運動神経系症状(Hot flush,発汗)及び腟萎縮症状に代表される更年期障害及び卵巣欠落症に伴う諸症状 に対して改善作用を示すこと.
2.
子宮内膜増殖作用の抑制効果:配合されたNETA
がE
2による子宮内膜増殖作用に対する抑 制効果を示すこと(NETAの配合意義).表 2.7.3-1に,有効性を評価した国内第
II
相臨床試験2
試験[Study 203,Study 205]及び第III
相臨床試験1
試験[Study 305]並びに海外第II
相臨床試験[Study 202(参考資料)]を示した.国内において有効性を評価した
3
試験のうち対照群を設定した試験は,第II
相臨床試験(プラ セボ対照二重盲検比較試験)[Study 203]及び第III
相臨床試験(実薬対照非盲検比較試験)[Study 305]が該当する.更に,海外で実施された第
II
相臨床試験(米国におけるE
2単独貼付 剤対照二重盲検比較試験)[Study 202]は,国内では倫理面の問題から実施できなかったE
2単 独投与群を設定して子宮内膜増殖作用の抑制効果を検討した主要な試験であり,参考として国内3
試験と併せて言及する.なお,国内外の女性では子宮内膜増殖に影響する可能性のある背景因 子のBMI
において,[Study 305]の本剤群の平均値が22.76 kg/m
2であるのに対して[Study202]の E
2単独投与群及び本剤群の平均値は27.07 kg/m
2及び27.86 kg/m
2と違いが見られた.し かしながら,日本人のHRT
施行例での子宮内膜増殖症のデータは見あたらないもののHRT
未施※新薬承認情報提供時に置き換えた E2貼付剤A※
行例集団での子宮内膜増殖症あるいは子宮体癌の発見率に近いと考えられる子宮体癌検診におけ る発見率は
0.23%(733
人/321,690人)14)であり,CPMPガイドラインではHRT
未施行例で約0
~1%15)とされていることから,背景に違いはあっても国内外で子宮内膜増殖症の発現率には大 きな違いはないと推察された.また,[Study 202]における各投与群の
BMI
にはバラツキがな く,背景に偏りのない被験者集団で群間比較されていることから,日本人における子宮内膜増殖 症の抑制効果を検討する上で[Study 202]を参考にすることは可能であると考えた.メノエイドコンビパッチ(
RPR1 065 22
)M2 .7
臨床概要- 34 -
表
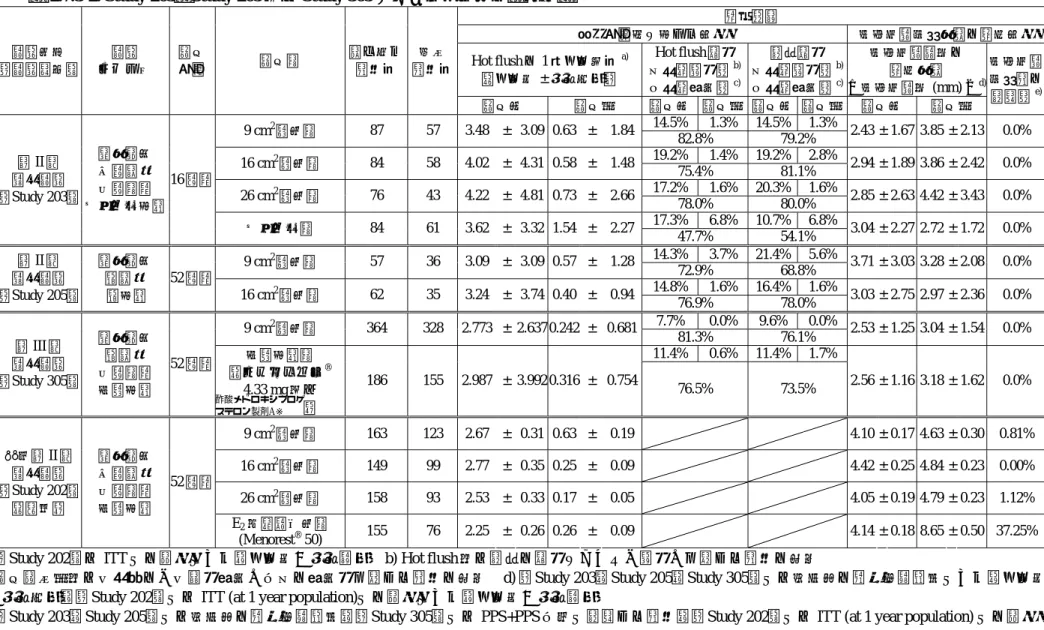
2.7.3-1.
有効性を評価した臨床試験一覧試験区分
[試験番号]
(国)
試験
デザイン 対象 用法・用量 組み入れ 症例数
投与
期間 目的 主要評価項目 添付資料
番号 各群で製剤1枚及びプラ
セボ1枚,又はプラセボ 2枚を,3又は4日ごと下 腹部に貼付する.
合計 331例 RPR106522 9 cm2製剤群 87例 RPR106522 16 cm2製剤群 84例 RPR106522 26 cm2製剤群 76例 第II相
臨床試験
[Study 203]
(日本)
無作為化 二重盲検 並行群間 プラセボ対照
更年期障害 又は卵巣 欠落症患者
プラセボ群 84例
16 週間
・有効性,安全性及び有用性をプラセボ と比較する.
・子宮内膜に対する影響,出血状況など を評価し,NETAの必要量を検索する.
最終全般改善 度
5.3.5.1-1
(評価資料)
各群で製剤1枚を,3又 は4日ごと下腹部に貼付 する.
合計 119例 RPR106522 9 cm2製剤群 57例 第II相
臨床試験
[Study 205]
(日本)
無作為化 非盲検 非対照
更年期障害 又は卵巣 欠落症患者
RPR106522 16 cm2製剤群 62例
52 週間
・52週間投与した際の子宮内膜に対する 影響及び出血状況などを中心に安全性を 検討する.
・更年期障害に対する有効性についても 検討する.
有害事象の発 現率,臨床検 査値異常変動 の発現率
5.3.5.2-1
(評価資料)
各群で製剤1枚を,3又 は4日ごと下腹部に貼付 する.更に,実薬対照群 では1日1回1錠黄体ホ ルモンを併用する.
合計 550例
RPR106522 9 cm2製剤群 364例 第III相
臨床試験
[Study 305]
(日本)
無作為化 非盲検 並行群間 実薬対照
更年期障害 又は卵巣 欠落症患者
実薬対照群
[HRT (フェミエスト® 4.33 mg及び )]
186例
52 週間
・子宮内膜増殖症発現率を主たる評価項 目として,既存HRTと比較する.
・更年期障害に対する有効性についても 検討する.
子宮内膜増殖 症発現率
5.3.5.1-2
(評価資料)
※新薬承認情報提供時に置き換えた
酢酸メドロ キシプロゲス テロン製剤A※
ドコンビパッチ(
RPR1 065 22
)M2 .7
臨床概要35 -
試験区分
[試験番号]
(国)
試験
デザイン 対象 用法・用量 組み入れ 症例数
投与
期間 目的 主要評価項目 添付資料
番号 各群で製剤1枚及びプラセ
ボ1枚を,3.5日ごと下腹 部に貼付する.
