2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
8.5.2 薬力学
8.5.2.1 主要解析項目 tmax時のΔQT/QTc の最小二乗平均値とその差を表 2.7.6.8.5-2 に示す。 tmax時のΔQTcF の最小二乗平均値は、レボフロキサシン群で 2.1 msec と延長、プラセボ群で−1.3 msec と短縮し、tmax時のΔΔQTcF は 3.4 msec(片側 95%CI 上限 = 5.2 msec)であった。
なお、ΔΔQTc の中では、ΔΔQTcB が最も大きく、次に ΔΔQTcP、ΔΔQTcF の順であった。 表2.7.6.8.5-2 tmax 時のΔQT/QTc の最小二乗平均値とその差 項目 推定値 片側95%信頼区間上限 QTcF 間隔 最小二乗平均 レボフロキサシン(L) 2.1 4.1 プラセボ(P) −1.3 0.6 平均値の差 L − P 3.4 5.2 QT 間隔 最小二乗平均 レボフロキサシン −8.8 −5.6 プラセボ −4.6 −1.4 平均値の差 L − P −4.2 −0.9 QTcB 間隔 最小二乗平均 レボフロキサシン 7.6 9.7 プラセボ 0.3 2.5 平均値の差 L − P 7.2 8.9 QTcP 間隔 最小二乗平均 レボフロキサシン 4.6 6.6 プラセボ −0.5 1.4 平均値の差 L − P 5.2 6.8 8.5.2.2 サブグループ解析 年齢別、男女別のtmax時のΔQTcF の最小二乗平均値とその差を表 2.7.6.8.5-3 に示す。 年齢別には、若年者、高齢者のいずれの年齢層でもプラセボ群と比較してレボフロキサシ ン群でΔQTcF は延長し、プラセボ群との差は若年者よりも高齢者の方で大きい傾向があった。 また、QTcB 間隔と QTcP 間隔でも同様に、プラセボ群との差は高齢者の方で大きい傾向があ った。 男女別には、男性、女性のいずれの性別でもプラセボ群と比較してレボフロキサシン群で ΔQTcF は増加し、プラセボ群との差は男性よりも女性の方で大きい傾向があった。また、QTcB 間隔とQTcP 間隔でも同様に、プラセボ群との差は女性の方で大きい傾向があった。 表2.7.6.8.5-3 年齢別、男女別の tmax時のΔQTcF の最小二乗平均値とその差 サブグループ 推定値 片側95%信頼区間上限 若年者 最小二乗平均 レボフロキサシン(L) 2.5 5.6 プラセボ(P) 0.2 3.3 平均値の差 L − P 2.3 4.6 高齢者 最小二乗平均 レボフロキサシン 1.7 4.1 プラセボ −2.8 −0.4 平均値の差 L − P 4.5 7.1 男性 最小二乗平均 レボフロキサシン 0.4 2.8 プラセボ −2.2 0.2 平均値の差 L − P 2.6 5.2 女性 最小二乗平均 レボフロキサシン 3.8 7.1 プラセボ −0.4 2.9 平均値の差 L − P 4.2 6.8
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 8.5.2.3 中心傾向の解析 ΔΔQTcF は、投与開始後 1 時間で最も大きく 3.4 msec となり、その後はより小さな値で推 移し、投与開始8 時間以降では群間の差がほとんどなかった。 8.5.2.4 カテゴリカル解析 QT 間隔の絶対値が 1 回以上 450 msec を超えた被験者数は、レボフロキサシン群が 6 名、 プラセボ群が7 名であった。QTcB 間隔が 450 msec を超えた被験者が 1 名認められたが、QTcF 間隔、QTcP 間隔では 450 msec を超えた被験者は認められなかった。また、QT/QTc 間隔が 480 msec を超えた被験者は認められなかった。 ΔQT が 1 回以上 30 msec を超えた被験者数は、レボフロキサシン群が 1 名、プラセボ群が 2 名であった。ΔQTcB が 1 回以上 30 msec を超えた被験者数は、レボフロキサシン群、プラ セボ群ともに2 名であった。ΔQTcF、ΔQTcP が 30 msec を超えた被験者は認められなかった。 また、ΔQT/QTc が 60 msec を超えた被験者は認められなかった。ΔQT/QTc が基準値を超えた 被験者数に、レボフロキサシン群とプラセボ群の差は認められなかった。 8.5.2.5 薬物動態パラメータと QT/QTc 間隔値の相関関係
レボフロキサシンのCmax及びAUC0-24hと、tmax時のΔQTcF、ΔQT、ΔQTcB 及び ΔQTcP と
の間に明らかな相関関係は認められなかった。 8.5.2.6 治験薬投与後の RR 間隔と QT/QTc 間隔値の相関 RR 間隔と QT/QTc 間隔の関係を示す回帰直線の傾き及び切片は、レボフロキサシン投与後 とプラセボ投与後とで同様であった。 8.5.2.7 心電図波形の形態学的変化 T 波の波形異常は、T 波平低下がレボフロキサシン群で 1 名に 11 件、プラセボ群で 1 名に 20 件発現したが、これらはすべて同一被験者での事象であり、投与開始前から発現していた ことから、治験責任医師は有害事象ではないと判断した。
8.6 安全性の結果
8.6.1 死亡
本治験では死亡は認められなかった。8.6.2 重篤な有害事象
本治験では重篤な有害事象及び中止に至った有害事象は認められなかった。2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
8.6.3 有害事象・副作用の分析
