Title
Acquired Tolerance to Oxygen Stress in Bifidobacterium longum
105-A by Heterologous Expression of Catalase Gene( 本文
(FULLTEXT) )
Author(s)
賀, 建龍
Report No.(Doctoral
Degree)
博士(農学) 甲第583号
Issue Date
2012-03-13
Type
博士論文
Version
publisher
URL
http://hdl.handle.net/20.500.12099/42969
※この資料の著作権は、各資料の著者・学協会・出版社等に帰属します。
Contents
1. Introduction---I--- 1 1. 1Bljidobacterium---
1 1.1.1 Description---1 1.1.2 History---4 1.1.3 Species---'---I---5 1.1.4 Ecology---6 1.2 Peroxidase---9 1.2.1 Description---i--i---9 1.2.2 GPX---i---10 1.2.3 Catalase---J---12 1.2.3. 1 Molecular mechanism---1 3 1.2.3.2 Cellular role---141.2.3.3 Distribution among organisms---1 5 1.3 Research the response of the
Bljidobacterium
to Oxygen---1 7 2. Materials and Methods---202. 1 Media and Buffer---i---20
2. 1. 1 Luria Broth media---20
2.1.2 MRS media---,---20
2.1.3 TE buffer---I---21
2.1.4
PEG---:---21
2. 1.5 PBS buffer--I---21
2.2 Isolate DNA from microorganisms---2 1 2.3 Extract plasmid---2 1 2.3.1 With
QIA
prep spin Mini prep kit---212.3.2 With 2-propano1---22 2.3.2. 1 The reagent---.---22 2.3.2.1. 1 Solution I---22 2.3.2. 1.2 Solution II---22 2.3.2. 1.3 Solution III---22 2.3.2.2 Protocol---22 2.4 Purify DNA---23
2.4.1 With Nucleo Spin Extract II kit---24
2.4.2 With Ethano1---24
2.4.2. 1 I The reagent---24
2.4.3 With PEG---25
2.4.3. 1 The reagent---25
2.4.3.2 Protocol---25
2.5 Preparation of competent cell---25
2.5.1 Electrocompetent cell of BiBdobacterium---25
2.5. 1. 1 The reagent---25
2.5.1.2 Protocol---26
2.5.2 Chemically competent cell of E.boli---26
2.5.2. 1 The reagent---26
2.5.2.2 Protocol---27
2.6 SDS-PAGE---i---27
2.6.1 The reagent---27
2.6.2
Protocol---i-I--29
2.7 Detection activity of catalase---32
2.7.1 Add H202 detect activity.f
catalas;---i---32
2.7.1.1 Thethe.,y-;---i---32
2.7. 1.2 Protocol---.---32 2.7.2 Assay ofcatalase---32 2.7.2. 1 The reagent---33 2.7.2.2 Protocol---33 2.8 Short-term H202 exposure---34 2.8.1 The reagent---34 2.8.2 Protocol---342.9 Long-term with aerated cultures---35
2.9.1 The reagent---35
2.9.2 Protocol---I""_"___"__"______"__""_MM___M""__"Mum""__"__35 2. 1 0 PCR---3 5 2.10.1 With po1ymerase KOD-PLUS---i---35
2.10.2 With po1ymerase GO Taq---35
2. 1 1 DNA extraction from qgarose
gels---36
2.12 Digestion with restriction enzyme---T---36
2. 12. 1 The restricti.n
enzyme---J-i---36
2. 12.2 Protocol---36 2. 1 3 DNA ligation---I---36 2. 1 4 Transformation---36 2. 14.1 Chemical transformation---37 2. 14. 1. 1 The reagent---37 2. 14. 1.2 Protocol---372. 14.2 Electroporation---38 2. 14.2. 1 The
reagent---;---38
2. 14.2.2 Protocol---i--38 2. 1 5 Gateway system---3 8 2.16 Flow cytometry---39 2. 16.1 The reagent---.---39 2. 1 6.2 The instrument---392. 1 7 Isolate RNA from microorganisms---I---39
2. 1 8 RT-PCR---39 2.19 qRT-PCR---39 2.19.1 The reagent---39 2. 1 9.2 The instrument---39 2.20 Assay of H202---40 2.20. 1 The
reagent---i---40
2.20.2 Protocol---40 2.2 1 Site-Directed Mutagenesis---402.22 The strains, the plasmids and primers---42
3. Results---44
3. 1 Isolate DNA &om microorganisms---i---44
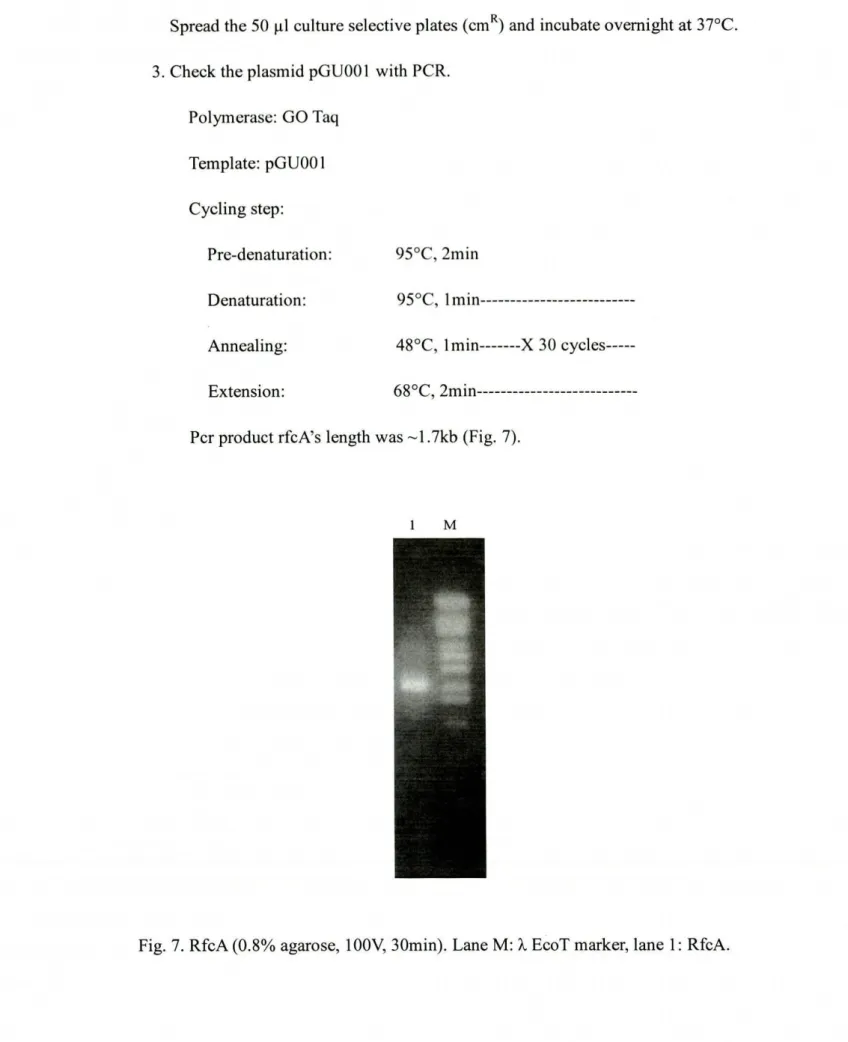
3.2 Get the PCR product---45
3.2.1 Design the prim'ers---45
3.2.2 PCR---45
3.2.2. 1 PCR of Hup-promoter---45
3.2.2.2 PCR
ofB-KatE---i_"MUM"___"__47
3.2.2.3 PCR of Hup-terminator---483.2.2.4 Overlap PCR---49
3.3 Construct destination vector---50
3.3.1 Digest pKKT427---50
3.3.2 Construct destination vector pGUOO1---51
3.4 Construct the plasmids---53
3.4. 1 BP reaction---53
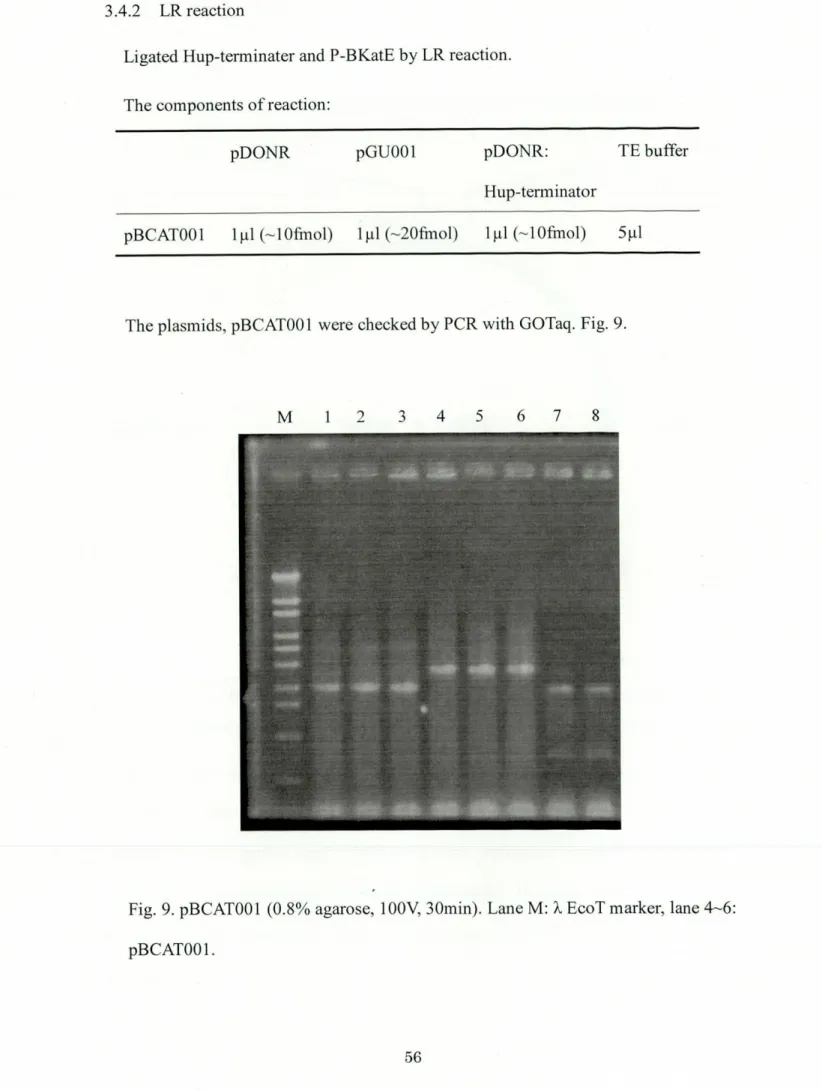
3.4.2 LR
,eacti.n---_M"_"_"____""""q_____"_:MM____56
3.5 Analyze the sequences---573.5.1 Hup-promoter---58
3.5.2 Hup-terminator---58
3.6 The catalase activity in E. coli UM255---58
3.7 The catalase activity in B. longum 1 05-A--.---59
3.7.1
pBCATOO1
(Heme-catalase)
activity---593.8 Short-term H202 exposure OfB.longum 105-A---63
3.9 Long-term with aerated cultures ofB.longum 105-A-i---64
3.9. 1 0D---64
3.9.2 CFU---65
3.9.3 LIVE/DEAD---66
3.9.4 H202 accumulation---67
4. Discussion---68
4. 1 Protect
Bljidobacterium
from oxidative stress by expression of catalase---684.2 Improvement of catalase expression level---7 1 4.2.1 Transcription---71
4.2.2 Translation---75
5. Conclusion---85
