授業展開
6/17 抹消神経作用薬
(プリント)
6/24 中枢神経作用薬
(第8章)
7/1
抗菌薬
(第14章)
7/8 消化性潰瘍薬
(第12章)
7/18 循環器作用薬
(第9章)
7/22 抗炎症薬
(第10章)
糖尿病薬
(第11章)
教科書:
ベーシック薬学教科書シリーズ 創薬科学・医薬化学
橘高敦史
[編] (化学同人)
化学構造と薬理作用 医薬品を化学的に読む
柴崎正勝ら 監修 (広川書店)
14章 抗菌薬
β—ラクタム系抗生物質
ペニシリン系
セファロスポリン系
ペネム系
合成抗菌薬
スルホンアミド系
キノロンカルボン酸系
マクロライド系抗生物質
クロムフェニコール
アミノグリコシド系抗生物質
グリコペプチド系抗生物質
選択毒性
細菌と動物細胞の違い
① 細胞壁
細胞壁合成酵素阻害剤
Ex) ペニシリン, バンコマイシン
② ビタミン合成酵素系
代謝拮抗薬
Ex) サルファ剤
③ リボソームの違い
タンパク質合成酵素阻害
Ex) マクロライド系抗生物質
④ 転写・複製系の違い
DNAジャイレースの阻害
Ex) キノロン系抗生物質
7
−3 ヒスタミン関連医薬品
ヒスタミンの生合成
102 チグミン rivastigmine が臨床で用いられている( 図7-2-10). いずれの薬剤もアルツハイマ ー病 症状の改善に有効であるが,あくまでも対症療法的であり 根本治療法薬の開発が急務である. m合
0〉O
へ
O
J b ZN CN
へ
o
r
、
C H3 H 3C 00三
D
。
へ
donepezil ドネペジル ICso 6.7 n M 図7-2-10 9H rデ/ ¥ 、/ ' ¥ 込γ / ¥、 ( " ';1 11 0 ‘、
h、 心 / 、ノ¥ ーチ¥ . / ¥ ¥ / "N'
...,. 卜1 3C O ¥/λ、./'"¥ tacrinel λj
タクリン galar1thamine-bH3 ガ 、ランタミン。
r
、
Q H3 C H3〈m-o
ハJ ヘγN、C H3 C H3 C H3 rivastigmine リパスチグミン!日
ヒスタミン関連医薬品
│
ヒスタミン histamine はイミダゾール環の 4位にメチレン基2個を隔ててアミノ基 (NH2) が 結合した構造をもっ塩基性化合物である 生体内生物活性アミンの一種であり,アミノ酸の lつ であるヒスチジン histidine が Lーヒスチジン脱炭酸酵素 L-histidine decarboxylase により脱炭酸されて生成する( 図 7-3-1) • 内 4 U H M H N O / O Yl c
j
N 夕 、 N / 4 H C O2 L-ヒスチジン)J見炭酸酵素¥
L -h istid ine L -ヒスチジン ヒスタミンhistamine 図7-3-1 ヒスタミンの生合成 体反応などの刺激によって貯蔵されていたヒスタミンが脱頼粒により 細胞外へ遊離される. 一方, 胃粘膜のエン テロ クロマフイン様 (ECL ) 細胞やヒスタミン作動' 性中枢神経系ニュ ーロンでは必 要に応じて生成され遊離される. 第7 章 化学構造と薬理作用 103 遊離されたヒスタミンは細胞膜表面に存在する特異的受容体と結合する. 4種類のヒスタミン 受容体,すなわち H I. H2. H3. H1受容体に結合したヒスタミンは,様々な生理および病態生理 作用を引き起こす( 図 7-3-2). これらの受容体はすべて. 7 回膜貫通型G タンパク質共役受容 体 (GPCR) である.山
│ ヒスタミン神経 │ C a2+ ↑ c AM P ↑ 気管支収縮(1111前息) 血管内皮剥IIJJ包N O産主E充進 (JfJl'i:;i'1IL: │削玉) I血管透過性充進( 炎症) 知覚神経終末興イ! f ( 発揃.j長捧) 11111時型( I 型) アレルギー¥
/
│ 神経輿管 ( 覚醒など)I
図 7-3-2 ヒスタミンとヒスタミン受容体 (ティ・エヌ・エス 薬理学 ・薬物治療学. p.l55) 種々の物理的, 化学的刺激や抗原抗体反応によ って肥満細胞や好塩基球中に貯蔵されていたヒ き起こす. 一方,胃粘膜の E C L 細胞から遊離されたヒスタミンは,胃の壁細胞のヒスタミン H2 受容体に作用して, 胃酸を分泌させる. また. JJ品ではヒスタミンを神経伝達物質とするヒス タミ ンニュ ーロ ンが存在し,シナプス後) 慌の H,およびH2受容体を介して覚醒や食欲抑制などに関 与 している この神経の シナプス前! 院にはヒスタミ ン遊離を抑制す るH3受容体が分布する (図 7-3-2). H4受容体は主 として骨髄と循環血液中の血球系細胞に見られる (表 7-3-1) • ヒトでは遊離されたヒ スタミンは,主に 2 つの経路によって代謝を受ける. 1つは,イミダゾ ール N -メチルトランスフエラーゼにより I 位窒素がメチル化され, その後モノアミンオキシダ ーゼにより N -メチルイミダゾリル酢酸に代謝される. もう Iつは,ヒスタミナーゼにより 4位 側鎖が酸化的脱アミノ化されイ ミダゾ リル酢酸となった後 最終的にイ ミダゾ リル酢酸リボシド として代謝される (図 7-3-3). ほとんどのヒスタミンは代謝を受け不活性化され,尿中に排j世 される.ヒスタミン