合計 625例 RPR106522 9 cm2製剤群 163例 RPR106522 16 cm2製剤群 149例 RPR106522 26 cm2製剤群 158例 海外第II相
臨床試験
[Study 202]
(米国)
無作為化 二重盲検 並行群間 実薬対照
閉経後 健康女性
対照群
[E2単独貼付剤 (Menorest® 50) ]
155例
52 週間
・エストロゲン誘発性の子宮内膜増殖症 発現率について,E2単独投与群との比 較により,E2に配合されたNETAの3用 量の効果を評価する.
・性器出血,血管運動神経系症状,脂質 プロファイル,代謝系及び凝固系パラメ ータ,骨代謝マーカー,皮膚忍容性,貼 付剤接着力,臨床検査及び臨床上の安全 性パラメータ,QOL及び血清中E1, E2,NET濃度に及ぼす効果を評価する.
子宮内膜増殖 症発現率
5.3.5.1-4
(参考資料)
メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 36 -
(1) 第II相臨床試験(プラセボ対照二重盲検比較試験)[Study 203] 5.3.5.1-1 国内で実施した第
I
相臨床試験(単回投与試験)[Study 108]及び第I
相臨床試験(反復投与 試験)[Study 109]に引き続き,19 年 月から19
年 月に実施した.更年期障害又は卵巣 欠落症患者を対象に,RPR106522 9 cm2製剤,16 cm2製剤及び26 cm
2製剤を用いて,有効性,安 全性及び有用性についてプラセボを対照として無作為化二重盲検並行群間比較法にて検討した.また,子宮内膜への影響も併せて評価し,NETAの必要量について探索した.
有効性の評価項目は,最終全般改善度,全般改善度,患者による評価及び臨床症状効果判定
(Hot flushの
1
日平均回数並びに各臨床症状の程度の改善度)とした.なお,更年期障害の臨床 症状については,Hot flushの1
日平均回数並びにHot flush,発汗,腟萎縮症状,睡眠障害,排尿
障害,冷感,精神神経系症状及び運動器系症状の程度を指標とした.安全性の評価項目は,有害 事象及び副作用の発現率,臨床検査値異常変動の発現率,乳房検診,概括安全度とした.子宮内 膜への影響の評価項目は,性器出血,経腟エコーによる子宮内膜厚の測定,子宮内膜組織診(実 施可能な被験者のみ実施)及び子宮細胞診とした.主要評価項目は最終全般改善度とした.最終 全般改善度及び全般改善度は,臨床症状(Hot flush,発汗,腟萎縮症状及び睡眠障害)の程度の 推移に基づき判定した.投与期間は更年期障害に伴う症状を評価するためには8
週間が必要14-15) と考えられるが,子宮内膜に対する安全性も評価する上で最低必要な期間として,16週間と設定 した.症例数は1
群につき解析可能例数として65
例とし,10% の中止・脱落を考慮して各群75
例,計300
例と設定した.(2) 第II相臨床試験(一般臨床オープン試験)[Study 205] 5.3.5.2-1
第
II
相臨床試験(プラセボ対照二重盲検比較試験)[Study 203]19
年 月から20
年 月まで実施した.更年期障害又は卵巣欠落症患者を対象に,RPR106522 9cm
2製剤及び16 cm
2製剤を用いて,52週間連続投与時の有効性,安全性及び子宮内膜への影響を 無作為化非盲検法にて検討した.なお,最高用量の26 cm
2製剤は海外臨床試験の結果より安全性 を考慮し,検討から除外した.安全性の評価項目は有害事象及び臨床検査値異常変動の発現率,乳房検診,臨床検査値,概括 安全度とした.子宮内膜への影響の評価項目は,性器出血,経腟エコーによる子宮内膜厚の測定,
子宮内膜組織診及び子宮細胞診とした.子宮内膜組織診の判定は盲検下にて第三者的立場である
3
名の病理医が独立して実施し,判定に不一致が生じた場合は子宮内膜判定委員会を開催し,最 終的な判定を行うことで判定の客観性を確保した.また,有効性の評価項目としては,最終全般 改善度,全般改善度及び患者による評価とした.なお,最終全般改善度及び全般改善度は,臨床 症状(Hot flush,発汗,腟萎縮症状,睡眠障害,排尿障害,冷感,精神神経系症状及び運動器系 症状)の程度の推移に基づき判定した.主要評価項目は有害事象及び臨床検査値異常変動の発現 率とした.投与期間は,本剤及び類薬の海外での試験から,子宮内膜に対する安全性を評価でき る期間であることが確認されている12
箇月間(52週間)とした.症例数は1
群につき解析可能 例数として45
例とし,10% の中止・脱落を考慮して各群50
例,計100
例と設定した.(3) 第III相臨床試験(実薬対照非盲検比較試験)[Study 305] 5.3.5.1-2
第
II
相臨床試験2
試験[Study 203,Study 205]の結果より,NETA用量の異なる3
種のRPR106522
製剤(9 cm2製剤,16 cm2製剤,26 cm2製剤)の有効性,安全性及び子宮内膜への影 響はほぼ同様であることが確認された.これらの結果に基づいて,20 年 月から20
年 月まで,更年期障害又は卵巣欠落症患者を対象にNETA
用量が最少であるRPR106522 9 cm
2製剤 を用いて,子宮内膜増殖症発現率について既存のHRT〔E
2単独貼付剤(フェミエスト®4.33 mg)と MPA2.5 mg(
)〕を対照として無作為化並行群間比較法により検討した.また,有効性,安全性及び子宮内膜への影響についても検討した.
有効性の評価項目は,臨床症状効果判定とした.安全性の評価項目は,有害事象及び臨床検査 値異常変動の発現率,乳房検診とした.子宮内膜への影響の評価項目は,性器出血,経腟エコー による子宮内膜厚の測定,子宮内膜組織診(子宮内膜増殖症発現率を含む)及び子宮細胞診とし,
子宮内膜増殖症発現率を主要評価項目として設定した.子宮内膜増殖症はエストロゲン優位な内 分泌環境を一因として発生し,その一部が子宮内膜癌へ移行すると考えられていることから,子 宮内膜癌を予見できる最も有用な指標であるとされており,参考とした欧州医薬品委員会
(CPMP: Committee for Proprietary Medicinal Products)ガイドライン16)でも,子宮内膜増殖症発現 率を子宮内膜に対する安全性の主要評価項目として採用している.また,評価方法としては,
RPR106522
製剤群と実薬対照群の子宮内膜増殖症発現率の差が1% 以内であれば,両群間の子宮
内膜増殖症発現率に臨床上問題となる差がないと判定することとしたが,CPMPガイドラインで は,子宮内膜増殖症発現率の片側95%信頼区間の上限が 2% 以下であることを確認することが求
められていることから,この要件についても検討した.対照薬としてフェミエスト®を選択した理由は,RPR106522製剤との共通点が多い市販品であ ることによる.すなわち,E2含有製剤であること,投与経路が経皮であること及び投与方法が週
2
回の貼替えであることが挙げられる.また, はHRT
施行時に最も使用頻度が高いこ とから選択した.更に,対照薬の用法用量に関しては,フェミエスト®は推奨用量である4.33 mg
を, は持続的投与法において一般的に使用されている1
日1
回2.5 mg
を経口投与する こととした.投与期間については,CPMPガイドラインでは子宮内膜への影響を評価するために 最低12
箇月間投与することが必要とされていること及び海外で実施されたRPR106522
製剤を用 いた第II
相臨床試験[Study 201,Study 202]で子宮内膜増殖症の発現の評価が12
箇月投与で可 能であったことから,12箇月間(52週間)とした.非盲検で実施した理由は,第三者的立場である
3
名の病理医で構成された子宮内膜判定委員会 が盲検下にて組織標本の判定を行うことから,主要評価項目である子宮内膜増殖症発現率の判定 にバイアスが混入する可能性は低いと判断したためである.また,子宮内膜増殖症発現率につい ては,PPSを解析対象としたが,PPS以外で子宮内膜増殖症が発現した場合(未投与例と評価対 象除外例は除く)には,それらの症例も含めて投与群ごとに集計,解析及び比較を行うこととし,発現率が過小評価とならないように配慮した.
