マイクロフロー系による医薬中間体の合成に関する
研究
著者
杉本 篤史
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(理学), 学位記番号:
論理第79号, 学位授与年月日: 2008-07-31, 指導教
員: 柳 日馨.
マイクロフロー系による医薬中間体の合成に関する研究
Studies on Synthesis of Key Intermediate of
Drugs by Microflow Systems
杉本 篤史
Atsushi Sugimoto
大阪府立大学大学院 理学系研究科
Graduate School of Science
Osaka Prefecture University
2009
- 1 -
目次
総論 ... 3 引用文献 ... 11 本論 ... 13 第1 章 マイクロフロー系による光バートン反応 ... 13 第1 節 水銀ランプを用いるマイクロフロー系による光バートン反応 ... 13 第2 節 低出力光源を用いるマイクロフロー系による光バートン反応 ... 26 第3 節 連続マイクロフロー型光バートン反応 ... 34 第4 節 結論 ... 38 実験の部 ... 39 引用文献 ... 43 第2 章 条件検索型マイクロリアクターシステムによる薗頭反応... 45 第1 節 条件検索型マイクロリアクターシステムによる反応条件検索 ... 45 第2 節 連続マイクロフロー型薗頭反応 ... 59- 2 -
第3 節 結論 ... 61 実験の部 ... 62 引用文献 ... 66 結論 ... 68 論文リスト ... 69 謝辞 ... 70- 3 -
総論
近年の医薬品産業においては、医薬品の創出から商用製造までの流れをいかに スピードアップして、新薬をいち早く世の中に提供するかが重要なカギとなって いる。医薬品開発において、合成すべき化合物数とそのスケールのモデル図を以 下に示す(Figure 1)。 Figure 1 医薬品開発において合成すべき化合物数とそのスケール 医薬品開発において、最も上流域にある「化合物探索」段階においては、HTS (High Throughput Screening)や、コンビケム(コンビナトリアル・ケミストリー) の技術が発達しており、開発候補化合物が大幅に増加している。しかし、その合 成法はスケールアップに向かないものが多く、後段で新たに製造法開発を一から 実施しなくてはならないことが少なくない。つまり、商用生産段階に早く到達で きるように、開発スピードを落とさないためには、「前臨床試験」段階や「臨床試 験」段階といった、mg ~ kg スケールの反応条件検索から製造の部分の省力化・ 短期化が非常に重要となる。 この段階での検討手段として、近年、活発に研究を進めているマイクロリアク ター1)を効率的に活用できないかと考え、反応条件のスクリーニングから、見出- 4 -
された最適条件での中~大量合成を一貫して行えるシステムを開発すべく研究を 行なった(Figure 2)。 Figure 2 本研究の位置づけ マイクロリアクターとは「微細加工技術を用いて製作された、幅、深さ、内径 などが数μm~数百μm の空間内で化学反応、物質生産を行なう装置」という定 義が一般的である。マイクロリアクターの流路は非常に微小であるため、容器内 の流体体積に対する、容器の表面積が、反応を行なうフラスコよりも格段に大き くなる。例えば、1 mL の反応液を 1 辺 1 cm の立方体のチューブ型容器に入れた 場合、その表(壁)面積は、4 cm2であるのに対し、断面が100 μm 四方、長さ 100 m のマイクロ空間に入れた場合、その表(壁)面積は 400 cm2に達する。 以下に挙げる、マイクロリアクターのメリットは全てこの特長に起因する。 ①温度制御が容易に効率よく行える 外部との熱交換の効率がフラスコに比べて格段に向上し、内部流体の温度制御 並列化 運転時間 延長 商用生産 Stage 化合物探索 前臨床 臨床 Scale μg mg g kg ton フラスコ パイロットプラント 製造プラント ロボット コンビケム HTS S SO2 H N N H CH3 CO2H HO HC O O O NOHThis Study
光反応 触媒反応 並列化 運転時間 延長 商用生産 Stage 化合物探索 前臨床 臨床 Scale μg mg g kg ton フラスコ パイロットプラント 製造プラント ロボット コンビケム HTS S SO2 H N N H CH3 CO2H HO HC O O O NOHThis Study
光反応 触媒反応- 5 -
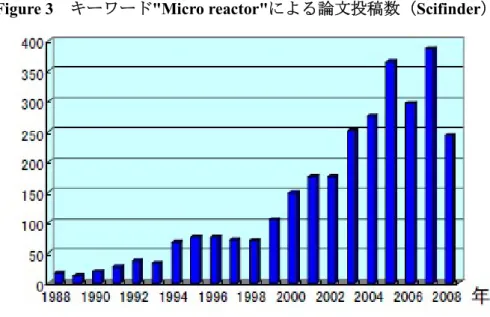
が容易となり、精密な温度制御や急激な加熱・冷却を必要とする反応も行なうこ とができる。また、フラスコで見られるような、試薬の滴下部分周辺の温度や試 薬濃度が局所的に上がるような現象が起こらないため、結果的に副生成物生成の 抑制も容易となる。 ②効率的な混合が行える マイクロリアクターでは、流路径が小さいため、反応液は、レイノルズ数が小 さい「層流」で流れる。つまり分子拡散による混合が支配的となる。分子拡散混 合に要する時間は分子拡散距離の二乗に比例することから、リアクターの内径を 1/10 にすれば、拡散時間(=反応時間)は 1/100 となる。つまり、フラスコでは不 可能な高速で効率的な混合を行える。 ③界面での反応が効率よく起こる 気液、液液、固液などの反応系では、界面積が飛躍的に向上し、効率よく反応 を行なうことができる。 上記のような特長を生かして、現在、世界中で数多くの有機合成への応用研究 が行われている1), 2) (Figure 3)。特徴的な例として、発熱の大きな反応や、不安 定な中間体を扱う反応、器壁表面を利用する反応、界面を利用する反応なども挙 げられる3)。- 6 -
著者の研究室においても、マイクロリアクターを用いたさまざまな研究実績を 残している4)。たとえば、イミダゾリウム系イオン液体である[bmim]PF6を反応媒 体とする薗頭反応をバッチ式反応装置で行なうと、通常、助触媒として必要な銅 塩の添加なしに反応が進行することを見出している。そこで、数十マイクロメー トルオーダーの流束を作り出すことで、高効率混合を実現する IMM 製マイクロ ミキサーを用い、ヨードベンゼンとフェニルアセチレンとの反応をフロー系で実 施した。結果、反応終了後にヘキサン抽出することで生成物が高収率で得られ、 また、副生するアンモニウム塩を水処理すると最下相にイオン液体相が分離する。 このイオン液体にはパラジウム触媒が固定化されており、再利用が可能である4 b)。 これまでに行なわれてきたマイクロリアクターを用いた研究は、マイクロ空間 での特異な反応現象を見出す研究が中心であったが、近年、各種の合成反応に応 用するための研究が世界で活発化するようになった。特に最近では物質製造のプ ロセスとして工業化を意識した研究が数多く見られるようになっている。アカデ ミアにおいてマイクロリアクターを用いて数十グラムから数百グラムの標的化合 物を合成する論文として、当研究室によるイオン液体を反応媒体としたマイクロ フロー型触媒反応を溝呂木-ヘック反応に応用し、触媒循環型システムを構築し た "Continuous Microflow Synthesis of Butyl Cinnammate by a Mizoroki-Heck Reaction Using a Low-Viscosity Ionic Liquid as the Recycling Reaction Medium" (Figure 4)や、京都大学の吉田らによる"Grignard Exchange Reaction Using aMicroflow System: From Bench to Pilot Plant"(Scheme 1)、大阪大学の深瀬らによる "Large-Scale Synthesis of Immunoactivating Natural Product, Pristane, by Continuous Microfluidic Dehydration as the Key Step"(Scheme 2)が報告されることで、製造プ ロセスとしての潜在力が立証されるようになった。現在では、化学工業や医薬品 製造業に至るまで、その取り組みは一般化に向かって急速に発展している5)。
- 7 -
Figure 4 イオン液体を用いた触媒循環型システムによる溝呂木-ヘック反応 Scheme 1 Scheme 2 CO2Bu Ph CO2Bu Ph-I, Pr3N Pd-cat. [bmim]NTf2 micromixer residence time unit CPC CYTOS Lab Systemtemp. control 130 °C Pd-cat. [bmim]NTf2 hexane recycle
Dual Microextraction System