6. References---89
Intro duction
1.1
Bljidobacterium
1.1.1- Description
At birth,the gastrointestinal tract is sterile and incapable of digesting food. Within
hours, bacteria ingested during the birthing process rapidly colonize the gut. The
gastrointestinal tract soon contains about 1 0 times as many bacteria as there are cells
in the body. Hundreds of species are present, many of which are uncultivable and
remain unidentified. It is these bacteria that are responsible for priming the
gastrointestinal immune system. This gut flora includes 100 trillion bacteria, some
three pounds, which are intimately linked to the bodyTs natural defense system.
(18)
probiotics are deflned as live microbial food ingredients that benefit human health.Most probiotics fall into the group of organisms known as lactic acid-producing
bacteria and are normally consumed in the form of yogurt, fermented milks or other
fermented foods. The concept ofprobiotics arose atthe turn of the 20th century from
a hypothesis flrSt Proposed by. Noble Prize winning Russian scientist Elie
Metchnikoff, who suggest that the long, healthy life of Bulgarian peasants resulted from their consumption of fermented milk products. He believed that when consumed, the fermenting Bacillus
(Lactobacillus)
positively influenced the micro flora of the colon, decreasing toxic microbial activities. The historical association of probioticswith fermented dairy products, stilltrue today, stems from these early observations.
Investigations in the probiotics field during the past several decades, however, have
expanded beyond bacteria isolated from fermented dairy products to those of
Possible health effects of probiotics
Intestinal
eHects
・ Relieve
effects, promote recovery from diarrhea
(rotavirus,
travelers' andantibioticinduced)
・ produce
lactase, alleviate symptoms of lactose intolerance and malabsorption
・ Relieve
constipation
・ Treat
colitis
Immune system
eHects
・ Enhance
specific and nonspeciflC immune response
・
Inhibit pathogen growth and translocation
・ Stimulate
gastrointestinal immunity
・ Reduce
chance of infection from common pathogens
(Salmonella,Shigella)
Other
eHects
・ Reduce risk of certain cancers
(colon,bladder)
・ Detoxifycarcinogens
・ Suppress tumors ・ Lower'
serum cholesterol concentrations
・ Reduce blood
pressure in hypertensives
・ Treat food
allergies
・ synthesize
nutrients
(folic
acid, niacin, riboflavin, vitamins B6 & B12)
・ Increase
nutrient bioavailability
・ Improve
urogenital health
・ Optimize
effects of vaccines
(e.g.
rotavirus vaccine, typhoid fevervaccine)
There is some debate about whether or not yogurt starter bacteria should beStreptotoccus thermophilus are used to ferment milk and turn it into yogurt. But these cultures are not very resistant to conditions in the stomach and small intestine and
generally do not reach the gastrointestinal tract in very high numbers. 'Therefore, they cannot mediate some probiotic effects. But these starter bacteria have been shown to
improve lactose digestion in people lacking lactase and have demonstrated some
itnmunity enhancing effects. For these reasons, they are often considered 'probiotic'.
Most gastrointestinal organisms are relatively benign. Some are potentially more
pathogenic; however, many are actually beneficial; it is these beneBcial organisms
that have attracted attention as.possible probiotics. The table below lists some
suggested health benefits of consuming probiotics. Those that have signiflCant
research to back up the claims are discussed in more depth later in this article.
(79)
Bljidobacterium,
one type of probiotics, are a natural part of the bacterial flora inthe human body and have a symbiotic bacteria-host relationship with humans. B.
longum promotes good digestion, boosts the immune system, and produces lactic and
acetic acid that controls intestinal pH. These bacteria also inhibit the growth of
Candida albicans, E. coli
(9),
and other bacteria that have more pathogenic qualitiesthan
Bljidobacterium. Bljidobacterium
are normal inhabitants of the human andanimal colon. Newborns, especially those that are breast-fed, are colonized with
bifldobacteria within days after birth.
