近畿大学学術情報リポジトリ
115
0
0
全文
(2) Doctoral Dissertation. Molecular Properties and Expression Mechanisms of Ascorbate Peroxidase Isoenzymes in Higher Plants. Kazuya Yoshimura Graduate School, Kinki University Division of Agricultural Science (Major: Agricultural chemistry).
(3) (英文題目) Molecular Properties and Expression Mechanisms of Ascorbate Peroxidase Isoenzymes in Higher Plants. Kazuya Yoshimura March, 2001 Graduate School, Kinki University Division of Agricultural Science Major: Agricultural chemistry (Advisor: Prof. Shigeru Shigeoka). (和文題目) 高等植物のアスコルビン酸ペルオキシダーゼアイソザイムの 分子特性と発現機構. 近畿大学大学院 農学研究科 農芸化学専攻 吉村和也 (指導: 重岡成教授). -------------------------------------------------------------------------------------------------------Submitted to the Graduate School, Kinki University, to fulfill the requirement for the Doctorate Degree..
(4) Acknowledgements I wish to express my science graduate to Dr. Shigeru Shigeoka, Professor of Faculty of Agriculture, Kinki University, for his kind guidance, valuable advice, stimulating discussion, and critical review thought the work including the manuscript of this thesis. Thanks are due to Dr. Ryutaro Utsumi and Dr. Tamo Fukamizo, Professors of Faculty of Agriculture, Kinki University, for reading the entire text in its original form. I am thankful to Dr. Takahiro Ishikawa, Faculty of Life and Environmental Science, Shimane University, for his valuable help, kind suggestion, and stimulating discussion throughout the work. I greatly thank to Dr. Yoshihisa Nakano, Professor of Applied Biochemistry, Osaka Prefecture University, for his technical support of the production of monoclonal antibody. I am greatly indebted to Dr. Hans J. Bohnert, University of Arizona, for generously. providing. the. cDNA. clones. of. APX. isoenzymes. from. Mesemnbryanthemum crystallinum. I wish to thank Dr. Toru Takeda and Dr. Masahiro Tamoi, Faculty of Agriculture, Kinki University, for their valuable helps and valuable discussions throughout the work. Finally, special thanks are due to Kosuke Sakai and Yukinori Yabuta, for their many helpful collaborations.. Thanks are also due to all the past and present members. of the laboratory of Nutrition Chemistry in the Faculty of Agriculture, Kinki University, for their kind cooperations. This research was supported by a Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists..
(5) ABBREVIATIONS AsA. reduced ascorbate. APX. ascorbate peroxidase. cAPX. cytosolic ascorbate peroxidase. chlAPX. chloroplastic APX. gAPX. glyoxisomal (microbody) -bound ascorbate peroxidase. mAPX. microbody -bound ascorbate peroxidase. sAPX. stromal ascorbate peroxidase. tAPX. thylakoid-bound ascorbate peroxidase. BSA. bovine serum albumin. CCP. cytochrome c peroxidase. DAR. dehydroascorbate reductase. DAsA. dehydroascorbate. GP. guaiacol peroxidase. GSH. reduced glutathione. GPX. glutathione peroxidase. H2O2. hydrogen peroxide. HEPES. N-2-hydroxyethylpiperazine-N‘-ethanesulfonic acid. IPTG. isopropyl--D-thiogalactopyranoside. LB. Luria-Bertani broth. mAb. monoclonal antibody. MES. 2-(N-morpholino)ethanesulfonic acid. MDAR. monodehydroascorbate reductase. MDAsA. monodehydroascorbate. 1. singlet oxigen. O2. O2-. superoxide. OH•. hydroxyl radical. PAGE. polyacrylamide gel electrophoresis. PBS. phospate buffered saline. Poly(A)+RNA polyadenylated RNA PVDF. polyvinylidene difluoride. RACE. rapid amplification of cDNA ends.
(6) RT-PCR. reverse transcription-PCR. SDS. sodium dodecyl sulfate. SOD. superoxide dismutase. Tris. tris(hydroxymethyl)methylglycine.
(7) CONTENTS CHAPTER I. Introduction ••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••. CHAPTER II. Molecular Characterization and Physiological Role of a. 1. Glyoxysome-Bound Ascorbate Peroxidase from Spinach ••••••••••• CHAPTER III. 7. Comparative Study on Recombinant Chloroplastic and Cytosolic Ascorbate Peroxidase Isoenzymes of Spinach •••••••••••••••••••••••••. CHAPTER IV. 26. Characterization of Monoclonal Antibodies against Ascorbate Peroxidase Isoenzymes: Purification and Epitope-mapping Using Immunoaffinity Column Chromatography ••••••••••••••••••••••••••••••. CHAPTER V. 40. Expression mechanism of Chloroplastic Ascorbate Peroxidase Isoenzymes in Spinach ••••••••••••••••••••••••••••••••••••••••••••••••••••••••. CHAPTER VI. Alternatively Spliced mRNA Variants of Chloroplastic Ascorbate Peroxidase Isoenzymes in Spinach ••••••••••••••••••••••••••••••••••••••••. CHAPTER VII. 49. 63. Regulation of Tissue-specific Alternative Splicing of Chloroplastic Ascorbate Peroxidase Isoenzymes in Spinach •••••• 77. REFERENCES ••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••. 91. PUBLICATIONS •••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••. 107.
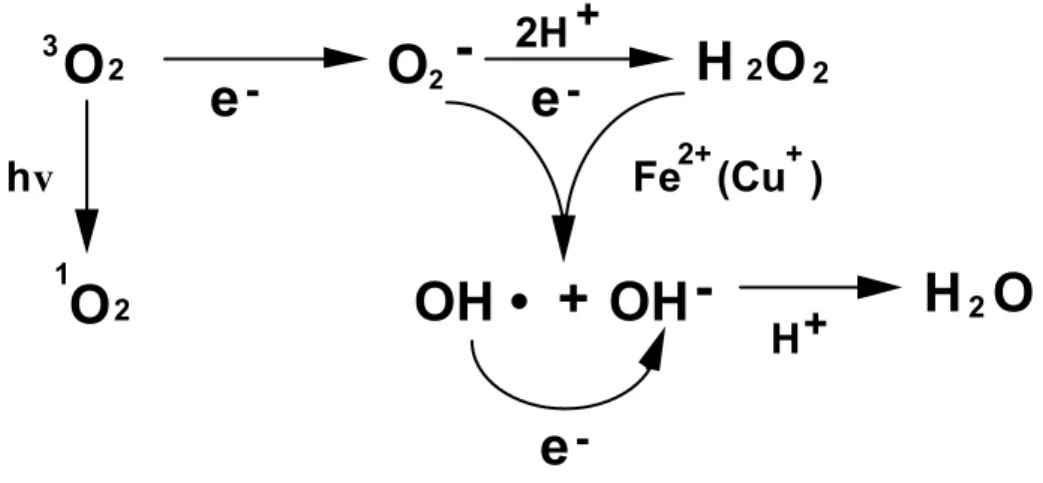
(8) CHAPTER I Introduction Generation of active oxygen species Many lines of geological and biological evidence sustain the view that most of the atmospheric dioxygen (O2) has been supplied by aerobic photosynthetic organisms. Then the atmospheric O2 has accumulated and its concentration has been increasing to 1% and 10% of the present atmospheric level 600 and 400 million years ago, respectively (Asada 1992).. As the O2 level of the atomosphere increased, life in O2. has led to the evolution of biochemical adaptations that exploit the reactivity of active oxygen species (AOS) (Fig. I-1). AOS, such as singlet oxygen (1O2), superoxide -. (O2 ), hydrogen peroxide (H2O2), and hydroxyl radicals (OH•), inactivate enzymes and damage important cellular components (Halliwell and Gutterridge 1985). The term AOS is generic, embracing not only free radicals but also H2O2 and 1O2.. While it is. generally assumed that OH• and 1O2 are so reactive that their production must be -. minimized (Asada 1999, Noctor and Foyer 1998), O2 and H2O2 are synthesized at very high rates even under optimal conditions.. They are involved in virtually all. major areas of aerobic biochemistry (e.g. respiratory and photosynthetic electron transport; oxigation of glycolate, xanthine, and glucose) and are produced in copious quantities by several enzyme systems [e.g. plasmalemma-bound NADPH-dependent superoxide synthase (Alvarez and Lamb 1997) and superoxide dismutase (SOD; 1.15.1.1) (Bowler et al. 1992)].. -. The chief toxicity of O2 and H2O2 is thought to. reside in their ability to initiate cascade reactions that result in the production of OH• and other destructive species such as lipid peroxides.. These dangerous cascades are. prevented by efficient operation of the cell‘s antioxidative defense systems.. In some. circumstances, however, the destructive power and signaling potential of AOS are utilized as an effective means of defense (Chen et al. 1993, Foyer et al. 1983, Levine et al. 1994). In photosynthetic organisms including higher plants, AOS producing processes are driven by or associated with light-dependent events. Photosynthetic cells are prone to oxidative stress because they contain an array of photosensitising pigments and they both produce and consume O2.. The photosynthetic electron transport. system is the major source of AOS in plant tissues, having the potential to generate 1.
(9) 1O2. -. and O2 (Asada 1994).. The extent to which these reactions occur under. non-stressed conditions is debatable but it is generally believed that the production of AOS is an unavoidable consequence of the operation of the photosynthetic electron transport chain.. The major oxygen-consuming processes associated with. photosynthesis are the oxygenase reaction of ribulose-1,5-bisphosphate carboxylase (Rubisco), which is the initiating reaction of the photorespiratory pathway and direct reduction of O2 by the photosystem I (PSI) electron transport chain or indirectly mediated by a stromal factor (Asada 1999). A possible candidate of stromal factor is MDAR.. In addition, certain photosystem II (PSII) components are capable of. reducing O2.. The O2 may also be reduced by a cyanide-insensitive respiratory. pathway in chloroplasts that competes for electrons with photosynthetic electron transport (Berger et al. 1993, Bennoun 1994).. Furthermore, even under optimal. conditions, other metabolic processes in microbody and mitochondrial, and plasma membrane-linked electron transport systems produce AOS (Foyer et al. 1994).. The. impression of biotic and abiotic stress conditions can give rise to excess concentrations of AOS, resulting in oxidative damage at the cellular level.. 3. O2. e-. O2 -. 2H + e-. hv. H 2O 2 2+. +. Fe (Cu ). OH • + OH -. 1. O2. H+. H2O. eFig. I-1. Generation of active oxygen species on the reduction of O2 to H2O2.. Scavenging system of active oxygen species in higher plants -. Efficient destruction of O2 and H2O2 requires the action of both nonenzymic and enzymic constituents. A highly efficient antioxidative defense system is present in all photosynthetic organisms (Asada 1999, Noctor and Foyer 1998). 2. The nonenzymic.