目標症例数として
RPR106522 9 cm
2製剤群333
例,実薬対照群167
例を設定した.CPMPガイ ドラインでは,子宮内膜増殖症発現率の片側95% 信頼区間の上限が 2% 以下であることを確認す
ることが求められ,このための必要症例数として1
年投与で300
例を推奨している.また,「致酢酸メドロキシ プロゲステロン 製剤A※
酢酸メドロキシ プロゲステロン 製剤A※
酢酸メドロキシ プロゲステロン 製剤A※
メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 38 -
命的でない疾患に対し長期間の投与が想定される新医薬品の治験段階において安全性を評価する ために必要な症例数と投与期間について」(薬審第
592
号,平成7
年5
月24
日)又は「外国臨 床データを受け入れる際に考慮すべき民族的要因について」(医薬審第672
号,平成10
年8
月11
日)では,「一般的に発生率1% の有害事象の検出には 300
症例の臨床試験が必要である」と 記されている.これらのことを勘案し,RPR106522 9 cm2製剤群の評価症例数を300
例として,10%の中止・脱落を考慮し,333
例を目標症例数として設定した.実薬対照群については子宮内 膜増殖症の発現が0
例であっても,子宮内膜増殖症発現率の95% 信頼区間の上限が 2% 以下であ
ることを担保できる症例数である150
例を評価症例数とし,10% の中止・脱落を考慮し,167例 を目標症例数として設定した.(4) 海外第II相臨床試験(米国におけるE2単独貼付剤対照二重盲検比較試験)[Study 202] 5.3.5.1-4(参考資料)
閉経後健康女性を対象として,米国にて
19
年 月から19
年 月まで実施された.RPR106522 9 cm
2製剤,16 cm2製剤及び26 cm
2製剤並びにE
2単独貼付剤を用いて52
週間連続投 与時の有効性及び安全性について検討した.主要評価項目は子宮内膜増殖症発現率とし,各
RPR106522
製剤とE
2単独貼付剤を比較するこ とにより,RPR106522製剤に配合されたNETA
の3
用量の効果を評価した.有効性の評価項目と しては,Hot flushの1
日平均回数,Hot flushの程度及び発汗の程度とした.安全性の評価項目と しては,有害事象及び臨床検査値異常変動の発現率,脂質プロファイルなどとし,併せて生活の 質(QOL)の評価も実施した.症例数は1
群につき解析可能例数として100
例とし,中止・脱落 例を考慮して各群150
例,計600
例と設定した.2.7.3.2 個々の試験結果の要約
2.7.3
項での解析対象は,「更年期障害に対する効果」についてはFAS
とし,「子宮内膜増殖 作用の抑制効果」については少なくとも1
回投与を受けたすべての被験者とした(Study 305の 子宮内膜増殖症発現率については,PPS + PPS以外で発現した子宮内膜増殖症発現数を対象とし た).個々の試験の有効性の結果を表 2.7.3-2に示した.なお,各国内臨床試験における「更年期障 害に対する効果」については下記の判定基準を用いて評価した.
臨床症状の程度は,下記の基準に従い分類した.
「高度」:日常生活に支障をきたす程度
「中等度」:気になるが日常生活にあまり支障をきたさない程度
「軽度」:あまり気にならない程度
「全くなし」:症状が全くない状態
臨床症状の程度の改善度は,評価時点における程度を投与前と比較し,下記の判定基準に従っ て判定した.
「著明改善」:「高度」又は「中等度」→「全くなし」
「中等度改善」:「高度」→「軽度」,「軽度」→「全くなし」
「軽度改善」:「高度」→「中等度」,「中等度」→「軽度」
「不変」:投与前に比べて変化なし
「悪化」:投与前に比べて
1
段階以上の悪化「投与前より症状なし」
全般改善度,最終全般改善度は下記の判定基準に従い判定した.
1.
著明改善:半数以上の症状が著明改善と判定され,悪化と判定された症状がないもの.あるいは著明改善が存在し,すべての症状が中等度改善以上の場合.
2.
中等度改善:半数以上の症状が中等度改善以上と判断され,悪化と判定された症状のな いもの.かつ著明改善の基準を満たさない場合.3.
軽度改善:少なくとも1
つ以上の軽度改善以上が存在し,その数が悪化と判定された数 より多い.又は中等度改善以上の数が悪化・不変の合計数以上の場合.4.
不変:1~3又は5
のいずれにも判定されない場合.5.
悪化:半数以上の症状が悪化と判定され軽度改善の基準にも満たない場合.6.