NaOH aq A B hexane 115 g in 11.5 h (80%, 10 g/h) inorganic salts in aq. solution "separation" "extraction" "reaction" pumps flow control
residence time unit
0.8 mL/min 1 mL/min I CO2Bu Pd-cat CO2Bu + ionic liquid EtMgBr F F F F Br F F F F F MgBr F EtBr F F F F H F PFB BPFB + + MeOH Bu2O OH p-TsOH・H 2O THF
- 8 -
マイクロリアクターの特長として、「研究開発から工業生産への移行が効率的 である」ということがよく挙げられる。確かに、バッチ生産におけるフラスコか らプラント(反応釜)へのスケールアップにおいては、しばしば反応条件の再検 討が必要となり、長い検討タスクを要するが、マイクロリアクターにおいては、 実験室レベルで反応条件を最適化すれば、そのリアクターを数多く並列化(ナン バリングアップ)すれば、容易に工業生産まで行なうことができると言われてい る。しかしながら、それを実現するためには、並列した多くのリアクターに均一 に流体を送液することができる高性能なポンプの設置や、それぞれのリアクター の温度、閉塞などを常時検知できるシステムの構築や、装置細部の洗浄性などの 多くの課題がある。特に医薬品製造においては、GMP(Good Manufacturing Practice)という非常に重要なルール、規制があり、製造時のパラメータの履歴、 設備の洗浄性など、より高いハードルが設けられている。一方で、コスト面では バッチプラントと比較して、マイクロプラントは投資費用が大幅に削減されると いうメリットもある。さらに安全面では、マイクロリアクターの反応器内の液量 は極めて少ないことから、高温・高圧などの極限操作を行なっても、爆発などの リスクは大幅に低減するメリットがある。 Table 1. バッチ式反応器とマイクロリアクターの特徴比較 本論文においては、マイクロリアクターを用いた有機合成の工業化達成の前段 階である、医薬品開発のスピードアップに貢献するためのシステム構築について混合性能
熱交換率
過反応の抑制
多目的性
バッチ
リアクター
マイクロ
リアクター
○
△
◎
◎
△
◎
×
◎
安全性
△
◎
コスト
(設備費用)
△
◎
- 9 -
述べる。これまでは医薬品開発の流れの中で、上流にあたる「化合物探索」や、 下流である「製造」の段階での使用が検討されていたが、本研究においてはその 2 つの段階の橋渡しをスムーズに行なう上で、マイクロリアクター利用の検証を 行なう。これが達成できれば、本来、多くの時間と労力を必要とするスケールア ップに伴う反応条件最適化という作業を大幅に短縮することが可能となり、スペ シャリティケミカルズの合成研究を効率的に行なう新しいモデルとなりうる。ま た、マイクロリアクターでの実施例が少ない光反応や均一系の触媒反応を研究対 象とし、それにより合成する化合物も医薬中間体などの極めて実例が少ない化合 物を選択した。 第1 章では、マイクロフロー系による光バートン反応に関する研究として、光 照射が可能なマイクロリアクターを用いた光バートン反応による脳血管攣縮抑制 薬の鍵中間体の合成について述べる。まず、基礎的な研究として、バッチ反応と 同様の水銀ランプを光源とした反応に関する検討結果と考察について述べる(第 1 章、第 1 節)。反応液量が非常に少ない系でマイクロリアクターを用いた場合に は、水銀ランプのような高出力の光源を用いずとも反応を行なうことが可能と考 え、反応に必要な波長のみを、必要な強度で与えられる低出力光源を用いて反応 を行い、より最適な反応系を構築した(第1 章、第 2 節)。続いて、内容積の大き な光反応用マイクロリアクターとブラックライトを組み合せて、長時間の連続運 転によりg スケールの合成を実施した。さらなる自動化・省力化を目指し、大日 本スクリーン㈱が製作した PC によって運転制御が可能な光反応用マイクロリア クタシステム(DS-AMS-1)を用いて、g スケールの合成を実施した結果について も述べる(第1 章、第 3 節)。 第2 章では、「反応条件検索型マイクロリアクターシステム」を使用し、医薬化 合物合成における反応条件検索から大量合成までを一貫して行えるシステムの構 築を行なったことについて述べる。従来、マイクロリアクターを用いて、反応の 滞留時間、温度、試薬量などのパラメータを最適化するためには、実験→分析→ 洗浄→条件設定→実験→…というサイクルでの操作が必要であり、多くの時間と 労力を要してきた。本研究で使用した「反応条件検索型マイクロリアクターシス- 10 -
テム」は、あらかじめ種々のパラメータをソフト上で入力することにより、連続 的に反応条件スクリーニングができ、短時間で効率的に条件最適化を実施するこ とができる。本研究では、タンパク質分解酵素阻害剤の原薬合成における臭化チ オフェン誘導体とトリルアセチレンとの薗頭反応の条件を当該システムを用いて スクリーニングしたところ、化合物の大量合成に資する反応条件を短時間で見出 すことができた。また、さらに大きな100 g スケールでの合成を行なうため、内 容積の大きな「ミリリアクター」を作成、運転した結果についても述べる。- 11 -
引用文献
1. For reviews, see: (a) Ehrfeld, W.; Hessel, V.; Lowe, H. Microreactors: New
Technology for Modern Chemistry; Wiley-VCH: Weinheim, 2000. (b) Hessel,
V.; Hardt, S.; Löwe, H. Chemical Micro Process Engineering; Wiley-VCH,
Weinheim, 2005. (c) Wirth, T. Microreactors in Organic Synthesis and
Catalysis; Wiley-VCH, Weinheim, 2008. (d) Hessel, V.; Renken, A.;
Schouten, J. C.; Yoshida, J. Micro Process Engineering; Wiley-VCH, Verlag,
2009. (e) Jahnisch, K.; Hessel, V.; Lowe, H.; Baerns, M. Angew. Chem. Int.
Ed. 2004, 43, 406. (f) Pennemann, H.; Watts, P.; Haswell, S. J.; Hessel, V.;
Lowe, H. Org. Process Res. Dev., 2004, 8, 422. (g) Watts, P.; Haswell, S. J.
Chem. Soc. Rev. 2005, 34, 235. (h) Doku, G. N.; Verboom, W.; Reinhoudt, D.
N.; Berg, A. V. D. Tetrahedron 2005, 61, 2733. (i) Yoshida, J.; Nagaki, A.;
Iwasaki, T.; Suga, S. Chem. Eng. Technol. 2005, 28, 259. (j) Geyer, K.;
Codée, J. D. C.; Seeberger, P. H. Chem. Eur. J. 2006, 12, 8434. (k)
Kobayashi, J.; Mori, Y.; Kobayashi, S. Chem. Asian J. 2006, 1, 22. (l)
deMello, A. J. Nature 2006, 442, 394. (m) Song, H.; Chen, D. L.; Ismagilov,
R. F. Angew. Chem. Int. Ed. 2006, 45, 7336. (n) Mason, B. P.; Price, K. E.;
Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107,
2301. (o) Ahmed-Omer, B.; Brandt, J. C.; Wirth, T. Org. Biomol. Chem. 2007,
5, 733. (p) Yoshida, J.; Nagaki, A.; Yamada, T. Chem. Eur. J. 2008, 14, 7450.
(q) Fukuyama, T.; Rahman, M. T.; Sato, M.; Ryu, I. Synlett 2008, 151. (r)
Yoshida, J. Flash Chemistry. Fast Organic Synthesis in Microsystems; Wiley:
Chichester, U.K., 2008.