Bljidobacterium
were first isolated &om thefeces of breast-fed infants. The population of these bacteria in the colon appears
relatively stable until advanced age, when itseems to decline. They are saccharolytic
organisms that produce acetic and lactic acids without generation of CO2, except during degradation of gluconate. They are also classified as lactic acid bacteria
Bindobacterium www. sciencephoto. com
1.1.2 History
Bljidobacterium
was Brst isolated as a bacteria in France in 1 899. Itwas found in ahealthy breast-fed baby, but the bacteria did not come from the breast milk. Itcame
B-om something the baby had consumed. The first name
bljidobacterium
was calledBacillus
bljidus
communis. There are 30 different types ofbljidobaclerium
speciesthat exist. When this was known, in 1960,
bljidobacterium
was accepted as its own1.1.3 Species
The table below lists the species ofbifidobacteria.
Bljidobacterium
angulatumBljidobacterium
animalisBljidobacterium
asteroidsBljidobacterium
bljidum
Bljidobacterium
boumBljidobacterium
breveBljidobacterium
catenulatumBljidobacterium
choerinumBljidobacteriumco7yneforme
Bljidobacterium
cuniculiBljidobacterium
dentiumBljidobacterium
gallicumBljido
bacterium gallinarumBljidobacterium
indicumBljidobacterium
longumBljidobacterium
magnumBljidobacterium
mefyCicumBljidobacterium
spBljidobacterium
pseudolongumBljidobacterium
psych;aeroph
ilumBljidobacterium
pullorumBljidobacterium
ruminantiumBljidobacterium
saeculareBljidobacterium
scardoviiBljidobacterium
simiaeBljidobacterium
subtileBljidobacterium
thermacidophilumBljidobacterium
thermophilumBljidobacterium
urinalisBljidobacterium
magnumBljidobacterium
m e7yCicumBljidobacterium
minimumBljidobacterium
pseudocatenulatumBljidobacterium
minimum1.1.4 Ecology
While
Bljidobacterium
infantis,
B. brevi, and B. longum are the largest group ofbacteria in the intestine of infants,
Bljidobacteria
are said to be only the 3rd or 4thlargest group in adults
(and
comprise only 3-6% of adult fecalnora).
The number-ofBljidobacteria
actually decline in the human body with age. In infants who arebreast-fed,
Bljidobacteria
constituteab6ut
90% of their intestinal bacteria;- however, this number is lower inbottle-fed
infants. When breast-fed infantsT diets are changedto cows milk and solid food,
Bljidobacteria
arejoined
by rising numbers of otherbacteria found ihthe human body such as Bacteroides and Streptococci lactobacilli.
The lower number
,ofBljidobacteria
in formula-fed babies might account for a higher
risk of diarrhea and allergies that is usually associated with babies who aren't breast-fed; in addition, because
Bljidobacteria
produces lactic acid instead of gas(like
E.colt),
infants and people in general with moreBljidobacteria
than other bacteria will have less gas and digestive problems. There is also a significantdifference in the incidence of antibiotic-associated diarrhea in the children receiving
probiotic-supplemented
(inriched
withBljidobacterium)
formula(16%)
than nonsupplemented formula(31%). (14,
68, 88,90)
Tissier described B.
bljidus
in the stools of breast-fed infants, itwas recognizedthat the organisms became predominant in the stools between days 4 and 7 after birth.
Although L. acidophilus was the most numerous lorganism in the stools of bottle-fed
infants, bifidobacteria were also found in smaller numbers. It was believed that
bifidobacteria were present in small numbers in stools of adults, but they were
difficult to isolate because of the lack of a suitable culture medium or method of
reducing oxygen tension. For the next half century little progress was made in
interest in the occurrence and distribution of bifidobacteria was revived. Mata et a1.
reported the average figures for the incidence of bifldobacteria in the feces of breast-fed infants to be approximately loll, in weanlings 1010, and in adults 101
organ.isms per g
(wet weight).
The figures expressed by these authors are in generalagreement with those expressed earlier by Gyllenberg and
Roin;,
Smith and Crabb, Zubrzycki and Spaulding,Weijers
and van de Kamer, and Werner. They aresomewhat higher than those reported by Kalser et a1. Werner and Seeliger, Mossel,
and Gorbach et a1. In a study ofintestina1 flora in a rural area ofGuatemala,
Mata
andurrutia reported that bill-dobacteria appeared on the flrSt day of life in only a few
infants. One-third of the babies they studied had these bacteria on the second day of
life. By the end of the flrSt Week all infants had them in concentrations ranging &om 101 to loll organisms per g of feces. These workers also reported that Bacteroides and Veillonellae were not frequently found in the stools of breast-fed neonates,
although their concentration ranged from 108 to 1011 when found. When 12 breast-fed infants were studied throughout the flrSt year Of life, bifldobacteria continued to be
the most numerous bacterium, amounting to 1010 to 1011 organisms per g of feces.
With food supplementation the anaerobic gram-negative bacilli became more
numerous and eventually outnumbered other bacterial groups. Mata and Urrutia
summarized their data by stating that nearly 100% of all bacteria cultured from the
stools of breast-fed infants were bifidobacteria. During weaning there was a decrease
by 1 log and a proliferation of Bacteroides. In adults, Bacteroides outnumbered all
other groups. Although this study was a valuable aid in estimating the relative
occurrence of various groups of intestinalorganisms and their variation with age and
other factors, no attempt was made at a hef classification of members of the genus
Bifidobacterium. These organisms were simply referred to as the bifidobacteria group.
classiflCation schemes. Miller reported on the fecal flora of seven eskimo children.
He reported B. adolescentis in concentration of 108 to loll per g in all the specimens
studied and noted that in one case members of the genus bifidobactera exceeded those
of the bacteroides group. In a study of the intestinal flora of adults, Moore et a1.
reported that in some cases members of bifldobactera outnumbered bacteroides, thus
confirming the observations of some earlier workers on this point. Significant
differences in incidence of bifidobacteria among individuals and possibly among
certain restricted populations are evident. An investigation of variation in the
incidence ofbifidobacteria among newborns was carried out in our laboratory. In the
course of investigations, we noted fewer bifidobacteria in newborns in a large, urban
universityl hospital than in a suburban hospital in the same area. In the past,
bifldobacteria could readily be cultured from the stools
6f
breast-fed infants at the Hospital of the University of Pennsylvania. Recently, difficulty was experienced inculturing these organisms from the stools of infants in the nursery. Gram-stained spreads of feces of
breast-fed
infants, 3 to 4 days old, were examined. Of 61breast-fed infants
(14,
35, 36, 37, 38,66)
Bljidobacteria
as well as other beneficial bacteria can be found in fermented dairyfoods, especially yogurt. Eating substances rich with these probiotics is a sort of
home remedy for diarrhea, vaginitis, and
_yeast infections because it promotes the growth of these as opposed to other bacteria. B. infantis has been proven to
dramatically reduce imitable bowel sydrome
(IBS)
in patients and if given alone canStzlePtOCOCCUS
S. thern70PJLj]usATCC l9258 S.8Lljb,ATCC437 65 1.2 Peroxidase 1.2.1 Description IFGran(-)
OeJ20CLmuS OeL7j JCM6 I 25Lactococcus
Genealogical treePeroxidases
(EC
number1.ll.1.A)
are a large family of enzymes that typicallycatalyze a reaction of the form:
ROORr +
electron donor
(2 e-)
+ 2H' - Roll + R'OI1For many of these enzymes the optimal substrate is hydrogen peroxide, but others
are more active with organic hydroperc.xides such as lipid peroxides. Peroxidases can
contain a heme co factor in their active sites, or redox-active cysteine or
selenocysteine residues.
