総 説
二値エンドポイントの単群臨床試験における
ベイズ流デザイン
Bayesian Design of Single-arm Clinical Trials
with Binary Endpoints: A Review
手良向 聡 Satoshi Teramukai
京都大学医学部附属病院探索医療センター検証部 Department of Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital
e-mail:[email protected]
The aim of single-arm clinical trials of a new drug is to determine whether it has suffi-cient promising activity to warrant its further development. For the last several years Bayesian statistical methods have been proposed and used. Bayesian approaches are ideal for earlier phase exploratory trials or proof-of-concept studies as they take into account information that accrues during a trial. Posterior and predictive probabilities are then updated and so become more accurate as the trial progresses. If the relevant external information is available, the decision will be made with a smaller sample size. The goal of this paper is to provide a review for statisticians who use Bayesian methods for the first time or investigators who have some statistical background. In addition, a clinical trial is presented as a real example to illustrate how to conduct a Bayesian approach for single-arm clinical trials with binary endpoints.
Key words: exploratory trial, proof-of-concept study, sample size, interim monitor-ing, stopping rule.
1. は じ め に 1900 年代中頃に臨床試験の方法論がほぼ確立して以来,統計的評価の方法として,フィッシャー 流あるいはネイマン・ピアソン流の頻度論(frequentist)に基づく仮説検定・推定が主に用いられて きた.ベイズ流(Bayesian)アプローチのデータ解析への応用は,物理学をはじめとする多くの自然 科学分野および社会科学分野ですでに広く行われている.医学・生物学分野においても,データ解 析にベイズ流階層モデルなどを適用した事例は数多く存在する.しかしながら,データ解析にでは なく,臨床試験のデザイン(実験の計画)とモニタリング(実験の逐次的な監視)にベイズ流アプロー チを適用した事例は,悪性腫瘍領域の第 I 相試験に用いられている CRM(continual reassessment method)を除いては,未だそれほど多くない(O’Quigley et al, 1990; Ishizuka and Morita, 2006).
大学等の研究機関で実施するトランスレーショナルリサーチおよび臨床試験には,対象疾患が 難治性かつ重篤であり,しかも患者数が限られているという特徴がある.このような状況におい ては,基礎研究で認められたコンセプトを実証するための POC 試験(proof of concept study)と 呼ばれる探索的な性格の強い試験が主であり,疾患の重篤性などを考えるとプラセボ対照は当然 のこと同時対照を設定すること自体が困難な場合が多い.また,被験者のリスクを最小にするた めに臨床試験の途中で結果をモニタリングしながら意思決定を行うというような柔軟な対応も必 要である.さらに,試験の被験者数を最小にするために,存在する証拠や情報(事前情報)を十分 に生かすことも重要となる.これらの目的を達成するためには,従来の頻度論的アプローチに基 づく推論は試験デザインに完全に依存するため,予期しない事態が発生して試験途中でのデザイ ン(標本サイズあるいは中間解析の時期や方法など)の変更を行った場合などに,柔軟性などの観 点からやや不十分であり,新規臨床試験デザインの開発が不可欠となる.ベイズ流アプローチは, 柔軟性と効率性の面から有望である(Berry, 2004; Berry 2005; Berry, 2006).臨床試験におけるベ イズ流アプローチの主な特長は以下の通りである. ⃝1 解釈が容易な「確率」だけを用いて整合性のある推測と意思決定を行うことができる ⃝2 標本サイズが小さくても事前分布を事後分布に更新して推測ができる ⃝3 予測分布を用いて試験結果を予測することができる また,ベイズ流臨床試験デザインの動作特性(operating characteristics)が頻度論的に評価可能で あることも 1 つの利点と考えられている. 同時対照群を置かない単群臨床試験は,探索的な臨床試験(exploratory study)の大部分を占め, その主目的は,治療効果に対する確定的な証拠を得ることではなく,さらに研究を継続すべき有望 な治療をスクリーニングすることである.単群試験デザインの多くは,効果が認められない場合は 試験を早期中止することが望ましい致死的な疾患(主に悪性腫瘍)の領域で開発されてきた(Green et al, 2003).悪性腫瘍領域の第 II 相単群臨床試験については,1960 年代から多くの頻度論的手 法が提案され,1990 年代以降ベイズ流アプローチを用いたデザインがいくつか提案されている (Thall and Simon, 1994; Heitjan, 1997; Tan and Machin, 2002).Thall and Simon (1994) は標 本サイズの決定法と事後分布に基づく多段階モニタリング法,Heitjan (1997) は事後分布に基づ く説得確率(persuasion probability)を用いたモニタリング法,Tan and Machin (2002) は事後分 布に基づく 2 段階モニタリング法とそれに基づく標本サイズ決定方式を提案している.その他に
も効用(損失)関数を明示的に用いるベイズ流決定理論に基づく手法の提案も数多く存在する(例え
ば,Sylvester, 1988; Stallard, 1998).しかしながら,決定理論に基づくアプローチは多くの不確 かなパラメータを設定する必要があり,実践上の有用性が低いと考えて本論文では取り扱わない. 従って,本論文では,Spiegelhalter et al. (2004) による Bayesian の流派に関する分類(empirical, reference,proper,decision-theoretic)に従うと,「Proper Bayesian の立場」,すなわち「利用で きる情報を事前分布に反映させた上で,事後分布あるいは予測分布に基づく推定および決定を行 うという立場」から実践的な手順を中心に解説する.