8.6.3.1 有害事象・副作用 8.6.3.1.1 有害事象 有害事象発現状況を表2.7.6.8.6-1 に示す。 レボフロキサシン群全体での有害事象発現率は77.1%(37/48)、プラセボ群全体で 35.4% (17/48)であり、レボフロキサシン群で高かった。年齢別及び男女別の有害事象発現率はい ずれの群でも明らかな差は認められなかった。 レボフロキサシン群で発現頻度の高かった有害事象(発現率5%以上)は、注射部位紅斑 62.5%(30/48)、注射部位そう痒感 33.3%(16/48)、鼻咽頭炎 6.3%(3/48)、そう痒症 6.3%(3/48) であり、注射部位反応に関する有害事象が多かった。注射部位反応はいずれも一過性で軽度 であり、処置を必要とせずに消失が確認され、投与継続に問題は認められなかった。 注射部位反応の有害事象を除くと、レボフロキサシン群での有害事象発現率は31.3% (15/48)、プラセボ群では 27.1%(13/48)となり、レボフロキサシン群とプラセボ群の有害 事象発現率に明らかな差はなかった。また、注射部位反応以外の有害事象の内容に、明らか な群間の違いは認められなかった。2.7.6 個々の試験のまとめ クラビット点滴静注バッグ 5 00 mg / 100 m L レボフロキサシン水和物 クラビット点滴静注 50 0 mg / 20 mL 84 表2.7.6.8.6-1 有害事象発現状況(レボフロキサシン群、ステップ別・全体) ステップ1 ステップ2 ステップ3 ステップ4 全体 評価被験者数 12 12 12 12 48 発現被験者数(%) 10(83.3) 10(83.3) 9(75.0) 8(66.7) 37(77.1) 95%信頼区間a) (51.6, 97.9) (51.6, 97.9) (42.8, 94.5) (34.9, 90.1) (62.7, 88.0) 発現件数 18 16 23 19 76 器官別大分類b) 基本語b) 発現 被験者数 (%) 件数 発現 被験者数(%) 件数 発現 被験者数(%) 件数 発現 被験者数 (%) 件数 発現 被験者数(%) 件数 感染症および寄生虫症 小計 2(16.7) 2 1(8.3) 1 1(8.3) 1 0(0.0) 0 4(8.3) 4 鼻咽頭炎 2(16.7) 2 0(0.0) 0 1(8.3) 1 0(0.0) 0 3(6.3) 3 上気道感染 0(0.0) 0 1(8.3) 1 0(0.0) 0 0(0.0) 0 1(2.1) 1 神経系障害 小計 0(0.0) 0 0(0.0) 0 1(8.3) 4 0(0.0) 0 1(2.1) 4 浮動性めまい 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 頭痛 0(0.0) 0 0(0.0) 0 1(8.3) 2 0(0.0) 0 1(2.1) 2 鎮静 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 心臓障害 小計 1(8.3) 1 0(0.0) 0 0(0.0) 0 1(8.3) 1 2(4.2) 2 動悸 1(8.3) 1 0(0.0) 0 0(0.0) 0 0(0.0) 0 1(2.1) 1 上室性頻脈 0(0.0) 0 0(0.0) 0 0(0.0) 0 1(8.3) 1 1(2.1) 1 胃腸障害 小計 0(0.0) 0 1(8.3) 1 2(16.7) 2 0(0.0) 0 3(6.3) 3 下痢 0(0.0) 0 1(8.3) 1 1(8.3) 1 0(0.0) 0 2(4.2) 2 悪心 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 a) F 分布による信頼区間 b) MedDRA/J V.10.0
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ 5 00 mg / 100 m レボフロキサシン水和物 クラビット点滴静注 50 0 mg / 20 mL 85 表2.7.6.8.6-1 有害事象発現状況(レボフロキサシン群、ステップ別・全体)(続き) ステップ1 ステップ2 ステップ3 ステップ4 全体 評価被験者数 12 12 12 12 48 発現被験者数(%) 10(83.3) 10(83.3) 9(75.0) 8(66.7) 37(77.1) 95%信頼区間a) (51.6, 97.9) (51.6, 97.9) (42.8, 94.5) (34.9, 90.1) (62.7, 88.0) 発現件数 18 16 23 19 76 器官別大分類b) 基本語b) 発現 被験者数(%) 件数 発現 被験者数(%) 件数 発現 被験者数(%) 件数 発現 被験者数(%) 件数 発現 被験者数(%) 件数 皮膚および皮下組織障害 小計 0(0.0) 0 2(16.7) 3 1(8.3) 1 3(25.0) 3 6(12.5) 7 多汗症 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(8.3) 1 2(4.2) 2 そう痒症 0(0.0) 0 1(8.3) 1 0(0.0) 0 2(16.7) 2 3(6.3) 3 発疹 0(0.0) 0 1(8.3) 1 0(0.0) 0 0(0.0) 0 1(2.1) 1 皮膚刺激 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 筋骨格系および結合組織障害 小計 1(8.3) 1 1(8.3) 1 0(0.0) 0 0(0.0) 0 2(4.2) 2 四肢痛 1(8.3) 1 0(0.0) 0 0(0.0) 0 0(0.0) 0 1(2.1) 1 重感 