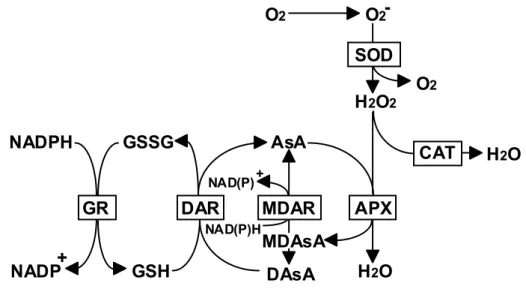
(10) antioxidants are generally small molecules. the destruction of AOS.. Ascorbate (AsA) plays a pivotal role in. In addition, the tripeptide glutathione (GSH) and lipophilic. antioxydant -tocopherol together with the carotenoid pigments fulfill essential antioxidant functions. The enzymic antioxidative components are a prerequisite for life in O2; they include superoxide dismutase (SOD), catalase, ascorbate peroxidase (APX; EC 1.11.1.11), electron donor-nonspecific peroxidases like guaiacol peroxidase (GP; EC 1.11.1.7), and the enzymes involved in the synthesis and regeneration of the reduced forms of antioxidants.. -. O2 produced in the different compartments of plant. cells is rapidly converted to H2O2 by the action of SOD (Bowler et al. 1992).. The. low concentration (10 µM) of H2O2 inhibited the thiol-modulated enzymes of the photosynthetic carbon reduction cycle (the Calvin cycle), since H2O2 readily oxidizes the reduced thiol groups (Kaiser 1979).. Therefore, it cannot be allowed to. accumulate H2O2 to excess in organelles such as chloroplasts where photosynthesis depends on thiol-modulated enzymes of the Calvin cycle. Catalases convert H2O2 to water and O2 and are located in microbodies and cytosol.. This enzyme has extremely. high maximum catalytic rates but low substrate affinities (Willekens et al. 1995). Furthermore, the absence of catalase in the chloroplast precludes a role in protection of the thiol-regulated enzymes.. An alternative mode of H2O2 destruction is via. peroxidases, which are found throughtout the cell and which have a much higher affinity for H2O2 than catalase (Asada 1999). reductant when they reduce H2O2 to H2O.. Peroxidases, however, require a. In animals, peroxidases that use reduced. GSH are important in the H2O2 detoxification (Meister 1994).. By contrast, the. enzyme activities of catalyzing GSH-dependent reduction of H2O2 has not been detected in higher plants.. Several plant genes have recently been isolated and. showed homology to mammalian phospholipid hydroperoxide GSH-dependent peroxidase (Criqui et al. 1992, Holland et al. 1993). In higher plant cells, the oxidative damage to the chloroplasts and other cellular compartments by H2O2 is minimized by the AsA-GSH cycle consisting of APX and the regeneration system of AsA (Fig. I-2) (Asada et al. 1999). In this cycle, APX reduces H2O2 to form water and monodehydroascorbate (MDAsA).. MDAsA. spontaneously disproportionates to AsA and dehydroascorbate (DAsA; 105 M-1 s-1 at pH 7.0).. MDAsA is also directly reduced to AsA by action of NAD(P)H-dependent. MDAsA reductase (MDAR; EC 1.6.5.4). 3. DAsA reductase (DAR; EC 1.8.5.1).
(11) utilizes GSH to reduce the DAsA and thereby regenerate the AsA.. The GSH is then. regenerated by GSH reductase (GR; EC 1.6.4.2), utilizing reducing equivalents from NADPH.. O2. O2SOD H2O2. NADPH. GSSG. AsA NAD(P). GR + NADP. DAR GSH. CAT. +. NAD(P)H. MDAR. O2. H2O. APX. MDAsA DAsA. H 2O. Fig. I-2. Ascorbate-glutathione Cycle. AsA, Ascorbate; MDAsA, Monodehydro ascorbate; DAsA, Dehydro ascorbate; GSH, Reduced glutathione; GSSG, Oxidized glutathione; SOD, Superoxide dismutase; CAT, Catalase; APX, Ascorbate peroxidase; MDAR, MDAsA reductase; DAR, DAsA reductase; GR, Glutathione reductase.. Ascorbate peroxidase isoenzymes APX is a H2O2-scavenging enzyme which uses AsA as a natural electron donor. 2AsA+H2O2 ------> 2MDAsA+2H2O APX is a hemeprotein, but it is clearly distinct from the GP in terms of enzymlogical and molecular properties (Foyer et al. 1991, Asada 1992). APX has been found in higher plants including spinach (Nakano and Asada 1987, Tanaka et al. 1991), pea (Gerbling et al. 1984, Mittler and Zilinskas 1991a), maize (Koshiba 1993), tea (Chen and Asada 1989) and legumes root nodule (Dalton et al. 1987) and also detected in eukaryotic algae include Euglena (Shigeoka et al. 1980) and Chlamydomonas (Yokota et al. 1988, Shigeoka et al. 1991) and certain cyanobacteria (Tel-Or et al. 1986, Miyake et al. 1991). In addition to photosynthetic organisms, APX has been found in. 4.
(12) the protozoan Trypanosoma cruzi (Boveris et al. 1980) and mammals (Wada et al. 1998).. In higher plants, APX isoenzymes are distributed in at least three distinct cell. compartments, stromal APX (sAPX) and thylakoid-bound APX (tAPX) in chloroplasts, and cytosolic APX (cAPX) (Asada 1992, Miyake and Asada 1992).. A second family. of cAPX has also reported in various plant species, such as spinach, Arabidopsis thaliana, soybean, and rice (Ishikawa et al. 1995, Santos et al. 1996, Jespersen et al. 1997, Caldwell et al. 1998).. More recently, it was reported a novel APX isoenzyme. is located in mitochondria, but the molecular characterization of the enzyme is still unknown (Jiménez et al. 1997, Leonardis et al. 2000). In chloroplasts of higher plants which lack catalase, chloroplastic APX (chlAPX) occurs in the stroma in a soluble form (sAPX) and in the thylakoids in a membrane-bound form (tAPX) (Miyake et al. 1993). tAPX binds preferentially to the stromal thylakoids on which the PSI complex is localized (Miyake and Asada 1992).. The thylakoidal scavenging system called the water-water cycle is composed. of CuZn-SOD attached on the thylakoids (in several plants, Fe-SOD), APX (tAPX), and ferredoxin (Asada 1999).. Ferredoxin reduces MDA directly to AsA.. The. stromal scavenging system is composed of CuZn-SOD, APX (sAPX), MDA reductase, DHA reductase, and GR. NAD(P)H for the reduction of either MDA of DHA is photogenerated via ferredoxin-NADP+ oxidoreductase.. By contrast, cAPX is. localized in cell compartments other than chloroplasts and also in non-photosynthetic tissues. In this study Considering the specific distributions and roles of the APX isoenzymes and the potential for AOS production in each organelle of higher plants, it seems likely that the APX isoenzymes are expressed by distinct regulatory mechanisms.. However, no. studies have presently the analysis of the molecular properties, the regulation mechanisms, and the physiological roles of all the APX isoenzymes in higher plants. In the present thesis, I studied the followings; (1) Molecular characterization and physiological role of a glyoxysome-bound ascorbate peroxidase from spinach (Plant Cell Physiol. 39, 23-34, 1998) (2) Comparative study on recombinant chloroplastic and cytosolic ascorbate peroxidase isoenzymes of spinach (Arch. Biochem. Biophys. 353, 55-63, 1998), 5.
(13) (3) Characterization. of. monoclonal. antibodies. against. ascorbate. peroxidase. isoenzymes: purification and epitope-mapping using immunoaffinity column chromatography, (4) Expression mechanism of chloroplastic ascorbate peroxidase isoenzymes in spinach (Biochem. J., 328, 795-800, 1997), (5) Alternatively spliced mRNA variants of chloroplastic ascorbate peroxidase isoenzymes in spinach (Biochem. J. 338, 41-48, 1999), and (6) Regulation of tissue-specific alternative splicing of chloroplastic ascorbate peroxidase isoenzymes in spinach.. 6.
(14) CHAPTER II Molecular Characterization and Physiological Role of a Glyoxysome-Bound Ascorbate Peroxidase from Spinach APX isoenzymes have been purified and characterized from many plant sources including spinach (Nakano and Asada 1987, Miyake et al. 1993), tea (Chen and Asada 1989), pea (Mittler and Zilinskas 1991a) and komatsuna (Ishikawa et al. 1996a). chlAPX isoenzymes differ from cAPX in the following aspects: molecular mass, substrate specificity, pH optimum, and stability (Asada 1992). The molecular genetics of APX have gained considerable attention in recent years. cDNAs encoding cAPX isoenzymes have been isolated, sequenced, and characterized from several plant species such as spinach (Mittler and Zilinskas 1991b, Kubo et al. 1992, Ishikawa et al. 1995). The crystal structure of the recombinant cAPX has been determined (Patterson et al. 1995). In the preceding report, I have reported the first complete cloning and molecular characterization of sAPX and tAPX isoenzymes from spinach, which differ from each other by only in the presence or absence of their C-terminal peptide, which constructs a hydrophobic thylakoid membrane binding domain (Ishikawa et al. 1996c). Yamaguchi et al. (1996) have also reported the nucleotide sequence of a cDNA encoding a tAPX of pumpkin. More recently, a novel type of APX isoenzyme was found to be localized on the membranes of microbodies (glyoxisome and peroxisome) in pumpkin (Yamaguchi et al. 1995b) and cotton (Bunkelmann and Trelease 1996). At first, the glyoxisomal-bound APX (gAPX) was recognized as a rich glyoxysomal membrane protein with a molecular mass of 31 kDa, which was retained in peroxisomes during the microbody transition from glyoxysome to peroxisome (Yamaguchi et al. 1995a). The partial amino acid sequence of the 31 kDa protein showed a high homology to those of already known cAPXs, and glyoxysomes retained the activity of APX. It thus became clear that a novel APX isoenzyme exists in the microbodies. The detailed properties of the enzyme have not been clarified, but its cDNA has been cloned from cotton cotyledons (Bunkelmann and Trelease 1996). Microbody is used as a general term which includes both peroxisomes and glyoxysomes (Huang et al. 1983). Peroxisomes in plant cells are known to contain enzymes related to the oxidative photosynthetic carbon metabolic cycle of photorespiration and the fatty acid -oxidation cycle; glyoxysomes, in addition, contain glyoxylate cycle enzymes and generally occur in germinating fatty seeds. Several H2O2-producing flavin oxidases such as glycolate oxidase and acyl CoA oxidase are well know to exist inside microbodies (del Río et al. 1992). Moreover, it has been reported that the superoxide radical is directly generated in the peroxisomal. 7.
(15) matrix and membranes (Sandalio et al. 1988a, del Río et al. 1989). As scavenging enzymes of these active oxygen species, peroxisomes typically possess catalase (Cat) and SOD (Sandalio and del Río 1988b, Droillard and Paulin 1990). These facts reported so far and the occurrence of a new type of the gAPX raise the question of how Cat and APX are functionally utilized as efficient scavengers of H2O2. In order to explore the physiological role of the gAPX isoenzyme in the active oxygen species metabolism of this organelle, I describe here the identification of a cDNA clone which encodes the gAPX from spinach cotyledon, the suborganellar distribution of the enzyme in glyoxysomes, and the enzymatic characterization of the recombinant protein overexpressed in E. coli.. Materials and Methods Materials Spinach seeds (Spinacia oleracea ) were germinated on moist gauze at 15˚C in the dark. The cotyledons from seedlings grown for 10-14 days in the dark were transferred to illumination (140 µE s-1 m-2) for 24 h to obtain the greening cotyledons as described previously (Ishikawa et al. 1996c). Restriction enzymes and modifying enzymes were purchased from Takara (Kyoto, Japan). E.coli strains Y1090 and DH5 were obtained from Amersham Pharmacia Biotech. (Buckinghamshire, UK). The expression vector pET-3a and its companion production strain BL21(DE3)pLysS were purchased from Novagen (WI, USA). All other chemicals were of analytical grade and were used without further purification. Enzyme assays The APX activity was assayed spectrophotometrically as previously described (Shigeoka et al. 1980). The assay mixture (2 ml) contained 50 mM potassium phosphate buffer, pH 7.0, 0.4 mM AsA, 0.1 mM H2O2, and the enzyme at 37˚C. The oxidation of AsA was followed by a decrease in the absorbance at 290 nm (=2.8 mM-1 cm-1). Oxidation of alternate electron donors was measured in the same assay mixture as that used for AsA, but AsA was replaced by 20 mM pyrogallol (430 nm, 2.47 mM-1 cm-1), 10 mM guaiacol (470 nm, 22.6 mM-1 cm-1), 0.4 mM D-iso-AsA (290 nm, 3.3 mM-1 cm-1), 0.15 mM NAD(P)H (340 nm, 6.22 mM-1 cm-1) and 40 µM reduced Cyt c (550 nm, 19 mM-1 cm-1). The activity of glutathione peroxidase was monitored spectrophotometrically by following the oxidation of NADPH in the presence of glutathione reductase (Shigeoka et al. 1980). MDAR was assayed using MDAsA generated by AsA and an AsA oxidase system (Shigeoka et al. 1987). The. 8.