判定不能メノエイドコンビパッチ(
RPR1 065 22
)M2 .7
臨床概要- 40 -
表
2.7.3-2. Study 203
,Study 205
及びStudy 305
における有効性の結果一覧評価項目
更年期障害に対する効果 子宮内膜増殖作用の抑制効果 Hot flushの1日平均回数a)
(平均値 ± 標準偏差)
Hot flush程度 上段:高度率b) 下段:改善率c)
発汗程度 上段:高度率b) 下段:改善率c)
子宮内膜肥厚の 抑制作用
〔子宮内膜厚 (mm) 〕d) 試験区分
[試験番号]
試験 デザイン
投与
期間 投与群 組み入れ 症例数
完了 症例数
投与前 投与後 投与前 投与後 投与前 投与後 投与前 投与後
子宮内膜 増殖症の 発現率e) 14.5% 1.3% 14.5% 1.3%
9 cm2製剤群 87 57 3.48 ± 3.09 0.63 ± 1.84
82.8% 79.2% 2.43 ± 1.67 3.85 ± 2.13 0.0%
19.2% 1.4% 19.2% 2.8%
16 cm2製剤群 84 58 4.02 ± 4.31 0.58 ± 1.48
75.4% 81.1% 2.94 ± 1.89 3.86 ± 2.42 0.0%
17.2% 1.6% 20.3% 1.6%
26 cm2製剤群 76 43 4.22 ± 4.81 0.73 ± 2.66
78.0% 80.0% 2.85 ± 2.63 4.42 ± 3.43 0.0%
17.3% 6.8% 10.7% 6.8%
第II相 臨床試験
[Study 203]
無作為化 二重盲検 並行群間 プラセボ対照
16週間
プラセボ群 84 61 3.62 ± 3.32 1.54 ± 2.27
47.7% 54.1% 3.04 ± 2.27 2.72 ± 1.72 0.0%
14.3% 3.7% 21.4% 5.6%
9 cm2製剤群 57 36 3.09 ± 3.09 0.57 ± 1.28
72.9% 68.8% 3.71 ± 3.03 3.28 ± 2.08 0.0%
14.8% 1.6% 16.4% 1.6%
第II相 臨床試験
[Study 205]
無作為化 非盲検 非対照
52週間
16 cm2製剤群 62 35 3.24 ± 3.74 0.40 ± 0.94
76.9% 78.0% 3.03 ± 2.75 2.97 ± 2.36 0.0%
7.7% 0.0% 9.6% 0.0%
9 cm2製剤群 364 328 2.773 ± 2.637 0.242 ± 0.681
81.3% 76.1% 2.53 ± 1.25 3.04 ± 1.54 0.0%
11.4% 0.6% 11.4% 1.7%
第III相 臨床試験
[Study 305]
無作為化 非盲検 並行群間 実薬対照
52週間 実薬対照群
(フェミエスト® 4.33 mg及び
)
186 155 2.987 ± 3.992 0.316 ± 0.754
76.5% 73.5% 2.56 ± 1.16 3.18 ± 1.62 0.0%
9 cm2製剤群 163 123 2.67 ± 0.31 0.63 ± 0.19 4.10 ± 0.17 4.63 ± 0.30 0.81%
16 cm2製剤群 149 99 2.77 ± 0.35 0.25 ± 0.09 4.42 ± 0.25 4.84 ± 0.23 0.00%
26 cm2製剤群 158 93 2.53 ± 0.33 0.17 ± 0.05 4.05 ± 0.19 4.79 ± 0.23 1.12%
海外第II相 臨床試験
[Study 202]
(米国)
無作為化 二重盲検 並行群間 実薬対照
52週間
E2 単独貼付剤群
(Menorest® 50) 155 76 2.25 ± 0.26 0.26 ± 0.09 4.14 ± 0.18 8.65 ± 0.50 37.25%
a) [Study 202]はITTでの結果であり,平均値±標準誤差 b) Hot flush又は発汗の程度において「高度」を示した症例の割合
c) 投与終了後又は中止時の「中等度改善」以上の改善度を示した症例の割合 d) [Study 203,Study 205,Study 305]では安全性の解析対象集団であり,平均値
±標準偏差,[Study 202]ではITT (at 1 year population)での結果であり,平均値±標準誤差
e) [Study 203,Study 205]では安全性の解析対象集団,[Study 305]ではPPS+PPS以外で発現した症例,[Study 202]ではITT (at 1 year population) での結果
※新薬承認情報提供時に置き換えた
酢酸メドロキシプロゲ ステロン製剤A※
(1) 第II相臨床試験(プラセボ対照二重盲検比較試験)[Study 203] 5.3.5.1-1 更年期障害又は卵巣欠落症患者
331
例を対象として,RPR106522製剤の16
週間連続投与によ る有効性及び安全性をプラセボ群を対照とした二重盲検並行群間比較法により検討した.RPR106522
製剤は9 cm
2製剤,16 cm2製剤及び26 cm
2製剤を用いた.1) 更年期障害に対する効果
最終全般改善度における「中等度改善」以上の症例の割合(改善率)はプラセボ群
54.1%
(40/74例)に対し,9 cm2製剤群
81.6%(62/76
例),16 cm2製剤群79.7%(59/74
例)及び26 cm
2製剤群75.4%(49/65
例)であった.投与16
週時又は中止時の改善率について,プラセボ群 と3
種類のRPR106522
製剤群のそれぞれとの2
群比較を行った結果,RPR106522製剤に含有さ れるNETA
の用量に依存せず,いずれもRPR106522
製剤群が有意に高かった(χ2検定:各々p <0.001,p = 0.001,p = 0.012).投与後 8
週時の全般改善度も同様な結果を示した.また,患者に よる評価では,投与後8
週時,16週時又は中止時の「良くなった」以上の症例の割合(好印象 率)は,いずれの評価時点においてもプラセボ群に比べRPR106522
製剤群で高かった.また,好印象率は投与期間が長くなるに従って増加する傾向が示された.
Hot flush
の1
日平均回数は,すべての投与群で投与前と比べて減少した.また,投与後4
週時,8
週時,12週時及び16
週時又は中止時において,RPR106522製剤群はいずれもプラセボ群に比 べ明らかな減少が見られ,投与後16
週時又は中止時では,プラセボ群が平均1.54
回であったの に対し,9 cm2製剤群,16 cm2製剤群及び26 cm
2製剤群ではそれぞれ0.63
回,0.58回,0.73回で あった.投与後16
週時又は中止時の各群のHot flush
の1
日平均回数について,プラセボ群を対 照に多重比較を行った結果,9 cm2製剤群,16 cm2製剤群でプラセボ群との間に有意差が認めら れた(Dunnett型多重比較:各々p = 0.021,p = 0.015,p = 0.067).Hot flushの1
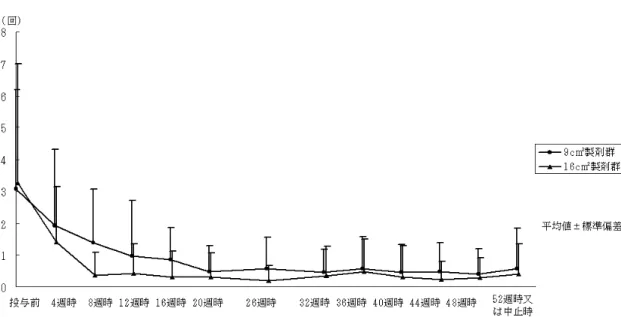
日平均回数の経 時的変化を図 2.7.3-1に示した.図 2.7.3-1. Study 203における
Hot flush
の1
日平均回数の経時的推移メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 42 -
Hot flush
の程度は,すべての投与群で投与前と比べて軽減した.また,いずれのRPR106522
製剤群も各評価時点でプラセボ群より程度は軽減していた.投与後16
週時又は中止時のHot flush
の程度について,プラセボ群と3
種類のRPR106522
製剤群のそれぞれとの2
群比較を行っ た結果,ではプラセボ群に比べ9 cm
2製剤群,16 cm2製剤群及び26 cm
2製剤群で有意差が見られ た(Wilcoxonの順位和検定:各々p < 0.001,p = 0.001,p = 0.001).投与後16
週時又は中止時に「高度」を示した症例の割合(高度率)はプラセボ群で
6.8%(5/74
例)であったが,9 cm2製剤 群,16 cm2製剤群及び26 cm
2製剤群ではそれぞれ1.3%(1/76
例),1.4%(1/72例),1.6%(1/64例)であった.また,Hot flushの程度の改善度においても,投与後
16
週時又は中止時の 改善率はプラセボ群47.7%(31/65
例)に比べ,9 cm2製剤群82.8%(53/64
例),16 cm2製剤群75.4%(46/61
例)及び26 cm
2製剤群78.0%(46/59
例)であり,投与後16
週時又は中止時の改善 率について,プラセボ群と3
種類のRPR106522
製剤群のそれぞれとの2
群比較を行った結果,いずれの
RPR106522
製剤群においても有意に高かった(χ2検定:各々p < 0.001,p = 0.002,p =0.001).