0(0.0) 0 1(8.3) 1 0(0.0) 0 0(0.0) 0 1(2.1) 1 全身障害および投与局所様態 小計 10(83.3) 14 9(75.0) 10 7(58.3) 14 7(58.3) 15 33(68.8) 53 胸部不快感 1(8.3) 1 0(0.0) 0 1(8.3) 1 0(0.0) 0 2(4.2) 2 熱感 0(0.0) 0 1(8.3) 1 0(0.0) 0 0(0.0) 0 1(2.1) 1 注射部位紅斑 10(83.3) 10 7(58.3) 7 7(58.3) 7 6(50.0) 6 30(62.5) 30 注射部位疼痛 0(0.0) 0 0(0.0) 0 0(0.0) 0 1(8.3) 1 1(2.1) 1 注射部位知覚異常 0(0.0) 0 0(0.0) 0 0(0.0) 0 2(16.7) 2 2(4.2) 2 注射部位そう痒感 3(25.0) 3 2(16.7) 2 5(41.7) 5 6(50.0) 6 16(33.3) 16 倦怠感 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 臨床検査 小計 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 白血球数増加 0(0.0) 0 0(0.0) 0 1(8.3) 1 0(0.0) 0 1(2.1) 1 a) F 分布による信頼区間
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 8.6.3.1.2 QT/QTc 関連有害事象 QT/QTc 関連有害事象として、浮動性めまいが 1 名に 1 件認められたが、レボフロキサシ ン投与日(発現時)の心電図、血圧・脈拍に異常は認められなかったことから、レボフロキ サシンがQT/QTc 間隔に影響したことによる症状ではないと考えられた。 8.6.3.1.3 臨床検査値の評価 すべての検査項目で、レボフロキサシン群とプラセボ群との間で平均値の推移に特記すべ き差は認められず、検査値の分布の推移に特記すべき傾向は認められなかった。 8.6.3.1.4 バイタルサイン、身体的所見及び安全性に関連する他の観察項目 レボフロキサシン群、プラセボ群ともにバイタルサイン及び体重の推移に特記すべき傾向 は認められず、投与群間に明らかな違いは認められなかった。 治験責任医師による心電図の評価で、臨床的に有意と判断された異常のうち、有害事象と されたものは軽度の上室性頻脈1 件であり、心電図の 10 秒間の測定途中で発現し、処置なく 測定中に自然に消失した。 8.6.3.1.5 副作用 レボフロキサシン群全体での副作用発現率は68.8%(33/48)、プラセボ群全体で 16.7%(8/48) であり、レボフロキサシン群で高かった。また、治験責任医師により重度又は中等度と判定 された副作用はなく、すべて軽度であった。注射部位反応を除いた副作用発現率は、レボフ ロキサシン群で12.5%(6/48)、プラセボ群では 12.5%(6/48)となり、レボフロキサシン群 とプラセボ群で差はなかった。 なお、「ほとんど関連なし」を含めた場合の副作用(副作用II)の発現率は、レボフロキサ シン群全体で72.9%(35/48)、プラセボ群全体で 27.1%(13/48)であり、レボフロキサシン 群で高かった。また、治験責任医師により重度又は中等度と判定された副作用はなく、すべ て軽度であった。注射部位反応を除いた副作用II 発現率は、レボフロキサシン群で 16.7% (8/48)、プラセボ群で 18.8%(9/48)であり、レボフロキサシン群とプラセボ群で差はなか った。 8.6.3.2 重症度別の有害事象・副作用 治験責任医師により重度又は中等度と判定された有害事象はなく、すべて軽度であった。 副作用もすべて軽度であった。
8.7 結論
レボフロキサシン群ではプラセボ群と比較してtmax時のΔQTcF が延長し、このときのΔΔQTcF は 3.4 msec(片側 95%CI 上限 = 5.2 msec)と最大の群間差を示したが、平均値と片 側95%CI 上限が共に、ICH-E14 ガイドラインの定義での「陰性」(ΔΔQT/QTc 平均値 5 msec
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 以下、片側95%CI 上限 10 msec 未満)に該当する結果であった。また、一般的に薬剤誘発性 QT/QTc 間隔延長のハイリスク集団とされている高齢者や女性においても、ΔΔQTcF は「陰性」 であった。QTcF 間隔が 450 msec を超える被験者及び ΔQTcF が 30 msec を超える被験者は認 められなかった。さらに、濃度依存性のQT/QTc 間隔の延長は認められず、レボフロキサシ ン投与に起因する異常T 波や U 波は認められなかった。これらの結果から、レボフロキサシ ン500 mg 静脈内単回投与が QT/QTc 間隔に及ぼす作用は弱く、臨床的に問題となる影響は小 さいと判断した。
2
.
7
.
6
個々の試験のまとめ レボフロキサシン水和物 クラビット点滴静注バッグ500mg/100 mL クラビット点滴静注500ma/20 mL 9. DR-3355注射剤の市中肺炎を対象とした無作為化群間比較による検証的試
験(第
111相)
…………・添付資料番号5
.
3
.
5
.
1
-
1
9.1治験方法
表2
.
7
.
6
.
9
.
1
-
1
に治験方法の概略を示す。 治験の目的 治験責任医師名 治験実施医療機 関 治験期間 対 象 表2
.
7
.
6
.
9
.