(16) reaction mixture (2 ml) was comprised of 50 mM phosphate buffer (pH 7.0), 1 mM AsA, 1 unit AsA oxidase, 0.1 mM NADH, and the enzyme. Marker enzymes, Cat (Mutsuda et al. 1996) and hydroxypyruvate reductase (Bogin and Wallace 1969) for microbodies and cytochrome c oxidase (Hodges and Leonard 1974) and fumarase (Walk and Hock 1977) for mitochondria, were assayed by the methods in the cited references. The protein was determined with Coomassie Brilliant Blue G-250 using bovine serum albumin as the standard according to Bradford (Bradford, 1976). Construction of spinach greening cotyledon cDNA library The construction of the cDNA library was performed as described previously (Ishikawa et al. 1996c). Total RNA was isolated from greening cotyledons of spinach seedlings (5.0 g FW) with guanidine-isothiocyanate, and poly(A)+RNA was then purified using the PolyATtract mRNA Isolation Systems (Promega, WI, USA). A cDNA was synthesized using a cDNA synthesis kit (Amersham), and a cDNA library was constructed in gt11 as described by the supplier (Amersham). Cloning of spinach gAPX cDNA The spinach cDNA library in gt11 was screened with a monoclonal antibody (mAb) raised against Euglena APX (EAP1) diluted 1 to 1,000 as described previously (Ishikawa et al. 1996b). E.coli strain Y1090 was infected with recombinant phages; then the plaques formed were imprinted on isopropyl--D-thiogalactopyranoside (IPTG)-impregnated nitrocellulose filters (Millipore, MA, USA). The filters were treated with the EAP1 and then with goat anti-mouse IgG-peroxidase conjugate (Bio-Rad Laboratories, CA, USA), followed by staining with 4-methoxy-1-naphthol (Aldrich, WI, USA). Positive plaques were rescued from the master plate, rescreened, and then amplified in Y1090. The insert was excised from the phage and subcloned into the plasmid vector pBluescript SK(+) (Stratagene, CA, USA). The nucleotide sequence was determined by the dideoxy method using a 373A DNA sequencer (Applied Biosystems, CA, USA). Overexpression of recombinant spinach gAPX in E.coli For the construction of the plasmid to express the cloned gAPX cDNA, the following oligonucleotide primers were used. The upstream 24-mer oligonucleotide was derived from the cDNA nucleotide sequence homologous to the non-coding and coding strands corresponding to the nucleotide positions 12 to 35. It included an Nde I restriction site (underlined), giving the following sequence: 5'-AAGCTCCAA CATATGGCGATGCCG-3'. The downstream 26-mer oligonucleotide was derived from the nucleotide sequence complementary to the non-coding strand of the cDNA. 9.
(17) nucleotide positions 1085 to 1110, which included a Bam HI restriction site (underlined), giving the following sequence: 5'-GCTCTTCATAGTTGGATCC ATTCTGA-3'. The PCR reaction was initiated directly with an aliquot of the plasmid containing the full-length cDNA of spinach gAPX. The plasmid was denatured by heating for 5 min at 94˚C. The sample was then subjected to the PCR in 100 µl reaction mixture containing 400 nM of each oligonucleotide, 200 µM dNTPs, 1.5 mM MgCl2, and 2.5 units Taq DNA polymerase. Thirty cycles (1 min 94˚C, 1 min 55˚C, 2 min 72˚C) were performed, followed by an elongation of 7 min at 72˚C. The double strand PCR product with the correct size (1099 bp) was purified, ligated into pT7Blue T-vector (Novagen), and then transformed into E. coli strain DH5. The sequence of the insert region of pT7Blue T-vector was verified by DNA sequencing. The plasmid obtained was digested with Nde I and Bam HI restriction enzymes; then the fragment was ligated into the Nde I-Bam HI sites of the pET-3a expression vector and introduced into the E.coli strain DH5. Plasmid DNA was prepared from the ampicillin-resistant transformants and tested by digestion using Nde I and Bam HI to see if the inserted DNA was indeed present. The resulting construct, named pET-SAP30, was introduced into E. coli strain BL21(DE3)pLysS. The recombinant E.coli cells were grown at 37˚C in Luria-Bertani medium [1% (w/v) bacto-tryptone, 0.5% (w/v) bacto-yeast extract, 1% (w/v) NaCl] supplemented with 50 µg/ml ampicillin and 34 µg/ml chloramphenicol. IPTG was added to a final concentration of 0.4 mM to midexponential phase cells (OD600 ~0.7), and the culture was shaken for an additional 3 to 4 h. Partial purification of recombinant gAPX All procedures were carried out at 4˚C. The recombinant E. coli cells (4.0 g FW) were harvested by centrifugation at 500 g for 10 min, resuspended in 10 ml of 50 mM sodium phosphate buffer (pH 7.0) containing 1 mM EDTA, 1 mM AsA, and 20% (w/v) sorbitol, and disrupted by sonication (10 kHz) for 3 min. This lysate was centrifuged at 100,000 g for 30 min. Approximately 10 mg of protein per ml of insoluble suspension was incubated with 0.5% Triton X-100 in the above phosphate buffer for 1 h. The solubilized enzyme was obtained by centrifugation at 105,000 g for 60 min. The supernatant was submitted to a HiLoad 26/10 Q Sepharose column equilibrated with the phosphate buffer. The active fractions were then chromatographed onto a HiLoad 16/60 Superdex 200 column equilibrated with the phosphate buffer.. 10.
(18) Isolation of intact glyoxysomes To avoid contamination by APX isoenzymes from chloroplasts, spinach cotyledons (100 g FW) grown under dark conditions were used for the isolation of glyoxysomes. They were chopped with razor blades in 200 ml of the grinding medium containing 25 mM MOPS (pH 7.8), 0.4 M mannitol, 1 mM EDTA, 0.1% (w/v) BSA, 8 mM cysteine, and 1 mM AsA. The homogenate was squeezed through four layers of cheesecloth. The homogenization of the residue was repeated with a further 200 ml of the grinding medium. The filtrates were combined and centrifuged at 1,000 g for 5 min to remove unbroken cells and cellular debris, followed by centrifuging for 15 min at 14,000 g. The obtained precipitate was suspended in 140 ml of the washing medium containing 5 mM MOPS (pH 7.5), 0.4 M mannitol, 1 mM EDTA, 0.1% BSA, and 1 mM AsA, centrifuged for 5 min at 1,000 g, and then centrifuged for 15 min at 14,000 g. The resultant 1,000-14,000 g pellets were then resuspended in 8 ml of the washing medium and subjected to a linear sucrose density gradient centrifugation (40-70% sucrose concentration) in 10 mM Tricine buffer (pH 7.5) containing 1 mM EDTA, 1 mM AsA described previously (Shigeoka et al. 1980). The tubes were centrifuged at 100,000 g for 3 h at 4˚C. Intact glyoxysomes were collected from the gradient by aspiration. SDS-PAGE and immunoblot analysis SDS-PAGE was performed in 12.5% slab gels, according to the method of Leammli (Leammli 1970). Coomassie staining of proteins was accomplished by an incubation gels fixative (20% methanol, 7% acetic acid) containing 0.2% of Coomassie Brilliant Blue R-250 for 30 min and destained overnight in fixative. For immunoblot analysis, gels were transferred to PVDF membranes (Millipore), using an electroblot apparatus (model 200/2.0, Bio-Rad Laboratories) at 15 V for 1 h. After transfer, the membrane was blocked for 1 h in 3% dry milk in phosphate-buffered saline (PBS). The mAb to Euglena APX (EAP1) (Ishikawa et al. 1996b), which reacted with cAPX and chlAPX isoenzymes from higher plants, was diluted 1:2,000 in TBS (10 mM Tris, pH 8.0, and 150 mM NaCl) containing 0.1% BSA, and the membrane was incubated at room temperature for 1 h. The membrane was washed in TPBS (PBS containing 0.05% Tween-20), followed by incubation for 30 min in anti-mouse-Igs peroxidase-conjugated secondary antibody (Boehringer Mannheim, Germany) diluted 1:5,000 in TBS containing 0.1% BSA. The membrane again was washed in TPBS, and the immunoreactive proteins were visualized using 4-methoxy-1-naphthol (Aldlich).. 11.
(19) N-terminal sequence analysis The resolved native and recombinant proteins were separated using SDS-PAGE and transferred to PVDF membranes (Millipore) as described above. The membrane was washed extensively with water, stained with 0.25% Coomassie R-250 in 5% aqueous methanol and 7.5% acetic acid for 5 min, and destained with 90% aqueous methanol for 10 min. The portion of the membrane containing the desired protein band was cut out, and the N-terminal sequence was performed by an automated pulse-liquid protein sequencer (model 492, Applied Biosystems) as described previously (Ishikawa et al. 1996b).. Results cDNA cloning of spinach gAPX In the preceding study, we have reported that mAb (EAP1) raised against Euglena APX cross-reacts with both cAPX and sAPX from spinach leaves (Ishikawa et al. 1996b) and becomes a good probe for isolating the four cDNA clones which encode two cAPX and two chlAPX isoenzymes (Ishikawa et al. 1995, 1996c). Herein, I isolated one positive clone (SAP30) with a nucleotide sequence different from those of the other four APX cDNA clones. The cDNA sequence of the SAP30 showed an open reading frame starting from nucleotide 24 up to 881 and coding for a protein containing 286 amino acids with a molecular mass of 31,507 Da (Fig. II-1A). The calculated overall identity of the sequence was 83.7% and 80.5% at the protein level with the sequences of cotton (Bunkelmann and Trelease 1996) and Arabidopsis gAPXs (GenBank accession No.X98003), respectively (Fig. II-2). A putative membrane-spanning region of the predicted protein was identified near the C-terminal sequence; it possessed a hydrophobic domain, as was the case for the cotton enzyme (Fig. II-1B). These data suggested that the SAP30 encodes a spinach gAPX. The deduced spinach gAPX showed relatively high homologies to those of cAPX and chlAPX isoenzymes from spinach with amino acid identities of 64.2% and 47.3%, respectively, indicating that the gAPX has a higher degree of homology in its amino acid sequence with cAPX than with chlAPX isoenzymes. However, the C-terminal of gAPX, which involved the putative transmembrane segment, was approximately 40 amino acids longer than that of cAPX. APX isoenzymes and yeast cytochrome c peroxidase (CCP) have been classified as members of the class I plant peroxidases from their amino acid sequences and have been part of the lineage of prokaryotic peroxidases (Welinder 1992). The deduced spinach gAPX showed a 34.8% identity over 230 amino acids with yeast CCP and had. 12.