Peroxidase can be used for treatment of industrial waste waters. For example, phenols, which are important pollutants, can be removed by enzyme-catalyzed
radicals, which participate in reactions where polymers and oligomers are produced
that are less toxic than phenols.
Furthermore, peroxidases can be an alternative option of a number of harsh
chemicals, eliminating harsh reaction conditions. There are many investigations
about
the use of peroxidase in many manufacturing processes like adhesives, computer
chips, carparts, and linings of drums and cans.
(10,
13, 20, 34, 43, 63,74)
The table below lists the types ofperoxidases.
I Glutathione peroxidase
(GPX)
・ Haloperoxidase ・ Myeloperoxidase(MPO)
・Catalase(Rat)
・ Hemoprotein ・ Peroxide I Peroxiredoxin I Animal heme-dependent peroxidases ・ Thyroid peroxidase I Vanadium bromoperoxidase ・ Lactoperoxidase 1.2.2 GPXGlutathione peroxidase
(GPX) (EC 1.ll.1.9)
is the general nameof an enzyme
family with peroxidase activity whose main biological role is to protect the organism from oxidative damage. The biochemica1
functioh
of glutathione peroxidase is to reduce lipid hydroperoxides to their corresponding alcohols and to reduce freehydrogen peroxide to water.
(2,
3, 6, 34,54)
GPX family is well studied in eukaryote, especially in animals, and act a key role
in H202
scavenging.
In the case of bacteria, very limited information about GPX wereavailable. Itwas reported in few of species, Neisseria meningitidis
(Aho
E. L. et a1.1995)
Streptococcuspyogenes(King
K. Y. et aL.2000)
and Eschericha colt(Arenas
F.A. et al
2010).
It is not commonly accepted that GPX plays an important role inbacteria, especially in anaerobic bacteria. No orthologue to these 3 bacterial GPX
(also
EukaryoticGPX)
has been found in the bifidobacterial genome. Glutathione synthesize pathway has not been found in bifidobacteria(figure
seebelow). (79, 80)
1.2.3 Catalase
Catalase is a common enzyme found in nearly all living organisms which are
exposed to oxygen, where it functions to catalyze the decomposition of hydrogen
peroxide to water and oxygen. Catalase has one of the highest turnover numbers of all
enzymes; one molecule of catalase can convert millions of molecules of hydrogen
peroxide to water and oxygen per second.
(1,
5, 10, 20, 22, 23, 45, 50, 63,72)
Catalase isa tetramer of four polypeptide chains, each over 500 amino acids long. It contains four porphyrin heme
(iron)
groups that allow the enzyme to react with the hydrogen peroxide. The optimum pH for catalase is approximately 7, while theoptimum temperature varies by species.
Catalase
(EC
1.11.1.6)
catalyzes disproportioning ofH202 tO H20 and 02 and thus protects the cells against the oxidative effect of H202. This enzyme is present in all aerobes and many aerotolerant anaerobes. Two types of phylogenetically remoteheme catalases are known: monofunctional catalases and bifunctional catalases
peroxidases, which
for
the catalase activity use H202(Km
- 2.5 6.5mM)
as anelectron donor and for the peroxidase activity
(Km
- 0.2 0.7mM)
use variousorganic compounds
byroga11o1,
diaminobenzidine, dimethoxybenzidine, dianizidine, NADH, NADPH,etc.).
Monofunctional catalases are found in all three empires ofliving nature, whereas the distribution of bifunctional heme catalases is limited
(with.
rare
exceptions)
to bacteria and archaea.Unlike mono- and dimeric bifunctional catalases-peroxidases, monofunctional
catalases are mainly tetrameric proteins characterized by higher temperature stability,
wide pH optimum
(5.5-16.5),
and lack of inactivation with ethan.I/ch1.r.f.rm. However, 3-amino-1In addition to heme catalases, there are Mn catalases with a unique primary structure and resistance to N3-, which have been found in some facultative anaerobes such as
the lactic acid bacterium Lactobacillus plantarum and the hyperthermophilic archaeon
Pyrobaculum calidifontis.
Catalase was first noticed as a substance in 1811 when Louis Jacques Th6nard,
who discovered H202
(hydrogen peroxide),
suggested that its breakdown is caused bya substance. In 1900, Oscar Loew was the firstto give it the name catalase, and found
its presence in many plants and animals. In 1937 catalase &om beef liver was
crystallised by James B. Sumner and the molecular weight worked out in 1938. In
1969 the amino acid sequence of bovine catalase was worked out. Then in 1981, the
3D structure of the protein was
revealedi
1.2.3.1 Molecular mechanism
The reaction of catalase in the decomposition of hydrogen peroxide is:
2H202 -+ 2H20+02
While the complete mechanism of catalase is not currently known, the reaction is believed to occur in two stages:
H202 +
Fe(III)-E.)
H20 +0-Fe(IV)-E(.+)
H202 +
0-Fe(IV)-E(.+)
- H20 +Fe(III)-E
+ 02Here
Fe()-E
represents the iron centre of the heme group attached to the enzyme.Fe(IV)-E(.+)
is a mesomeric form ofFe(V)-E,
meaning that iron is not completely oxidized to +V but receives some T'supporting electron'' A.om the heme ligand. ThisAs hydrogen peroxide enters' the active site, it interacts with the amino acids
Asn147
(asparagine
at position147)
and His74, causing a proton(hydrogen ion)
to transfer between the oxygen atoms. The free oxygen atom coordinates, freeing the newly-formed water molecule andFe(IV)-0. Fe(IV)-0
reacts with a secondhydrogen peroxide molecule to reform
Fe(III)-E
rand produce water and oxygen. The reactivity of the iron center may be improved by the presence of the phenolate ligand of Tyr357 in the fifth iron ligand, which can assist in the oxidation of theFe(III)
toFe(IV).
The efflCiency of the reaction may also be improved by the interactions of His74 and Asn147 with reaction intermediates. In general, the rate of the reaction canbe determined by the Michaelis-Menten equation.
Catalase can also oxidize different toxins, such as formaldehyde, formic acid,
phenols, and alcohols. In doing so, it uses hydrogen peroxide according to the
following reaction:
H202+ H2R -> 2H20 + R
Again, the exact mechanism of this reaction isnot known.
Any heavy metal ion
(such
as copper cations incopper(II)sulfate)
will act as anoncompetitive inhibitor on catalase. Also, the poison cyanide is a competitive
inhibitorl of catalase, strongly binding to the heme of catalase and stopping the enzymeTs action.
Three-dimensional protein structures of the peroxidated catalase intermediates are
available at the Protein Data Bank. This enzyme is commonly used in laboratories as
a tool for leaming the effect of enzymes upon reaction rates.
Hydrogen peroxide is a harmful by-product of many normal metabolic processes:
To prevent damage, it must be quickly converted into other, less dangerous
substances. To this end, catalase is frequently used by cells to rapidly catalyze the
decomposition of hydrogen peroxide into less reactive gaseous oxygen and water
molecules.
The true biological signiflCanCe Of catalase isnot always straightforward to assess:
Mice genetically engineered to lack catalase are phenotypically normal, indicating
that this enzyme is dispensable in animals under some conditions.
Some human beings have very low levels of catalase
(acatalasia),
yet show few ill effects. It is likely that the predominant scavengers of H202 in normal mammalian cells are peroxiredoxins rather than catalase.Catalase works at an optimum temperature of 37 oC, which is approximately the
temperature of the human body.
Catalase is usually located in a cellular organelle called the peroxisome.
Peroxisomes in plant cells are involved in photorespiration
(the
use of oxygen andproduction of carbon
dioxide)
and symbiotic nitrogen fixation(the
breaking apart of diatomic nitrogenP2)
tO reactive nitrogenatoms).