1
-
1
治験方法の概略:日本比較試験 市中肺炎におけるレボブロキサシン注射剤の有効性について、 セブドリアキソンナトリクム水和 物 (gjl名セアトリアキソンナトリウム、以降セアトリアキソン)注射剤を対照に非劣性を検証す る。併せてレボアロキサシン注射剤の安全性を評価する。 2O.年 . 月 . 日 (最初の被験者の同意取得日)~20.年.月.日 (最終観察日) l 選 択 基 準 1)同意取得時の年齢が20歳 以 上 79歳以下の男性及び女性 2)病院外で日常生活をしていた人に発症し、細菌性肺炎と診断され、治験責任医師又は治験分担 医師が入院加療を必要と判断した患者 3)治験薬投与開始日又はその前日に下記の症状・所見基準を満たす患者 (1)胸都X線あるいは胸部CT検査で、急性に新たに出現した浸潤影が認められる (2)体 温 37.0C以 よ (肢窺) (3)以下の2項目中l項目以上を満たす患者 i)CRP増加 (1.0mg/dL以上) ii)白血球数増多 (9000/mm3以 上) 4)咳殿、日客疾(膿性主主)、胸痛、呼吸困難などの呼吸器症状、文は湿性ラ音を認める 2除 外 基 準 1)推定原因菌に対して、レボブロキサシン文はぞアトリアキソンの有効性が明らかに期待できな い患者 既にM RSA、Paeruginosa、マイコプラズマ、クラミジア、 Q熱コクシエラ、ウイルス、真菌も しくは抗酸菌が病原微生物として確認されている。 L.pneumophila は「投与前迅速検査」が陽性 であるものを含む。 2)非定型肺炎が強く疑われる愚者 なお、非定型肺炎の鑑別は「非定型肺炎の鑑別基準」を参考に判断する。 3)誤喰性肺炎を繰り返す患者、又は院内肺炎 、 肺化 膿 症 (肺膿湯)及び膿胸の患者 4)キノロン系抗菌薬文はs-ラクタム系抗生物質に起因すると考えられるアレルギー歴のある愚者 5)てんかんなどの痘盤性疾患を合併する又はとれらの既往歴を有する患者 6)妊婦文は妊娠している可能性がある文は妊娠を希望している女性、授乳中の女性 なお、妊娠の疑いがある場合、妊娠検査にて確認する。 7)重度の心機能障害が認められている患者(心不全、虚血 性 心疾患など) 8)重度の肝機能障害が認められている患者 (AST又はALTが施設正常値上限の3倍 以上) 9)中等度以上の腎機能障害が認められている患者 (血清クレアチエン値が 2mg/c札以上) 10)集中治療室管理もしくは人工呼吸器装着を必要とする重篤な患者、重症文は進行性の基礎疾 患・合併症(穆原病、白血病、進行癌などの悪性疾患、うっ血性 心 不 全、呼吸不全を伴う慢性 気道疾患など)を有し、試験の安全な遂行又は効果の妥当な判定が困難な患者 なお、基礎疾患・合併症の重症度は 「基礎疾愚・合併症の重症度判 断基準」を参考に判断する。 11)本治験の併用禁止薬の投与及び併用禁止療法を必要とする患者 また、 マクロライド少量投与及び届IJ腎皮質ステロイドの全身・吸入投与を、治験薬投与開始前 14日間に新規投薬開始及び投与量の変更を行なった、あるいは治験薬投与期間中に行う可能性 のある患者は登録不可とする。 12)治験薬 投与開始前l週間以内に内版文は注射で使用する他の 抗 菌 薬 (マクロライド少量投与は 除く)が投与され、既に症状が改善しつつある患者 ただし、他の抗 菌薬 の 投与が72時間未満の患者は、症状の改善の有無にかかわらず組み入れ不 可とする。 13) 治験薬投与開始前1週間以内にレポフロキサシン (クラビット~)又はセフ トリアキソン . など)を使用している愚者2.7.6個々の試験のまとめ クラビット点滴静注バッグ500mg/100 mL クラビット点滴静注500ma/20 mL レボフロキサシン水和物 対象(続き) 被験者数 ン ィ ザ デ 一 薬 験 一 験 ム ロ -ム ロ 用 法・用 量 表2.7.6.9.1・1 治験方法の概略:日本比較試験(続き) 14)治験薬投与開始前1週 間 以 内 に ア ジ ス ロ マ イ シ ン (・・・・・・・圃)を使用している患者 ただし、マクロライド少量投与療法で使用している場合は登録可とする。 15)2003年以降のレボブロキサシン開発治験 (注射剤・経口剤)への参加歴がある患者 16)過去に、開発中の薬弗jの治験に組み入れられた場合、同意取得時点で当該薬剤lの投与終了日か ら90日を経過していない愚者 lηその他、治験責任医師又は治験分担医師が本治験参加に不適当と判断した患者 計画された被験者 240名 (レボブロキサシン群 120名、ぞア トリアキソン群 120名) 登録被験者 260名 割付被験者 260名 投与被験者 259名 完了被験者 217名 中止被験者 43名 解析対象被験者 安全性解析対象集団 259名(レボブロキサシン君事 136名、セアトリアキソン群 123名) 最大の解析対象集団 (FAS) 219名(レボブロキサシン群 118名、ぞアトリアキソン群 101名) 治験実施計画書に適合した集団 (pPS) 200名(レボフロキサシン群 108名、 セフトリアキソン群 92名) 多施設共同、 無作為化(中央登録方式)、オープンラベル、非劣性検証試験 レポフロキサシン注射劃) (ロット番号 060551)