発汗の程度は,すべての投与群で投与前に比べて軽減した.また,いずれの
RPR106522
製剤 群も各評価時点でプラセボ群より程度は軽減していた.投与後16
週時又は中止時の発汗の程度 について,プラセボ群と3
種類のRPR106522
製剤群のそれぞれとの間で2
群比較を行った結果,プラセボ群に比べ
9 cm
2製剤群,16 cm2製剤群,26 cm2製剤群で有意差が見られた(Wilcoxonの 順位和検定:各々p = 0.006,p = 0.004,p = 0.003).投与後16
週時又は中止時の高度率はプラセ ボ群で6.8%(5/74
例)であったが,9 cm2製剤群,16 cm2製剤群及び26 cm
2製剤群ではそれぞれ1.3%(1/76
例),2.8%(2/72例)及び1.6%(1/64
例)であった.また,発汗の程度の改善度に おいても,投与後16
週時又は中止時の改善率はプラセボ群54.1%(33/61
例)に比べ,9 cm2製剤 群79.2%(42/53
例),16 cm2製剤群81.1%(43/53
例)及び26 cm
2製剤群80.0%(36/45
例)であ り,投与後16
週時又は中止時の改善率について,プラセボ群と3
種類のRPR106522
製剤群のそ れぞれとの2
群比較を行った結果,いずれのRPR106522
製剤群においても有意に高かった(χ2検 定:各々p = 0.007,p = 0.003,p = 0.008).腟萎縮症状の程度の改善度において,投与後
16
週時又は中止時に改善率はプラセボ群60.0%
(15/25例)に比べ,9 cm2製剤群
74.2%(23/31
例),16 cm2製剤群82.8%(24/29
例)及び26 cm
2製剤群84.6%(22/26
例)と,いずれのRPR106522
製剤群においても高かったが,投与後16
週時又は中止時の改善率について,プラセボ群と3
種類のRPR106522
製剤群のそれぞれとの2
群比較を行った結果,プラセボ群との間に有意な改善は認められなかった(χ2検定:各々p =0.266, p = 0.076, p = 0.060).
睡眠障害の程度の改善度において,投与後
16
週時又は中止時の改善率はプラセボ群47.7%
(21/44例)に比べ,9 cm2製剤群
59.0%(23/39
例),16 cm2製剤群70.0%(21/30
例)及び26
cm
2製剤群58.8%(20/34
例)と,いずれのRPR106522
製剤群においても高かったが,投与後16
週時又は中止時の改善率について,プラセボ群と3
種類のRPR106522
製剤群のそれぞれとの2
群比較を行った結果,プラセボ群との間に有意な改善は認められなかった(χ2検定:各々p =0.324, p = 0.074, p = 0.345).
そのほか,排尿障害,冷感,精神神経系症状及び運動器系症状の程度の改善度において,投与 後
16
週時又は中止時の改善率は,プラセボ群との間にいずれの症状も有意な改善は認められな かった.2) 子宮内膜増殖作用の抑制効果
経腟エコーにより測定した子宮内膜厚は,投与後
16
週時又は中止時ではRPR106522
製剤群で 投与前に比べ平均0.92~1.57 mm
増加し,肥厚傾向が見られたが,プラセボ群ではその傾向は見 られなかった.また,投与期間が16
日と短く治験薬との因果関係が否定された子宮内膜癌の1
例を除き,プラセボ群を含むすべての投与群で,子宮内膜増殖症及び子宮内膜癌は観察されなか った.(2) 第II相臨床試験(一般臨床オープン試験)[Study 205] 5.3.5.2-1
更年期障害又は卵巣欠落症患者
119
例を対象として,RPR106522製剤の52
週間連続投与時の 子宮内膜に対する影響を検討し,併せて有効性及び安全性も検討した.RPR106522製剤は9 cm
2 製剤及び16 cm
2製剤を用いて実施した.1) 更年期障害に対する効果
最終全般改善度における改善率は,9 cm2製剤群では
48.1%(26/54
例),16 cm2製剤群では54.1%(33/61
例)と両群共に高い改善率を示し,群間差は認められなかった(χ2検定:p =0.524).また,投与後 26
週時及び52
週時又は中止時における全般改善度の改善率は最終全般改 善度と同程度であり,長期間投与によっても作用の減弱を生じないことが示唆された.Hot flush
の1
日平均回数は両群共に投与後4
週時より平均発現回数に減少が見られ,投与後12
週時以降,52週時又は中止時まで各評価時点において1
日1
回未満であった(図 2.7.3-2).9cm
2製剤群の投与初期の1
日平均回数は16 cm
2製剤群よりも多かったが,これは,投与前の血清 中E
2濃度が,16 cm2製剤群(29.616 pg/mL)よりも9 cm
2製剤群(42.645 pg/mL)で高かったこ とが影響していると考えられる.しかし,投与後52
週時又は中止時では9 cm
2製剤群と16 cm
2 製剤群のHot flush
の1
日平均回数に群間差は見られなかった(Wilcoxonの順位和検定:p =0.534).Hot flush
の1
日平均回数の経時的変化を図 2.7.3-2に示した.また
Hot flush
及び発汗などの各臨床症状は,排尿障害を除いて,両群共に投与前と比較して顕 著な改善が見られた.メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 44 -
図 2.7.3-2. Study 205における
Hot flush
の1
日平均回数の経時的推移2) 子宮内膜増殖作用の抑制効果
経腟エコーにより測定した子宮内膜厚は,9 cm2製剤群では投与前に
3.71 mm
であったが,投 与後26
週時に3.91 mm
とやや増加し,投与後52
週時又は中止時では3.28 mm
と投与前と比較し て減少した.16 cm2製剤群では,投与前に3.03 mm
であったが,投与後26
週時に2.95 mm
とや や減少し,投与後52
週時又は中止時では2.97 mm
と投与後26
週時と比較して変動が見られなか った.また,子宮内膜増殖症及び子宮内膜癌は両群共に観察されなかった.(3) 第III相臨床試験(実薬対照非盲検比較試験)[Study 305] 5.3.5.1-2
更年期障害又は卵巣欠落症患者
550
例を対象に,RPR106522 9 cm2製剤の52
週間連続投与によ る安全性(NETA配合意義)を既存のHRT
を対照として検討した.主要評価項目として子宮内 膜増殖症発現率を設定し,Hot flush,発汗などの更年期障害及び卵巣欠落症に伴う諸症状に対す る有効性も併せて検討した.1) 更年期障害に対する効果
Hot flush
の1
日平均回数は,投与前では9 cm
2製剤群2.773
回及び実薬対照群2.987
回であった が,投与後12
週時にはそれぞれ0.480
回及び0.498
回と大きく減少し,投与後24
週時以降52
週 時又は中止時まで両群共に0.3
回前後で推移し効果の持続が認められた.なお,投与後24
週時及 び52
週時又は中止時におけるHot flush
の1
日平均回数について群間比較を行ったところ,いず れも両群間に有意な差は認められなかった(Wilcoxonの順位和検定:各々p = 0.108,p = 0.144).Hot flush
の1
日平均回数の経時的変化を図 2.7.3-3に示した.図 2.7.3-3. Study 305における
Hot flush
の1
日平均回数の経時的推移Hot flush
の程度の高度率は,9 cm2製剤群及び実薬対照群は投与前にそれぞれ7.7%(28/364
例)及び11.4%(21/184
例)であったが,投与後52
週時又は中止時ではそれぞれ0.0%(0/362
例)及び0.6%(1/181
例)であった.また,各群の投与前と投与後24
週時及び52
週時又は中止 時の前後比較を行った結果,両群共に投与後24
週時及び52
週時又は中止時において有意な差が 認められた(1標本Wilcoxon
検定:すべてp < 0.001).Hot flush
の程度の改善率は,投与後52
週時又は中止時に,9 cm2製剤群で81.3%(278/342
例),実薬対照群で76.5%(130/170
例)であ り,投与後52
週時又は中止時のHot flush
の程度の改善率について群間比較を行った結果,両群 間に有意な差は認められなかった(χ2検定:p = 0.202).発汗の程度の高度率は,9 cm2製剤群では投与前