Hydrogen peroxide is used as a potent antimicrobial agent when cells are infected
with a pathogen. Pathogens that are catalase-positive, such as
Mycoba;terium
tuberculosis, Legionella pneumophila, and Campylobacter
jejuni,
make catalase in order to deactivate the peroxide radicals, thus allowing them to survive unharmed within the host.All known
animals
use catalase in every organ, with particularly highconcentrations occuming in the liver. One unique use of catalase occurs in
bombardier beetle. The beetle has two sets of chemicals ordinarily stored separately
in its paired glands. The larger of the pail, the storage chamber or reservoir, contains
hydroquinones and hydrogen peroxide, whereas the smaller of the pair, the reaction
chamber, contains catalases and peroxidases. To activate the
spray;
the beetle mixes the contents of the two compartments, causing oxygen to be liberated from hydrogenperoxide. The oxygen oxidizes the hydroquinones and also acts as the propellant.
Catalase is also universal among plants, but not among fungi, although some
species have been found to produce the enzyme when growing in an environment
with a low pH and warm temperatures.
Very few aerobic microorganisms are known that do not use catalase.
Streptococcus species are an example of aerobic bacteria that do not possess catalase.
Catalase has also been observed in some anaerobic microorganisms, such as
Methanosarcina barkeri.
Catalase is used in the food industry for removing hydrogen peroxide &om milk
prior to cheese production. Another use is in food wrappers, where it prevents food
from oxidizing. Catalase is also used in the textile industry, removing hydrogen
peroxide from fabrics to make sure the material is peroxide-free. A minor use is in
contact lens hygiene - a few lens-cleaning
products disinfect the lens using a
hydrogen peroxide solution; a solution containing catalase is then used to decompose
the hydrogen peroxide before the lens is used again. Recently, catalase has also begun to be used in the aesthetics industry. Several mask treatments combine the enzyme with hydrogen peroxide on the face with the intent of increasing
cellular oxygenation
1.3 Research the response of the
Btjidobactert'um
to OxygenIn de Vries et al. study, bifldobacteria were divided into three groups
(84).
The fact that none of theBljidobacterium
strains used in the present investigation grew onagar
plates under aerobic conditions, is in accordance with the.observations ofDehi T. and S. Bald et al.. It is evident that bifldobacteria cannot tolerate the 0- tension of air.Accumulation ofH202 Seems tO be a minor factor in explaining the high
sensitivity of
ttvo other
Bljidobacterium
strains tor02(S
822 andi
328, groupIII).
Despite of the fact that small amounts ofH202 Were formed by cell suspensions and cell-free extractsof these strains, no H202 could be detected in aerated cultures. The partial
inactivation of fructose-6-phosphate phosphoketolase observed in aerated cultures
was presumably caused by traces of intracellular H202. Even in the absence of 02,
concentrated cell suspensions of these strains do not succeed in establishing the oxidation-reduction potential required for fermentation of glucose. Addition of
cystcine or ascorbic acid lowers the oxidationreduction
potehtial
to the required value. This is true for both anaerobically-grown cells as cells from aerated cultures. In theabsence of cysteine, anaerobic growth of these strains is slow. Cells do not succeed in
bringing the oxidation-reduction potential of the medium to a value suitable for more
rapid growth until the concentration of the cells exceeds a given value. The1
oxidation-reduction potential obtained finally in such a culture is very low. From the
results, it is evident that does not exert a lethal action on these strains
(group III),
butprevents growth by establishing a too high oxidation-reduction potential.
(1
8, 38, 46,47,77,89)
In Kawasaki S. et al. study, B. boum and B. thermophilum show growth
conditions, so they are reasonably classified as anaerobes. The accumulation of H202
in 02-Sensitive species must be the end product of 02 reduction. NADH-dependent
oxidase activities were detected as part of an 02 reduction system. Although the total
activity of NADH dependent oxidase in the CFE was similar among species, the
activity profiles differed between 02-Sensitive and microaerophilic species. The
growth inhibition ofB.
bljidum
and B. longum under 20% 02 conditions was partiallyreversed when catalase was added to the medium. The inability of exogenously added catalase to decompose intracellular H202 might be a reason for the
failure
to obtaincomplete growth recoveries. These results indicate that the primary factor in aerobic inhibition is the production ofH202 derived from 02 reduction.
(39,40)
Oxidative stress can be defined as an excess of reactive oxygen species
(ROS)
that have strong oxidizing potential for tells. ROS cause damage to macromolecularconstituents such as DNA, RNA, proteins and lipids. Toxicity occurs when the degree of oxidative stress exceeds the capacity of cell defence systems. ROS originate &om partial.reduction of molecular oxygen
(02)
tO SuPerOXide(02-),
hydrogen peroxide(H202)
and hydroxyl radical(OH. ).
The biological sources ofROS are numerous, e.g.they can be generated in aerobiosis by flavoproteins, and by macrophages during
inflammatory reactions. Thus, oxidative stress plays an important role in pathologies
of the gastrointestinal tract ofhumans such as inflammatory bowel diseases
and in the
radio-induced tissue
injury
that may occur during radiotherapy.As probiotics, some species of
Bljidobacterium
are widely used in varioustherapeutics and food products. However, as bifidobacteria are obligate anaerobic
bacteria, their sensitivity to 02 limits their manufacturing and storage. Bifidobacteria have an oxidase function that uses 02 aS an electron acceptor to reduce to H20 and
aerobic growth inhibition in bifidobacteria. Although NADH peroxidase was found in
bifldobacteria and some types of peroxidases were predicted, including thiol
peroxidase, alkyl hydroperoxide reductase, and peptide methionine sulfoxide
redhctase,
these peroxidases are unable to decompose the H202 Produced bybifldobacteria under aerobic conditions. In addition, we could not detect H202
scavenging activity in B. longum 105-A.
Superoxide dismutase, catalase, and glutathione peroxidase
(GPX)
constitute the enzymatic antioxidant system. GPX and catalase are the enzymes that can decomposeH202 tO H20. GPX was mainly studied in eukaryotic organisms; however, little is
known about GPX activity in prokaryotic organisms, and a GPX gene has not been
detected in the genomic sequences ofbifidobacteria. In addition, bifidobacteria do not
carry glutathione synthesize pathway. Catalase is an enzyme commonly found in
aerobic bacteria but is absent in almost all anaerobic bacteria
lincluding
Bljidobacterium.
B. subtilis KatE(heme-dependent catalase)
isa well-known catalasethat is used to improve the viability of some species of bacteria via heterologous
expression
(13,
50,63).
In this study, we investigated the effects of expressing B. subtilis heme-dependent catalase on theoxidative
stress resistance of B. longum1 05-A. For comparison, the effects ofAexogenously added cafalase on B. longum were
Materials and Methods
2.1 Medium and Buffer
E. coli and B. subtilis were grown in LB medium at 37oC. For anaerobic culture, B.
longum 105-A was grown in MRS medium
(Difco,
Franklin, NewJersey)
containing0.34% L-ascOrbic acid sodium salt
Papalai
Tesque, Kyoto,Japan),
0.02% L-cysteinepacalai Tesque),
and 50 mM sucrose at 37oC. For aerobic culture,I B. longum 105-Awas grown in MRS medium containing 50 mM sucrose and 10 LIM hemin
(Sigma-Aldrich,
St. Louis,MO)
at 37oC. When necessary, antibiotics were used at the following concentrations: chloramphenico1(Cm;
25LLg/mL),
kanamycin(h;
50pg/mL)
and spectinomycin(Sp;
75LLg/mL).
Aerated cultures(10 mL)
ofBljidobacterium
were grown in 25-mL tubes using silicone plugswith shaking at 120
rpm. Anaerobic cultures
(10 mL)
were grown in 12-mL test tubes with screw caps.CFUs ofB. longum 105-A were counted as follows: appropriate sample dilutions were
prepared in sucrose solution
(50
mM sucrose and 1 mM triammonium citrate, pH6.0),
plated on MRS agar, and incubated for 48 h at 37oCunder anaerobic conditions using
the AnaeroPack system
(Mitsubishi.