(20) A. B. AGTAACCAGTGAAGCTCCAATCAATGGCGATGCCGGTCGTCAACACAGAATACCTCAAGG M A M P V V N T E Y L K E. 60 13. AAATCGACAAAGCTCGTCGCGATCTTCGCGCTCTCATCTCTAATCGCAATTGCGCTCCTC I D K A R R D L R A L I S N R N C A P L. 120 33. TCATGCTCCGCCTCGCGTGGCATGATGCAGGGACTTATTGCGCGAAGACGAAGACCGGCG M L R L A W H D A G T Y C A K T K T G G. 180 53. GCCCTAATGCTTCAATTCGGAACGAGGAAGAGTGCGCTCATGGTGCCAATAATGGTTTGA P N A S I R N E E E C A H G A N N G L K. 240 73. AGAAAGCTATTGATTGGTGTGAGGAGGTGAAGTCTAAACACCCAAAGATTACATATGCAG K A I D W C E E V K S K H P K I T Y A D. 300 93. ACCTCTATCAGCTTGCTGGTGTTGTTGCAGTTGAGGTCACTGGAGGACCTACTGTTGACT L Y Q L A G V V A V E V T G G P T V D F. 360 113. TTGTTCCTGGGAGAAAGGACTCAAATGCGTGTCCCAAGGAAGGGCGACTTCCAGATGCTA V P G R K D S N A C P K E G R L P D A K. 420 133. AACAAGGTGCACCACATCTGAGGGACATATTTTACAGGATGGGTCTCACTGACAAAGACA Q G A P H L R D I F Y R M G L T D K D I. 480 153. TCGTAGCACTGTCAGGGGGTCATACCCTGGGAAGGGCACATCCAGAGAGATCAGGCTTTG V A L S G G H T L G R A H P E R S G F D. 540 173. ATGGCCCATGGACCCAGGAGCCACTCAAGTTTGATAATTCCTATTTTGTGGAACTCTTGA G P W T Q E P L K F D N S Y F V E L L K. 600 193. AAGGAGAATCAGAAGGACTGTTGCAACTTCCTACTGATAAAACTTTGGTGGAGGACCCTG G E S E G L L Q L P T D K T L V E D P A. 660 213. CATTTCGCCCTTTCGTGGAACTGTATGCTAAGGATGAGGATGTCTTCTTCCGTGACTATG F R P F V E L Y A K D E D V F F R D Y A. 720 233. CTGTCTCACACAAGAAACTTTCGGAATTAGGATTTACTCCAAGCGGAGGCAAGTCAAGCT V S H K K L S E L G F T P S G G K S S C. 780 253. GCAAAGACAGCACTATACTGGCTCAGAGTGCAGTCGGAGTTTTAGTTACTGCAGCAGTGG K D S T I L A Q S A V G V L V T A A V V. 840 273. TCATTTGTAGCTACATCTATGAAGTCCGCAAACGCTCAAAGTGAAGAGATTACACAAGTA I C S Y I Y E V R K R S K *. 900 286. ACGTCATTCATATCCATTACATACCGTCTGTATAATAAACTAGTAACATCATATATCTGA GATTCATCCTCCTCATACTATCTCCTACATCTAGGTATATATGTCACTATCATTATCAAG TATCAACTAACTTTCTATCTCGAATTTAAGTGGTTGTTTCAACTTGCTAGAAACCAGGAG TGGATCAGAATGGTACGAACTATGAAGAGCCAAATAAGTTTTACTAATATGAATATTTTC ATTTGTTCAAAAATAAAAAAAAAAAAAAAAAAAAAA. 960 1020 1080 1140 1176. 3. 0. -3. 1. 58. 11 5. 17 2. 22 9. 28 6. Fig. II-1 Nucleotide and predicted amino acid sequence of the cDNA for spinach gAPX and hydropathy profile of its amino acid sequence. (A) The amino acid sequences deduced from an open reading frame are shown below the nucleotide sequences. The amino acid sequences of the N-terminal of the native gAPX are single-underlined. Hydrophobic sequences representing the putative membrane binding domain in gAPX are double-underlined. The distal and proximal His residues are shown by heavy dots. The boxes show amino acid residues that correlate with active sites. (B) Hydrophobicity was analyzed by the GENETYX software program for a window size of nine amino acid residues. Positive hydropathy indicates hydrophobicity. The arrow indicates a putative membrane-binding domain in gAPX.. 13.
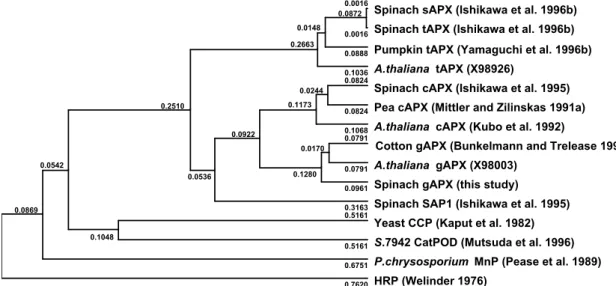
(21) less homology with the classical plant peroxidases such as horseradish peroxidase. This is also the case for the other previously described APX isoenzymes (Ishikawa et al. 1995, 1996c). A phylogenic tree was constructed according to Hein's alignment algorithm (Hein 1990). As illustrated in Fig. II-3, gAPX was more closely related to. S.gAPX C.gAPX A.gAPX S.SAP1 S.cAPX S.sAPX S.tAPX. MAM--PVVNTEYLKEIDKARRDL--RALISNRNCAPLMLRLAWHDAGTY----CAKTKTGGPNASIRN MAF--PVVDTEYLKEIDKARRDL--RALIALKNCAPIMLRLAWHDAGTY----DVSTKTGGPNGSIRN MAA--PIVDAEYLKEITKARREL--RSLIANKNCAPIMLRLAWHDAGTY----DAQSKTGGPNGSIRN MGRV-PIVNENYRRVIEAARRDLHRSLVQDNNNSAPILLRLSFHDAVDY----DAATKRGGANGSVRL MGKSYPTVSENYQKSIEKARRKL--RGLIAEKQCAPLMLRLAWHSAGTF----DCTSKTGGPFGTMKH --------YASDPAQLKNAREDI--KELLQSKFCHPIMVRLGWHDAGTYNKDIKEWPQRGGANGSLSF --------YASDPAQLKNAREDI--KELLQSKFCHPIMVRLGWHDAGTYNKDIKEWPQRGGANGSLSF. S.gAPX C.gAPX A.gAPX S.SAP1 S.cAPX S.sAPX S.tAPX. EEECAHGANNGLKKAIDWCEEVKSKHPKITYADLYQLAGVVAVEVTGGPTVDFVPGRKDSNA---CPK EEEFTHGANSGLKIAIDFCEEVKAKHPKITYADLYQLAGVVAVEVTGGPTIDFVPGRKDSNI---CPR EEEHTHGANSGLKIALDLCEGVKAKHPKITYADLYQLAGVVAVEVTGGPDIVFVPGRKDSNV---CPK AQELNRTPNKGIETAVRFCEPIKRRHPDITYADLYQLAGIVAVEVTGGPAIDADVADQDNIP-----QAELAHGANNGLVIAVRLLEPIKEQFPEITYADFYQLAEFVAVEVTGGPEVPFHPGREDKPEPPQ--DVELKHGANAGLVNALKLLQPIKDKYSGVTYADLFQLASATAIEEAGGPTIPMKYGRVDATGPEQCPE DVELKHGANAGLVNALKLLQPIKDKYSGVTYADLFQLASATAIEEAGGPTIPMKYGRVDATGPEQCPE. S.gAPX C.gAPX A.gAPX S.SAP1 S.cAPX S.sAPX S.tAPX. EGRLPDAKQG--APHLRDIF-YRMGLTDKDIVALSGGHTLGRAHPERSGF---------DGP-----EGRLPDAKRG--APHLRDIF-YRMGLSDKDIVALSGGHTLGRAHPERSGF---------DGP-----EGRLPDAKQG--FQHLRDVF-YRMGLSDKDIVALSGGHTLGRAHPERSGF---------DGP------------NPRRGADHLRTVF-YRMGLNDKDIVVLSGAHALGGAHKDRSGF---------DGD-----EGRLPDATKG--CDHLRDVFIKQMGLTDQDIVALSGGHTLGRCHKDRSGF---------EGA-----EGRLPDAGPPSPAQHLRDVF-YRMGLDDKDIVALSGAHTLGRSRPERSGWGKPETKYTKDGPGAPGGQ EGRLPDAGPPSPAQHLRDVF-YRMGLDDKDIVALSGAHTLGRSRPERSGWGKPETKYTKDGPGAPGGQ. S.gAPX C.gAPX A.gAPX S.SAP1 S.cAPX S.sAPX S.tAPX. -WTQEPLKFDNSYFVELLKGESEGLLQLPTDKTLEDPAFRPFVELYAKDEDVFFRDYAVSHKKLSELG -WTNEPLKFDNSYFLELLKGESEGLLKLPTDKALLDDPEFRKYVELYAKDEDAFFRDYAESHKKLSEL -WTQEPLKFDNSYFVELLKGESEGLLKLPTDKTLLEDPEFRRLVELYAKDEDAFFRDYAESHKKLSEL -FTRNPLTFDNSYFVELLRGDTPGLVKFPTDKALLTDPRFRPFVDLYARDQRAFFRDYAESHKKMSLL -WTTNPLVFDNTYFKELLSGEKEGLLQLPSDKALLSDPVFRPLVEKYAADEDAFFADYAEAHLKLSEL SWTAEWLKFDNSYFKDIKEKRDADLLVLPTDAALFEDPSFKVYAEKYAADQEAFFKDYAEAHAKLSNQ SWTAEWLKFDNSYFKDIKEKRDADLLVLPTDAALFEDPSFKVYAEKYAADQEAFFKDYAEAHAKLSNQ. S.gAPX C.gAPX A.gAPX S.SAP1 S.cAPX S.sAPX S.tAPX. FTPSG--GKV SSCKDSTILAQSAVGVLVTAAVVICSYIYEVRKRSK GFTPTSARSKVMVKDSTVLAQGAVGVAVAAAVVILSYFYEVRKRMK GFNPNSSAGKAVA-DSTILAQSAFGVAVAAAVVAFGYFYEIRKRMK GLNHPESNLYESNSCSTRLSVVLNPTLSKTEAVQCNTDMLDPMQLEMVAAQAATDTYNMPIYTAVNCN GFADA GAKFDPAEGITLNGTPAGAAPEKFVAAKYSSNKD GAKFDPAEGITLNGTPAGAAPEKFVAAKYSSNKRSELSDSMKEKIRAEYEGFGGSPNKPLPTNYFLNI. S.SAP1 S.tAPX. SLRD MIVIGVLAVLSYLAGN. Fig. II-2. Comparison of the deduced amino acid sequences of spinach gAPX with other APX isoenzymes. The deduced sequence of spinach gAPX (S.gAPX) is aligned with that of cotton gAPX (C.gAPX) (Bunkelmann and Trelease 1996), Arabidopsis gAPX (A.gAPX) (GenBank accession No.X98003), spinach SAP1 (Ishikawa et al. 1995), spinach cAPX (S.cAPX) (Ishikawa et al. 1995), spinach sAPX (S.sAPX) (Ishikawa et al. 1996c), and spinach tAPX (S.tAPX) (Ishikawa et al. 1996c) using a single-letter code. The gaps are introduced to optimize the alignment. Residues found at the same position as spinach gAPX are shaded.. the APXs and yeast CCP than to other peroxidases. The class I peroxidase shares the common features of the distal histidine site (R-L-A-W-H). The spinach gAPX had a completely conserved residue (His40) in the sequence. The other heme ligand site,. 14.