9.6%(35/364
例)が投与後52
週時又は中止時 で0.0%(0/362
例),同様に実薬対照群では11.4%(21/184
例)が1.7%(3/181
例)に低下した.また,各群の投与前と投与後
24
週時及び52
週時又は中止時の高度率について前後比較を行った 結果,投与前との群内比較では両群共に投与後24
週時及び52
週時又は中止時において有意な差 が認められた(1標本Wilcoxon
検定:すべてp < 0.001).発汗の程度の改善率は,投与後 52
週 時又は中止時に,9 cm2製剤群で76.1%(248/326
例),実薬対照群で73.5%(114/155
例)であり,投与後
52
週時又は中止時のHot flush
の程度の改善率について群間比較を行った結果,両群間に 有意な差は認められなかった(χ2検定:p = 0.548).更年期障害に伴うその他の臨床諸症状(腟 萎縮症状,睡眠障害,排尿障害,冷感,精神神経系症状及び運動器系症状)の程度の改善率につ いて,投与後52
週時又は中止時における群間比較を行った結果両群間で有意な差が認められた ものはなかった(χ2検定:各々p = 0.994, p = 0.771, p = 0.525, p = 0.461, p = 0.640, p = 0.764).メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 46 -
2) 子宮内膜増殖作用の抑制効果
子宮内膜増殖症発現率を表 2.7.3-3に示した.9 cm2製剤群及び実薬対照群共に,子宮内膜増殖 症及び子宮内膜癌を発現した症例は認められず,子宮内膜増殖症の発現率は
0.0% であり,両群
間の発現率に差は見られなかった.表
2.7.3-3. Study 305
における子宮内膜増殖症発現率投与群 評価対象例数a) 発現例数 発現率b) (%) 片側95%
信頼区間上限 発現率の差 (%)
9 cm2製剤群 331 0 0.0 0.81
実薬対照群 153 0 0.0 1.74
0.0
a) 評価対象症例:PPS + PPS
以外の子宮内膜増殖症発現数.ただし,未投与例と対象外例は除く.b) 発現率={[子宮内膜増殖症発現数(未投与例と対象外例を除く)]
/[PPS + PPS以外の子宮内膜増殖症発現数(未投与例と対象外例を除く)]}×100
CPMP
ガイドラインによれば,子宮内膜の安全性評価においては,投与期間が1
年,症例数が 約300
例の条件下で,子宮内膜増殖症発現率の片側95% 信頼区間の上限が 2% 以下であることと
規定されている.この規定は市販のHRT
を受けた女性の子宮内膜増殖症発現率が1~2% と推測
されていることに由来する.本試験におけるRPR106522
製剤群の子宮内膜増殖症発現率の片側95% 信頼区間上限は 0.81% であり,この要件を十分満たしていた.これらのことから,
RPR106522 9 cm
2製剤は,市販のHRT
と同様に,子宮内膜増殖症の発現を十分に抑制していると 考えられた.経腟エコーにより測定した子宮内膜厚は,9 cm2製剤群では投与前に
2.53 mm
であったが,投 与後24
週時に3.18 mm
とやや増加し,投与後52
週時又は中止時には3.04 mm
と投与後24
週時 と比較してほとんど変動がなかった.実薬対照群では,投与前に2.56 mm
であったが,投与後24
週時に3.42 mm
とやや増加し,投与後52
週時又は中止時には3.18 mm
と投与後24
週時と比較 してやや減少した.いずれも臨床上問題となる変化ではなく,両群間における子宮内膜厚の変化 量にほとんど違いは認められなかった.(4) 海外第II相臨床試験(米国におけるE2単独貼付剤対照二重盲検比較試験)[Study 202] 5.3.5.1-4(参考資料)
閉経後健康女性
625
例を対象として,RPR106522製剤の52
週間連続投与による有効性及び安 全性をE
2単独投与群を対照とした二重盲検並行群間比較法により検討した.RPR106522製剤は9 cm
2製剤,16 cm2製剤及び26 cm
2製剤を用いた.1) 更年期障害に対する効果
ITT
を解析対象集団としたHot flush
の1
日平均回数は,投与前ではE
2単独投与群が平均2.25
回,RPR106522製剤群(9 cm2製剤群,16 cm2製剤群及び26 cm
2製剤群)ではそれぞれ2.67
回,2.77
回,2.53回であったが,投与後52
週時又は中止時ではE
2単独投与群が平均0.26
回,RPR106522
製剤群(9 cm2製剤群,16 cm2製剤群及び26 cm
2製剤群)ではそれぞれ0.63
回,0.25 回,0.17回であり,いずれも投与前に比べ減少していた.Hot flush
の程度については,「全くなし」を0,「軽度」を 1,「中等度」を 2,「高度」を 3
と分類した.投与前ではE
2単独投与群の平均が0.59,RPR106522
製剤群(9 cm2製剤群,16 cm2 製剤群及び26 cm
2製剤群)ではそれぞれ0.64,0.62,0.64
であった.投与後52
週時又は中止時 にはE
2単独投与群が平均0.10,RPR106522
製剤群(9 cm2製剤群,16 cm2製剤群及び26 cm
2製剤 群)ではそれぞれ0.16,0.08,0.07
であり,各投与群共に程度が軽減していた.発汗の程度についても,同様に分類した.投与前では
E
2単独投与群の平均が0.48,
RPR106522
製剤群(9 cm2製剤群,16 cm2製剤群及び26 cm
2製剤群)ではそれぞれ0.59,0.57
及 び0.64
であった.投与後52
週時又は中止時にはE
2単独投与群が平均0.07,RPR106522
製剤群(9 cm2製剤群,16 cm2製剤群及び
26 cm
2製剤群)ではそれぞれ0.15,0.07
及び0.09
であり,各 投与群共に程度が軽減していた.2) 子宮内膜増殖作用の抑制効果
子宮内膜増殖症の発現率については,1年投与例(ただし
1
年未満で子宮内膜増殖症と診断さ れた症例を含む)を解析対象とした.子宮内膜増殖症の発現率はE
2単独投与群で37.25%
(38/102例)であったのに対し,9 cm2製剤群で
0.81%(1/123
例),16 cm2製剤群で0.00%
(0/97例)及び
26 cm
2製剤群で1.12%(1/89
例)であり,いずれのRPR106522
製剤群もE
2単独 投与群と比較し有意に抑制した(Fisherの直接確率計算法:各々p < 0.001).経腟エコーにより 測定した子宮内膜厚については,投与前はE
2単独投与群及びRPR106522
製剤群共に平均4 mm
前後であった.投与後52
週時又は中止時の時点においては,E2単独投与群では子宮内膜厚が平 均8.65 mm
と著しく肥厚したが,RPR106522製剤群(9 cm2製剤群,16 cm2製剤群及び26 cm
2製 剤群)ではそれぞれ4.63 mm,4.84 mm,4.79 mm
であり,大きな変化は認められなかった.メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 48 -
2.7.3.3 全試験を通しての結果の比較と解析
RPR106522
製剤は,有効成分としてE
2とNETA
を含有する配合剤である.配合された有効成 分のうち,E2に由来する「更年期障害に対する効果」については,Hot flushの1
日平均回数並び にHot flush,発汗,腟萎縮症状,睡眠障害,排尿障害,冷感,精神神経系症状及び運動器系症状
の程度を指標とした比較及び解析を行った.また,NETAの配合理由である「子宮内膜増殖作用 の抑制効果」については,子宮内膜増殖症の抑制作用及び子宮内膜肥厚の抑制作用を指標とした 比較及び解析を行った.2.7.3.3.1
試験対象集団有効性を評価した国内
3
試験の試験対象集団一覧及び患者背景をそれぞれ表 2.7.3-4及び表2.7.3-5
に示した.3試験間において選択基準などで細かな違いはあるものの,ほぼ同一の対象集 団であると考えられた.試験間の人口統計学的特性のうち,[Study 205]の血清中E
2濃度が他 の試験と比べてやや高値(9 cm2製剤群 42.645 pg/mL,16 cm2製剤群29.616 pg/mL)であったが,
これは選択基準の違いにより,[Study 205]のみ閉経前の患者が含まれていたためと考えられる。
その他の項目では顕著な差は見られなかった.