2. For recent examples of organic synthesis using microreactors, see: (a)
Kobayashi, J.; Mori, Y.; Okamoto, K.; Akiyama, R.; Ueno, M.; Kitamori, T.;
Kobayashi, S. Science 2004, 304, 1305. (b) Kawaguchi, T.; Miyata, H.; Mae,
K.; Yoshida, J. Angew. Chem. Int. Ed. 2005, 44, 2413. (c) Comer, E.; Organ,
M. G. J. Am. Chem. Soc. 2005, 127, 8160. (d) Nagaki, A.; Togai, M.; Suga,
S.; Aoki, N.; Mae, K.; Yoshida, J. J. Am. Chem. Soc. 2005, 127, 11666. (e)
Uozumi, Y.; Yamada, Y. M. A.; Beppu, T.; Fukuyama, N.; Ueno, M.;
Kitamori, T. J. Am. Chem. Soc. 2006, 128, 15994. (f) He, P.; Watts, P.;
Marken, F.; Haswell, S. J. Angew. Chem. Int. Ed. 2006, 45, 4146.
(g) Usutani,
- 12 -
H.; Tomida, T.; Nagaki, A.; Okamoto, H.; Nokami, T.; Yoshida, J. J. Am.
Chem. Soc. 2007, 129, 3046. (h) Nagaki, A.; Tomida, Y.; Usutani, H.; Kim,
H.; Takabayashi, N.; Nokami, T.; Okamoto, H.; Yoshida, J. Chem. Asian J.
2007, 2, 1513-1523. (i) Sahoo. R. H; Kralj, J. G.; Jensen, K. F. Angew. Chem.
Int. Ed. 2007, 46, 5704. (j) Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka,
S.; Fukase, K. Org. Lett. 2007, 9, 299. (k) Carrel, F. R.; Geyer, K.; Codée, J.
D. C.; Seeberger, P. H. Org. Lett. 2007, 9, 2285. (l) Bogdan, A, R.; Mason, B.
P.; Sylvester, K. T.; McQuade, D. T. Angew. Chem. Int. Ed. 2007, 46, 1698.
(m) Miller, P. W.; Long, N. J.; de Mello, A. J.; Vilar, R.; Audrain, H.; Bender,
D.; Passchier, J.; Gee, A. Angew. Chem. Int. Ed. 2007, 46, 2875. (n) Hornung,
C. H.; Mackley, M. R.; Baxendale, I. R.; Ley, S. V. Org. Process Res. Dev.,
2007, 11, 399.
3. (a) Chambers, R. D. ; Spink, R. C. H. Chem. Commun. 1999, 883. (b)
Krummdract, H.; Koop, U.; Stold, J. GIT Labor-Fachz. 1999, 43, 590.
4. (a) Fukuyama, T.; Rahman, M. T.; Sato, M.; Ryu, I. Synlett 2008, 151. (b)
Fukuyama, T.; Shinmen, M.; Nishitani, S.; Sato, M.; Ryu, I. Org. Lett. 2002,
4, 1691. (c) Liu, S.; Fukuyama, T.; Sato, M.; Ryu, I. Org. Process Res. Dev.
2004, 8, 477. (d) Rahman, M. T.; Fukuyama, T.; Kamata, N.; Sato, M.; Ryu, I.
Chem Commun. 2006, 2236. (e) Fukuyama, T.; Hino, Y.; Kamata, N.; Ryu, I.
Chem Lett. 2004, 33, 1430. (f) Fukuyama, T.; Kobayashi, M.; Rahman, M.
T.; Kamata, N.; Ryu, I. Org. Lett. 2008, 10, 533. (g) Wienhofer, I. C.; Studer,
A.; Rahman, M. T.; Fukuyama, T.; Ryu, I. Org. Lett. 2009, 11, 2457.
5. (a) Liu, S.; Fukuyama, T.; Sato, M.; Ryu, I. Org. Process Res. Dev. 2004, 8,
477. (b) Wakami, H.; Yoshida, J. Org. Process Res. Dev. 2005, 9, 787. (c)
Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka, S.; Fukase, K. Org. Lett.
2007, 9, 299. (d) Bayer, Forschungszentrum Karlsruhe: EP903174 (Mar 24,
1999) ; 特開平 11-171857. (e) Chemie Technik 1998, 27 (10), 124. (f)
Schwalbe, T.; Autze, V.; Wille, G.; Chimia 2002, 56, 636. (g) 西澤恵一郎,
福田三壽, 化学装置, 2003 年 9 月号, 79. (h) Method for producing imido
ether compound, PCT Int. Appl. (2006), WO 2006077854. (i) Kawaguchi, T.;
Miyata, H.; Mae, K.; Yoshida, J. Angew. Chem. Int. Ed. 2005, 44, 2413.
- 13 -
本論
第
1 章 マイクロフロー系による光バートン反応
第
1 節 水銀ランプを用いるマイクロフロー系での光バートン反応
光バートン反応は、亜硝酸のアルキルエステルが光照射されると、NO 基が酸 素上からδ位炭素上に転位し、ニトロソ化合物あるいは互変異性体のオキシムに 変換される反応である 1)。その反応機構は、まず O-N 結合が光によりホモリテ ィック開裂を起こし、アルコキシラジカルとNO ラジカルが発生するとされてい る。次いで、アルコキシラジカルがδ位上の水素ラジカルを引き抜いて、炭素ラ ジカルを発生させ(1,5-radical translocation)2)、その炭素ラジカルがNO ラジカル と再結合して生成物を与える(eq. 1-1)ものと考えられているが、最近、連鎖型 の可能性が示された3)。 (eq. 1-1) 本研究において、モデル反応として採用したのは、脳血管攣縮抑制薬の鍵中間 体合成における光バートン反応である。本脳血管攣縮抑制薬は、植物成分である オレアノール酸を原料とし、それを化学修飾することで得られる、非ペプチド性 のETA(エンドセリン A)受容体選択的拮抗薬として、急性期の虚血性脳血管攣 縮抑制薬、具体的には脳浮腫やクモ膜下出血後の脳血管攣縮を抑制する薬として、 現在開発研究が進められている(scheme 1-1)4)。 OH NO + ONO O NO + OH N O OH N HO- 14 -
Scheme 1-1 脳血管攣縮抑制薬合成ルート その大量合成法についても長年研究が進められており、出発原料であるオレア ノール酸から原薬を得るまでの全ての工程において、安全な製造法が構築されて おり 5)、すでにフラスコでの大量製造も複数回実施されている。本研究で取り上 げる光バートン反応は、ヒドロキシラクトン5 にピリジン中、ニトロシルクロラ イド(NOCl)を作用させ、ナイトライト型化合物 1 を得る。次いで、この化合物 1 に光を照射し、オキシム型化合物 2 を得る工程で実施される(Scheme 1-2)。 Scheme 1-2 ONO O O O h 1 -NO O O O O 3 1,5-radical translocation HO O O O 4 NO HO HC O O O 2 NOH HO O O O 5 NOCl O O O ONO HO CO2H HO O O O CH=NOH OH HN CO2H O O O O CO2H oleanolic acid 脳血管攣縮抑制薬h