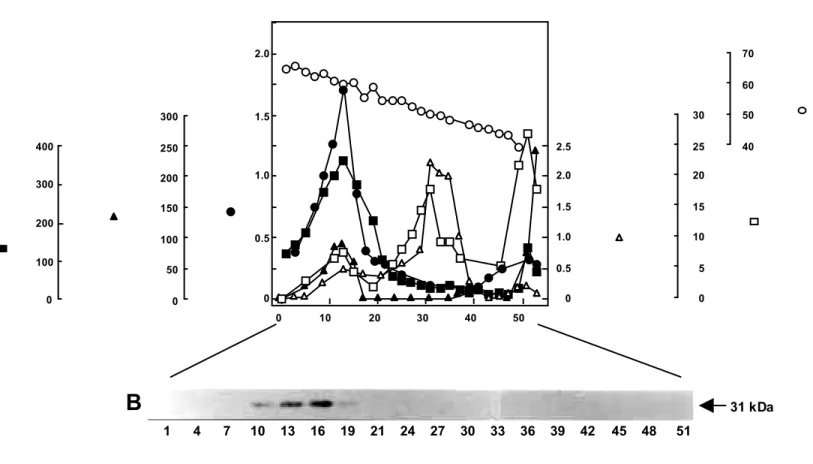
(22) the proximal histidine of the gAPX, was also conserved at the amino acid position of His160. The Asp184 residue corresponding to the active site of CCP was conserved in the gAPX as well as APX isoenzymes. The Trp residue, which was corresponding to the storage of oxidizing equivalents in Compound I (Fishel et al. 1991), was present at the amino acid position of 176. These results clearly support the idea that gAPX belongs to the class I peroxidases as do the other APX isoenzymes and that the class I peroxidase genes might have evolved from the same ancestral gene. Suborganellar distribution of gAPX Fig. II-4A shows the subcellular distribution of the APX activity together with marker enzymes (Cat, hydroxy pyruvate reductase, cytochrome c oxidase, fumarase) in a linear sucrose density gradient centrifugation. The APX activity was present only in the glyoxysome fraction; no activity was detected in the other fractions. Furthermore, in order to explore the occurrence of APX in spinach glyoxysomes, each fraction in a linear sucrose density gradient centrifugation was subjected to SDS-PAGE for immunoblot analysis using the mAb (EAP1) raised against Euglena APX. The EAP1 cross-reacted with a 31 kDa protein band in glyoxysomal fractions, which 0.0016 0.0872 0.0148 0.2663. 0.0244 0.1173. 0.2510. 0.0170 0.0542 0.0536. 0.0016. Spinach tAPX (Ishikawa et al. 1996b). 0.0888. Pumpkin tAPX (Yamaguchi et al. 1996b). 0.1036 0.0824. 0.0824. 0.0922. 0.1280. 0.1068 0.0791. 0.1048. A.thaliana tAPX (X98926) Spinach cAPX (Ishikawa et al. 1995) Pea cAPX (Mittler and Zilinskas 1991a) A.thaliana cAPX (Kubo et al. 1992) Cotton gAPX (Bunkelmann and Trelease 1996). 0.0791. A.thaliana gAPX (X98003). 0.0961. Spinach gAPX (this study). 0.3163 0.5161. 0.0869. Spinach sAPX (Ishikawa et al. 1996b). Spinach SAP1 (Ishikawa et al. 1995) Yeast CCP (Kaput et al. 1982). 0.5161. S.7942 CatPOD (Mutsuda et al. 1996). 0.6751. P.chrysosporium MnP (Pease et al. 1989). 0.7620. HRP (Welinder 1976). Fig. II-3 Phylogenic tree for APX isoenzymes and other peroxidases. The dendrogram is generated by comparison of the known amino acid sequences according to Hein (1990). Numbers indicate branch length as proportional genetic divergence. Numbers in parenthese show the cited references and accession numbers. The enzymes are as follows: CCP, chytochrome c peroxidase; S.7942 CatPOD, Synechococcus PCC 7942 Cat-peroxidase; MnP, manganese-dependent peroxidase; HRP, horseradish peroxidase.. 15.
(23) A 2.0. 70 60. 400. 250 1.0. 200. 300. 150 200 0.5. 100 100. 50. 0. 30. 50. 2.5. 25. 40. 2.0. 20. 1.5. 15. 1.0. 10. 0.5. 5. 0. 0. 1.5. 300. 0. 0. 0. 10. 20. 30. 40. 50. B. 31 kDa 1. 4. 7. 10. 13. 16. 19. 21. 24. 27. 30. 33. 36. 39. 42. 45. 48. 51. Fig. II-4 Subcellular distribution of APX and marker enzymes following linear sucrose density gradient centrifugation (A) and immunoblot analysis using the mAb (EAP1) raised against Euglena APX (B). (A) The 1,000-14,000 g pellets obtained by differential centrifugation from spinach cotyledon homogenate were resuspended in 8 ml of the washing medium containing 5 mM MOPS (pH 7.5), 0.4 M mannitol, 1 mM EDTA, 0.1% BSA, and 1 mM AsA and were then subjected to linear sucrose density gradient centrifugation (sucrose concentration from 40% to 70%). (B) Aliquots of the fractions (20 µl of each fraction) were treated with 1% SDS at 100˚C for 3 min and subjected to SDS-PAGE and immunoblot analysis. The arrow indicates the position of the gAPX (31 kDa). Detailed procedures are described in the Materials and Methods section..
(24) correlated well with the calculated molecular mass of SAP30; no cross-reactivity was found in any other fraction such as mitochondria (Fig. II-4B). The N-terminal amino acid sequence of the 31 kDa protein was determined to be M-A-M-P-V-V-N-T-E-Y-, which was completely identical with that of the deduced amino acid sequence of the SAP30 clone. These findings clearly indicated that SAP30 is the cDNA clone encoding APX isoenzyme which localizes in glyoxysomes of spinach. The presence of the C-terminal hydrophobic membrane-spanning domain of gAPX suggested that the gAPX exists as a glyoxysome membrane-bound form. The suborganellar distribution of the APX in glyoxysomes was confirmed. Most of the APX activity was detected in the KCl-insoluble membrane fraction of osmotically disrupted glyoxysomes unlike Cat, a typical soluble matrix enzyme of glyoxysomes (Table II-1). Furthermore, the latency experiment using Triton X-100 showed a high latent Cat activity in glyoxysomes. However, intact glyoxysomes showed no latent APX activity (Table II-2). The treatment of intact glyoxysomes with trypsin resulted in an irreversible inactivation of the APX; it had no effect on the Cat activity (Table II-3). The MDAR activity was also observed in the membrane fraction (Table II-1). The latent activity and tryptic treatment of the MDAR resembled those of gAPX (Table II-3). The MDAR activity was present only in the glyoxysome fraction; no activity was detected in any other fraction in a linear sucrose density gradient centrifugation (data not shown). The MDAR activity detected in glyoxysome fraction does not seem to be due to contamination by other organelles. These results clearly imply that both gAPX and MDAR are bound to the external side of the membrane of spinach glyoxysomes.. Table II-1. Suborganellar distribution of activities of APX, MDAR, and Cat in spinach glyoxisomes Enzyme activity APX (nmol/min) Crude extract KCl-soluble fraction KCl-insoluble fraction. MDAR (nmol/min). 915 ± 33 nil 638 ± 22. 108 ± 7.5 nil 99.4 ± 4.2. Cat (µmol/min) 174 ± 12 177 ± 15 1.5 ± 0.1. The intact glyoxisomes (0.6 mg of protein) were treated with 50 mM MES buffer (pH 6.0) containing 0.2 M KCl in a final volume of 500 µl. Preparations were incubated on ice and vortexed every 10 min. After 30 min of incubation, each preparation was centrifuged at 100,000 g for 30 min at 4 ˚C. The resuspended pellet and the soluble fraction were assayed for enzyme activity as described in the Materials and Methods section. Data are mean values ± SD from three assays.. 17.
(25) Table IV-2. Effect of Triton X-100 on enzyme activities of APX, MDAR, and Cat in whole glyoxisomes Enzyme activity Treatment None + 0.2 % Triton X-100. APX (nmol/min). MDAR (nmol/min) Cat (µmol/min). 1760 ± 146 1470 ± 125. 380 ± 31 325 ± 27. 5.0 ± 0.2 165 ± 11. The intact glyoxisomes were treated with Triton X-100 in 50 mM MES buffer (pH 6.0) containing 0.3 M sucrose at detergent-to-protein ratios (w/w) of 10 to 1. The glyoxisome protein (1.21 mg) was used in a final volume of 0.5 ml. The total enzyme activities were measured in the presence of 0.3 M sucrose as described in Materials and Methods section. Each value represents the mean of three assays ± SD.. Expression and partial purification of recombinant gAPX The expression systems using E. coli for the two cAPX and one sAPX isoenzymes reported in the literature yielded soluble proteins (Patterson and Poulos 1994, Dalton et al. 1996, Ishikawa et al. 1996c). These APXs exist as a soluble form in vivo but not as a membrane-bound form. I examined the optimum conditions for the expression of our recombinant gAPX which was subcloned into the pET-3a expression vector. The initiation of induction after growth saturation (optical density of 0.7 at 600nm) and the continuation of induction for 3 h resulted in a high yield of recombinant gAPX. The recombinant enzyme protein prepared from the insoluble fraction correlated with the deduced molecular mass from SAP30 (Fig. II-5A). The immunoblots of the soluble and insoluble fractions using the EAP1 raised against Euglena APX also revealed a predominant band at 31 kDa (Fig. II-5B). More than 95% of the recombinant gAPX was accumulated in the insoluble membrane fraction as active enzyme. I therefore developed the solubilization of the enzyme using some detergents and partial purification. Sodium deoxycholate (6 mM), Triton X-100 (0.5%) and CHAPS (0.2%) in 50 mM potassium phosphate (pH 7.0) containing 1 mM AsA solubilized about 76, 78, and 69%, respectively, of the recombinant gAPX from the bacterial membrane. Triton X-100 was thus a useful detergent for the membrane-bound recombinant gAPX because of the high yield and enzymatic stability. When using CHAPS or octyl-glucoside, the solubilized enzyme lost its activity within one day (data not shown). The procedure yielded a recombinant enzyme preparation purified approximately 2.2-fold over the crude enzyme, giving a final 32.0% recovery of the activity. The SDS-PAGE of the partially purified recombinant gAPX showed one major and a few faint minor protein bands; the major band contained. 18.