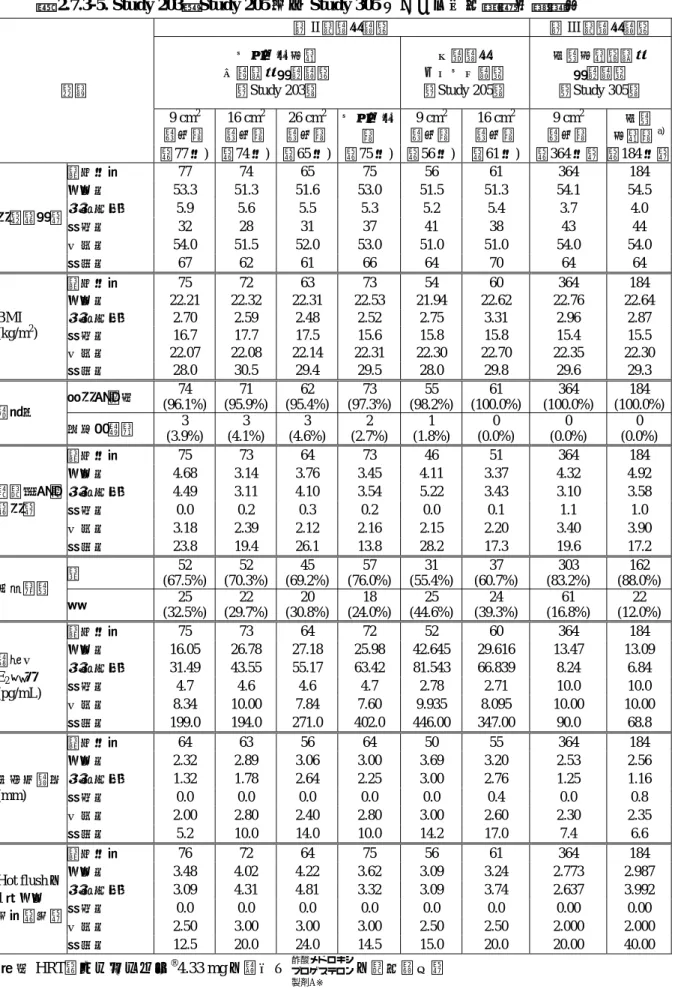
表 2.7.3-4. 試験対象集団一覧
第II相臨床試験 第III相臨床試験 試験名
項目
プラセボ対照 二重盲検比較試験
[Study 203]
一般臨床 オープン試験
[Study 205]
実薬対照 非盲検比較試験
[Study 305]
対象 更年期障害又は卵巣欠落症患者
年齢 20歳以上65歳未満
子宮の有無 子宮を有する患者
更 年 期 障 害 又 は 卵 巣 欠 落 症 に 伴 う 症 状
a) Hot flush b) 発汗
c) 腟萎縮症状(性交障害,乾燥感,腟炎など)
のうち少なくとも1つの症状を有する患者
a) Hot flush(平均 1日 1 回以上)
b) 発汗 c) 腟萎縮症状
のうち少なくとも 1つの 症状を有する患者
前治療薬など
投与開始の 4 週間以内に ホ ル モ ン 薬 〔 た だ し , Hot flush が強い場合(5 回/日以上)2週間でも可 とする],1 週間以内に 自律神経調整薬,向精神 薬,睡眠薬及び更年期障 害の適応を有する漢方薬 を服薬していない患者
投与開始の 2 週間以内に ホルモン剤(ただし,デ ポ ー 剤 は 4 週 間 と す る),1 週間以内に自律 神経調整薬,向精神薬,
睡眠薬及び更年期障害の 適応を有する漢方薬を服 薬していない患者
投与前検査(子宮内膜組 織診,子宮細胞診,経腟 エコー,血清中ホルモン 濃度)の2週間(14日)
以内に性ステロイドホル モン剤を服薬していない 患者〔ただし,デポー剤 は 4 週間(28日)とす る〕
閉経後期間
最終月経後 6 箇月以上,
又は両側卵巣摘出後 1 箇 月以上経過した患者
[規定なし]
自然閉経患者(最終月経 後 1年以上経過している 患者)又は両側卵巣摘出 患者(摘出後 1箇月以上 経過している患者)
血清中FSH濃度 [規定なし] FSH 30 mIU/mL以上の患 者
選択基準
子宮内膜厚 [規定なし]
経腟エコーにて子宮内膜 の厚さが 8 mm 未満であ ることが確認できた患者
メノエイドコンビパッチ(
RPR106522
)M2.7
臨床概要- 50 -
表 2.7.3-5. Study 203,Study 205及び
Study 305
における人口統計学的特性第II相臨床試験 第III相臨床試験 プラセボ対照
二重盲検比較試験
[Study 203]
一般臨床 オープン試験
[Study 205]
実薬対照非盲検 比較試験
[Study 305]
9 cm2 製剤群
16 cm2 製剤群
26 cm2 製剤群
プラセボ 群
9 cm2 製剤群
16 cm2 製剤群
9 cm2 製剤群
実薬 対照群a) 項目
(77例) (74例) (65例) (75例) (56例) (61例) (364例) (184例)
算出例数 77 74 65 75 56 61 364 184
平均値 53.3 51.3 51.6 53.0 51.5 51.3 54.1 54.5
標準偏差 5.9 5.6 5.5 5.3 5.2 5.4 3.7 4.0
最小値 32 28 31 37 41 38 43 44
中央値 54.0 51.5 52.0 53.0 51.0 51.0 54.0 54.0
年齢(歳)
最大値 67 62 61 66 64 70 64 64
算出例数 75 72 63 73 54 60 364 184 平均値 22.21 22.32 22.31 22.53 21.94 22.62 22.76 22.64 標準偏差 2.70 2.59 2.48 2.52 2.75 3.31 2.96 2.87
最小値 16.7 17.7 17.5 15.6 15.8 15.8 15.4 15.5
中央値 22.07 22.08 22.14 22.31 22.30 22.70 22.35 22.30 BMI
(kg/m2)
最大値 28.0 30.5 29.4 29.5 28.0 29.8 29.6 29.3
更年期障害 74 (96.1%)
71 (95.9%)
62 (95.4%)
73 (97.3%)
55 (98.2%)
61 (100.0%)
364 (100.0%)
184 (100.0%) 診断名
卵巣欠落症 3 (3.9%)
3 (4.1%)
3 (4.6%)
2 (2.7%)
1 (1.8%)
0 (0.0%)
0 (0.0%)
0 (0.0%) 算出例数 75 73 64 73 46 51 364 184 平均値 4.68 3.14 3.76 3.45 4.11 3.37 4.32 4.92 標準偏差 4.49 3.11 4.10 3.54 5.22 3.43 3.10 3.58
最小値 0.0 0.2 0.3 0.2 0.0 0.1 1.1 1.0
中央値 3.18 2.39 2.12 2.16 2.15 2.20 3.40 3.90 閉経後期間
(年)
最大値 23.8 19.4 26.1 13.8 28.2 17.3 19.6 17.2
無 52
(67.5%) 52 (70.3%)
45 (69.2%)
57 (76.0%)
31 (55.4%)
37 (60.7%)
303 (83.2%)
162 (88.0%) 前治療薬
有 25
(32.5%) 22 (29.7%)
20 (30.8%)
18 (24.0%)
25 (44.6%)
24 (39.3%)
61 (16.8%)
22 (12.0%)