H1受容体拮抗薬
第一世代ヒスタミン
H1受容体拮抗薬(抗ヒスタミン薬)
1067-3-1-1
第一世代ヒスタミン
Hl
受容体措抗薬 ヒスタミン Hl 受容体措抗薬は,ヒスタミンと H l受容体との相互作用を可逆的かつ競合的に 阻害する薬物である. 1937 年に最初の Hl 受容体措抗作用を示す化合物が発見され 1940 年代に臨床的に用いられ る薬物が登場 した. 第一世代 Hl 受容体措抗作用を示す薬物のいくつかを表 7-3-2 に示した Hl 受容体措抗薬の構造は,図 7-3-4 の一般式の ように表す こと ができる.ぐ対抗〈
く〉-汁片
Nく
n = 2 または 3x =
N, 0 または省略 y = c または N A r = 芳香族化合物 図 7-3-4 ヒスタミンの構造( 左) と Hl 受容体措抗薬 ( 右) の一般式 ヒスタミンと第一世代 Hl 受容体措抗薬の構造的類似性と相違を以下にまとめた 1) ヒスタミンにおける側鎖アミンは第一級であるが,第一世- 代の薬物は,ジメチルアミノ基ま たはピ ロリ ジン,ピベ リジンやピペラジンのような環構造の一部となっている第三級アミン である( 表中太線で示した一部) 2) ヒスタミンにおける メチ レン基は 2個であるが,第一世代の薬物は, 2個または 3個である ( 表中太線で示 した一部)• 3) ヒスタミンにおいてはエチル基 と複素環式芳香族化合物であるイミダゾール環は直結 して い るが,第一世代の薬物では,エチル基やプロピル基が炭素原子 (C) ,酸素原子 (0) や窒素 原子 (N) を介 して芳香族化合物と結合している. 4) ヒスタミンにおける芳香環はイミダゾールであるが,第一世代の薬物では,複素環式化合物 を含む芳香環を 2 個もつジア リル メチル基やそれらジア リルメチル基が結合 し三環性構造を 形成したもの,また,ジアリルメチル基の炭素原子が窒素原子(N)
に置き換わったものな ど様々である( 表中四角で示した部分)• しかし,すべての化合物において芳香族化合物を有 しているという点は共通 している. 第一世代 H l受容体措抗薬は,主に炎症反応やアレルギー反応にかかわる症状の藤和に用いら れるが,一方, 中枢神経での作用として鎮静,認知能力の低下,反応時間延長,食欲充進作用, 抗めまい作用,パーキ ンソン病の筋固縮減少がある . このため副作用として眠気,意欲減退や疲 労感を感 じることがあるが,逆にこれ らの作用は,不眠症,動揺病( 乗り 物酔い) の予防,メ ニ エール症候群に対する鎮量薬,初期のパーキ ンソン病の治療などに用いら れる. その他,抗セロトニ ン,抗ブラジキニン,抗コリンや局所麻酔作用をもつものもあり,それは おそらく H l受容体措抗薬の構造が それらの受容体に作用する薬物の構造と類似するためであ ると考えられている( 図 7-3-5). 第三級アミン ジアリールメチル基脂溶性が高く血液
−脳関門を通過しやすい。
そのため、中枢作用として鎮静、認知能力の低下、反応時間の延長な
どを引き起こす。
副作用として眠気、意欲減退、疲労感
ヒスタミン
H1受容体拮抗薬
第一世代ヒスタミン
H1受容体拮抗薬(抗ヒスタミン薬)
第7 準 化学構造と薬理作用105
2
、に伴う痘痔, 花粉症,ア レルギー性鼻炎などの症状が現れるH l
受容体措抗薬は,こ れらの 症状を緩和する 目的で使用さ れ, 副作用の強弱に より 第一 世代および第二世代 に分類される. 表7-3-2
第一世代ヒスタミンH l
受容体措抗薬の構造的類似性による分類口一州
Nく
口 一 ,一,-¥ -Ar/ Aヘ
y - X = Nn = 2
,またはO または省略3y = c
または N A r = 芳香族化合物X 1
n 構造と 化合物名 2/9)
H3)
(;ri'
O" ,,-N¥CH
3 diphenhydramine ジフェン ヒ ドラミン O 3 N 1 2λ
c
ベ
R
二
Y L
L
h o m ochlorcyc lizine ホモクロル シクリジンベ
F
hydroxyzine ヒド ロキシ ジン 2CAZYviH3
省 111告 chlorpheniramine クロルフェ ニラミ ン トリプロ リジンtriprolidine promethazine プロメタジン 3F
O
K
内三