これまでの報告の多くは,事前分布の選択,標本サイズの決定,中止規則など試験デザインの ある一側面に焦点を当てている.しかしながら,ベイズ流アプローチが期待されるほど普及して
いない現状を鑑みると,デザインから解析・報告まで一連の手順についての指針が必要とされて いる.また,計算上の困難さも普及の妨げになっていると考えるため,本論文では基本的な統計 ソフトウェアで実施できる単純な方法を中心に記述する.赤池 (1989) は「ベイズ的方法の有効性 は,なんらかの公理によって保証されるものではなく,現実の問題に対して著しい有効性を示す ような適用事例の集積を通じてはじめて,広く一般に受け入れられるようになるものである」と 述べている.適用事例を増やすために簡便さは重要な要素である.第 2 節では,ベイズ流枠組み, 事前分布の選択,標本サイズの決定,中止規則,および臨床試験の実施手順について述べる.第 3 節では,ベイズ流臨床試験を理解できるように 1 つの事例を与える.第 4 節に実践的な指針を 与え,最後にまとめを述べる. 2. 方 法 2.1 ベイズ流枠組み ベイズ流統計学の基礎理論については,繁桝 (1985),渡部 (1999),中妻 (2007),松原 (2008) に詳しい.臨床試験に関する参考書としては,Spiegelhalter et al (2004) が卓絶している.単群臨 床試験の統計解析は,個々の患者あるいは患者集団において観察された二値のエンドポイント(以 下,「成功」または「失敗」と呼ぶ)から,ベータ分布を事前分布としたベイズ流アプローチを用 いて成功確率を推定することにより実施できる.ベータ分布 Beta(a, b) の確率密度関数は,区間 [0, 1] 上に定義され, f (x; a, b) = 1 B(a, b)x a−1 (1 − x)b−1 である.ここで,B(a, b) はベータ関数と呼ばれ, B(a, b) = Z 1 0 ta−1(1 − t)b−1dt と定義される.ベータ分布を用いると,パラメータ a (a > 0) と b (b > 0) の選び方によって多様 な分布を表現することができる.ベータ分布 Beta(a, b) の平均と分散は,それぞれ a a + b と ab (a + b)2(a + b + 1) で与えられる.また,一様分布 Beta(1, 1) を特殊型として含んでいる.さらに,この場合にベー タ分布は自然共役事前(natural conjugate prior)分布,すなわちある尤度に対して事前(prior)分 布と事後(posterior)分布が同一の分布族に属するという性質を持つ事前分布であり,ベイズ更新 に関する積分に数値計算を必要としないため,事後分布の導出が簡単である. 臨床試験において n 人の患者に独立した介入が行われたと仮定できるとき,関心のあるパラ メータである真の成功確率を π とすると,成功数 s は二項分布 Bin(n, π) に従う.ベイズ流の枠 組みにおいては,試験開始時に π はあるベータ分布 Beta(a, b) に従うと考える.これを事前分布 と呼び,事前分布のパラメータを超パラメータ(hyper parameter)と呼ぶ.n 人の患者に治療が実
施され,s 人に成功が観察された場合,ベータ分布が共役事前分布であるという性質から,ベイ ズの定理により成功確率 π の分布は Beta(a + s, b + n − s) に更新される.ある区間内にパラメー タ π が存在する確率が 1 − α のとき,その区間を 100(1 − α)% 信用区間または確信区間(credible interval,または credibility interval)と呼ばれる.本論文では,分布の両側に等確率 α/2 の端領 域をもつ区間(two-sided equi-tail-area intervals)を 100(1 − α)% 信用区間と定義する.