- 15 -
原料のナイトライト体1 は不安定な中間体であり、結晶状態、室温下では数日 間、また、アセトン溶液中、室温下では数十分間で、元の中間体であるヒドロキ シラクトン体5 に戻ってしまう。このような不安定な化合物を基質とし、さらに 制御が困難な光反応によって、満足のいく収率を得ることは非常に困難であった が、過去の基礎研究と大量合成法研究の結果、バッチ反応容器での最適な反応条 件はすでに見出され、報告されている 5)。工業的には、600 L の反応釜に原料 1 の溶液を入れ、そこに高圧水銀ランプの光を照射する。この時、高圧水銀ランプ の出力は、10 kW という非常に大きなものとなる。また、スケールが大きくなる ほど、光を長時間照射する必要があり、その間に、安定性の低い原料が分解して しまうなどの問題があった。このため、これ以上のスケールアップには限界があ ると考えられていた。 そこで著者は、この光反応をフロー系で連続的に行なうことができないかと考 えた。フロー系で行なう場合、サイズの大きな反応釜や高出力の光源を必要とせ ず、反応容量の大小に関わらず、常に安定した品質で目的物を得ることができる。 また、反応液に光照射を行なう上で、その光強度はBeer-Lambert 則(eq. 1-2) により、その透過距離が長くなるほど弱まっていくため、光の透過距離をできる だけ短くする必要がある。log(I0 / I) = εc ℓ (eq. 1-2)
I0 : 媒質に入射する前の光強度, I : 媒質中をℓ 移動したときの光強度, ε : モル吸光係数, c : 媒質のモル濃度 これらの観点から、効率的な工業的光反応を目指すためには、「小型のフロー 系反応装置」がベストであると考えた。最終的には、目的とする化合物の必要量 や生産性、光照射のエネルギー効率などを考えて、最適なサイズを見極める必要 があるが、まずはフロー系での光反応の基礎的な研究が必要と考え、脳血管攣縮 抑制薬の鍵中間体合成における光バートン反応について、マイクロリアクターを 用いて検討することとした。なお、マイクロリアクターを用いた光反応の実施例 は、最近になって数多く報告されている6,7)。
著者の研究室においては、cyclohexen-2-one と vinyl acetate の混合液をミクロガ ラス社製のマイクロリアクターに送液し、そこに高圧水銀ランプを照射すると、2
- 16 -
時間の滞留時間で、[2 + 2] 環化付加生成物を 88%の収率で得られることを報告し ている8)。この反応をPyrex 製 10 mL のナス型フラスコを用いて行なうと、2 時間、 4 時間の反応で、それぞれ 8 %、22 %の収率しか得られず、マイクロリアクター を用いることで大幅な反応時間の短縮を実現している。さらにg スケールの反応 を行なうために、2 つのリアクターを直列に並べ、処理量を 2 倍にし、45 時間の 連続運転で、2.9 g(収率 85 %)の生成物を得ることにも成功している(Scheme 1-3)。 Scheme 1-3 ミクロガラス社製マイクロリアクターを用いた[2+2]光環化付加反応 OOAc 0.5 mL / h microreactor h (Hg, 300 W) OAc O 88 % residence time 2 h 20 ℃ + O OAc 1.0 mL / h micro reactor OAc O 2.9 g (after 45 h) 85 % isolated yield total residence time 2 h
20 ℃
+ microreactor
- 17 -
本研究に用いたマイクロリアクターは、大日本スクリーン製造㈱が製作した光 反応用マイクロリアクターである。Figure 1-1 に示すように金属の上板、下板で 光照射窓となるガラス板と流路プレートを挟み込む構造になっている。随時、分 解して洗浄が可能であり、マイクロリアクターを用いた場合の大きな課題である、 閉塞時の処理や、様々な反応を行なう場合のコンタミネーションの問題点は解消 される。反応液は、下板につけられた流入口から送液され、流路を流れた後に、 再度下板の流出口から払い出される。流路を流れる際、上部がガラス板であるこ とから、上部から光照射が可能である。 Figure 1-1 大日本スクリーン社製マイクロリアクター <ガラス板について> 本研究では、Quartz ガラス(石英:厚さ 10 mm)、Pyrex ガラス(パイレック ス:厚さ10 mm)、Lime-soda ガラス(ライムソーダ:厚さ 12 mm)の 3 種類の ガラス板を検討に用いた。検討結果の詳細については後述する。 <流路プレートについて> 大日本スクリーン社の得意技術であるフォトエッチング技術によって、厚さ 0.08 ~ 0.25 mm のプレート上に目的に応じた、様々な深さ、幅、長さの流路を 形成させることができる(Figure 1-2)。 フォトエッチングとは、目的の加工形状にマスキング(表面を部分的に被覆保 護すること)による防食処理を施した上で、エッチング液などの腐食剤によって 上板 下板 ガラス板 流路 プレート- 18 -
不要部分を除去することで所望の形状を得るエッチング加工技術に、精密な写真 技術・精密画像技術をマッチさせた精密加工技術がフォトエッチングと呼ばれ、 エッチング液による腐食加工(化学切削)から材料表面を保護するマスクとして、 一般にレジスト(フォトレジスト・エッチングレジスト)が用いられる。精密写 真と腐食技術の応用であるフォトエッチング加工は、エレクトロニクス製品など における極小部品・極薄板製品や特殊・複雑形状の製品など、金属加工における 超精密な加工要求にも対応が可能な加工方法である。 Figure 1-2 マイクロリアクターの流路構造 <リアクターのサイズについて> 本研究では、2種類のサイズのマイクロリアクター(以下、マイクロリアクタ ーA,B)を用途によって使い分ける。マイクロリアクターA は、流路の Hold-up volume は 0.1 ~ 0.2 mL 程度であり、少ない容量で反応を行えるため、新しく検 討を行なう際の反応条件スクリーニングなどを行なうことができる(Figure 1-3)。 一方、マイクロリアクターBは流路のHold-up volume が 2 ~ 4 mL 程度であり、 同一の反応条件(滞留時間)で反応を行なう場合、マイクロリアクターA の 20 倍ほどの処理能力を持つ。よって、マイクロリアクターA で決定した反応条件で、 g スケールの合成を行なうことを目的としたマイクロリアクターである(Figure 1-4)。- 19 -
Figure 1-3 マイクロリアクターA
Figure 1-4 マイクロリアクターB
各種ガラス板の選択や、光強度、反応温度など、光バートン反応の条件検討を マイクロリアクターA(流路:幅 1000 m、深さ 107 m、長さ 2.2 m、Hold-up volume 0.2 mL)と 300 W の高圧水銀ランプを用いて実施した結果について以下に述べる。 まず、Quartz ガラス(石英:厚さ 10 mm)、Pyrex ガラス(パイレックス:厚 さ10 mm)、Lime-soda ガラス(ライムソーダ:厚さ 12 mm)の中から、最適な ガラス材の選択検討を実施した。
- 20 -
原料が残存する条件で反応を実施することで、反応速度の差も評価できると考 え、滞留時間を6 min と短く設定して反応を行なった。3 種のガラス材の使用結 果を比較したところ、Lime-soda ガラスを用いた際に最も反応が効率よく進行し た。Pyrex ガラスを用いると、目的物も得られるが、原料がほとんど無くなった 上に、分解物と見られるピークが多数見られた。また、Quartz ガラスを用いた場 合には、原料が消失する上、生成物も見られず、非常に反応系が複雑となった (Table 1-1)。 Table 1-1. 各種ガラス材を用いた光バートン反応 上記の結果の原因について、本研究で用いた高圧水銀ランプの発光スペクトル と、ガラスの透過能から考察した。 Figure 1-5 のように、本バートン反応に必要な 365 nm 付近の光の相対強度が強 いため、本研究では光源として高圧水銀ランプを選択しているが、その他にも多 くの波長の光が発せられており、特に 300 nm 以下の短波長の光を多く発してい ることがわかる。a) Microreactor type A, [1] : 9 mM in acetone., pyridine 0.2 equiv., Distance between light and the microreactor surface: 15 cm.
b) HPLC yield.