- 第三級アミン部分は太線で,芳香族環の音1¥分は凶角で囲 っている . 第7 準 化学構造と薬理作用105
2
、に伴う痘痔, 花粉症,ア レルギー性鼻炎などの症状が現れるH l
受容体措抗薬は,こ れらの 症状を緩和する 目的で使用さ れ, 副作用の強弱に より 第一 世代および第二世代 に分類される. 表7-3-2
第一世代ヒスタミンH l
受容体措抗薬の構造的類似性による分類口一州
Nく
口 一 ,一,-¥ -Ar/ Aヘ
y - X = Nn = 2
,またはO または省略3y = c
または N A r = 芳香族化合物 X 1 n 構造と 化合物名 2/9)
H3)
(;ri'
O" ,,-N¥CH
3 diphenhydramine ジフェン ヒ ドラミン O 3 N 1 2λ
c
ベ
R
二
Y L
L
h o m ochlorcyc lizine ホモクロル シクリジンベ
F
hydroxyzine ヒド ロキシ ジン 2CAZYviH3
省 111告 chlorpheniramine クロルフェ ニラミ ン トリプロ リジンtriprolidine promethazine プロメタジン 3F
O
K
内三
- 第三級アミン部分は太線で,芳香族環の音1¥分は凶角で囲 っている . 第7 準 化学構造と薬理作用 1052
、に伴う痘痔, 花粉症,ア レルギー性鼻炎などの症状が現れる H l受容体措抗薬は,こ れらの 症状を緩和する 目的で使用さ れ, 副作用の強弱に より 第一 世代および第二世代 に分類される. 表 7-3-2 第一世代ヒスタミン H l受容体措抗薬の構造的類似性による分類口一州
Nく
口 一 ,一,-¥ -Ar/ Aヘ
y - X = Nn = 2,またはO または省略3 y = c または N A r = 芳香族化合物 X 1 n 構造と 化合物名 2/9)
H3)
(;ri'
O" ,,-N¥CH
3 diphenhydramine ジフェン ヒ ドラミン O 3 N 1 2λ
c
ベ
R
二
Y L
L
h o m ochlorcyc lizine ホモクロル シクリジンベ
F
hydroxyzine ヒド ロキシ ジン 2CAZYviH3
省 111告 chlorpheniramine クロルフェ ニラミ ン トリプロ リジンtriprolidine promethazine プロメタジン 3F
O
K
内三
- 第三級アミン部分は太線で,芳香族環の音1¥分は凶角で囲 っている .その他、表7
−3−2を参照。
脂溶性が高く、血液
−脳関門を通過しやすい。
第二世代型
H1受容体拮抗薬
第一世代との比較: アミノ基上の置換基(極性官能基の追加)
・ 生理的条件でイオン化されやすい。
・ アルブミンと結合しやすい。
血液
−脳関門の透過性の低下
第 7 章 化学構造と薬理作用 109 この中で,エパスチンは,カレパスチンのプロドラッグであり 肝初回通過効果により活性代 謝物に変化して,その作用を示す( 図7-3-6) .
エパスチン カレノ可スチン ( プロドラッグ) (Hl受容体指抗薬) 図7-3-6
工パスチンの代謝7-3-2
ヒスタミン H2
受容体括抗薬
消化器官は食物の消化,栄養の吸収,老廃物の排池を行う一連の器官であり,胃や十二指腸は 主 4こ食物剥自北に関係している . このため,通常,これらの器官では食物を消化するために胃酸 やペプシンなどが分泌される . 胃酸は一連の複雑な経路を経て最終的に壁細胞から分泌される. 旦酸分泌に関わる壁細胞内シグナルとしては, ① C a2+ に依存する経路, ② サイクリック A M P民
ム
M P
l.
に依存する経路の 2 つの経路が存在する. ガストリンとアセチルコリンは ① を介して作用を発現する . ガス トリ ンあるいはアセチルコ リンが壁細胞のそれぞれの受容体に結合すると細胞内の C a2+濃度を上昇させ,アデニル酸シク ラーゼの活性化を介して細胞内 c A M P レベルの上昇を引き起こす. c A M P は H +/K + ATPase を活性化し, 胃酸分泌を促進させる また, Ca2+濃度の上昇は,プロテインキナ ーゼの活性化, H + /K + ATPase の活性化を促し胃酸を分泌させる. ヒスタミンは ② を介 して作用 を発現する. ガスト リンあるいはアセチルコリンの受容体は, 壁細胞の近傍にある E C L様細胞にも存在する. E C L 様細胞はヒスタミンの主たる分泌細胞であ り,ガストリンあるいはアセチルコリンが. E C L 様細胞のそれぞれの受容体に結合するとヒス タミンが遊離され,壁細胞の H2 受容体に結合 し,アデニル酸シクラ ーゼの活性化を介し て細胞 内 c A M P レベルの上昇を引き起こす c A M P はH +/K + ATPase を活性化し 胃酸分泌を促進させ る( 図7-3-7).