Gas Chemical, Tokyo,Japan).
2.1.1
LuriaBrothmedia(LB)
Trypto neYeast extract
Agrose
2.1.2 MRSmedium
Difco Lactobacilli MRS Broth
...0.5%
*Approximat6 Fomul Per Liter Protesose Peptone Beef Extract Yeast Extract Dextrose Polysorbate 80 Ammonium Citrate Sodium Acetate Magnesium sulfate Manganese sulfate Dipotassium phosphate
L-Ascorbic Acid Sodium Salt
(Vc)
L- Cysteine Agrose 2.1.3 TEbufferTris-HC1(pH8)
EDTA(pH
8)
2.1.4 PEG Polyethylene glycol 6000 *Use a brown bottle in one month.
2.1.5 PBSbuffer KC1 0.34% 0.02% 1.5%
(ifnecessary)
1 0mM 0.02%Na2HPO4
KH2PO4
0.144%
0.024%
*Adjust
the pH to 7.4 with HC1.Because that the solution can't be stored, so the concentration be 10 x to stored.
2.2 Isolate DNA from microorganisms
Using UltraClean Microbial DNA Isolation kit.
2.3 Extractplasmid
2.3.1 With
QIA
prep spin Mini prep kit UsingQIA
prep spin Mini prep kit.2.3.2 With 2-propano1 2.3.2.1 Thereagent 2.3.2.1.1 SolutionI
Tris-HC1(pH8)
EDTA(pH8)
50mM 25mM 1 0mM*Confect the lM ofTris-HCl to store.
Ajust
pH with lM ofNaOH. Confect the lM ofEDTA to store.Ajust
pH with lM ofNaOH.NaOH
*Because that the solution can't be stored, so the concentration be 10 x to
stored. When useing it, provisional confect. SDS is the last one added into
the tube.
SDS :sodium dodecyl sulfate
2.3.2.1.2 SolutionIII Potassium
Acetate(5M)
Acetate 60mL ll.5mL 28.5mL 2.3.2.2 Protocol1. Transfer the microbial culture to a microcentrifuge tube.
2. Centrifuge for 2 min at 12,000 rpm
(-12,000
xg),
discard the supematant. 3. Add 200 pl Solution I and mix thoroughly by vortexing.4. Add 400 Lil Solution II and mix thoroughly by inverting the tube 4-6 times.
5. Add 300 pl Solution III and mix thoroughly by inverting the tube 4-6 times.
6. Wait 5 min.
7.
C)entrifuge
for 10 min at 12,000 rpm(-12,000
xg),
transfer the supernatant toa new microcentrifuge tube.
8. Add lLtl of 10 mg/mL RNAse into the microcentrifuge tube.
9. Incubate at 37oC for 30-60 min.
times.
ll. Centrifuge for 15 min at 12,000 rpm
(-12,000
xg),
transfer the supernatantto a new microcentrifuge tube.
12. Add 500 lil of 70% ethanol and mix thoroughly by inverting the tube 2
times.
13. Centrifuge for 10 min at 1'2,000 rpm
(-12,000
xg),
discard the supernatant. 14. Centrifuge for an additional 1 min at 12,000 rpm(-12,000
xg)
to removeresidual ethano1.
15. Dry the pellet in the incubator or in the aspirator.
16. About 15 min later, dissolved in 50 lilofTE buffer. Store p.1asmid fi.ozen
(-20oC).
2.4 PurifyDNA
2.4.1 With Nucleo Spin Extract II kit
using PCR clean-up Gel extraction NucleoSpin@ Extract II. MACHEREY-NAGEL
kit.
2.4.2 With Ethano1
2.4.2.1 Thereagent
lI3M Sodium Acetate buffer, pH 5.2
(stole
at 4oC)
lICold 100% Ethano1(-20oC)
tCold 70% Ethanol in sterile dH20
(-20oC)
)DNA sample4 oC Microc'entrifuge
(normal
microcentrifuge in cold room worksfine).
Allcentrifugations should be on "soft''
(no brake)
setting.2.4.2.2 Protocol
1. Transfer DNA to a container where it f111sone fourth the total volume.
(a
500pl tube should have no more than 125 Lil ofDNA solution, for
example)
2.I Add one tenth volume of Sodium Acetate buffer to equalize ion
concentrations.
3. Add at least two volumes ofcold 100% ethano1; let stand in
-20oC freezer for
at least one hour.
4. Centrifuge sample for 15 minutes at highest speed in a 4oC microcentrifuge.
5. Remove as much supernatant as possible with a 1 mL micropipet; recentrifuge,
then remove the rest with a 200
pl pipet.
6. Add 200 pl ofcold 70% ethano1; centrifuge for 5 minutes in a 4 oC centrifuge.
7. Remove supernatant with a 200 Lil pipet; evaporate remaining ethanol in a
37 oC water bath.
8. Resuspend pellet in desired volume of water or TE buffer.
2.4.3 With PEG
2.4.3.1 The reagent
LPEG
A70% Ethanol in sterile dH20
(-20oC)
IIDNA sample1. Mix 1 volume ofDNA sample with 1 volumes of PEG.
2. Incubate at 4oC for 30 min.
3. Centrifuge for 15 min at4oC with 15,000 rpm, discard the supematant.
4. Add 500 Lil 70% ethanol and don't mix.
5. Centrifuge for 15 min at4oC with 15,000 rpm, discard the supematant.
6. Centrifuge for 1 min at4oC with L15,000 rpm to remove the residual ethano1.
7. Dry the pellet in the incubator or in the aspirator.
8. About 1 5 min later, resuspend pellet in desired volume ofddH20.
2.5 Preparation of competent cell
2.5.1 Electrocompetent cell ofBifidobacterium
2.5.1.1 Thereagent
Sucrose buffer
Sucrose
1 00mM Ammonium citrate
bH6.0)
*
Ajust
pH&ith
lM citrate. Autoclave 121oC for 20 min. MRS medium1.712%
2.5.1.2 Protocol
1. Incubate the Bifidobacteria at 37oC with anaerobic culture by MRS medium.
3. Transfer 10 mL culture to the tube.
4. Centrifuge for 10min at4oC with 6,000 rpm, discard the supernatant.
5. Add 10 mL sucrose buffer and mix thoroughly by vortexing.
6. Centrifuge for 10min at4oC with 6,000 rpm, discard the supernatant.
7. Add 5 mL sucrose buffer and mix thoroughly by vortexing.
8. Centrifuge for 10min at 4oC with 6,000 rpm, discard the supernatant
remaining 1 mL.
9. Transfer 50 lilto a new microcentrifuge.
10. Transfer the tube to ice. Use it in 1 hour.
2.5.2 Chemically competent cell ofE.coli
2.5.2.1 Thereagent
0.1 M CaC12
LB medium
2.5.2.2 Protocol
1. Incubate E. coli at37oC with 120 rpm shaking.
2. When OD660-0.5, stop culture.
3. Transfer 1.5 ml culture to a microcentrifuge tube, and chill on ice for 10
minutes.
4. Pellet the cells by centrifugation at 3000 x g for 5 minutes at 4oC, discard the
Sup ernatant.
6. Incubate tubes on ice for 20 minutes.
7. Pellet the c-ells
-as
before(3000
x g, 5 minutes,4oC),
discard the supernatant.8. Add 100 pl cold 0.1 M CaC12 and mix throughly by mild vortexing.
Store the competent cells frot2en
(-80oC).