entry reactor top flow rate(mL/h) residence time(min) 2 (%)b
1
2
3
pyrex glass
soda lime glass quartz 2.0 6 7 2.0 6 46 2.0 6 trace 21 19 trace 1 (%)b 2 h acetone, rt. pyridine (0.2 equiv) microreactor: type A ONO O O O OH HC O O O NOH 1
- 21 -
Figure 1-5 高圧水銀ランプの分光分布 また、高圧水銀ランプの光を各ガラスに透過させたスペクトルをFigure 1-6 に 示す。 Figure 1-6 高圧水銀ランプの光を各種ガラス材に透過させた際のスペクトル Lime-soda ガラスを用いると 320~330 nm 以下の光はすべてカットされるが、 反応に必要な 365 nm 付近の光も少しカットしてしまう。Pyrex ガラスは、 Lime-soda ガラスに比べて、365 nm 付近の光をよく透過する一方、300 nm 以下の 短波長の光もいくらか透過する。また、Quartz ガラスは、200 nm 付近の光までを よく透過するため、高圧水銀ランプから発せられた光はほぼ全て透過させる。 300 320 340 360 380 400 wave length (nm) E n e rg y Lime Soda Pyrex Quartz None- 22 -
これらのデータから、反応が分解系に入ってしまう原因は、300 nm 以下の光が 原因であると考えられる。よって、高圧水銀ランプを光源として本バートン反応 を実施する場合は、ガラスはLime-soda ガラスを用いる必要があると結論付けた。 <光強度の検討> 次に、光源からの光の強度が反応に与える影響について検討した。高圧水銀ラ ンプの光を各ガラスに透過させた際の、バートン反応に必要な 365 nm の光強度 を比較する(Figure 1-7)。今回用いる、Lime-Soda ガラスは、Quartz ガラス、Pyrexガラスと比べて、反応に必要な光もカットしていることがわかる。また、距離を 7.5 cm から 15 cm に遠ざけると、強度が約半分になっていることもわかる。 Figure 1-7 高圧水銀灯の光を各ガラス材を透過させた際の光強度(365 nm) 前項でガラス材を検討した際の滞留時間(6min)を固定し、光源とリアクター の距離を縮めながら反応を実施した結果、15 → 10 → 7.5 cm と、光源とリアク ターの距離を縮める(=光の強度を上げる)につれて、バートン反応の収率は徐々 に向上し、最高で約60 %に達した。ところが、さらに距離を縮めた(5 cm)時、 原料は消失するものの、分解物ピークが増え、目的物の収率は大きく低下した (Figure 1-8)。 0 2 4 6 8 10 12 14 16 None (7.5cm) Quartz (7.5cm) Pyrex (7.5cm) Lime soda (7.5cm) Lime soda (15cm) E n er gy ( u W / c m 2 )
- 23 -
Figure 1-8 光強度の影響評価 この原因を追究するため、リアクター内部の様子を観察したところ、流路内に は多くの気泡が発生していることが確認された。これは溶媒であるアセトンが沸 騰しているものと考え、各条件でのリアクターの表面温度を確認した(Figure 1-9)。 Figure 1-9 光源からの距離と収率、リアクター表面温度の関係 各距離でのリアクター表面温度を測定した結果、光源とリアクターの距離が近 づくにつれて、表面温度は大きく上がり、距離が5 cm の時には、その温度は 50℃ を越えていた。このことから、流路内で発生していた気泡がアセトンが沸騰した ものであるという仮説は支持される。 10% 20% 30% 40% 50% 60% 70% 0 5 10 15 20 Distance(cm) H P L C y ie ld (% ) 10% 20% 30% 40% 50% 60% 70% 0 5 10 15 20 Distance (cm) H P L C y ie ld ( % ) 0 10 20 30 40 50 60 T e m p . o f R e a c to r S u rf a c e (℃ )- 24 -
<反応温度の検討> 上記の検討においても、反応温度を変えて反応を行っているが、光源の距離(= 光強度)も同時に変わっており、反応温度のみをパラメータとした検討ではない。 そこで、Figure 1-10 のような装置を組み、反応温度が本反応に及ぼす影響を検討 した。 Figure 1-10 マイクロリアクターを用いる光バートン反応の実験モデル 図のように、マイクロリアクターを恒温槽に漬けることにより、リアクター表 面温度を所望の温度に調整できるようにし、光源の距離を変えても、一定の温度 で反応を行えるようにした。これにより、光強度と反応温度を独立して操作する ことができる。 まず最初に、光源の距離を15 cm とし、0 ~ 50 ℃で反応を実施し、目的物の 収率と原料の残存率を確認した結果、0 ~40 ℃では、目的物収率、原料残存に大 差は無かったが、50 ℃で反応を行なうと、収率が大きく低下し、原料残存もほと んど確認されなかった(Figure 1-11)。原料の分解が起こっているものと考えら れ、HPLC 上でも多くの分解物ピークが確認された。送液用
シリンジポンプ
反応液
回収部
光源(高圧水銀灯)
マイクロ
リアクター
- 25 -
Figure 1-11 光バートン反応における反応温度の影響 このことからマイクロリアクターで光反応を行なう場合、光源からは光だけで なくかなりの熱も発せられるために、リアクターの熱を効率的に除去する必要が ある。また、流路自体が非常に浅く、反応液量が非常に少ないマイクロリアクタ ーを使用する上では、今回用いたような光強度の強い光源を用いずとも、反応を 行なうことが可能となるのではないかと考えられる。そこで、反応に必要な波長 を、必要なだけの強度で与えられる光源を検索した。 hacetone, pyridine (0.2 equiv) microreactor: type A 0% 10% 20% 30% 40% 50% 60% 0 10 20 30 40 50 Reaction temp. (degC)
1 2 ONO O O O HO HC O O O NOH 1 2
- 26 -
第
2 節 低出力光源を用いるマイクロフロー系による光バートン反応
前節でも述べたように、高圧水銀ランプなどの高出力の光源で光反応を行なう 場合、多くの熱も発せられるために、リアクターの熱を効率的に除去する必要が ある。また、原料の分解や、副生成物の生成を引き起こす波長の光も発しており、 それらをカットするために、フィルターを設ける必要がある。そこで、反応に必 要な波長を、必要な強度で与えられる光源としてブラックライトを検討した。 ブラックライトとは、わずかに眼で見える長波長の紫外線を放射するライトで あり、蛍光物質が通常の蛍光灯とは異なり、そのピーク波長は350 ~ 370 nm の 光源である。また、蛍光管には深い青紫のガラスを用いて波長 400 nm 以上の可 視光線をカットしている。可視光線より長い波長の光をカットしているため、熱 の発生も非常に少ない(Figure 1-12)。 Figure 1-12 ブラックライト(15 W) 一方で、先の検討で用いた高圧水銀ランプから放出される光の大部分は赤外線 から可視光線の領域であり、そのエネルギー効率は極端に低い。また、電力のほ とんどが熱になってしまうために、光源の温度が上昇し、リアクターを加熱して しまう難点もある。 今回、検討で用いたブラックライト(東芝:ネオボール15 W)と、高圧水銀ラ ンプ(300 W)の波長と強度の比較を分光光度計を用いて行なった。Figure 1-13 に示すように、ブラックライトは高圧水銀ランプに比べて、強度こそ弱いものの、- 27 -
バートン反応に必要な 365 nm 付近の光しか発しておらず、原料の分解を引き起 こすと考えられる 300 nm 以下の光、また熱の原因となる可視から赤外の光は全 てカットされている。 Figure 1-13 高圧水銀ランプとブラックライトのスペクトル比較 また、光源の立ち上がり時間と、安定状態での出力も安定しており、長時間の 使用にも問題がない(Figure 1-14)。これにより、消費電力と光源コストの大幅 な削減はもとより、実験や大量合成での作業性、安全性は大幅に向上する。 Figure 1-14 各種光源の安定化に要する時間 0 200 400 600 300 320 340 360 380 400wave length (nm)
E
n
e
rg
y
(
W
/
c
m
2/
n
m
)
Hg lamp (300 W, 7.5 cm)Black light (15 W, 5 cm)0%
20%
40%
60%
80%
100%
120%
0
5
10
15
20
25
時間[min]
光
強
度
(対
安
定
状
態
)
高圧水銀灯(300 W) ブラックライト(15 W)- 28 -