このため,通常,胃や十二指腸では食物を消化するための攻撃因子( 胃酸やペプシンなど) と 置腸主制莫を守る防御因子( 粘液と重炭酸イオンの分泌,プロスタグランジン, 血流,細胞障害後 の修復と再生) のバランスが保たれている. しかし,ストレスなどの原因によりよれらのパラン るj)!.崩れるとL渓撃因ヨカヰ目対的に強まり潰蕩が形成される. 潰蕩の発生には胃酸が最も重要な因 子であるため. . r u 産分泌抑制 比胃 二十二指腸潰湯治療法のひとつとなる .表7
−3−3 を参照。
ヒスタミン
H2受容体拮抗薬
消化性潰瘍(第8章)で取り扱う。
H2ブロッカーは、ヒスタミンの化学構造をもとに開発された。
110/ ¥
G(CCK-B) (CC附 M] C a2+ ¥ と進効果 ヒスチ ジン 一一+ ヒス│タミン ECL 細胞 壁細胞 図 7-3-7 胃酸分泌の機構 ( 医科薬理学( 改訂凹版) , p.l16,南山堂) H+ /K+ -A TPase これまで述べたヒスタミン Hl 受容体括抗薬は胃酸分泌に影響 しない Hl 措抗薬では抑制さ れない胃酸分泌充進を抑制する薬物は,ヒスタミンの化学構造をもとに開発された. ヒスタミ ンの構造を少しずつ変化させ,構造一活性相聞を検討し最初の H2 括抗薬ブリマミド burimamide が見出された. その後,さらなる活性増強をめざして構造変換が行われメチアミド metiamide へと誘導され,ついに広く臨床的に用いられる 乙ぷJ 二シふムcimetidine の開発に成功した( 図 7-3-8) .ぐ
Y
JH2
ー
ヒスタミンぐ
X;x y¥
町 一 プリマミド: R = H , X = C H2 メチアミ ド・ R = C H3, X = S H H fl.1 ヘ / ¥ ,N、 ,N、マ
γ
、S ' v I
℃ H3 /N C H 3 川 ¥ C N H シメチジン 図7-3-8 ヒスタミンの構造変換によるシメチジンの開発経路 シメチジンにはアンドロゲン受容体への結合 エストラジオールの代謝阻害やプロラクチン分 泌刺激などの作用が見られる . これらの作用は,男性では女性化乳房,性機能障害,女性では乳 汁分泌過多を 引き起こす. また,チト クロムP450 (CYP)
を阻害して 他の薬物の代謝に影響を 及ぼす可能性がある. これらの副作用の軽減と更なる作用増強をめざし H2受容体措抗薬の開発 が続けられ,ラニチジン,ファモチジン,ニザチジン ラフチジン ロキサチ ジンアセタ ー トが 開発された. ファモチジンやラフチジンの場合には若干異なるが,これらの薬物は下に示す一般 式で表すことができ,共通する点が多い( 図 7-3-9)ヒスタミン
第7章 化学構造と薬理作用H2受容体拮抗薬
111匹件目イ;:;
物 基 合 換 化 置 族 い 香 き 芳 大 ﹁ l l J 1 ,J -﹁ 1 1﹂ ﹁ t L。
+& 4 た ま フ -Q u n × :=---l H H「ロケ「
svtH3i
川 U 2 仏 兵4 ジLンH216
十
μ
日刊
ranitidine ラニチジン 一 目 H 「 H 1均
叩
「
1
C仏l:
、コ
J
:X
工
合
yr
SNYIγγN
札、-N0
2 famotidine ユ.エヰ牛 込ン ニザチジンnizatidine一
l'W
し
lafutidine ラフチジン roxatidine acetate ロキサチ ジンアセター ト 図7-3-9 ヒスタミン H2受容体措抗薬 次にこ れ ら薬物 に共通する点を 以下に示 した. 1) X は硫黄または酸素原子であり, X と窒素原子の間に 2 - 4個のメチレン基をもっ. 2) 窒素原子上にかさ高い極性置換基を有する( 点線四角部分) . この点はメチル基などの小さな 置換基である第一世代 HI措抗薬 とは異なる . 3) X に直接あるいはメチレン基を介して置換芳香族化合物 I個と結合している (四角部分) • ロキサチジンア セタートを 除き,それら芳香族化合物は複素環式化合物である. この点は 2 個の芳香族置換基あるいは三環性芳香族置換基を有する HI 措抗薬とは異なる. H z受容体措抗薬は,ヒ スタミンの Hz受容体への結合を可逆的かつ競合的に阻害 し, 胃酸分泌 を抑制し,かつガストリンやアセチルコリンによって誘導される胃酸分泌も低下させる. このよ うに Hz受容体措抗薬は胃液分泌を特異的に抑制するため,比較的副作用の少ない 胃 ・十二指腸 潰傷の治療薬と して広く用い られている. 芳香族へテロ環が1個 かさ高い極性基βーラクタムの基本的な構造
250 抗 菌 薬 の違い→タンパク質合成阻害薬 クロラムフェニコール,マ クロラ イド系抗生物質,アミノグ リコ シド系抗生物質,テトラサ イク リン系 抗生物質,最近ではオキサゾリジノン系合成抗菌薬.5.