2.6 SDS-PAGE
2.6.1 Thereagent
Acrylamide / bisacrylamide monomer stock solution
(30%
/0.8%)
Acrylamide monomer
N,N-methylenebi sacrylamide
4 x Running gel buffer
(1.5
M Tris-HCl,pH8.8)
Tris-HC1*Ajust
pH with HC1. Store at4oC.4 x Stacking gel buffer
(500
mM Tris-HC1,pH6.8)
Tris-HC1*Ajust
pH with HC1. Store at4oC.5 x Sample Buffer Glycerol Tris-HCl
bH6.8)
Bromophenolblue 29.2% 18.15% 0.2mM 0.05%SDS stock solution
(10%)
APS-Ammonim persulfate initiator solution
(10%)
Ammonium persulfate... TEMED-N, N, NT, NT-tetramethylethylenediamine 1 x Running buffer Tris-HCl Glycine CBB stain solution CBB-R250 Methanol Acetic acid 25mM 200mM 0.25%Protein MW Maker
(Daiichi) from
top to bottom1. 97,400; 2. 66,267; 3. 42,400; 4. 30,000; 5. 20,100; 6. 14,400
CBB destain solution
Acetic acid
Methanol
2.6.2 Protocol
1x Running Gel Solution H20 1.5 M Tris-HCl, pH 8.8 10% (w/v)SDS AcrylamideBis-acrylamide (30%/0.8% w/v) 7 0/o 10 0/o 15.3 ml 12.3 ml 7.5 ml 7.5 ml 0.3 m1 0.3 ml 6.9 ml 9.9 ml 120/o 150/o 10.2 m1 7.2 m1 7.5 ml 7.5 m1 0.3 ml 0.3 ml 12.0 ml 15.0 ml
10% (w/v)ammonium persulfate (APS) 0.15 m1 0.15 m1 0.15 ml 0.15 ml
TEMED 0.02 m1 0.02 ml 0.02 m1 0.02 m1
Stacking Gel Solution
(4% Acrylamide)
II200.5 M Tris-HCl, pH 6.8
20% (w/v)SDS
Acrylamidemis-acrylamide (30%/0.8% w/v)
10% (w/v)ammonium persulfate (APS)
TEMED 3.075 ml 1.25 ml 0.025 m1 0.67 m1 0.025 m1 0.005 ml
Choose a percentage acrylamide based on the molecular weight range of proteins
you wish to separate:
% Gel M.W. Range
50 kDa- 500kDa
12 10 kDa- 200 kDa
15 3kDa- 100kDa
2. Preparing Sample
Mix protein :5 x samplebuffer :2-mercaptoethano1 - 20
:5 :1.
Heat sample by boiling for 5-10 minutes.
Cool down the temperature on ice.
Centrifuge the sample and use the supernatant.
3. Loading samples on gel
Pull the combs out- straight upwards.
Wash-out the wells and fill the wells with 1 x Running buffer.
Add the samples and protein MW marker into the wells
4. Electrophorese
When the samples in the stacking gel,
adjust
20 mA for -30 min.The samples into the running gel,
adjust
to 30 mA until the samples move to the bottom of the gel5. Finished Gel: Remove from Rig
6. Staining the gel with CBB stain solution for 30 min.
7. Destaining
Add CBB destain solution and shaking ovemight.
Remove de-staining solution, rinse gels twice with dH20.
8. Take the photo.
2.7 Detection activity of catalase
2.7.1 Add H202 tO detect activity ofcatalase
2.7.1.1 Thetheory
Catalase
(EC
1.11.1.6),
present in the peroxisomes of nearly all aerobic cells,serves to protect the cell from the toxic effects of hydrogen peroxide
by
catalyzing its decomposition into molecular oxygen and water without the
production of fi:ee radicals. The mechanism of catalysis is not fully elucidated, but the overall reaction is as
follows:
2H202--2H20+02
The presence of catalase activity leads to bubble formation resulting from the
transformation ofH202 tO H20 and gaseous 02.
2.7.1.2 Protocol
1. Incubate the bacteria until-OD660 -1.0.
2. Transfer 1 5 mL of the bacteria culture to a microcentrifuge tube.
3. Centrifuge for 10 min at 12,000 g and discard the supernatant.
4. Add lmL PBS buffer and mix thoroughly by vortexing.
6. Resuspend the pellet with 30 pl PBS buffer.
7.
Samples(20-pl)
ofPBS resuspended cells were mixed with 10 mlH202(8.8 M)
2.7.2 Assayofcatalase 2.7.2.1 Thereagent Dichromate solution Potassium dichromate Acetic acid PBS buffer H202 0n Various concentrations 1.67% 6.67% 0.1mM -1mM 2.7.2.2 Protocol
Draw the standard curve
1. Transfer 900 pl dichromate solution to a microcentrifuge tube.
2. Add 300 pl H202 0fvarious concentrations and mix thoroughly by vortexing.
3. Heat sample by boiling for 5-10 minutes.
4. Down the temperature to -25oC.
5. Measure the sample at OD570.
6. Drawing the standard curve.
Assay of the samples
1. Transfer 900 Lil dichromate solution to a microcentrifuge tube.
3. Bacteia samples
(109c.f.u.)
are mixed with 0.88 mM H202 in PBS buffer.4. Transfer 300 lil of the mixture to the tube and mix thoroughly by vortexing.
5. Heat sample by boiling for 5-10
minLtes.
6. Down the temperature to -25oC.7. Measure the sample at OD570.
8. Calculate the data base on the standard curve.
2.8 Short-term H202 exposure 2.8.1 Thereagent MRS medium PBS buffer Hemin H202 C atalase Sucrose buffer Sucrose 1 00mM Ammonium citrate
bH6.0)
*Ajust
pH with lM citrate. Autoclave 121oC for 20 min....10U
1.712%
2.8.2 Protocol
1. Incubate the Bifldobacterium with MRS + 10 LIM hemin.
2. Transfer 1 mL culture to a microcentrifuge tube.
4. Add 1 mL PBS buffer and mix thoroughly by vortexing.
5. Centrifuge for 10 min at 12,000 x g and discard the supernatant.
6. Add 1 mL MRS medium and mix thoroughly by vortexing.
7. Incubate at 37oC with 4.4 mM H202 for 1 h. *Negative: With 0 M H202.
8. Remove the H202 by addition ofcatalase
(10
UmL-1).
9. Spread desired dilution volume of culture on MRS plate and incubate for 48
hours.
10. Take count of the colonies.
2.9 Long-term with_aerated cultures
2.9.1 Thereagent
MRS medium
Hemin
2.9.2 Protocol
1. Anaerobic incubate the Bifidobacterium with MRS.
2. Inoculate 160 pl Bifidobacterium to 8 mL MRS not contain Vc with aerobic method.
3. Incubate at 120 rpm and spread desired dilution volume on MRS plate at 12-h
intervals during 2 days.
2.10 PCR
2.10.1 With po1ymerase KOD-PLUS
Using KOD
-Plus- kit.
2.10.2 With po1ymerase GOTaq
using GoTaq@ DNA kit.
2.ll DNA extraction from agarose gels
using PCR clean-up Gel extraction NucleoSpin@ Extract II. MACHEREY-NAGEL
kit.
2.12 Digestion with restriction enzyme
2.12.1 The restriction enzyme
The restriction enzymes are applied by TAKARA or NEB.
2.12.2 Protocol
Combine the following in a microfuge tube in order :
1LL1 10 x Buffer
6.5 lilH20 2 lilDNA 0.5 LilEnzym9
2.13 DNAligation
Using DNA Ligation kit < Mighty Mix > kit.
2.14 Transformation 2.14.1 Chemical transformation 2.14.1.1 Thereagent Competent cells Plasmid S.0.C medium or LB medium Selective plate Ampicillin
(AmpR)
Kanamycin(KmR)
spectinomycin(SpR)
chloramphenico1(CmR)
1 00pg/mL 50pg/mL 75 pg/mL 25 pg/mL 2.14.1.2 Protocol1. Thaw, on ice, one vial of chemically competent cells for each transformation.