セブト日アキソンナトリクム(ロット番号 Ad3355XO-07T01、Ad3355XO-07T02) lレボブロキサシン群の投与量及び投与方法 レポフロキサシン l回 500mgを1日 1回、約 60分聞かけて点滴静脈内投与した。 2セア トリアキソン群の投与量及び投与方法 セアトリアキソンは1回1g (カ価)を添付の溶解液に溶解し、 1日 2田(斬・タ)、約30分 聞 か けて点滴静脈内投与した。 (レボフロキサシン群 136名、 セアトリアキソン群 124名) (レボブロキサシン君事 136名、セアトリアキソン群 123名) (レボアロキサシン群 118名、セアトリアキソン群 99名) (レボブロキサシン群 18名、セアトリアキソン群 25名) 7~14 日間 1有 効 性 主要評価 評価判定委員会判定による投与終了/中止時の臨床効 果 (日本化学療法学会 「呼吸器感染症にお ける新規抗微生物薬の臨床評価法(案)Jの臨床効果判定基準を参考に「有効」、「無効」、文は 「判 定不能」に分類) 副次評価 1)治験責任医師文は治験分担医師に よ る 投 与 終 了 /中止時の臨床効果(有効率) 2)評価判定委員会判定による投与開始 3日後、投与開始 7日後、最終観察時の臨床効果(有効率) 3)評価判定委員会判定による投与終了/中止時の微生物学的効果(陰性化率) 4)評価判定委員会判定による投与終了/中止 時の微生物学的効果(菌消失率) 項目 計 算 式 有 効 率 = r有効被験者数J/ r解析対象被験者数J (投与終了/中止時、投与開始 3日後、及び投与開始 7日後では、 PPS での判定不能は分母から除き、 FASの判定不能は含める。) (最終観察時では、 PPSの判定不能及び観察中止は分母から除き、FAS の判 定不能は含める。) 陰 性化率 = r菌消失又は推定菌消失被験者数J/ r解析対象被験者数」 (判定不能は分母から除く) 臨床効果 (有効率) 微生物学的効果 (陰性化率) 微生物学的効果 菌消失率 = r消失菌株数J/ r解析対象菌株数J(判定不能は分母から (菌消失率) 除く) 2安 全 性 1)有害事象発現率及び面1)作用発現率 2)主要背景因子31Jの有害事象発現率及び副作用発現率 3)臨床検査値の推移 評価スケジュール 表 2.7.6.9.1-2参照
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 表2.7.6.9.1-1 治験方法の概略:日本比較試験(続き) 統計解析手法 1.有効性 主要評価項目として、評価判定委員会判定による投与終了/中止時のレボフロキサシン群とセフト リアキソン群の臨床効果(有効率)の差の点推定値を求め、正規近似に基づく両側95%信頼区間を 算出した。両側95%信頼区間の下限値が−10%以上の場合、レボフロキサシン群のセフトリアキソ ン群に対する非劣性が検証されたと判定した。 副次評価項目は、投与群ごとに点推定値及びその両側95%信頼区間を求めた。 2.安全性 安全性解析対象集団を対象として、投与群ごとに有害事象及び副作用を発現した被験者数、被験者 の割合とその両側95%信頼区間、及び件数を示し、器官別大分類・基本語別の集計も併せて行った。
2.7.6 個々の試験のまとめ
クラビット点滴静注バッグ500 mg/100 mL
レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
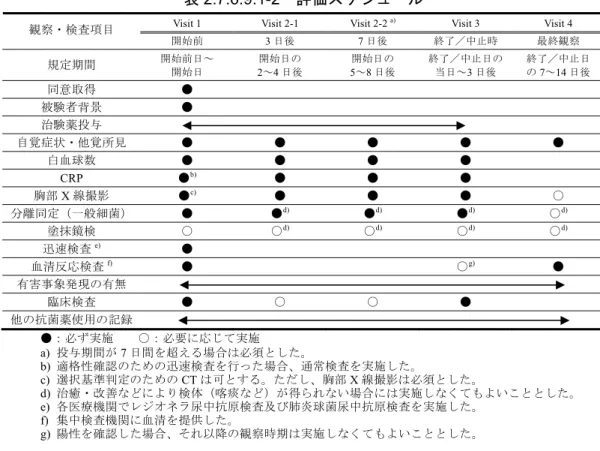
表2.7.6.9.1-2 評価スケジュール
Visit 1 Visit 2-1 Visit 2-2 a) Visit 3 Visit 4 観察・検査項目 開始前 3 日後 7 日後 終了/中止時 最終観察 規定期間 開始前日~ 開始日 開始日の 2~4 日後 開始日の 5~8 日後 終了/中止日の 当日~3 日後 終了/中止日 の7~14 日後 同意取得 ● 被験者背景 ● 治験薬投与 自覚症状・他覚所見 ● ● ● ● ● 白血球数 ● ● ● ● CRP ●b) ● ● ● 胸部X 線撮影 ●c) ● ● ● ○ 分離同定(一般細菌) ● ●d) ●d) ●d) ○d) 塗抹鏡検 ○ ○d) ○d) ○d) ○d) 迅速検査e) ● 血清反応検査f) ● ○g) ● 有害事象発現の有無 臨床検査 ● ○ ○ ● 他の抗菌薬使用の記録 ●:必ず実施 ○:必要に応じて実施 a) 投与期間が7 日間を超える場合は必須とした。 b) 適格性確認のための迅速検査を行った場合、通常検査を実施した。 c) 選択基準判定のためのCT は可とする。ただし、胸部 X 線撮影は必須とした。 d) 治癒・改善などにより検体(喀痰など)が得られない場合には実施しなくてもよいこととした。 e) 各医療機関でレジオネラ尿中抗原検査及び肺炎球菌尿中抗原検査を実施した。 f) 集中検査機関に血清を提供した。 g) 陽性を確認した場合、それ以降の観察時期は実施しなくてもよいこととした。
9.1.1 被験者数の設定根拠
治験実施計画書に適合した集団(PPS)による主要評価項目の解析に必要な被験者数は 1 群101 名となる。中止、脱落例を考慮し、各群 120 名、合計 240 名を目標被験者数とした。9.1.2 用法・用量の設定根拠