核酸の転写や複製にかかわる酵素の違い→DNA
ジャイレ ース(DNA
ギラーゼともいう) の匝害 . キノロン系抗菌薬,ニューキノロン系合成 抗菌薬.β
ーラクタム系抗生物質
細菌の細胞壁合成を阻害する医薬品の一つで、ある 日一ラクタム系抗生物質(
s
-lactam antibiotics)
は, 四員環ラクタム( 同一ラクタム.s-lactam)
骨格をもっ そのほとんどは,四員環ラクタム環がもう一つの環と縮環構成をと る
F
ラクタム系抗生物質はペニシリン(penicillin)
類に見られる ような四 員 環 ラ ク タ ム + 五 員 環 の 縮 環 構 成 の も の と セ フ ァ 口 ス ポ リ ン(
cephalosporin)
類に見られるような四員環ラクタム + 六員環の縮環構成の ものの,大きく二つに分類される 四員環ラクタム+ 五員環の縮環構成のものにはさらに 4 種類あり,ペニシ リンの ようなペナム骨格のもの,二重結合を含むペネム,硫黄原子が酸素原 子に置き換わったオキサベナム,あるいは炭素原子に置き換わりかつ二重結 合をもっカルパペネムに分けられる( 図14
,1) ,四員環ラクタム+ 六員環の 縮環構成のものには,セ フェム,オキサセフェム ,カルパセフェムの3
種類 がある 一方,二環性ではないモノパクタムとよばれるs-
ラクタム骨格だ けからなる誘導体もあるので,基本的な構造は8
種類である βーラクタム ラクタムは環状アミドの総 1'ir., sーラクタムではアミド 窒素原子がF位にあるので 四員環となるRJH
ヲ
;;:
;討会
R1
』
:;R1:1L 1
ベネム オキサペナム カルパペネム山
市
川
γ
o
wl
J
o
日
川
市
o
山l
J
S
NH
γ
o
RJP3
セフェム( セ7 7 ' 口スポリン) オキサセフェム カJレ1¥セフェム 笹翠m β
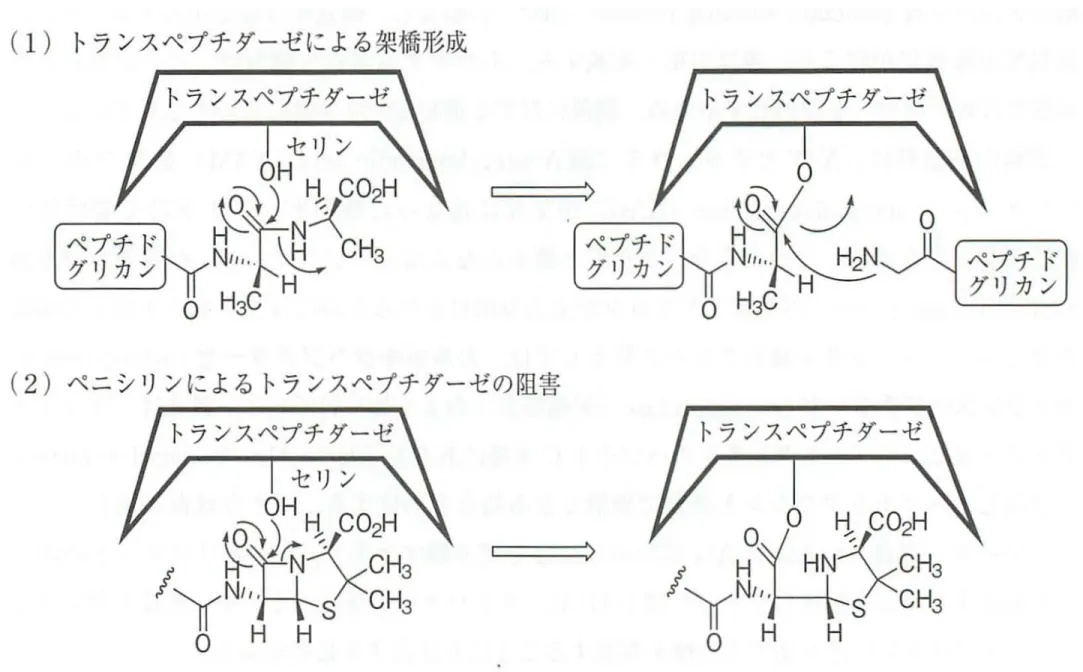
ラクタム系抗生物質の基本構造 細菌の細胞壁 はペプチドグリカン構造である. すなわち. N
ーアセチルム ラ ミ ン 酸(N-acetylmuramic acid ;
N A M) とN-
ア セ チ ル グ ル コ サ ミ ン細胞壁の生合成
。ーラクタム系抗生物質 (N-acetylglucosamine ; N A G)からなる糖鎖パックボーンがあり,ペプチド 鎖が N A M糖に結合している この細胞壁を合成する最終段階では,ムレイ ントランスペプチダーゼとよばれる酵素により,ペプチ ド鎖末端の 2 連続し たDーアラニンのうち最終の D アラニンと ,別のペプチド鎖の中盤に位置す るL - リシンの側鎖からペプチド結合で伸びた 5連続のグリシンの N 末端と が付け替わる さらに,別の糖鎖パックボーンと連結していく これを繰り 返して細胞壁の網目構造が構築される( 図 14.2) . . N V 帆N ( N A G Y N A M y帆N 帆P IL-Alal D-Ala ゼ ダ チ プ ペ ス 、 / -7 'h ド 、J J t レ ム D宇Ala D-Ala 曜翠I D
細胞壁ペプチドグリカン形成の最終ステップの模式図 ペニシリン類やセファロスポリン類が阻害するのは,この最終段階である ムレイントランスペプチダーゼの働きが阻害される と,細胞壁の網目状構造 を正しく築くことができない その結果,塩濃度の高い細胞内部へ浸透圧に より水が浸入し細胞は膨i
問 ・破裂する. なぜペニシリンやセファロスポリンはムレイントランスペプチダーゼの作 用を阻害できるのか ムレイントランスペプチダーゼが本来取り込むはずの ペプチド鎖末端の O-Ala-O-Ala-COOI- I がとる構造と,ペニシリンの 6 位側 鎖のアミド結合F
ラクタム環-2
位のカルボキシ基までがとるコンフォメー ションは非常に似ている そのため,ペプチ ドを転移させるために D-Ala D - Ala - C O O H が入る酵素の基質ポケッ トにペニシリンが代わりにはまり込 み,酵素の求核性基( ヒドロキシ基) がs-
ラクタム環を開瑞しながら反応し てアシル化 される この酵素は トランスペプチダーゼ (transpeptidase)であ るのでアシル化されたのち,活性中心付近には水分子が入れないようになっ 251 D-Ala D-Ala トランスペプチダーゼ ペプチド転移反応を触媒する 醇素 中間にアシル化酵素を 形成する N-アセチルムラミン酸 N-アセチルグルコサミンβーラクタムの作用機序
186
トラ ンス ペプチダーゼ
N A G : N-acetylglucosamine,トJ A M : N-acetylmuramic acid
図
7-11-3
ブドウ球菌細胞壁の生合成一
長
s
比
一
。
penicillin ペニシ リン守
主
fJ
0 -Ala-D-Ala 図7-11-4