ベイズ流の方法では,現時点の観察データ(患者数 n と成功数 s)に基づいて,将来の結果(将来 の m 人の患者において成功数が k (= 0, . . . , m) になるであろう確率)を予測することが可能であ る.この場合にその確率はベータ二項分布と呼ばれる分布に従い,次の式から計算できる. Pr(K = k | s, n, m) = Γ(a 0+ b0) Γ(a0)Γ(b0) m k ! Γ(a0+ k)Γ(b0+ m − k) Γ(a0+ b0+ m) ここで,a0= a + s,b0= b + n − s であり,Γ(.) はガンマ関数と呼ばれる関数である.この予測分 布は,臨床試験の中間モニタリングにおいて試験を早期中止する規則(中止規則)を構築する際に 特に有用である. 2.2 事前分布の選択 ベイズ流枠組みにおいては,試験開始前に事前分布 Beta(a, b) の超パラメータ a と b の特定が 要求される.事前分布は,関心のあるパラメータ(真の成功確率)に関する知識をデータの観察に 先立って定量化する確率分布である.ベータ分布の超パラメータ a と b を決定する主な方法を以 下に示す. 1) 過去に実施された類似の臨床試験が存在するとき,その総対象者数を a + b(a を成功数,b を失敗数)とする.ここでは,過去の試験データと今から実施しようとする試験データが対等とみ なせるという条件が必要である.その上で,これらの証拠が同等の重みをもつとみなすことがで きないときは,過去の試験データの情報量 a + b を一定の割合で割り引くことを考慮しなければ ならない. 2) 事前情報に基づく成功確率の平均 M と分散 V から以下の式を用いて a と b を導出する. a =M [M (1 − M ) − V ] V b =(1 − M )[M (1 − M ) − V ] V この成功確率の平均および分散は,過去に実施された類似の臨床試験やそれらの要約,あるいは 専門家の意見から得られた情報に基づいて決定される. 3)利用できる事前情報が存在しないため,a = b = 1,すなわち一様(uniform)分布とする. 結果の頑健性を評価するために,懐疑的(sceptical)事前分布(治療効果について懐疑的あるいは 悲観的な態度を表わす)や熱狂的(enthusiastic)事前分布(治療効果について熱狂的あるいは楽観的な 態度を表わす)のような操作的な事前分布を用いることも可能である(Tan et al, 2002; Spiegelhalter et al, 2004).上記の方法 2)において,専門家の意見を導出(elicitation)するための様々な技法が提 案されている.それらは以下の 4 種類に分類される(Chaloner, 1996; Chaloner and Rhame, 2001; Tan et al, 2003; Spiegelhalter et al, 2004).
⃝1 略式の議論に基づく導出 ⃝2 構造化されたインタビューと正式な意見の集約 ⃝3 構造化された質問票を用いる導出 ⃝4 コンピュータに基づく導出 しかしながら,どのような技法を用いても専門家の選択,導出の時期などによるバイアスの影響を完 全には除去できないため,専門家による事前分布の導出については多くの批判がある(Spiegelhalter et al, 2004). 2.3 標本サイズの決定 ベイズ流本来の枠組みでは,試験開始前に標本サイズを決定する必要はなく,意思決定に十分 な情報が得られたと判断した時点で臨床試験を終了すればよい(Berry, 1993).しかしながら,ほ とんどの臨床試験においては,リソースの確保などの実際的な理由により試験開始前に最大標本 サイズ N を決定する必要がある.二値変数をエンドポイントとする単群臨床試験については,成 功確率の事後分布に基づく標本サイズの決定方法がいくつか提案されている(Thall and Simon, 1994; Tan and Machin, 2002; Mayo and Gajewski, 2004; Gajewski and Mayo, 2006; Whitehead, et al, 2008).Thall and Simon (1994) は精度に基づく方法と検出力(真に効果があるときに有効 と判断される確率)に基づく方法を提案している.Tan and Machin (2002),Mayo and Gajewski (2004),および Gajewski and Mayo (2006) は,検出力に基づく方法の改良版を提案しているが, 付加的なパラメータ(彼らの論文中の記法では ε )の設定が必要であり,実用的にやや問題がある