上記のようなブラックライトの特性から、短波長をカットするガラス材を用い る必要はなくなる。そこで、ガラス材の検討をもう一度実施した結果、ブラック ライトを用いた際には、高圧水銀ランプと比較して、2 倍の反応時間を要したが、 Pyrex ガラスとの併用により最も高い収率で目的物が得られた。これは、ブラッ クライトが副反応を引き起こす短波長の光を発していないために、高圧水銀ラン プで見られた原料の分解に起因する収率低下が抑制されたためである。また、高 圧水銀ランプを用いたときに結果の良かった Lime-soda ガラスは、反応に必要な 波長(365 nm 付近)の光もカットしてしまっているため、原料が残存し、目的物 の収率が低下した(Table 1-2)。 Table 1-2 高圧水銀ランプ、ブラックライトでの反応実施結果の比較 <UV-LED(紫外発光ダイオード)について>発光ダイオード(LED : Light Emitting Diode)は、順方向に電圧を加えた際に発 光する半導体素子であり、発光色は用いる材料によって異なり、赤外線領域から Hg lime-soda Black light Black light Black light quartz pyrex lime-soda
6 min 9 min 12 min 24 47 60 29 55 71 15 29 39 56 -
-yield of 2 (%) light source reactor top residence time
光源からリアクターの距離:7.5cm 2 h acetone, rt. pyridine (0.2 equiv) microreactor: type A ONO O O O OH HC O O O NOH 1
- 29 -
可視光域、紫外線領域で発光するものまで製造することができる。寿命は非常に 長く、構造が簡単なため大量生産が可能で、安価である。
本研究では、35 mW でピーク波長が 365 nm の UV-LED(紫外発光ダイオード) を48 個備えた光源(SEOUL OPTO DEVICE Co., Ltd.: 1.7 W)を使用した(Figure
1-15)。この光源は、マイクロリアクターA のサイズに合わせて作成されており、 光源とリアクターとの距離を限界(1.5 cm)まで近づけることができる。 Figure 1-15 UV-LED この光源を用いて光バートン反応を行なった結果、ブラックライトを用いた時 と同じ滞留時間(12 min)で反応が完結し、HPLC 収率も同等の結果が得られた。 しかし、UV-LED 光源の出力は、ブラックライトの 1/10 程度であることから、そ のエネルギー効率は非常に高い。 以下に、各光源とガラスの組合せによる収率とエネルギー効率評価を表にまと める。エネルギー効率の指標として、HPLC 収率を、光源が滞留時間の間に発す るエネルギーで除した値(yield / Wh)で比較した(Table 1-3)。
- 30 -
Table 1-3 各種光源とガラス材の組み合わせによる光バートン反応 以上のように、ブラックライト、UV-LED は、安価で消費電力も小さいため、 マイクロリアクターでの光反応において、非常に効率の良い省エネ型の光源とな ることを見出した。 <副生成物の制御> 引き続き、反応で生成する副生成物とその制御についての検討を実施した。本 反応において、生成する副生成物は主に、ケトラクトン体 5 と硝酸エステル体 7 の2 つであり、以下のような反応機構で生成すると考えられている(Scheme 1-4)。 ケトラクトン型化合物5 は、本バートン反応の基質であるナイトライト型化合物 1 の原料である。バッチ反応、マイクロリアクターのどちらで反応を実施したと きも、数%オーダーで生成し、その制御方法は確立できていない。entry light source/reactor top flow rate(mL/h) residence time(min) yield of 2(%)b Wh yield/Wh
1 2 5 3 300 W Hg lamp/ Pyrex glass 300 W Hg lamp/ lime soda glass
15 W black light/ Pyrex glass 15 W black light/ lime soda glass
2.0 6 56 2.0 6 21 1.0 2.0 12 6 71 15 30 30 3 1.5 1.89 0.70 23.7 10.0 4 15 W black light/Pyrex glass 2.0 6 29 1.5 19.3
6 1.7 W UV-LED/Pyrex glass 1.0 12 70 0.34 206
a) Microreactor type A, [1] : 9 mM in acetone., pyridine 0.2 equiv., Distance between light and the microreactor surface: 7.5 cm (Hg), 3.0 cm (black light), 1.5cm (UV-LED). b) HPLC yield.
2 h acetone, rt. pyridine (0.2 equiv) microreactor: type A ONO O O O OH HC O O O NOH 1
- 31 -
Scheme 1-4 光バートン反応における副生成物の生成機構 一方で、硝酸エステル型化合物7 は、推定反応機構にもあるように、溶媒や気 相中の酸素が生成の原因であると考えられる。それを確認するためにバッチ反応 系とマイクロリアクターを用いた系で、酸素の影響と溶存酸素量低減による副生 成物の抑制検討を実施した。結果、バッチ反応の場合は、反応溶媒の脱気も重要 であるが、反応器内の気相の酸素を除去することが重要であることがわかった。 一方、マイクロリアクターを用いた場合には反応器内に気相は存在しないため、 溶媒の脱気のみで良好な結果が得られた(Table 1-4)。 ONO O O O acetone, r.t. h 1 -NO O O O O 3 1,5-radical translocation HO O O O 4 NO HOO O O 2 NOH HO O O O pyridine 5 NOCl O2 HOO O O 6 O HO O O O 7 ONO2 NO O- 32 -
Table 1-4 バートン反応における酸素の影響 また、バートン反応においては、NO ラジカルを発した分子以外、つまり系外 からの NO も反応に影響を与えるという報告もなされており(Scheme 1-5)8)、 NO の発生を光量、流速、圧力、温度で制御すれば、収率向上が可能と考えた。 Scheme 1-5 そこで、NO ガスを直接溶解させた溶媒を用いて反応を行えば、反応時間短縮 や収率向上が図れるのではないかと考えた。常温で NO を飽和させたアセトンを 溶媒に用いた場合、脱気処理を施したアセトンを用いた場合と、マイクロリアク acetone, r.t. h ONO O O O 1 HO O O O 7 ONO2 HO HC O O O 2 NOH +a) Microreactor type A, [1] : 9 mM in acetone., pyridine 0.2 equiv., flow rate: 2.0mL/h, residence time: 6 min., Distance between light and the microreactor surface: 7.5 cm. b) Batch, [1] : 9 mM in acetone., pyridine 0.2 equiv., reaction time: 3 h.
c) HPLC yield. entry reactor 2 (%)c 1 2 3 Batch air open Microreactor type A Batch N2 atmosphere 1 trace 12 56 58 1 (%)c 7 (%)c 30 9 8 trace ON14O H AcO + ON15O H AcO hν OH H AcO + OH H AcO N14+15 OH N14+15 OH
- 33 -
ターA を用いたバートン反応を行い、比較、検証した。結果、NO を飽和させた アセトンを用いた場合、反応はほとんど進行せず、ほぼ原料回収となった(Table 1-5)。これは、系中の NO 量増加により、原料への戻りの反応速度が増加してい ると考えられる。さらに、NO を飽和させたアセトンにブラックライトの光を当 て、その透過光の強度を測定すると、通常アセトンの約20 %しか光を通さないこ とがわかった。つまり、NO が溶解したアセトン自体が、反応に必要な光を吸収 するために、反応が進行しにくくなっているものと考えられる。 Table 1-5 NO 添加系でのバートン反応 <溶媒の検討> ここまでの検討で得られたマイクロリアクターによるバートン反応の最適条件 で、連続運転によるg スケールの合成を行なうためには、かなりの時間を要する。 その原因として、アセトン溶媒中の基質濃度が9 mM と極めて低いことが挙げら れる。スループット向上のために、基質が高い溶解度を持つ DMF に変更するこ とを検討した。これにより、基質濃度をアセトン使用時の4 倍まで上げることが でき、反応もアセトンと同様に、問題なく実施することを見出した。よって、連 続運転によるg スケールの合成は DMF を溶媒として実施することした。a) Microreactor type A, [1] : 9 mM in acetone., pyridine 0.2 equiv., light source: black light (15W)., reactor top: pyrex glass., Distance between light and the microreactor surface: 3.0 cm., flow rate: 2.0 mL/h., residence time: 6 min.