1) レボフロキサシン群 キノロン系抗菌薬の治療効果に相関する主要なPK/PD パラメータは、一般的に血中 24 時 間AUC と MIC の比(AUC/MIC)であり、肺炎の代表的な原因菌である肺炎球菌に対してレ ボフロキサシンが効果を示す条件はAUC/MIC ≥30 とされている。肺炎球菌の臨床分離株に 対するレボフロキサシンのMIC90は1 µg/mL であり、表 2.7.6.9.1-3 に示すように、第 I 相単回投与試験の500 mg 投与時の AUC0-infからAUC/MIC は 52.09 となり、肺炎球菌に対して効
果を示す条件を満たしていることから、500 mg 1 日 1 回は妥当と考えた。 表2.7.6.9.1-3 レボフロキサシンの PK/PD からの臨床推奨用量の検討 レボフロキサシンの静脈内投与量(単回投与) PK パラメータ 250 mg 500 mg 750 mg 1000 mg AUC0-inf(µg・h/mL) 23.45 52.09 91.12 127.85 下線はAUC/MIC ≥ 30 であり効果を示す AUC AUC0-infは健康成人での第I 相単回投与試験より引用
レボフロキサシン水和物 2.7.6個々の試験のまとめ クラビット点滴静注バッグ500mg/1口
o
mL クラビット点滴静注500ma/20 mL 一方、キノロン系抗菌薬への肺炎球菌の耐性化を防止するためには、 Cm置と MICの比 (Cm~ふ1IC) を 5~10 以上とすることが必要とされている。表 2.7.6.9.1 -4に示すように、肺 炎球菌に対するレポブロキサシンのMICがl同ImLの場合、 500mg 1日l回投与では Cm, 表2.7.6.9.1-4 肺炎球菌に対するレボフロキサシンの MICと500mg 1日1回投与の Cmax/MICとの関係 B一
4 -2 parC &gyrA 二 重 変 異 以 上 レボブロキサシン500mg l日 l回静脈内投与 C町叫品lIC 1.23 2.45 4.90 9.79 レボフロキサレ/ンMIC ( μg/mL) 肺 炎 球 菌 の 標 的 変 異E F 4
ごと
05 無(野生株) よ♀」呈 下線は耐性化抑制が期待される CmaxlMIC Cm~ は健康成人での第 I 相単回投与試験より引用 上記の PKIPDの考え方とモンテカノレロ・シミュレーションの手法を用い、日本人の肺炎球 菌性肺炎患者の臨床効果を予測したところ、表2.7.6.9.1-5に示すように、 500mg 1日l回 静 脈内投与の有効率は95%以上であった。 表2.7.6.9.1-5 モンテカル口・シミュレーションによる臨床効果の予測 l日投与量 250 mg 1日 l回 92.43% 500 mg 1日 l回 97.61% 750 mg 1日 l回 98.06% 有 効 率 以上より、レポブロキサシンの用法・用量を5ωmg1日l回点滴静脈内投与とした。 2) セフトリアキソン群 セフト リアキソン注射部作用添付文書に基づき投与量及び投与方法を設定した。承認されている通常 の用法・用量のうち、P
ラクタム系抗菌薬の有効性に相関がある timeabove MICが増大する l 日 2 回投与を選択し、 l 日用量が 1~2g であることから、 l 回 1 g (力価)1日2回(朝、 タ)点滴静脈内投与を選択した。 9.2被験者の内訳
本治験に登録された被験者は260名であり、無作為に割り付けた被験者は、レポブロキサシン 群は 136名、セアトリアキソン群は124名であった。治験薬が投与された被験者は、レポブロキ サシン群は136名、セアトリアキソン群は末投与の l名を除く 123名であった。セアトリアキソ ン群の末投与の l名は、登録後に判明した臨床検査結果 (ALT、ASTの上昇)が除外基準に抵触*
新薬承認情報提供時に置き換え2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL し、不適格となったため治験薬の投与を行わず、中止した。 レボフロキサシン群では136 名中 118 名が治験を完了し、18 名は未完了であった。レボフ ロキサシン群の未完了理由の内訳は「効果不十分」が8 名、「有害事象発現」が7 名、「選択・ 除外基準に抵触」が2 名、「併用禁止治療が必要」が1 名であった。セフトリアキソン群では 124 名中 99 名が治験を完了し、25 名は未完了であった。未完了理由の内訳は、「効果不十分」 が11 名、「選択・除外基準に抵触」が 4 名、「中止の申し出」、「医師判断で中止」が各 3 名、 「有害事象発現」、「併用禁止治療が必要」が各2 名であった。
9.3 解析対象
レボフロキサシン群では、登録された136 名すべての被験者を安全性解析対象集団とした。 FAS の採用被験者数は、安全性解析対象集団 136 名のうち FAS 解析不採用の 18 名を除く 118 名であった。不採用理由は、いずれも「対象外疾患」であり、その内訳は「細菌性肺炎 + マ イコプラズマ肺炎」、「肺化膿症」、「マイコプラズマ肺炎否定できず」、「非結核性好酸菌症 + 二次感染」、「肺気腫 + 二次感染」が各 2 名、「クラミジア肺炎」、「細菌性肺炎 + クラミジ ア肺炎」、「小細胞肺癌」、「感染性肺のう胞」、「膿胸」、「間質性肺炎 + 二次感染」、「感染性 ブラ」、「非定型肺炎否定できず」が各1 名であった。 