と考える.以下,p0を最小成功確率閾値(以下,対照値),p∗を最大成功確率閾値(以下,目標値)
として,Thall and Simon (1994) の精度に基づく方法と Whitehead et al. (2008) の事後分布に基
づく方法について述べる.p0は,もし真ならば評価をそれ以上続けるに値しない成功確率の上限 を示し,通常は試験対象での標準治療における成功確率を規準に設定する.一方,p∗は,もし真 ならば評価を続けるに値する成功確率の下限を示し,通常は p0より 0.05 ∼ 0.30 程度大きい値を 選択する. 1) 精度に基づく方法 試験終了時に,成功確率の事後分布が特定の精度を達成するように N を決定する.具体的に は,ある望ましい被覆確率(coverage probability)ω とある望ましい区間幅 δ を与えた下で,π の 事後分布(事後平均は p∗にほぼ等しいと仮定する)がその区間内に存在する確率が ω を超える,す なわち区間の下限 L = p∗− δ/2,区間の上限 U = p∗+ δ/2 として, Pr(L < π < U | data) > ω となるベータ事後分布のパラメータ a0と b0から N を決定する.すなわち,ベータ事前分布の超 パラメータを a, b とすると,N は (a0− a) + (b0− b) を超える最小の整数として決定される.通常, ω は 0.8 ∼ 1.0 の範囲,δ は 0.1 ∼ 0.3 の範囲で設定される. 2) 事後分布に基づく方法 試験終了時に,成功確率の事後分布に基づいて,「成功確率が対照値を超える確率がある値より も大きいので有効」,または「成功確率が目標値を超えない確率がある値よりも大きいので無効」
という二者択一の決定が行えるように N を決定する.具体的には,結果の確かさの程度を表す 2 つの値 η と ζ を与えた下で,π が p0を超える事後確率が η より大きい,すなわち Pr(π ≥ p0| data) ≥ η ならば「有効」と判断し,π が p∗を超えない事後確率が ζ より大きい,すなわち Pr(π < p∗| data) ≥ ζ ならば「無効」と判断する.より具体的には,標本サイズ n における成功数の棄却値(「有効」と 「無効」を分ける値)を unとすると, Pr(π ≥ p0| un) ≥ η かつ Pr(π < p∗| un− 1) ≥ ζ を満たす全ての整数 n と unの組合せから最小の n として N を決定する.通常,η と ζ は 0.8 ∼ 1.0 の範囲で設定される. 2.4 中 止 規 則 最大標本サイズ N に達する前に試験結果を評価することを中間モニタリング(interim monitoring) という.主に悪性腫瘍領域の第 II 相試験で用いられている多段階デザイン(通常は,2 段階デザイ ン)と呼ばれる頻度論的アプローチの特徴を以下に示す(Green, 2006).特に,⃝1 の特徴は「多重 性」というベイズ流の立場から見ると奇異に感じる問題に関連している. ⃝1 中間モニタリングの実施回数によって最終時点での推測( P 値と信頼区間)を調整しなけれ ばならない ⃝2 計画の変更(例えば,中間時点あるいは最終時点での解析が計画した標本サイズで実施でき ないなど)による棄却値の変更が容易ではなく,それが最終時点の推測にも影響する(Koyama and Chen, 2008) 試験途中で個々の患者あるいは患者集団において観察された結果(成功または失敗の組)につい ての情報を得て,関心のあるパラメータの分布を逐次更新するベイズ流の枠組みは,中間モニタ リングと基本的に相性がよい.現在まで,いくつかのベイズ流の中止規則(stopping rule)が提案 されている.それらは,中間時点における成功確率の事後分布(Tan et al, 2002),試験を最後ま
で継続すると仮定したときの予測成功数(すなわち,総成功数の予測分布)(Lee and Liu, 2008),
あるいはある人数を追加したときの推定精度の予測増加量に基づいている(Zohar et al, 2008).ま た,これらの方法をいくつか組み合わせて用いることも可能である. 1) 事後分布に基づく方法(有効中止) 中間時点での事後成功確率が目標値を超える確率が大きい場合に「有効」として臨床試験を中 止する.すなわち, Pr(π > p∗| data) > τS