b) HPLC yield. entry solvent 1 2 1 29 yield of 2(%)b acetone
(saturate with NO) acetone 2 h acetone, rt. pyridine (0.2 equiv) microreactor: type A ONO O O O OH HC O O O NOH 1
- 34 -
第
3 節
連続マイクロフロー型光バートン反応
これまでに見出された、マイクロリアクターを用いたバートン反応の最適反応 条件でg スケールの合成を実施するために、光反応用マイクロリアクターB (内 容積:4 mL、2 台)とそのサイズに見合う光源として、ブラックライト(出力: 20 W、8 本)を用いた。流速(滞留時間)は、さまざまな条件で試運転を実施す ることで設定した(Table 1-6)。 Table 1-6 g スケール合成の運転条件検討 結果、上記3 条件いずれの場合も閉塞などのトラブルは無く、Run 3 に示すよ うに、滞留時間を32 分で最も良好な結果が得られた。5.4 g のナイトライト体を 原料とし、20 時間の連続運転を終えて得られた反応液をバッチで晶析、精製を行 い、3.1g(単離収率 60 %)のオキシム体を得ることができた(Figure 1-16)。 Figure 1-16 連続運転による g スケール合成Run 原料(g) 滞留時間(min) 運転時間(hr) 処理液量(mL) yield (%)HPLC 収量(g) 1 5.8 12 8 320 20 0.96 2 4.3 16 8 240 40 1.2 3 5.4 32 20 300 60 3.1 15 mL/ h 1 microreactor Type B 2 3.1 g (after 20 h)
(purified by silica gel column chromatography)
black light (20 W x 4)
DMF solution (300 mL)
total residence time : 32 min
ONO O O O HO HC O O O NOH 5.4 g microreactor Type B black light (20 W x 4)
- 35 -
先に示した連続運転は、マニュアルでの操作が多く、将来の連続運転の省力化 を目指す上では、自動化などのさらなる改良が必要である。そこで、大日本スク リーン社に光反応用マイクロリアクタシステム(DS-AMS-1)の製作を依頼した (Figure 1-17)。 Figure 1-17 連続フロー型光反応用マイクロリアクタシステム(DS-AMS-1) ①マイクロリアクターユニット マイクロリアクターB 1台と光源(ブラックライト)を有する部分で、マイク ロリアクターの下板(SUS)には、冷熱媒が流せるように設計されており、リア クターの表面温度を室温~-20℃の範囲で任意に設定できる。マイクロリアクター 各部には熱伝対が設置されており、PC のソフトウェア上にて各部の温度を確認で きる。また、マイクロリアクター自体を少し傾けることができるようになってお り、運転開始時のエア抜きをしやすい構造となっている。 ②PC ユニット システム運転条件設定や、運転状態の監視が行えるソフトウェアが入っている。①
②
③
- 36 -
③薬液タンク、ポンプ、チラーユニット 3段構成となっており、上段には原料および反応後の液を溜めるタンク、中断 には原料液をリアクターに送液するHPLC ポンプ、下段にはリアクターに送液す る熱媒を供給するチラーがそれぞれ設置されている。 さらに、リアクターを無人で連続運転することを目指す上で、さまざまな安全 機構を充実させている。まず、リアクターと光源部分を防爆カバーで包み込み、 予期しない反応が起こった際の爆発等に備える。また、非常時に被害を最小限に 抑えられるよう、リアクター全体をドラフト内に設置し、ドラフト外に設置され ている薬液タンク、ポンプ、チラーユニットについては、そのユニット全体を排 気し、作業者が溶剤雰囲気に暴露されないようにしている。さらに、HPLC ポン プユニット、リアクターユニットには、液漏れ感知センサーが設置されており、 液漏れを感知した際には、システムが自動でシャットダウンするように設計され ている。 本研究においては、マイクロリアクターB(流路:幅 1000 m、深さ 500 m、 長さ0.5 m、16 レーン、内容積:4 mL)、ブラックライト(15 W、6 本)をリア クターユニット内に設置し、連続運転を実施した。8.64 g のナイトライト体を仕 込み、試運転により見出された最適流速12 mL / h で、システムを 40 時間運転し た結果、トラブル無く運転を完了することができ、得られた反応液をバッチで晶 析、精製を行い、5.3 g(単離収率 61 %)のオキシム体を得た(Figure 1-18)。 Figure 1-18 DS-AMS-1 を用いた光バートン反応による g スケール合成 Black light (15 W × 6) residence time : 20 min 12mL/h 1 ONO O O O 2 HO HC O O O NOH 8.6g (17mmol) / 480mL DMF 5.3 g (after 40 h) 60% yield Black light (15 W × 6) residence time : 20 min 12mL/h 1 ONO O O O 2 HO HC O O O NOH 8.6g (17mmol) / 480mL DMF 5.3 g (after 40 h) 60% yield- 37 -
DS-AMS-1 では 40 時間で 5 g 程度の合成であったが、さらなる大量合成を目指 し、mm スケールの流路を持つチューブリアクターによるバートン反応を検討を 行なうこととした。光反応をチューブリアクターで行なう例は、すでに Hook ら によって報告されていたが 9)、この場合は中圧水銀ランプを水冷用ジャケットで 覆い、その周りに内径1~3 mm 程度の FEP(Fluorinated EthylenePropylene)チュ ーブを巻きつけ、そこに反応液を送液する装置を用いて、2 種の化合物について 数100 g オーダーの合成を実施することに成功している(Figure 1-19)。 Figure 1-19 チューブリアクターを用いた光反応実施例 本研究においては、熱を発しにくいブラックライトを用いることで、光源周囲 にガラス板だけを設け、できるだけ光の強度が落ちないような形とした。また、 内径1~3 mm 程度のチューブを巻きつけても、先述した DS-AMS-1 と比べても、 合成スケールに大差がないため、より内径の大きい(8 mm)テフロンチューブを 巻きつけた装置を作成し(Figure 1-20)、使用を試みたが、送液の際にチューブ 内のエアの噛み込みにより、層流状態を作り出すことができず、検討を断念した。 Figure 1-20 チューブリアクター- 38 -
第
4 節
結論
本章では、脳血管攣縮抑制薬の鍵中間体合成における光バートン反応を、マイ クロリアクターで実施し、省エネルギー型の光源と組み合せることにより、効率 的に実施できることを見出した。また、小型のマイクロリアクターA を用い、少 量のサンプルで最適な反応条件を決定した後、20 倍の内容積を持つ長尺型のマイ クロリアクターB で g スケールの合成を行なうことができた。さらに、g スケー ル合成の省力化、自動化を測るために光反応用マイクロリアクタシステム (DS-AMS-1)を用い、40 時間の連続運転を行なった結果、5.3 g の目的物を合成 することに成功した。 マイクロリアクターを用いて、生理活性物質の反応条件検索からg スケールの 合成を迅速化できることは、医薬品開発の観点から、極めて有意義であり、今後 のプロセス化学において、反応条件スクリーニングから、g スケールの合成をシ ームレスに行なうことができるマイクロフロー系の反応モデルを提供することが できた。- 39 -
実験の部
共通項
核磁気共鳴(1H NMR)スペクトルは、CDCl3溶媒を用いてVARIAN OXFORD(300
MHz)で測定した。この際にリファレンスは TMS の 0.00 ppm とした。ケミカル シフト( )はparts per million (ppm)で測定した. 核磁気共鳴(13C NMR)スペ
クトルはCDCl3溶媒としてVARIAN OXFORD (75 MHz)で測定した。リファレ
ンスはCDCl3の77 ppm とした。HPLC は Shimadzu の LC-10ADvp を使用し、カラ
ムは WATERS Symmetry RP8 (4.6×150 mm)を用い、移動相は MeCN/H2O/H3PO4
(80/20/0.1)で流速 1 mL/min とした。検出波長は 197 nm で、HPLC 収率は別途 調製された標準品サンプルのピークエリアとの比較で計算した。溶媒、試薬は和 光純薬株式会社から購入した。生成物はMerck、 Silica Gel 60、70 - 230 mesh を 使用してフラッシュカラムクロマトグラムにより精製した。