(26) approximately 95% of the total protein (Fig. II-5A). The specific activity of the recombinant gAPX was 81.0 ± 2.5 µmol of AsA/min per mg of protein. N-terminal Table II-3. Effect of trypsin on enzyme activities of APX, MDAR, and Cat in whole glyoxisomes Enzyme activity Treatment None + 5 µg Trypsin + 5 µg Trypsin and Trypsin inhibitor. APX (nmol/min). MDAR (nmol/min). Cat (µmol/min). 1493 ± 87 176 ± 14. 403 ± 33 29.3 ± 1.0. 100 ± 8.2 85.1 ± 5.0. 1365 ± 95. 390 ± 25. 95 ± 7.5. The intact microbodies (1.2 mg of protein) were treated with trypsin in 50 mM MES buffer (pH 6.0) containing 0.3 M sucrose at a trypsin-to-protein ratio (w/w) of 1 to 250. After 15 min incubation at room temperature, the reaction was stopped by trypsin inhibitor in an eight-fold excess relative to the weight of trypsin. As a control the reaction was incubated for the same time with trypsin and trypsin inhibitor mixed prior to addition to the organelles. Both preparations were immediately used for the enzyme assays. The enzyme activities were measured in the absence of 0.3 M sucrose as described in Materials and Methods section. Each value represents the mean of three assays ± SD.. amino acid sequence analysis of the recombinant gAPX revealed the sequence M-A-M-P-V-V-N-T-E-Y-L-K-E-I-D-K-, identical to that deduced from the cDNA. Molecular mass analysis In cotton oilseed glyoxysomes, gAPX was found to be a homodimer, because the antiserum raised from 67-kDa peroxisomal membrane protein recognized a 31 kDa protein (Bunkelmann and Trelease 1996). The authors, however, did not identify the native gAPX activity. Judging from SDS-PAGE and immunoblot analysis (Fig. II-5), the subunit molecular mass of both the native and recombinant gAPXs was found to be 31 kDa, which correlated with the deduced molecular mass from SAP30 open reading frame. The E.coli soluble fraction corresponding to a few % of the total recombinant gAPX activity was subjected to a HiLoad 16/60 Superdex 200 column equilibrated with a 50 mM sodium phosphate buffer (pH 7.0) containing 1 mM EDTA, 1 mM AsA, and 20% (w/v) sorbitol. The soluble recombinant enzyme had a molecular mass of 31 kDa, indicating that the glyoxysome enzyme exists as a monomeric form in its native state. I failed to determine the exact molecular mass of the Triton X-100- solubilized recombinant enzyme because it aggregated during solubilization; this might have been caused by its C-terminal hydrophobic region. Enzymatic properties of gAPX. 19.
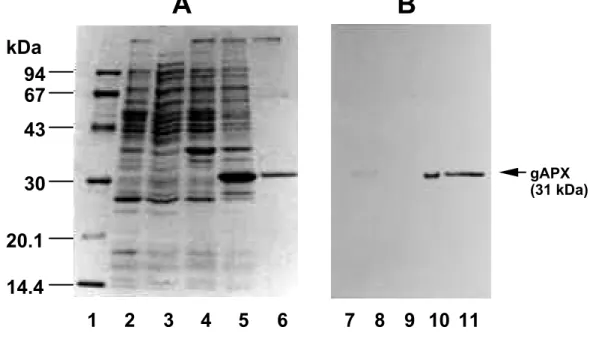
(27) The enzymatic properties of the almost purified recombinant gAPX were consistent with those of the native enzyme from intact glyoxysomes. Both recombinant and native gAPXs utilized AsA as the most effective natural electron donor; GSH and NAD(P)H could not substitute for AsA. They catalyzed the oxidation of pyrogallol at a rate one third lower than that with AsA. With respect to its donor specificity, gAPX resembled the chlAPX isoenzymes more than the cAPX isoenzymes (Nakano and Asada 1987, Chen and Asada 1989, Mittler and Zilinskas 1991a, Miyake et al. 1993, Ishikawa et al. 1996a). Km values for AsA and H2O2 were determined using Lineweaver-Burk plots with substrate concentrations of 0.1-0.5 mM for AsA and 20-350 µM for H2O2. The substrate-velocity curves with recombinant and native gAPXs showed Michaelis-Menten type kinetics with AsA and H2O2. The recombinant cAPXs from pea and soybean failed to obey Michaelis-Menten kinetics because of the complication of the monomer/dimer equilibrium (Dalton, et al. 1996, Patterson and Poulos 1994). From Lineweaver-Burk plots, the apparent Km values for AsA of the native and recombinant gAPX at 0.1 mM H2O2 were determined to be 1.82 ± 0.05 mM and 1.89 ± 0.05 mM, respectively. These. A. B. kDa 94 67 43 gAPX (31 kDa). 30 20.1 14.4 1. 2. 3. 4. 5. 6. 7 8 9 10 11. Fig. II-5 Overexpression and partial purification of recombinant gAPX in E.coli. The overexpression of pET-SAP30 in E.coli and partial purification of the recombinant gAPX were carried out as described in the Materials and Methods section. (A) Coomassie Brilliant Blue staining of SDS-PAGE. (B) Immunoblot analysis using the Euglena APX mAb, EAP1. Positions and sizes in kilodaltons of marker proteins are shown on the left side of the panel. Lane 1, molecular mass markers; lanes 2 and 7, a soluble fraction of the cells without. 20.
(28) pET-SAP30; lanes 3 and 8, a soluble fraction of the cells harboring pET-SAP30; lanes 4 and 9, an insoluble fraction of the cells without pET-SAP30; lanes 5 and 10, an insoluble fraction of the cells harboring pET-SAP30; lanes 6 and 11, partially purified recombinant gAPX.. values were approximately 3.5-7-fold higher than those of other APX isoenzymes (Chen and Asada 1989, Mittler and Zilinskas 1991a, Ishikawa et al. 1996a). The Km values for H2O2 of the native and recombinant gAPX were 80 ± 5.3 µM and 74 ± 4.0 µM, respectively, when the concentration of AsA was 0.4 mM. The partially purified recombinant gAPX was completely inhibited by 0.6 mM azide and 50 µM cyanide, indicating that the spinach gAPX, like other APX isoenzymes, is a typical hemeprotein. The enzyme was also sensitive to the thiol-modifying agent p-chloromercuric benzoate at 0.1 mM. The optimum pH and temperature of the recombinant gAPX were 7.0 and 37˚C, respectively. Previous reports with respect to cAPX and chlAPX isoenzymes from various sources indicate optimum pH ranges of 6.0-8.0 (Chen and Asada 1989, Mittler and Zilinskas 1991a, Ishikawa et al. 1996a). The enzyme retained its full activity up to 45˚C between pH 6.3 and 8.0 and lost its activity completely at 53˚C. One of the specific properties of APXs is rapid inactivation in an AsA-depleted medium. This is especially true for chlAPX, whose half-inactivation time was only 15 s (Miyake and Asada 1996). cAPX isoenzymes are more stable than the chlAPX isoenzymes (Asada 1992). When the partially purified recombinant gAPX was diluted with the. 100. gAPX cAPX. 80. Residual APX activity (%). 60. 40. sAPX. 20 0 0. 60. 120. 180. Time (min). Fig. II-6 Effect of the AsA-depletion on APX activities. The recombinant cytosolic APX (cAPX) and recombinant stromal APX (sAPX) from spinach were prepared as described previously (Ishikawa et al. 1995, 1996c) and partially purified. Each APX protein (1.2 mg/ml). 21.
(29) suspended in 10 mM potassium phosphate buffer (pH 7.0) containing 0.1 mM AsA and 20% (W/V) sorbitol was diluted with 10 mM potassium phosphate buffer (pH 7.0) to a final AsA concentration of less than 10 µM. After incubation for the indicated time, 1 mM AsA was added, and the enzyme activity was assayed.. AsA-depleted medium, the activity was stable over 180 min (Fig. II-6), which was in agreement with pea cAPX (Mittler and Zilinskas 1991a). Miyake and Asada (1996) have reported that the inactivation of sAPX isoenzyme in an AsA-depleted medium is caused by the instability of Compound I to H2O2 when AsA is not available for Compound I. When spinach recombinant gAPX is incubated with 2 µM H2O2 under anaerobic conditions, no inactivation is observed (data not shown). Accordingly, one of the reasons for the stability mechanism in spinach gAPX may be the insusceptibility of Compound I to H2O2. The exact reason for the difference in stability among APX isoenzymes is not clear at present.. Discussion In spinach, we have demonstrated the molecular characteristics of many APX isoenzymes, including the one in this study (Ishikawa et al. 1995, 1996c). This novel APX isoenzyme, gAPX, has some characteristic properties distinguishing it from other APX isoenzymes. The hydropathy analysis of its primary structures indicated that a putative significant hydrophobic segment is present in the C-terminal region (Fig. II-1B). A similar membrane-spanning region at the C-terminal region is present in the tAPX isoenzyme, although no other similarity is observed in the C-terminal region (Ishikawa et al. 1996c). As far as I know, no other reports exist of such a unique membrane spanning system. It is of interest whether the C-terminal hydrophobic region of the gAPX serves as a novel targeting motif of the microbody outer-membrane and the anchor of the protein. Based on the latency of gAPX activity by solubilization assay with Triton X-100 and the tryptic digestion in intact glyoxysomes (Table II-2), the catalytic domain of APX on glyoxysome membranes was found to be exposed to the cytosol. This was also true in an APX from pumpkin microbodies (Yamaguchi et al. 1995b). In cotton oilseed, however, the majority of gAPX, including the active site, was predicted to be on the matrix side of the glyoxysomes (Bunkelmann and Trelease 1996). SODs in peroxisomes from many plant species were detected as isoenzymes differing in their suborganellar localization. In watermelon cotyledons, a Cu/Zn-SOD has been detected in a soluble matrix and a Mn-SOD on the external side of the peroxisomal membrane as a binding form. On the other hand, in pea leaves, a Mn-SOD has been located only in the peroxisomal matrix (Sandalio and del Río 1988). Similarly, the. 22.