PPS の被験者数は、FAS 解析対象集団 118 名のうち PPS 解析不採用の 10 名を除く 108 名であ り、不採用理由は、レボフロキサシン群では「副腎皮質ステロイド投与(除外基準違反の被験者)」、 「投与期間不足」が各3 名、「重度/進行性の基礎疾患・合併症」、「β-ラクタム系アレルギー歴 あり」、「原因菌(緑膿菌)」、「副腎皮質ステロイド剤(併用薬違反の被験者)」が各1 名であった。 セフトリアキソン群では、登録された124 名のうち治験薬未投与の 1 名を除く 123 名を安 全性解析対象集団とした。FAS の被験者数は、安全性解析対象集団 123 名のうち FAS 解析不 採用の22 名を除く 101 名であった。不採用理由はレボフロキサシン群と同様、いずれも「対 象外疾患」であった。 PPS の採用被験者数は、FAS 解析対象集団 101 名のうち PPS 解析不採用の 9 名を除く 92 名であった。不採用理由は、「投与期間不足」、「副腎皮質ステロイド剤」、「併用禁止輸液投与」 が各2 名、「服薬用法違反」、「投与回数不足」、「併用禁止注射剤投与」が各 1 名であった。9.4 被験者背景
PPS の主な人口統計学的及びその他の基準値の特性を表 2.7.6.9.4-1 に示す。 PPS の人口統計学的及びその他の基準値の特性にレボフロキサシン群とセフトリアキソン 群で大きな差は認められなかった。FAS でも明らかな差は認められず、完了被験者と未完了 被験者の特性にも明らかな差は認められなかった。なお、原因菌の分離頻度にもレボフロキ サシン群とセフトリアキソン群で大きな差は認められなかった。2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 表2.7.6.9.4-1 主な人口統計学的及びその他の基準値の特性(解析対象集団:PPS) 投与群 レボフロキサシン群 CTRX 群 項目 評価被験者数 108 92 軽症 20(18.5) 15(16.3) 中等症 76(70.4) 66(71.7) 重症 9(8.3) 8(8.7) 感染症重症度1a) 判定不能 3(2.8) 3(3.3) 軽症 56(51.9) 44(47.8) 中等症 47(43.5) 40(43.5) 重症 5(4.6) 8(8.7) 感染症重症度2b) 超重症 0(0.0) 0(0.0) 軽度 14(13.0) 9(9.8) 中等度 82(75.9) 72(78.3) 重度 9(8.3) 8(8.7) 患者重篤度 判定不能 3(2.8) 3(3.3) 男 68(63.0) 51(55.4) 性別 女 40(37.0) 41(44.6) 年齢(同意取得時)(歳) mean ± SD 58.5 ± 15.7 58.9 ± 16.1 体重(kg) mean ± SD 55.332 ± 10.494 55.528 ± 14.128 BMI mean ± SD 21.31 ± 3.54 21.28 ± 4.33 CLcr c)(mL/min) mean ± SD 82.57 ± 28.83 80.14 ± 28.56 なし 23(21.3) 15(16.3) 基礎疾患・合併症 あり 85(78.7) 77(83.7) 0 0(0.0) 0(0.0) 1 1(0.9) 1(1.1) 2 11(10.2) 8(8.7) 3 36(33.3) 24(26.1) 4 18(16.7) 19(20.7) 5 11(10.2) 16(17.4) 6 20(18.5) 14(15.2) 7 4(3.7) 3(3.3) 8 4(3.7) 4(4.3) 9 0(0.0) 0(0.0) 評価判定委員会 胸部X 線点数 (点) d) 10 0(0.0) 0(0.0) 体温( C) mean ± SD 38.25 ± 0.85 38.27 ± 0.82 白血球数(/µL) mean ± SD 12776.9 ± 4899.3 13264.2 ± 5020.5 CRP(mg/dL) mean ± SD 14.0159 ± 8.8254 14.0970 ± 9.5187 なし 93(86.1) 77(83.7) 治験薬投与開始前 7 日以内の 抗菌化学療法の有無 あり 15(13.9) 15(16.3) なし 15(13.9) 9(9.8) 併用薬の有無 あり 93(86.1) 83(90.2) なし 36(33.3) 23(25.0) 併用療法の有無 あり 72(66.7) 69(75.0) a) 日本化学療法学会「呼吸器感染症における新規抗微生物薬の臨床評価法(案)」判定基準による分類 b) 日本呼吸器学会「成人市中肺炎診療ガイドライン」判定基準による分類 c) CLcr は Cockcroft and Gault 式を用いて算出
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
9.5 有効性の結果
9.5.1 主要評価項目:評価判定委員会判定による投与終了/中止時の臨床効果
(有効率)