となったときに中止する.通常,τSは 0.8 ∼ 1.0 の範囲で設定される. 2) 事後分布に基づく方法(無効中止) 中間時点での事後成功確率が対照値を超えない確率が大きい場合に「無効」として臨床試験を 中止する.すなわち, Pr(π < p0| data) > τF となったときに中止する.通常,τF は 0.8 ∼ 1.0 の範囲で設定される. 3) 予測分布に基づく方法(有効中止) 中間時点( n 人のうち,s 人が成功という結果が得られた時点)で,m 人を追加したときに,累積 予測成功数が uN(最大標本サイズ N における成功数の棄却値)を超える確率が大きい場合に「有 効」として臨床試験を中止する.すなわち, m X i=uN−s Pr(K = i | s, n, m) > τSP となったときに中止する.通常,τSP は 0.8 ∼ 1.0 の範囲で設定される. 4) 予測分布に基づく方法(無効中止) 中間時点( n 人のうち,s 人が成功という結果が得られた時点)で,m 人を追加したときに,累 積予測成功数が uN− 1(最大標本サイズ N における成功数の棄却値 −1)を超えない確率が大き い場合に「無効」として臨床試験を中止する.すなわち, uNX−s−1 i=0 Pr(K = i | s, n, m) > τF P となったときに中止する.通常,τF P は 0.8 ∼ 1.0 の範囲で設定される. 5) 信用区間幅の予測変化量に基づく方法 中間時点( n 人の結果が得られた時点)での成功確率の 1 − α% 信用区間の幅を W1−α,n(π) とす る.その時点以降に m 人を追加したときの成功確率の精度の最大予測増加量(信頼区間幅の最大 予測減少量)がある値より小さい場合,すでに十分な推定精度があるとして試験を中止する.すな わち, max |Wα,n+m(π) − Wα,n(π)| < ξ となったときに試験を中止する.通常,ξ は 0.02 ∼ 0.15 の範囲で設定される. 2.5 実 施 手 順 試験開始前: 1) 臨床研究者と試験統計家は事前情報を収集し,ベータ事前分布の特性について決定する(2.2 節参照). 2) 最大標本サイズ N を計算により決定する(2.3 節参照),あるいは実施医療機関の登録可能数 から決定する. 3) 臨床研究者が試験に中止規則を導入したい場合,試験統計家は試験の特徴に合った利用できる 中止規則を臨床研究者に説明し,中止規則の設定に必要ないくつかの値を決定する(2.4 節参照).
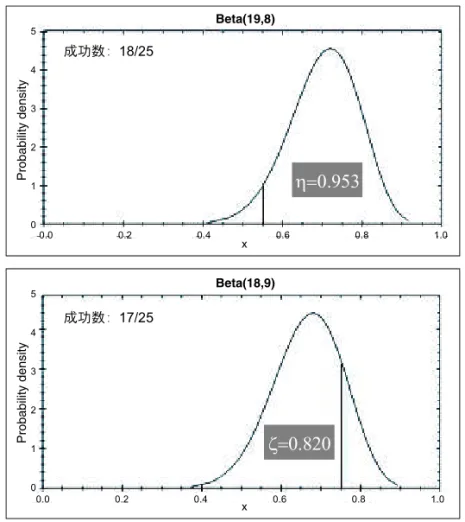
4) 試験統計家は試験の動作特性をシミュレーションにより評価する. 試験開始後: 1) 患者が試験に逐次的に登録される. 2) 中止規則が設定されている場合,患者あるいは患者グループの結果が明らかになった時点で, 試験統計家は逐次的に中間モニタリングを行い,中止規則に基づいて中止あるいは継続の判断を 行う. 3) 全ての患者の結果が明らかになった後,試験統計家は最終的な成功確率の代表値とその信用 区間を推定する. 3. 事 例 京都大学医学部附属病院で 2004 年から実施中の生体肝移植患者に対するドナー血輸血の効果 を評価する臨床試験を例として試験開始までの手順を説明する.本試験は,単群試験であり,主 要エンドポイントは移植後 6 か月以内の拒絶反応の有無(二値変数)である.ここで,拒絶反応が なかった場合を「成功」,拒絶反応があった場合を「失敗」と定義する.今回説明のために,実際 の試験デザインを一部変更したものを示すが,デザインの基本的枠組みは変更していない. 1) 事前分布の選択 試験治療について過去の客観的情報が存在しなかったため,一様事前分布 Beta(1, 1) を選択し た(2.2 節 3)参照). 2) 標本サイズの決定 試験実施医療機関における過去 5 年間の生体肝移植患者(n = 155)の成功確率が 0.55(95% 信頼 区間: 0.47 − 0.63)と推定されたことから,最小成功確率閾値(対照値)p0を 0.55 と設定した.最 大成功確率閾値(目標値)p∗は,試験を統括する臨床研究者の意見により 0.75 と設定した.最大 標本サイズは事後分布に基づく方法(2.3 節 2)参照)を用いて決定した.