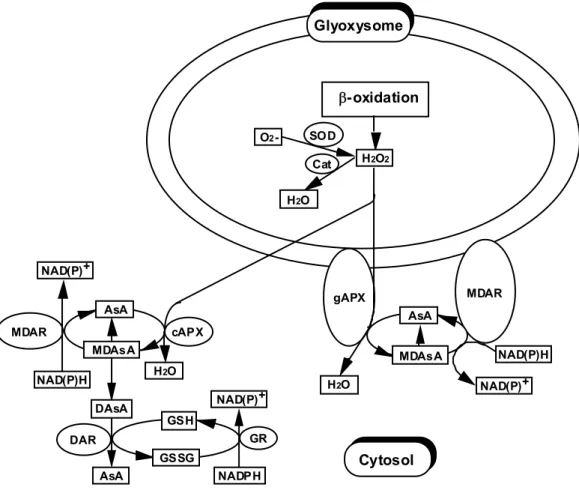
(30) suborganellar localization of APX in glyoxysomes may also depend on the tissue origin. In pumpkin, the gAPX had been recognized as a rich glyoxysomal membrane protein with a molecular mass of 31 kDa, which was retained in peroxisomes during the microbody transition from glyoxysomes to peroxisomes (Yamaguchi et al. 1995a). The fact that the SDS-PAGE analysis detected a main band which corresponds to the 31 kDa gAPX protein in intact glyoxysomes from spinach cotyledons also suggests that the APX commonly plays an essential role in these organelles (data not shown). In recent years, it has become clear that microbodies carry out essential functions in almost all eukaryotic cells and have an essentially oxidative type of metabolism (van den Bosch et al. 1992). The acyl CoA oxidase in -oxidation and the glycolate oxidase in photorespiration directly produce H2O2. Furthermore, there are two sites of O2 production in peroxisomes; one is the generating system of xanthine oxidase in the matrix and the other is the NADH-dependent electron transport system in the membrane. Some xenobiotics, such as clofibrate, stimulate the production of O2 and H2O2 inside microbodies (del Río et al. 1992). Taking into account these data, it seemd important to determine the extent to which APX and Cat are involved in the detoxification of H2O2 generated in microbodies. The Cat localized in their matrix appears to be rather unsuited to the natural state of leaves because it suffers from a light-induced inhibition of function accompanied by a loss of the enzyme protein (Feierabend et al. 1992). In addition, Cat is very inefficient at removing low concentrations of H2O2, because the Km value for H2O2 is high (~1 M) (Huang et al. 1983). The Km value for H2O2 of spinach gAPX was 74 ± 4.0 µM. The Km value for AsA of spinach gAPX was 1.89 ± 0.05 mM, which is approximately 3.5-7-fold higher than those of other APX isoenzymes (Chen and Asada 1989, Mittler and Zilinskas 1991a, Ishikawa et al. 1996a). It has been reported that the concentration of AsA in apoplasts of spinach leaves is estimated to be between 0.1 and 0.5 mM and that the average intracellular AsA concentration is far higher than the apoplastic AsA concentration (Takahama and Oniki 1992). Accordingly, the spinach cytosolic AsA concentration seems to be at least several mM, allowing gAPX to remove H2O2 in situ. Based on our present data and other data so far, I propose the H2O2-scavenging system involving spinach glyoxysomes shown in Fig. II-7. The MDAsA is the primary product of the APX reaction, and rapid regeneration of AsA is an absolute necessity for the scavenging of H2O2 by APX (Asada 1992). In chloroplasts, AsA is regenerated from the oxidation products in the sAPX and tAPX reaction by MDAR using NAD(P)H as the electron donor and direct reduction by ferredoxin photoreduced in PS-I (Miyake and Asada 1994). In caster bean and cotton glyoxysomes, MDAR has been found to be membrane-associated and appears to be bound to the matrix side of the organelles (Bowditch and Donaldson 1990, Bunkelmann and Trelease 1996).. 23.
(31) In contrast, the MDAR of spinach glyoxysomes was bound to the external side of the membrane of glyoxysomes, in analogy with APX. It seems conceivable that the MDAsA which is generated by the peroxidase reaction is effectively regenerated to AsA by utilizing cytosolic NAD(P)H via glyoxysome outer-membrane-bound MDAR. Since the microbody membrane is sparsely permeable to NAD(P)H (Liang et al. 1984), the reduction of MDAsA radical by the enzyme seems not to be affected by permeation of the NAD(P)H from the cytosol to the glyoxysome matrix site. Furthermore, in caster bean, Bowditch and Donaldson (1990) assessed the suitability of MDAsA as a physiological acceptor for the glyoxysomal membrane-bound NADH dehydrogenase. H2O2 diffuses readily through biological membranes (Takahashi and Asada 1983). In NaCl-sensitive pea cultivars, the Cat activity was inhibited, and glycolate oxidase was stimulated by NaCl (Corpas et al. 1993). The authors thus have suggested that H2O2 can diffuse out of peroxisomes into the cytosol as a result of NaCl-induced leakage of the peroxisomal membranes. We have demonstrated that. Glyoxysome. -oxidation O2 -. SOD H2O2. Cat H2O. NAD(P) +. MDAR. AsA. cAPX MDAsA. MDAsA. H2O. NAD(P)H DAsA. MDAR. gAPX. AsA. NAD(P) +. H2O. NAD(P) +. NAD(P)H. GSH GR. DAR. Cytosol. GSSG AsA. NADPH. Fig. II-7 Possible model of the functional organization of the H2O2-scavenging system in glyoxysomes. AsA, ascorbate; cAPX, cytosolic ascorbate peroxidase; gAPX, glyoxysome-bound ascorbate peroxidase; Cat, catalase; SOD, superoxide dismutase; MDAsA,. 24.
(32) monodehydroascorbate; MDAR, monodehydroascorbate reductase; DAsA, dehydroascorbate; DAR, dehydroascorbate reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; GR, glutathione reductase.. H2O2 formed in Euglena organelles, including chloroplasts and mitochondria diffuses from them into the cytosol (Ishikawa et al. 1993). The excretion of H2O2 has been reported from many photosynthetic organisms, including green microalgae (Zepp et al. 1987). It seems likely that some of the H2O2 generated in the microbody matrix diffuses from the organelle toward the cytosol and then is decomposed by the mAPX binding to the outside of the membrane. A set of the enzymes responsible for the AsA-GSH cycle located in the cytosol may serve as a partialy H2O2-scavenging system. Thus, the APX isoenzymes and AsA regeneration systems may function to protect microbodies from intoxication by H2O2 produced during some physiological activites.. Summary cDNAs encoding two cytosolic and two chloroplastic ascorbate peroxidase (APX) isoenzymes from spinach have been cloned recently (Ishikawa et al. 1995, 1996). I herein report the cloning of the fifth cDNA of an APX isoenzyme which localizes in spinach gAPX. The open reading frame of the 858-base pair cDNA encoded 286 amino acid residues with a calculated molecular mass of 31,507 Da. By determination of the latency of APX activity in intact glyoxysomes, the enzyme, as well as MDAR, was found to be located on the external side of the organelles. The cDNA was overexpressed in E.coli. The enzymatic properties of the partially purified recombinant gAPX were consistent with those of the native enzyme from intact glyoxysomes. The recombinant enzyme utilized AsA as its most effective natural electron donor; GSH and NAD(P)H could not substitute for AsA. The substrate-velocity curves with the recombinant enzyme showed Michaelis-Menten type kinetics with AsA and H2O2; the apparent Km values for AsA and H2O2 were 1.89 ± 0.05 mM and 74 ± 4.0 µM, respectively. When the recombinant enzyme was diluted with AsA-depleted medium, the activity was stable over 180 min. I discuss the H2O2-scavenging system maintained by APX and the regeneration system of AsA in spinach glyoxysome.. 25.
(33) CHAPTER III Comparative Study on Recombinant Chloroplastic and Cytosolic Ascorbate Peroxidase Isoenzymes of Spinach One of the characteristic properties of APX isoenzymes is that they are very labile in AsA-depleted medium; especially, chloroplastic isoenzymes (sAPX and tAPX) lost their activities within several minutes compared to the cytosolic forms (Asada 1997). These facts indicate one of the reasons why it is difficult to obtain large amounts of the highly purified chlAPX isoenzymes. chlAPX isoenzymes have therefore been purified only from spinach and tea leaves (Chen and Asada 1989, Nakano and Asada 1987). On the other hand, the cAPX has been purified from many plant species (Chen and Asada 1989, Mittler and Zilinskas 1991a, Tanaka et al. 1991, Ishikawa et al. 1996a, Elia et al. 1992). cDNAs for cAPX encoded by nuclear genes have been isolated and characterized from several plant sources including spinach (Mittler and Zilinskas 1991b, Kubo et al. 1992, Ishikawa et al. 1995). To date, the recombinant pea cAPX has been used for structural study, and the crystal structure of its recombinant protein has been refined to an R = 0.19 for data between 8.0 and 2.2 Å resolution (Patterson and Poulos 1994, 1995). In the previous study, we have demonstrated the first complete cloning of cDNAs and nuclear gene encoding sAPX and tAPX from spinach leaves. The molecular characterization indicates that both chlAPX isoenzymes arise from a common pre-mRNA by alternative splicing of two 3'-terminal exons (Ishikawa et al. 1996c, 1997, Chapter V). Advances in the expression of the recombinant APX isoenzymes utilizing these cDNA clones, as shown in Fig. III-1, would therefore provide new approaches to the structure and function of each APX isoenzyme of higher plants. In this chapter, I have introduced the cDNA clones encoding spinach chlAPX and cAPX isoenzymes in a pET expression vector to overexpress these proteins in E. coli and obtained the purified recombinant sAPX and cAPX relatively in quantity by conventional column chromatography. The recombinant APX isoenzymes obtained were used for comparative studies with the native APX isoenzymes from spinach leaves.. 26.
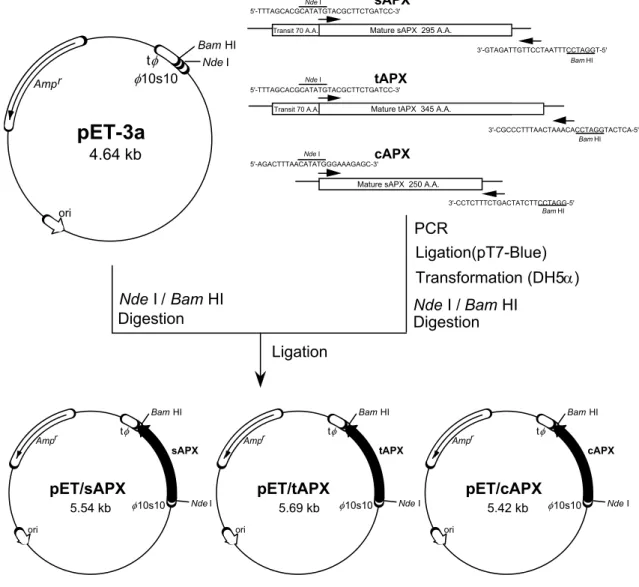
(34) sAPX tAPX. MASFTTTTAAAASRLLPSSSSSISRLSLSSSSSSSSSLKCLRSSPLVSHLFLRQRGGSAYV MASFTTTTAAAASRLLPSSSSSISRLSLSSSSSSSSSLKCLRSSPLVSHLFLRQRGGSAYV. 61 61. sAPX tAPX cAPX. TKTRFSTKC--------YASDPAQLKNAREDIKELLQSKFCH-PIMVRLGWHDAGTYNKDI TKTRFSTKC--------YASDPAQLKNAREDIKELLQSKFCH-PIMVRLGWHDAGTYNKDI MGKSYPTVSENYQKSIEKARRKLRGLIAEKQC-APLMLRLAWHSAGTFD---. 113 113 48. sAPX tAPX cAPX. KEWPQRGGANGSLSFDVELRHGANAGLVNALKLLQPIKDKYSGVTYADLFQLASATAIEEA KEWPQRGGANGSLSFDVELRHGANAGLVNALKLLQPIKDKYSGVTYADLFQLASATAIEEA -CTSKTGGPFGTMKHQAELAHGANNGLVIAVRLLEPIKEQFPEITYADFYQLAEFVAVEVT. 174 174 108. sAPX tAPX cAPX. GGPTIPMKYGRVDATGPEQCPEEGRLPDAGPPSPAQHLRDVF-YRMGLDDKDIVALSGAHT GGPTIPMKYGRVDATGPEQCPEEGRLPDAGPPSPAQHLRDVF-YRMGLDDKDIVALSGAHT GGPEVPFHPGREDKPEPPQ---EGRLPDATKG--CDHLRDVFIKQMGLTDQDIVALSGGHT. 234 234 164. sAPX tAPX cAPX. LGRSRPERSGWGKPETKYTKDGPGAPGGQSWTAEWLKFDNSYFKDIKEKRDADLLVLPTDA LGRSRPERSGWGKPETKYTKDGPGAPGGQSWTAEWLKFDNSYFKDIKEKRDADLLVLPTDA LGRCHKDRSG----------------FEGAWTTNPLVFDNTYFKELLSGEKEGLLQLPSDK. 295 295 209. sAPX tAPX cAPX. ALFEDPSFKVYAEKYAADQEAFFKDYAEAHAKLSNQGAKFDPAEGITLNGTPAGAAPEKFV 356 ALFEDPSFKVYAEKYAADQEAFFKDYAEAHAKLSNQGAKFDPAEGITLNGTPAGAAPEKFV 356 ALLSDPVFRPLVEKYAADEDAFFADYAEAHLKLSELGFADA 250. sAPX tAPX. AAKYSSNKD AAKYSSNKRSELSDSMKEKIRAEYEGFGGSPNKPLPTNYFLNIMIVIGVLAVLSYLAGN. 365 415. Fig. III-1 Comparison of amino acid sequence between APX isoenzymes from spinach. A sequence alignment is shown for sAPX, tAPX, and cAPX from spinach. The gaps are introduced to optimize the alignment. Residues found at the same position as sAPX are shaded. The asterisks show the consensus amino acids. The arrow indicates the processing site of chlAPXs.. Materials and Methods Materials The cDNAs encoding spinach APX isoenzymes were originally cloned into plasmid pBluescript SK(+) (Ishikawa et al. 1995, 1996c). Plasmid pET-3a and its companion production E. coli strain BL21(DE3)pLysS were obtained from Novagen. The E. coli strain DH5 was from Gibco BRL (MD, USA). The molecular biology reagents and enzymes were of analytical grade and were purchased from commercial sources. Construction of spinach APX isoenzymes expression plasmid The complete coding sequences of spinach APX isoenzymes were derived from a cDNA library constructed from greening cotyledons (Ishikawa et al. 1995, 1996c). For the construction of plasmid to express the APX isoenzymes, the DNA fragments encoding mature form were amplified by PCR. The oligonucleotide primers used for. 27.