立体構造の類似性 (1) トランスペプチダーゼによ る梨僑形成高幹
:::H
(2 ) ペニシリ ンに よるトランスペプチダーゼの阻害 トラ ンスペプチダーゼ/品琵
-P:了
京
5iγphJ
、
人
>
現
oh
一
。
図7-11-5
β—ラクタムはD-Ala-D-Alaのミミック構造
トランスベプチダーゼのはたらきとペニシリンによる阻害
ペニシリンの歴史
1877年 PasteurとJoubert
カビ類が最近を殺す物質を生産することが発見される。
1928年 Fleming
青カビのコロニーのまわりは、バクテリアが死滅。
1938年 FloreyとChain
ペニシリンの単離に成功
1944年 アメリカで大量生産
1945年 Hodgkin 構造決定(X線構造解析)
1957年 Sheehan ペニシリンの全合成
1958年 Beechams社 6-アミノペニシラミン酸の単離
ペニシリン
G
252
*
1章 6 i;i:も参照 チアゾリジン チアゾールを完全水素化した 次のような鱗造ふ
う
抗 菌 薬 ている 水分子がないので アシル化 された酵素が加水分解されて復活する 速度は非常に遅く,ムレイントランスペプチダーゼは事実上失活する14.2.1
ペニシリン
1877
年,Pasteur
とJoubert
は,あるカビ類が細菌を殺す毒性物質を産生 フレ E ン グ することを発見した* .1928
年にF le ming
は青カビ(Penicillium
の一種) の コロニーのまわりではバクテリアのコロニーが死滅していることに気づい フローリー チ ェ イ ン た.1938
年にF lorey
とC h
釦n
が凍結乾燥法を利用してペニシリンを単離し ホ ジ キ ン1944
年にはアメリカで大量生産に至った1945
年にD.
C.
Hodgkin
によりX
線構造解析がなされ,構造が確定した1957
年にはJ
C.
Sheehan
による ペニシ リンの全合成が達成された 翌年B eech a ms
社がペニシリン生合成中間体
6-
アミノペニシラン酸(6-aminopenicillanic acid ;
6-A P A )
を単離した( 図
14.3) . 6 - A P A
のアミノ 基をアシル化することでさまざまな半合成ペ ニシリンが合成できる この優れた誘導体研究の出発原料を得たことがその 後の広範囲なペニシリン誘導体研究へとつながった.げ科学
:: h M:::
ペニシリンG ペニシリンV ペニシリンG
およびペニシリンV
色アミで示した部分は共通するかアミノベニシラン酸(6-APA) ペニシリンはF
ーラクタム環とチアゾリジン(thiazolidine)
環が縮環した二 環性の構造で,システインとパリンから生合成される 側鎖のアシル基は発 酵培地の組成で変化する アメリカにおける最初の大量生産で、は, トウモロ コシ由来のフェニル酢酸(C
6H
5C H
2C O O
H ) が培地に多く含まれていたため, 側鎖R
はベンジル基 (ペニシリンG
とよばれる ) であった. こ れは発酵培地 に フ エ ノ キ シ カ ル ボ ン 酸 (C
6H
50 C H
2C O O
H ) を 添 加 す れ ば , 側 鎖R
がC
6H
50 C H
2 のペニシリンV
を生産で、きる ことを意味する さらに,より効率 のよい方法として前述のペニシリン生合成中間体の6-A P A
までを発酵で生 産し酸塩化物によるアシル化反応でさまざまなペニシリン誘導体が合成さ れた( 匡114.4)
また,ペニシリンG
やV
からペニシリンアシラーゼとよば れる酵素で加水分解する方法もある 現在では,より効率のよい方法として 化学的な脱アシル化によりかA P A
を合成している( セファ ロスポリ ンC
か らの7 - A C A
の合成法,図14.8
を参照) •6
位アミド側鎖の構造変換は,図14 .4
の方法でいろいろ検討できる 抗 菌活性の発現には ,s-
ラクタム環と遊離のカルボキシ基は必須で、あるが,252
*
1章 6i;i:も参照 チアゾリジン チアゾールを完全水素化した 次のような鱗造ふ
う
抗 菌 薬 ている 水分子がないので アシル化 された酵素が加水分解されて復活する 速度は非常に遅く,ムレイントランスペプチダーゼは事実上失活する14.2.1
ペニシリン
1877
年,Pasteur
とJoubert
は,あるカビ類が細菌を殺す毒性物質を産生 フレ E ン グ することを発見した* .1928
年にF le ming
は青カビ(Penicillium
の一種) の コロニーのまわりではバクテリアのコロニーが死滅していることに気づい フローリー チ ェ イ ン た.1938
年にF lorey
とC h
釦n
が凍結乾燥法を利用してペニシリンを単離し ホ ジ キ ン1944
年にはアメリカで大量生産に至った1945
年にD.