算出例数 75 73 64 72 52 60 364 184
平均値 16.05 26.78 27.18 25.98 42.645 29.616 13.47 13.09 標準偏差 31.49 43.55 55.17 63.42 81.543 66.839 8.24 6.84
最小値 4.7 4.6 4.6 4.7 2.78 2.71 10.0 10.0
中央値 8.34 10.00 7.84 7.60 9.935 8.095 10.00 10.00
血清中 E2濃度 (pg/mL)
最大値 199.0 194.0 271.0 402.0 446.00 347.00 90.0 68.8
算出例数 64 63 56 64 50 55 364 184
平均値 2.32 2.89 3.06 3.00 3.69 3.20 2.53 2.56
標準偏差 1.32 1.78 2.64 2.25 3.00 2.76 1.25 1.16
最小値 0.0 0.0 0.0 0.0 0.0 0.4 0.0 0.8
中央値 2.00 2.80 2.40 2.80 3.00 2.60 2.30 2.35
子宮内膜厚 (mm)
最大値 5.2 10.0 14.0 10.0 14.2 17.0 7.4 6.6
算出例数 76 72 64 75 56 61 364 184
平均値 3.48 4.02 4.22 3.62 3.09 3.24 2.773 2.987
標準偏差 3.09 4.31 4.81 3.32 3.09 3.74 2.637 3.992
最小値 0.0 0.0 0.0 0.0 0.0 0.0 0.00 0.00
中央値 2.50 3.00 3.00 3.00 2.50 2.50 2.000 2.000
Hot flushの 1日平均 回数(回)
最大値 12.5 20.0 24.0 14.5 15.0 20.0 20.00 40.00
a)
既存HRT
(フェミエスト®4.33 mg
の貼付と の経口投与)※新薬承認情報提供時に置き換えた
酢酸メドロキシ プロゲステロン 製剤A※


![表 2.7.3-4. 試験対象集団一覧 第 II 相臨床試験 第 III 相臨床試験 試験名 項目 プラセボ対照 二重盲検比較試験 [ Study 203 ] 一般臨床 オープン試験 [Study 205] 実薬対照 非盲検比較試験 [Study 305] 対象 更年期障害又は卵巣欠落症患者 年齢 20 歳以上 65 歳未満 子宮の有無 子宮を有する患者 更 年 期 障 害 又 は 卵 巣 欠 落 症 に 伴 う 症 状 a) Hot flush b) 発汗 c) 腟萎縮症状(性交障害,乾燥感,腟](https://thumb-ap.123doks.com/thumbv2/123deta/7594369.2535668/18.892.106.790.114.732/試験対象集団一覧相臨床試プラセボ一般臨床オープン歳以上有する.webp)
![表 2.7.3-5. Study 203,Study 205 及び Study 305 における人口統計学的特性(続き) 第 II 相臨床試験 第 III 相臨床試験 プラセボ対照 二重盲検比較試験 [Study 203] 一般臨床 オープン試験 [Study 205] 実薬対照非盲検 比較試験 [Study 305] 9 cm 2 製剤群 16 cm 2 製剤群 26 cm 2 製剤群 プラセボ群 9 cm 2 製剤群 16 cm 2 製剤群 9 cm 2 製剤群](https://thumb-ap.123doks.com/thumbv2/123deta/7594369.2535668/20.892.109.792.125.1158/における統計学プラセボ二重盲一般臨床オープン製剤群プラセボ.webp)
![表 2.7.3-6. Study 203,Study 205 及び Study 305 における Hot flush の 1 日平均回数 第 II 相臨床試験 第 III 相臨床試験 プラセボ対照 二重盲検比較試験 [Study 203] 一般臨床 オープン試験 [Study 205] 実薬対照非盲検 比較試験[Study 305] 評価 時期 9 cm 2 製剤群 16 cm 2 製剤群 26 cm 2 製剤群 プラセボ群 9 cm 2 製剤群 16 cm 2 製剤群 9 cm 2](https://thumb-ap.123doks.com/thumbv2/123deta/7594369.2535668/22.892.109.786.121.776/StudyStudy及びにおける日平均プラセボ二重盲一般臨床オープンプラセボ.webp)
![表 2.7.3-7. Study 203,Study 205 及び Study 305 における Hot flush の程度の改善率 第 II 相臨床試験 第 III 相臨床試験 プラセボ対照 二重盲検比較試験 [ Study 203 ] 一般臨床 オープン試験 [Study 205 ] 実薬対照非盲検比較試験 [Study 305] 評価時期 9 cm 2 製剤群 16 cm 2 製剤群 26 cm 2 製剤群 プラセボ群 9 cm 2 製剤群 16 cm 2 製剤群 9](https://thumb-ap.123doks.com/thumbv2/123deta/7594369.2535668/24.892.111.790.123.581/におけるプラセボオープン製剤群製剤群製剤群プラセボ製剤群.webp)