マイクロリアクターA, B を用いた光バートン反応(第 1 節、第 2 節)
原料(1)の合成 5) ケトラクトン5(615.7 g)をピリジン(6.16 L)に懸濁し、別途調製器で発生さ せたNOCl ガスを発生と同時に導入する。導入は、-30 ℃以下で約 40 分で行なっ た。このガス発生は、濃塩酸(1.01 L)に,室温撹拌下,亜硝酸ナトリウム(186.5 g)を水(253 mL)に溶かした水溶液に約 40 分で滴下して発生させる。ガス導入 後、-40~-43 ℃で撹拌熟成 30 分を行い、水(9.24 L)を流入してスラリー化し、 濾過、水洗(5.25 L)を行い、窒素による通気乾燥を 5 時間実施し、白色の結晶 を得た。 マイクロリアクターを用いたバートン反応 マイクロリアクター(大日本スクリーン製造㈱)を用いたバートン反応は、ナ イトライト 1 (45 mg, 0.09 mmol)をピリジン (0.2 equiv)を加えたアセトン(10- 40 -
mL)に溶解させたものを、シリンジポンプまたは HPLC ポンプを用いて、流速 1.0~2.0 mL / h (滞留時間 6 ~12 min)で送液し、高圧水銀ランプ(300 W)、 ブラックライト(15 W)、UV-LED(35 mW ×48:SEOUL OPTO DEVICE Co., Ltd.) をガラスカバー越しに照射することにより行なった。この時カバーガラスは、 Lime-soda ガラス、Pyrex ガラス、Quartz ガラスの三種類を用いた。2 の収率は HPLC により分析した。 バッチ反応容器でのバートン反応 バッチ反応容器でのバートン反応は、ナイトライト 1 (45 mg, 0.09 mmol)をピ リジン (0.2 equiv)を加えたアセトン(10 mL)に溶解させたものを 20 mL の Pyrex ガラス製の丸底フラスコに入れ、窒素もしくは空気存在下で、高圧水銀ランプ(300 W)、ブラックライト(15 W)を照射しながら撹拌する。生成物の収率は HPLC により分析した。
連続マイクロフロー型光バートン反応(第
3 節)
マイクロリアクターB を 2 台用いた連続運転 基質(5.4 g, 10.8 mmol)とピリジン(0.2 equiv.)を DMF(300 mL)に溶解させ た溶液を遮光した容器内に入れ、その液をHPLC ポンプを用いて、試運転により 見出された最適流速15 mL / h で、マイクロリアクターB(流路:幅 1000 m、深 さ500 m、長さ 0.5 m、16 レーン)を 2 台直列に繋いだもの(総内容積:8 mL) に送液する。得られた反応液に水600 mL を冷却しながら加え、目的物オキシム 体の晶析を行い、粗結晶を得た。その結晶をろ過、洗浄した後に、シリカゲルに よるカラムクロマトを実施し、3.1gのオキシム体を得ることができた(単離収率 60%)。1H NMR (CDCl3, 300 MHz) : 0.90 (s, 3H), 0.95 (s, 3H), 0.98 (s, 3H), 1.03 (s, 3H), 1.07 (s, 3H), 1.26 (s, 3H), 1.2-1.8 (m, 13H), 1.8-2.2 (m, 8H), 2.2-2.4 (m, 2H), 3.88 (d, 1H), 7.62 (s, 1H); 13C NMR (CDCl 3, 125 MHz) 16.7, 18.1, 18.9, 21.0, 24.2, 24.6, 26.9, 27.1, 28.4, 31.4, 33.1, 33.8, 34.0, 35.4, 36.4, 39.0, 39.5, 42.9, 43.5, 44.2, 45.0, 47.3, 48.8, 50.2, 54.6, 75.1, 88.6, 156.8, 179.2, 218.1. mp 285-289 °C.- 41 -
DS-AMS-1 を用いた連続運転 基質(8.64 g, 17.3 mmol)とピリジン(0.2 equiv.)を DMF(480 mL)に溶解させ た溶液を遮光した容器内に入れ、試運転により見出された最適流速12 mL / h で、 マイクロリアクターB(流路:幅 1000 m、深さ 500 m、長さ 0.5 m、16 レーン) を備えたシステムを40 時間自動運転した。 連続運転中には大きなトラブルは無く、得られた反応液に水 600 mL を冷却し ながら加え、目的物オキシム体の晶析を行い、粗結晶を得た。その結晶をろ過、 洗浄した後に、シリカゲルによるカラムクロマトを実施し、5.3 g のオキシム体を 得ることができた(単離収率 61 %)。 Lactone Nitrite (1 : ナイトライト体) 5)White powder; (Rf = 0.68, CH2Cl2 : EtOAc = 8 : 1). IR(CHCl3)
cm-1 : 2955, 1760, 1699. 1H NMR (CDCl3) 0.75 (s, 3H), 0.93 (s, 3H), 0.99 (s, 3H), 1.04 (s, 3H), 1.09 (s, 3H), 1.16 (s, 3H), 1.26 (s, 3H), 1.2-1.7 (m, 12H), 1.7-2.3 (m, 8H), 2.3-2.5 (m, 2H), 5.64 (dd, 1H, J = 3.5 Hz,2.0Hz), Mp. 254-258 ℃. HPLC (MeCN / H2O / AcOH = 80 / 20 / 0.1) tR = 27.1min. Oxime (2 : オキシム体) 5)
White powder; (Rf = 0.16, CH2Cl2 : EtOAc = 8 : 1). IR(CHCl3)
cm-1 : 3574, 3308, 3016, 2955, 2869, 1768, 1699. 1H NMR (CDCl3) 0.90 (s, 3H), 0.95 (s, 3H), 0.98 (s, 3H), 1.03 (s, 3H), 1.07 (s, 3H), 1.26 (s, 3H), 1.2-1.8 (m, 13H), 1.8-2.2 (m, 8H), 2.2-2.4 (m, 2H), 3.88 (d, 1H, J = 6.0 Hz), 4.20 (d, 1H, J = 6.0 Hz), 7.62 (s, 1H), 8.12 (s, 1H) ; 13C NMR (CDCl3) 16.7, 18.1, 18.9, 21.0, 24.2, 24.6, 26.9, 27.1, 28.4, 31.4, 33.1, 33.8, 34.0, 35.4, 36.4, 39.0, 39.5, 42.9, 43.5, 44.2, 45.0, 47.3, 48.8, 50.2, 54.6, 75.1, 88.6, 156.8, 179.2, 218.1. ONO O O O HO HC O O O NOH
- 42 -
12α-Hydroxy-3-oxooleanano-28,13-lactone (5 : ケトラクトン体) 5)
White powder;(Rf = 0.34, CH2Cl2 : EtOAc = 8 : 1). IR(CHCl3)
cm-1 : 3619, 2955, 1760, 1699. 1H NMR (CDCl 3) 0.91 (s, 3H), 0.99 (s, 6H), 1.05 (s, 3H), 1.10 (s, 3H), 1.19 (s, 3H), 1.32 (s, 3H), 1.2-1.8 (m, 14H), 1.8-2.2 (m, 8H), 2.44-2.55 (m, 2H), 3.91 (brs, 1H) ; 13C NMR (CDCl 3) 16.3, 18.2, 18.4, 19.1, 21.1, 21.2, 23.9, 26.7, 27.5, 28.0, 29.2, 31.6, 33.3, 33.4, 34.0, 34.1, 36.2, 39.5, 39.6, 42.2, 43.8, 44.7, 47.4, 51.2, 54.8, 76.2, 90.6, 179.8, 217.7. Nitrate (7 : 硝酸エステル体) 5)
White powder;IR(CHCl3) cm-1 : 3410, 1767, 1699, 1631, 1278. 1H NMR (CDCl 3) 0.92 (s, 3H), 0.98 (s, 3H), 1.00 (s, 3H), 1.05 (s, 3H), 1.10 (s, 3H), 1.24 (s, 3H), 1.2-2.0 (m, 20H), 2.51 (m, 2H), 3.95 (t, 1H, J = 2.6 Hz), 4.96 (d, 2H, J = 13.0 Hz), 5.07 (d, 2H, J = 13.0 Hz) ; 13C NMR (CDCl 3) 16.7, 18.2, 18.9, 19.8, 20.9, 22.2, 23.8, 26.8, 27.1, 28.4, 31.7, 33.1, 33.8, 34.0, 36.3, 38.2, 39.6, 42.7, 44.4, 44.5, 45.8, 47.3, 51.1, 54.6, 71.6, 75.6, 88.3, 179.1,217.2. HO O O O HO O O O ONO2