(35) the amplification of the spinach APX isoenzyme gene into pET3a vector were shown as follows (Fig. III-2): P-1; 5'-TTTAGCACGCATATGTACGCTTCTGATCC-3' P-2; 5'-TGGATCCTTTAATCCTTGTTAGATG-3' P-3; 5'-ACTCATGGATCCACAAATCAATTTCCCGC-3' P-4; 5'-AGACTTTAACATATGGGAAAGAGC-3' P-5; 5'-GGATCCTTCTATCAGTCTTTCTCC-3' sAPX. Nde I 5'-TTTAGCACGCATATGTACGCTTCTGATCC-3'. Mature sAPX 295 A.A.. Transit 70 A.A.. t 10s10. Amp r. Bam HI. 3'-GTAGATTGTTCCTAATTTCCTAGGT-5' Bam HI. Nde I. tAPX. Nde I 5'-TTTAGCACGCATATGTACGCTTCTGATCC-3'. Mature tAPX 345 A.A.. Transit 70 A.A.. pET-3a. 3'-CGCCCTTTAACTAAACACCTAGGTACTCA-5' Bam HI. 4.64 kb. cAPX. Nde I 5'-AGACTTTAACATATGGGAAAGAGC-3'. Mature sAPX 250 A.A. 3'-CCTCTTTCTGACTATCTTCCTAGG-5' Bam HI. ori. PCR Ligation(pT7-Blue) Transformation (DH5) Nde I / Bam HI Digestion. Nde I / Bam HI Digestion Ligation. Bam HI Ampr. sAPX. pET/sAPX 5.54 kb. ori. Bam HI. t. 10s10. Ampr. tAPX. pET/tAPX. Nde I. Bam HI. t. 5.69 kb. ori. 10s10. t. Ampr. cAPX. pET/cAPX. Nde I. 5.42 kb. 10s10. Nde I. ori. Fig. III-2 Construction of plasmid pET/APX for the expression of spinach APX isoenzymes. Shown are the steps performed to ligate the mature APX isoenzymes-encoding PCR-amplified fragments into the expression vector pET-3a, yielding the pET/APX isoenzymes. The details are described under Materials and Methods section. Ampr, ampicillin-resistance gene; ori, E. coli replication origin; 10s10, T7 promoter and transcription initiation site; t T7 transcription terminator.. The sequence was homologous to the spinach APX isoenzyme cDNA except for the replacement of the original nucleotides, which introduced the desired restriction sites (underlined). The DNA fragments coding the mature form of sAPX and tAPX were 28.
(36) amplified using the oligonucleotide P-1 and P-2, and P-1 and P-3, respectively. The DNA fragment encoding cAPX was amplified using the oligonucleotide P-4 and P-5. The PCR amplification was carryed out in a 100 µl reaction mixture containing 10 µl of 10 x PCR Buffer (Mg2+ free), 1.5 mM MgCl2, 200 µM each of dNTPs, 2.5 units of Recombinant Taq DNA polymerase (Takara), 1.0 µM of each oligonucleotides, and 5 ng of template cDNA. The conditions of the PCR were as follows: 30 cycles at 94˚C for 1 min, 60˚C for 1 min and 72˚C for 2 min. The DNA fragments generated by PCR were purified by gel electrophoresis and ligated into pT7Blue T-vector (Novagen). Following the transformation of E. coli strain DH5 cells, clones of candidate mutants were isolated and sequenced across the region of interest by the dideoxy chain-primer method in order to establish the fidelity of all APX isoenzyme construction. From these plasmids digested with Nde I and Bam HI, a 0.90-kbp DNA fragment encoding the mature form of sAPX, a 1.05-kbp DNA fragment encoding the mature form of tAPX, and a 0.78-kbp DNA fragment encoding cAPX were isolated and integrated into the pET-3a expression vector treated with the same restriction enzymes and then introduced into the E. coli strain DH5, respectively. Plasmid DNA was prepared from the ampicillin-resistant transformants and verified by digestion with the restriction enzymes Nde I and Bam HI. The resulting constructs, designed pET/sAPX, pET/tAsA, and pET/cAPX, respectively, were introduced into the E. coli strain BL21(DE3)pLysS to test each one's ability to direct the synthesis of recombinant spinach APX isoenzymes (Fig. III-2). Production of spinach recombinant APX isoenzymes in E. coli E. coli BL21(DE3)pLysS transformed pET/sAPX, pET/tAPX, and pET/cAPX, respectively, was grown in 50 ml LB medium supplemented with ampicillin at 37˚C for overnight. The cultures were then inoculated with the final 1.0 l LB culture. When the culture reached an absorbance of 0.6 at 600 nm, 400 µM IPTG was added, and the bacteria were grown for a further 6 h at 37˚C. Cells were harvested by centrifugation at 6,000g for 10 min, and the pellets were kept frozen at –20˚C for analysis of the accumulation by SDS-PAGE and for the purification of the enzymes. Enzyme assays The APX activity was assayed as described in chapter II. The protein content was determined by the method of Bradford (Bradford, 1976). Purification of recombinant APX isoenzymes All purification steps were carried out at 4˚C. The recombinant E. coli cells (3.8 g FW) transformed with pET/sAPX were resuspended in 20 ml of 10 mM. 29.
(37) potassium phosphate buffer, pH 7.0, containing 20% (w/v) sorbitol, 1 mM AsA, 0.1% PMSF, and 1 mM EDTA (buffer A) and disintegrated by passing them through a cooled French pressure cell at 15,000 psi (SLM Aminco, Inc.). This lysate was centrifuged at 15,000 g for 20 min. The supernatant, designated crude extract, was subjected to ultracentrifugation at 100,000 g for 30 min. The obtained supernatant was then loaded onto a DEAE-Sephacel column (1.6 x 12 cm) equilibrated with buffer A. The column was washed with 50 ml of buffer A and eluted with a 400-ml linear gradient NaCl (0-0.3 M) at a flow rate of 0.7 ml min-1. The activities of sAPX were eluted as a single peak at 0.08 M of NaCl. The active fractions were subjected to (NH4)2SO4 precipitation at 30% saturation. The precipitate was removed by centrifugation, and the supernatant was loaded onto a HiLoad 16/10 Phenyl Sepharose column equilibrated with 30% saturated (NH4)2SO4 in buffer A. The enzyme activity was eluted with a descending gradient of (NH4)2SO4 from 30 to 0% saturation in 144 ml of buffer A at an elution rate of 0.8 ml min-1. Active fractions were combined and fractionated with (NH4)2SO4, and the pellet precipitating at 70% saturation was dissolved in 2 ml of buffer A. The enzyme solution was loaded onto a HiLoard 16/60 Superdex 200 column equilibrated with buffer A containing 0.15 M NaCl. The active fractions were combined and stored at –20˚C. The recombinant E. coli cells (5.2 g FW) transformed with pET/cAPX were resuspended in 20 ml of buffer A and disintegrated by passing them through a cooled French pressure cell. This lysate was centrifuged at 15,000 g for 20 min. The crude extract was subjected to ultracentrifugation at 100,000 g for 30 min. The obtained supernatant was then loaded onto a DEAE-Sephacel column (1.6 x 12 cm) equilibrated with buffer A. The column was washed with 50 ml of buffer A and eluted with a 400-ml linear gradient NaCl (0-0.3 M) at a flow rate of 0.7 ml min-1. The activities of cAPX were eluted as a single peak at 0.12 M of NaCl. Active fractions were combined and fractionated with (NH4)2SO4, and the pellet precipitating at 70% saturation was dissolved in 2 ml of buffer A. The enzyme solution was loaded onto a HiLoard 16/60 Superdex 200 column equilibrated with buffer A containing 0.15 M NaCl. The active fractions were combined and adjusted to 30% saturation with (NH4)2SO4. The precipitate was removed by centrifugation, and the supernatant was chromatographed on a 5/5 Phenyl Superose column equilibrated with 30% saturated (NH4)2SO4 in buffer A. The column was eluted with a 11-ml linear gradient of 30-0% (NH4)2SO4 at a flow rate of 0.15 ml min-1. The active fractions were combined and stored. SDS-PAGE and immunoblot analysis SDS-PAGE was performed as described in chapter II, using a 12.5% resolving gel. Immunoblot analysis was carried out using the mAb to Euglena APX (EAP1). 30.
図

+7


Outline
関連したドキュメント
Second, it was revealed that ADAR1-mediated RNA editing positively regulates DHFR expression in human breast cancer-derived MCF-7 cells by destroying miR- 25-3p and miR-125a-3p
NELL1 (a) and NELL2 (b) mRNA expression levels in renal cell carcinoma cell lines OS-RC-2, VMRC-RCW, and TUHR14TKB and control HEK293T cells were analyzed using quantitative
RNA polymerase II subunit 5 (RPB5) is part of the lower jaw of RNAPII , and the exposed domain of RPB5 serves in interactions with transcriptional regulators including Hepatitis B
Quantitative analysis by real-time Reverse transcription-polymerase chain reaction (RT-PCR) of chronological change in the expression of hepatocyte growth factor (HGF),
東京大学 大学院情報理工学系研究科 数理情報学専攻. [email protected]
情報理工学研究科 情報・通信工学専攻. 2012/7/12
Then optimal control theory is applied to investigate optimal strategies for controlling the spread of malaria disease using treatment, insecticide treated bed nets and spray
As in the previous case, their definition was couched in terms of Gelfand patterns, and in the equivalent language of tableaux it reads as follows... Chen and Louck remark ([CL], p.