C.
Hodgkin
によりX
線構造解析がなされ,構造が確定した1957
年にはJ
C.
Sheehan
による ペニシ リンの全合成が達成された 翌年B eech a ms
社がペニシリン生合成 中間体6-
アミノペニシラン酸(6-aminopenicillanic acid ;
6-A P A )
を単離した( 図
14.3) . 6 - A P A
のアミノ 基をアシル化することでさまざまな半合成ペ ニシリンが合成できる この優れた誘導体研究の出発原料を得たことがその 後の広範囲なペニシリン誘導体研究へとつながった.げ科学
:: h M:::
ペニシリンG ペニシリンV ペニシリンG
およびペニシリンV
色アミで示した部分は共通するかアミノベニシラン酸(6-APA) ペニシリンはF
ーラクタム環とチアゾリジン(thiazolidine)
環が縮環した二 環性の構造で,システインとパリンから生合成される 側鎖のアシル基は発 酵培地の組成で変化する アメリカにおける最初の大量生産で、は, トウモロ コシ由来のフェニル酢酸(C
6H
5C H
2C O O
H ) が培地に多く含まれていたため, 側鎖R
はベンジル基 (ペニシリンG
とよばれる ) であった. こ れは発酵培地 に フ エ ノ キ シ カ ル ボ ン 酸 (C
6H
50 C H
2C O O
H ) を 添 加 す れ ば , 側 鎖R
がC
6H
50 C H
2 のペニシリンV
を生産で、きる ことを意味する さらに,より効率 のよい方法として前述のペニシリン生合成中間体の6-A P A
までを発酵で生 産し酸塩化物によるアシル化反応でさまざまなペニシリン誘導体が合成さ れた( 匡114.4)
また,ペニシリンG
やV
からペニシリンアシラーゼとよば れる酵素で加水分解する方法もある 現在では,より効率のよい方法として 化学的な脱アシル化によりかA P A
を合成している( セファ ロスポリ ンC
か らの7 - A C A
の合成法,図14.8
を参照) •6
位アミド側鎖の構造変換は,図14 .4
の方法でいろいろ検討できる 抗 菌活性の発現には ,s-
ラクタム環と遊離のカルボキシ基は必須で、あるが,s-
ラクタム系抗生物質σJ
工
i33::
二H2:L1331:
E222LRJR2::
ペニシリン G の側鎖アミドの加水分解で得られる 6 - A P A 6-APAのアシル化でさまさまなペニシリン誘導体が合成できる 硫黄原子は必須ではないこと ,s-
ラクタム環の5
,6-
シスの立体化学は抗菌 活性の発現にとって重要で、あること,などがわかった ペニシ リンG
は酸に弱く,胃酸で、加水分解されるので,経口投与はでき ない 一般のアミ ド結合の窒素と異なり ,ペニシリンのs-
ラクタム環では 窒素の非共有電子対は窒素原子上に局在化 している このアミドのカルボニ ル基はアミド結合では例外的に求核攻撃を受けやすい,二環性のひずみのた めに,環のつなぎ目が Sp2 窒素構造を含む共rj!言寄与構造をとれないことが理 由であるs-
ラクタム環が求核攻撃を受け開環すると ,s
ーラクタム環部分 のひずみは解消され安定な化合物になる。 このF
ーラク タム環の開裂には,6
位 の ア ミ ド 結 合 が 隣 接 基 関 与(n
巴ighboring-gro
L1p participation)
する すなわち,図14.5
のようにアミド カルボニル基がs-
ラクタム環のアミドカルボニル基を分子内で求核攻撃し そののち数ステップを経て,ペニシラン酸とペニシレン酸に分解する そこで,この分子内反応を起こりにくくするには,アシル基の R 部分に電 子求引性の置換基を設定し アミドのカルボニル基の酸素上の電子密度を低 減させるとよい,と推測できる実│ 段ペニシリンV
は電気陰性度の大きな酸 素原子が6
位のアミド側鎖上 α位に存在するため,隣接基関与を抑えること ができるその結果,経口投与も可能であるしかしペニシリンV
もG
同様 ペニシリン分解酵素で分解されやすく,またア レルギ一反応を起こす人もい ることから,さらなる改良が研究された半合成ペニシリンであるアンピシリ ンの側鎖α
位のアミノ基や,クロキサシリンのイソキサゾール( isoxazo!e)
環 などは,電子求引性基として作用している その結果,いずれも6
位アミド 側鎖のカルボニル基の酸素上の電子密度を低減し隣接基関与を抑えて,胃 酸による加水分解を遅らせるので,いずれも経口投与が可能である253
隣接基関与 反応点の近傍の官能基が分子 内で脱出世基を事11しだすのを助 けている場合などを指す 隣 接基補助(neighboring-group assistance. anchimeric assist-ance)。隣接碁効果 (neighbor-ing-group effect. anchimeric巴汀ecr ) なども類語 たとえ ば アスピリンが中性,ある いはアルカリ性で容易に加水 分解を受けサリチル酸に変換 される過程は 0 -位に位置す るカルポキシ基( カルボキシ イオン) の隣接基関与による ものである イソキサゾール 次のような五員環の芳香族複 素 環