η を 0.95,ζ を 0.80 と設 定すると,最大標本サイズ N は 25,成功数の棄却値 uNは 18 と計算された.ちなみに,成功数 が 18 という結果が得られたときの η は 0.953,成功数が 17 という結果のときの ζ は 0.820 とな る(図 1). 3) 中止規則 最小標本サイズを 10 と設定し,被験者の登録速度などを考慮して 10 例から 5 例おきに中間モ ニタリングを行う,すなわち 10 例,15 例,20 例,25 例の結果が蓄積された時点で最大 4 回の 解析を行うこととした.中止規則は,「有効中止」と「無効中止」の両方を考慮する予測分布に基 づく方法(2.4 節 3), 4)参照)を用いて設定した.パラメータ τSP と τF P を 0.90 と設定すると図 2 に示す中止境界が得られた. 4) 動作特性 設定した試験デザインの動作特性を評価するためにシミュレーション(10000 回)を行った結果 を表 1 に示す.真の成功確率が 0.55(対照値)のとき「有効」という結果が得られる割合(頻度論 的方法の第 I 種の過誤確率に相当するもの)は 0.073 (728/10000),真の成功確率が 0.75(目標値)
図 1. 最大標本サイズの決定における各パラメータの関係
のとき「無効」という結果が得られる割合(頻度論的方法の第 II 種の過誤確率に相当するもの)は 0.287 (2870/10000) であった.期待標本サイズはそれぞれ 14.4, 17.7 であり,最大標本サイズ 25 よりも 7∼10 例程度小さい値であった.ちなみに,3 回(10 例,15 例,20 例)の中間モニタリン グを実施しない場合の動作特性は,真の成功確率が 0.55 のとき「有効」という結果が得られる割 合は 0.064,真の成功確率が 0.75 のとき「無効」という結果が得られる割合は 0.273 であり,中 間モニタリングを加えることによって動作特性は大きく変化しなかった. 表 1. シミュレーションによる動作特性 真の成功確率 結果 解析 期待標本 サイズ 1 回目 2 回目 3 回目 最終 計 (10 例) (15 例) (20 例) (25 例) 0.55 有効 240 30 99 359 728 14.4 無効 4869 2578 630 1195 9272 0.75 有効 2530 659 1567 2374 7130 17.7 無効 732 855 164 1119 2870 4. 実践的指針 4.1 留 意 点 臨床試験を計画する研究者は,その試験特有の事情に合ったデザインを選択しなければならな い.また,仮定がデザイン特性にどの程度影響を与えるかについて感度分析(sensitivity analysis) を行うべきである.少なくとも,対照値と目標値,および事前分布(具体的にはベータ分布のパラ メータ a と b)の選択によって最大標本サイズがどの程度異なるかを知っておくことは重要である. 2.3 節 2)に示した事後分布に基づく最大標本サイズの決定方式は,事前情報の量(ベータ事前分 布のパラメータ a と b の和)と算出される標本サイズ N の和,すなわち a + b + N がほぼ一定に なるという特徴を持つ.しかしながら,事前情報の量が一定の値を超えると,棄却数が un= 1, あるいは un= n(標本サイズ)で頭打ちとなり,計算不能に陥るという欠点を持つので,注意が 必要である. 中間モニタリングの導入は任意であるが,難治性の重篤な疾患を対象とする場合は,少なくと も期待した効果が得られそうにないときの「無効中止」を考慮すべきである(2.4 節 2)あるいは 4)の方法).実際に悪性腫瘍領域の第 II 相試験においては,中間モニタリングで効果が明らかに なったとしても,より精度の高い効果に関する推定値を得ること,また安全性プロファイルなど の追加情報を得ることが優先されるため,「有効中止」が正当化されることは稀である(Green et al, 2003).しかしながら,非常に有望な新規治療法を評価するときには「有効中止」を考慮した くなる場合もあると考える.いずれにしても,中間モニタリングを導入する際には,効果および 安全性を含めて評価可能な標本サイズの下限として最小標本サイズを決めておくことを推奨する. 中間モニタリングの回数,設定したパラメータによって動作特性がどの程度変化するかについて の感度分析も重要である.