2. Add 1 lilplasmid
(-50ng)
into a vial of chemically competent cells and mixgently. Do not mix by pipetting up and down. 3. Incubate the
vial(s)
on ice for 30 minutes.4. Heat-shock the cells for 30 seconds at 42oC without shaking.
6. Add 250 pl of room temperature S.0.C. medium
(or
LBmedium)
to each vial. 7. Cap thevial(s)
tightly and shake horizontally(120 rpm)
at37oC for 1 hour. 8. Spread 20 pl and 100 plkom
each transformation on a prewarmed selectiveplate and incubate ovemight at 37oC. We recommend plating 2 different
volumes to ensure that at least 1 plate has well-spaced colonies.
2.14.2 Electroporation 2.14.2.1 The reagent MRS medium Competent cells Plasmid Selective plate * Th'e same as 2.14.1.1. 2.14.2.2 Protocol
1. Add 1 pl plasmid
(-50ng)
into a vial of competent cells(-50p1)
and mixgently. Do not mix by pipetting up and down. 2. Incubate the
vial(s)
on ice for 15 minutes.3. Prepare the tube added 1 mL of MRS medium.
4. Transfer the mixed competent cells to a 2-mm cuvette.
5. Electroporation was camied out with
EasyjecT (EQUIBIO),
set at 12kV/cm(time
constants obtained between 3.9 and 4.2ms).
6. Transfer the cells to the tube added MRS medium. 7. Incubater the tube ar 37oC for 3 hours.
incubate at 37oC for 2-3 days. We recommend plating 2 different volumes to
ensure that at least 1 plate has well-spaced colonies.
2.15 Gateway system
using MultiSite Gateway@ Pro kit.
2.16 Flow cytometry
2.16.1 Thereagent
Using LIVE/DEAD@ BacLightTM Bacterial Viability and Counting kit.
2.16.2 Theinstrument
cell Lab
QuantaTM
SC MPL.Betkman
c.ulter.2.17 Isolate NA from microorganisms
Using RNeasy Mini kit.
2.18 RT-PCR
Using TURBO DNA-free kit; High Capacity CDNA Reverse Transcription kit.
2.19 qRT-PCR
Using SYBR Premix Ex Taq kit.
2.19.2 The instrument
StepOnePlus Real-Time PCR system. Applied Biosystems.
2.20 AssayofH202 2.20.1 Thereagent Triton X-1 00 Horseradish peroxidase o-dianisidine dihydrochloride PBS buffer 2.20.2 Protocol 0.01% 0.63mM
Culture supernatants were diluted 3:2 with a solution of 0.2% Triton X-100, 0.01%
horseradish peroxidase, and 0.63 mM o-dianisidine dihydrochloride in 50 mM acetate
buffer, pH 5.0. The accumulation of H202 in the culture medium was assayed
spectrophotometrically at 460 nm by detecting the oxidation of o-dianisidine
dihydro chloride.
using KOI) plus Mutagenesis kit.
(Process
seebelow)
prCA:mLASE )Patabsq&--1
atalastfLD5
i
Dpn.I.P7c:T:01
i_
pB CATOO1/
L-LL Ligation//I
Tl
pBCATOO1 ::FtBSn Catala/
・-GCTT
RBSn
1
ATGAG...
Cata[ase
2.22 The strains, the plasmids and primers
Strains, plasmids or primers Characteristic(s)
Bacterial strains B. longum 105-A
Bacillus subtilis GTCO 1672 E. coli DH5a
E. coli One Shot MachlTM TIR E. coli One Shot ccdB SurvivalTM TIR E. colt UM255 Plasmids pDONRTM22 1-P 1-P5r pDONRTM22 1-P5-P2 pDOm1 5r: :HpkatE pDONR52: :hupT pKKT427 pGU100 Primers kat- f kat-r hup-f hup-r attB 5 -hupTf attB 2-hupTr prRB Sh9 prRBSh8 prRB Sh7 prRB Sh6 prRB Sh5 prRB Sh4 prRB Sh3 prRB Sh2 prWS1 1 prRBS 10 Wild type Wild type
F- endAl glnV44 thi-1 recAl relAl gy7A96 deoR nupG p80dlacZ4M15
A PacZYA-argF) U1 69, hsdR1 7jrK-mK'),
L-
F-p80PacZ)AM15 AlacX74 hsdR(rKmK+) ArecA1398 endAl tonA
F- mcrA A(mrr-hsdRMS-mcrBC) 80lacZAM15 AlacX74 recAl araA139
D(ara-leu)7697 galUgalK rpsL (StrR)endAl nupG tonAI:Ptrc -ccdA
pro leu rpsL hsdM hsdR endl lacy kalG2 katEI:Tn10 recA
pDONRTM221; CmrKmr; ccdB; attP1; attP5r pDONRTM22 1;Cmr Kmr; ccdB; attP5; attP2
PDO-TM221-P 1-P5r; Kmr; carrying theB. subtilis katE ;hup promoter PDO-TM221-P5-P2; Kmr; ca-rrying the hup terminator
Spr; A shuttle vector between E. coli and Bljidobacterium,3.9 kb modiflCation of pBRATA1 0 1
pKKT427; Cmr, Spr; ccdB; Reading Frame Cassette A
atgagtgatgaccaaaaca
ggggaca acttttgta tacaa agttgttc aaattc gtc tatcc caat
I
gggga caagtttgta ca aa aaagcaggcttttc cgccactttg ct ttggtcatcactcataaaagcatccttcttggg
gggga ca actttgta taca aa agttgc cttctgctcgtagcg atta gggga cca ctttgta ca aga aagctgggta tggaag cgctgaac tagtc c
AATAAAGCATCCTTCTTGG AAAAAGCATCCTTCTTGGG AAJuG CATCCTTCTTGGGT AAAGCATCCTTCTTGGGTC AAGCATCCTTCTTGGGTCA AG CATCCTTCTTGGGTCAG GCATCCTTCTTGGGTCAGG CATCCTTCTTGGGTCAGG G AAGCCTCCTTGGGTCAGGGGACAAGCACTT AAGCACTCCTTGGGTCAGGGGACAAGCACTT
prRBS9 prRBS8 prRB S 7 prRBS6 prRBS5 prRB S4 prRB S3 prRB S2 prRBS 1 prCATALASE juGCATCTCCTGGGTCAGGGGACAAGCACTT AAGCATTCCTTGGGTCAGGGGACAAGCACTT AAGCATCTCCTTGGGTCAGGGGACAAGCACTT AAGCATCTCCTTTGGGTCAGGGGACjuGCACTT AAGCATCCTCCTTTGGGTCAGGGGACAAGCACTT AAGCATACCTCCTTTGGGTCAGGGGACAAGCACTT AAGCATCACCTCCTTTGGGTCAGGGGACAAGCACTT AAGCATACCTCCTTTCGGGTCAGGGGACAAGCACTT juGCATACCTCCTTTCTGGGTCAGGGGACAAGCACTT ATGAGTGATGACCAAAACAAACGTGTAAATGAACACTCAA a
cmr ,Kmr and Spr, resistance to chloramphenicol, kanamycin, and spectinomycin, respectively. b
Results
3.1 Isolate DNA froJn miCrOOrgamiSmS
Isolate DNA &om microorganisms:
strain No. Biosafety level
Bacillus subtilis GTCO1672 BLSI
Bljidobacterium
longum 1 05-A BLS 1Bacillus subtilis was grown in LB at 37oC.
Bljidobacterium
longum 1 05-A was grown in MRS at 37oC.Isolated DNA of Bacillus subtilis