。
6-アミノペニシラミン酸 抗菌活性のファーマコフォア ・βーラクタム ・遊離カルボン酸 ・5,6位のシス配置 ・硫黄原子は活性に影響しない。ペニシリン
Gの化学的性質
ペニシリン
G は加水分解されやすい。経口投与できない。
理由その 1:共鳴構造がとれない。
NH O N+ -O O N O -N+理由その
2: 隣接基関与
s-
ラクタム系抗生物質σJ
工
i33::
二H2:L1331:
E222LRJR2::
ペニシリン
G
の側鎖アミドの加水分解で得られる
6 - A P A
6-APA のアシル化でさまさまなペニシリン誘導体が合成できる硫黄原子は必須ではないこと ,
s-
ラクタム環の
5
,
6-
シスの立体化学は抗菌
活性の発現にとって重要で、あること,などがわかった
ペニシ リン
G
は酸に弱く,胃酸で、加水分解されるので,経口投与はでき
ない 一般のアミ ド結合の窒素と異なり ,ペニシリンの
s-
ラクタム環では
窒素の非共有電子対は窒素原子上に局在化 している このアミドのカルボニ
ル基はアミド結合では例外的に求核攻撃を受けやすい,二環性のひずみのた
めに,環のつなぎ目が
Sp2窒素構造を含む共
rj!言寄与構造をとれないことが理
由である
s-
ラクタム環が求核攻撃を受け開環すると ,
s
ーラクタム環部分
のひずみは解消され安定な化合物になる。
この
F
ーラク タム環の開裂には,
6
位 の ア ミ ド 結 合 が 隣 接 基 関 与
(n
巴ighboring-gro
L1p participation)
する すなわち,図
14.5
のようにアミド
カルボニル基が
s-
ラクタム環のアミドカルボニル基を分子内で求核攻撃し
そののち数ステップを経て,ペニシラン酸とペニシレン酸に分解する
そこで,この分子内反応を起こりにくくするには,アシル基の
R
部分に電
子求引性の置換基を設定し アミドのカルボニル基の酸素上の電子密度を低
減させるとよい,と推測できる実│ 段ペニシリン
V
は電気陰性度の大きな酸
素原子が
6
位のアミド側鎖上
α
位に存在するため,隣接基関与を抑えること
ができるその結果,経口投与も可能であるしかしペニシリン
V
も
G
同様
ペニシリン分解酵素で分解されやすく,またア レルギ一反応を起こす人もい
ることから,さらなる改良が研究された半合成ペニシリンであるアンピシリ
ンの側鎖
α
位のアミノ基や,クロキサシリンのイソキサゾール( i
soxazo!e)
環
などは,電子求引性基として作用している その結果,いずれも
6
位アミド
側鎖のカルボニル基の酸素上の電子密度を低減し隣接基関与を抑えて,胃
酸による加水分解を遅らせるので,いずれも経口投与が可能である
253
隣接基関与
反応点の近傍の官能基が分子 内で脱出世基を事11しだすのを助 けている場合などを指す 隣 接基補助(neighboring-group assistance. anchimeric assist-ance)。隣接碁効果 (neighbor-ing-group effect. anchimeric巴汀ecr ) なども類語 たとえ ば アスピリンが中性,ある いはアルカリ性で容易に加水 分解を受けサリチル酸に変換 される過程は 0 -位に位置す るカルポキシ基( カルボキシ イオン) の隣接基関与による ものである
イソキサゾール
次のような五員環の芳香族複 素 環。
6位アミドのカルボニル基の電子密度を低下させると
…
経口投与可能な
βーラクタム類
252
*
1章 6i;i:も参照 チアゾリジン チアゾールを完全水素化した 次のような鱗造ふ
う
抗 菌 薬 ている 水分子がないので アシル化 された酵素が加水分解されて復活する 速度は非常に遅く,ムレイントランスペプチダーゼは事実上失活する14.2.1
ペニシリン
1877
年,Pasteur
とJoubert
は,あるカビ類が細菌を殺す毒性物質を産生 フレ E ン グ することを発見した* .1928
年にF le ming
は青カビ(Penicillium
の一種) の コロニーのまわりではバクテリアのコロニーが死滅していることに気づい フローリー チ ェ イ ン た.1938
年にF lorey
とC h
釦n
が凍結乾燥法を利用してペニシリンを単離し ホ ジ キ ン1944
年にはアメリカで大量生産に至った1945
年にD.
C.
Hodgkin
によりX
線構造解析がなされ,構造が確定した1957
年にはJ
C.
Sheehan
による ペニシ リンの全合成が達成された 翌年B eech a ms
社がペニシリン生合成 中間体6-
アミノペニシラン酸(6-aminopenicillanic acid ;
6-A P A )
を単離した( 図