4.2 ソフトウェア 1) 事前分布の選択 ベータ分布のパラメータ a と b から形状を評価したり,平均と分散から a と b を求めたりする際に は,テキサス大学 MD アンダーソン癌センターの生物統計・応用数学科が無償で提供する計算ツール 「Parameter Solver」が有用である(http://biostatistics.mdanderson.org/SoftwareDownload/). 2) 標本サイズの決定 精度に基づく方法については,前述の「Parameter Solver」が利用できる.事後分布に基づく 方法については,Whitehead et al (2008) に SAS プログラムが提供されている. 3) 中止規則の設定および中間モニタリングの実施 Zohar et al (2008) が提供する R プログラムを用いれば,ベータ事前分布と中間時点のデータ から事後分布,予測分布の計算などが実行できる.本論文の著者(手良向)に連絡いただければ本 プログラムは提供可能である. 5. ま と め 本論文では,二値変数をエンドポイントとする単群臨床試験におけるベイズ流アプローチにつ いて事例を用いて解説するとともに,いくつかの実践的な指針を与えた.臨床試験のエンドポイ ントとして有益な作用を例にとって説明したが,有益でない作用(有害事象など)をエンドポイン トとした場合においても,例えば有害事象の発生確率として許容できない閾値を考えれば同様の アプローチが可能である(Chen et al, 2006).成功確率ではなく有害事象の発生確率を考える場合, 頻度論的な意味での第 I 種の過誤と第 II 種の過誤の解釈および相対的重要度が異なってくるので 注意が必要である.また,ここで解説した単純な設定における手法を,複数群あるいは異なる型 のエンドポイントに拡張した手法については,悪性腫瘍の領域ですでに多くの提案がある(Thall et al, 1995 ほか多数). ベイズ流アプローチで実施された試験は罰則なしでいつでも情報を更新することができる(Berry, 2006).また,予測確率についての推論を行うことが可能である.しかしながら,探索的な臨床試 験では利点とみなされるこれらの特徴が,試験デザインとデータ解析の整合性(integrity)を強く 要求する検証的な臨床試験においては欠点とみなされることがある.従って,本論文で解説した 方法は,探索的試験,あるいは十分な標本サイズを確保することが困難な希少疾患における臨床 試験などに限定して用いた方が当面の理解を得やすいと考える. ベイズ流アプローチを臨床試験に用いる際には,臨床研究者と試験統計家の真の協同が重要で ある.臨床研究者は,既存あるいは外部情報の確かさ,成功確率閾値の見積もり,試験治療の臨 床試験を継続するかどうかを決定する確率などについて,専門家としての見解を有している.試 験統計家は情報の確かさを含めた事前情報の内容について臨床研究者と議論し,実施可能な標本 サイズや被験者の集積率について議論しなければならない.結果を報告する際には,成功確率の 推定が試験開始時から終了時までどのように推移したかを示す必要がある.中止規則の詳細や事 後成功確率の精度(信用区間)のような他の重要な情報も報告すべきである.近年,臨床試験にベ イズ流アプローチが有用であるという報告が増えつつある.米国 FDA の医療機器・放射線保健
センター(Center for Devices and Radiological Health)は,企業と FDA スタッフ向けに臨床試験 におけるベイズ流統計学の利用に関するガイドライン案を公表した(FDA, 2006).今後,このよ うな新しい臨床試験デザインの開発は,医薬品や医療機器の承認申請にインパクトを与える可能 性がある.特に,資源を有効に活用するという観点から効率よく臨床試験を行うことが今後ます ます重要になるであろう. 謝 辞
本総説の内容について,Dr. Sarah Zohar(Inserm, France)と交わした議論はたいへん有益でし た.また,貴重なご意見を頂きました査読者の先生方に厚く御礼申し上げます.
参考文献
赤池弘次. (1989). 事前分布の選択とその応用. In: 鈴木雪夫,国友直人編. ベイズ統計学とその応 用, 東京大学出版会, pp81-98.
Berry, D. A. (1993). A case for Bayesianism in clinical trials. Statistics in Medicine 12, 1377-1393; discussion 1395-1404.
Berry, D.A. (2004). Bayesian statistics and the efficiency and ethics of clinical trials. Statistical Science 19, 175-187.
Berry, D. A. (2005). Introduction to Bayesian methods III: Use and interpretation of Bayesian tools in design and analysis. Clinical Trials 2, 295-300.
Berry, D. A. (2006). Bayesian clinical trials. Nature Reviews – Drug Discovery 5, 27-36. Chaloner, K. (1996). Elicitation of prior distribution. In: Bayesian Biostatistics, Berry, D. A.
and Stangl, D. K. (eds.), 141-156. New York: Marcel Dekker.
Chaloner, K. and Rhame, F. (2001). Quantifying and documenting prior beliefs in clinical trials. Statistics in Medicine 20, 581-600.
Chen, C. and Chaloner K. (2006). A Bayesian stopping rule for a single arm study: With a case study of stem cell transplantation. Statistics in Medicine 25, 2956-2966.
FDA. (2006). Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials – Draft Guidance for Industry and FDA Staff Rockville, MD.
http://www.fda.gov/cdrh/osb/guidance/1601.html
Gajewski, B. J. and Mayo, M. S. (2006). Bayesian sample size calculations in phase II clinical trials using a mixture of informative priors. Statistics in Medicine 25, 2554-2566.
Green, S. (2006). Overview of phase II clinical trials. In: Handbook of Statistics in Clinical On-cology, 2nd edn, Crowley, J. and Ankerst, D. P. (eds.), 119-129. Boca Raton: CRC press. Green, S., Benedetti, J. and Crowley, J. (2003). Clinical Trials in Oncology, 2nd edn. Boca Raton:
Chapman and Hall/CRC.
Heitjan, D. F. (1997). Bayesian interim analysis of phase II cancer clinical trials. Statistics in Medicine 16, 1791-1802.
Ishizuka, N. and Morita S. (2006). Practical implementation of the continual reassessment method. In: Handbook of Statistics in Clinical Oncology, 2nd edn, Crowley, J. and Ankerst, D. P. (eds.), 97-116. Boca Raton: CRC press.
Koyama, T. and Chen H. (2008). Proper inference from Simon’s two-stage design. Statistics in Medicine 27, 3145-3154.
Lee, J. J. and Liu, D. D. (2008). A predictive probability design for phase II cancer clinical trials. Clinical Trials 5, 93-106.
松原 望 (2008). 入門ベイズ統計. 東京図書.
Mayo, M. S. and Gajewski, B. J. (2004). Bayesian sample size calculations in phase II clinical trials using informative conjugate priors. Controlled Clinical Trials 25, 157-167.
中妻照雄 (2007). 入門ベイズ統計学. 朝倉書店.
O’Quigley, J., Pepe, M. and Fisher, L. (1990). Continual reassessment method: a practical design for phase I clinical trials in cancer. Biometrics 46, 33-48.
繁桝算男 (1985). ベイズ統計入門. 東京大学出版会.
Spiegelhalter, D. J., Abrams, K. R. and Myles, J. P. (2004). Bayesian Approaches to Clinical Trials and Health-Care Evaluation. Chichester: John Wiley & Sons Ltd.
Stallard, N. (1998). Sample size determination for phase II clinical trials based on Bayesian decision theory. Biometrics 54: 279-294.
Sylvester, R. J. (1988). A Bayesian approach to the design of phase II clinical trials. Biometrics 44: 823-836.
Tan, S. B., Chung, Y. F. A., Tai, B. C., Cheung, Y. B. and Machin, D. (2003). Elicitation of prior distributions for a phase III randomized controlled trial of adjuvant therapy with surgery for hepatocellular carcinoma. Controlled Clinical Trials 24, 110-121.
Tan, S. B. and Machin, D. (2002). Bayesian two-stage designs for phase II clinical trials. Statistics in Medicine 21, 1991-2012.
Tan, S. B., Machin, D., Tai, B. C., Foo, K. F. and Tan, E. H. (2002). A Bayesian re-assessment of two phase II trials of gemcitabine in metastatic nasopharyngeal cancer. British Journal of Cancer 86, 843-850.
Thall, P. F. and Simon, R. (1994). A Bayesian approach to establishing sample size and moni-toring criteria for phase II clinical trials. Controlled Clinical Trials 15, 463-481.
Thall, P. F., Simon, R. and Estey, E. H. (1995). Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Statistics in Medicine 14, 357-379.
渡部 洋. (1999). ベイズ統計学入門. 福村出版.
Whitehead, J., Valdes-Marquez, E., Johnson, P. and Graham, G. (2008). Bayesian sample size for exploratory clinical trials incorporating historical data. Statistics in Medicine 27, 2307-2327. Zohar, S., Teramukai, S. and Zhou, Y. (2008). Bayesian design and conduct of phase II single-arm clinical trials with binary outcomes: A tutorial. Contemporary Clinical Trials 29, 608-616.