PPS での投与終了/中止時の有効率は、レボフロキサシン群で 88.5%(92/104)、セフトリ アキソン群で88.8%(79/89)であり、群間差(レボフロキサシン群−セフトリアキソン群) は−0.3%(95%CI:−9.3~8.7)であった。群間差の 95%信頼区間の下限値が−10%を上回った ため、レボフロキサシン注射剤のセフトリアキソンに対する非劣性が検証された。 表2.7.6.9.5-1 評価判定委員会判定による投与終了/中止時の臨床効果(有効率)(解析対 象集団:PPS) 投与群 有効 無効 判定不能 合計 有効率(%) a) (95%信頼区間)c) 有効率の差(%)b) (95%信頼区間)c) LVFX 群 92(85.2) 12(11.1) 4(3.7) 108(100.0) 88.5(82.3、94.6) CTRX 群 79(85.9) 10(10.9) 3(3.3) 92(100.0) 88.8(82.2、95.3) −0.3(−9.3、8.7) a: 有効被験者数/解析対象被験者数(判定不能は分母から除く) b: LVFX 群の有効率 − CTRX 群の有効率 c: 正規近似による信頼区間。括弧内の数値は%9.5.2 副次評価項目
9.5.2.1 治験責任医師又は治験分担医師判定による投与終了/中止時の臨床効果(有効率) PPS での投与終了/中止時の有効率はレボフロキサシン群で 93.5%(101/108)、セフトリ アキソン群で91.2%(83/91)、群間差は 2.3%(95%CI:−5.1~9.8)であり、群間差の 95%信 頼区間の下限値が−10%を上回った。 9.5.2.2 最終観察時(投与終了/中止 7~14 日後)の臨床効果(有効率) 評価判定委員会判定によるPPS での最終観察時の有効率は、レボフロキサシン群で 88.9% (80/90)、セフトリアキソン群で 83.8%(62/74)、群間差は 5.1%(95%CI:−5.5~15.7)であ り、投与終了/中止後も十分な効果を示していた。 9.5.2.3 投与終了/中止時の微生物学的効果(陰性化率) 評価判定委員会判定によるPPS での投与終了/中止時の微生物学的効果(陰性化率 = 「菌 消失又は推定菌消失被験者数」/「解析対象被験者数」)は、レボフロキサシン群で96.7% (59/61)、セフトリアキソン群で 97.8%(44/45)であり、群間差は−1.1%(95%CI:−7.3~5.1) であった。 9.5.2.4 投与終了/中止時の微生物学的効果(菌消失率) 評価判定委員会が判定した原因菌のPPS での微生物学的効果(菌消失率 = 「消失菌株数」 /「解析対象菌株数」)は、レボフロキサシン群で97.2%(69/71)、セフトリアキソン群で 98.0%2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL (50/51)であり、群間差は−0.9%(95%CI:−6.3~4.6)であった。 レボフロキサシン注射剤は、呼吸器感染症の主要な原因菌であるStreptococcus pneumoniae、 Haemophilus influenzae に対し、それぞれ 96.3%(26/27)、100.0%(26/26)の高い除菌効果を 示した。
9.6 安全性の結果
9.6.1 有害事象発現率・副作用発現率
有害事象発現率は、レボフロキサシン群で72.8%(99/136、340 件、95%CI:65.3~80.3)、 セフトリアキソン群で71.5%(88/123、275 件、95%CI:63.6~79.5)であり、両群の発現率 の差は1.2%(95%CI:−9.7~12.2)であった。 レボフロキサシン群での主な有害事象(5%以上発現)は、注射部位紅斑が 20.6%(28/136、 53 件)、ALT 増加が 13.2%(18/136、18 件)、注射部位そう痒感が 11.0%(15/136、36 件)、 AST 増加が 10.3%(14/136、14 件)、不眠症、好酸球数増加が各 9.6%(13/136、13 件)、注射 部位腫脹、γ-GTP 増加が各 8.1%(11/136、11 件)、便秘が 6.6%(9/136、9 件)、下痢が 5.9% (8/136、9 件)、血中アルカリホスファターゼ増加が 5.9%(8/136、8 件)、注射部位疼痛が 5.1%(7/136、8 件)であった。 セフトリアキソン群での主な有害事象(5%以上発現)は、下痢が 13.8%(17/123、18 件)、 ALT 増加が 13.8%(17/123、17 件)、AST 増加、好酸球数増加が各 12.2%(15/123、15 件)、 注射部位紅斑が8.9%(11/123、17 件)、注射部位疼痛が 7.3%(9/123、10 件)、不眠症、便秘 が各7.3%(9/123、9 件)、血中乳酸脱水素酵素増加が 6.5%(8/123、8 件)、注射部位腫脹が 5.7%(7/123、8 件)であった。 副作用発現率は、レボフロキサシン群で53.7%(73/136、223 件、95%CI:45.3~62.1)、セ フトリアキソン群で56.9%(70/123、150 件、95%CI:48.2~65.7)であり、群間差は−3.2% (95%CI:−15.4~8.9)であった。 レボフロキサシン群での主な副作用(5%以上発現)は、注射部位紅斑が 17.6%(24/136、 48 件)、注射部位そう痒感が 11.0%(15/136、36 件)、ALT 増加が 11.0%(15/136、15 件)、 AST 増加が 8.1%(11/136、11 件)、γ-GTP 増加が 6.6%(9/136、9 件)、下痢が 5.9%(8/136、 9 件)、注射部位腫脹、好酸球数増加が各 5.1%(7/136、7 件)であった。 セフトリアキソン群での主な副作用(5%以上発現)は、下痢、ALT 増加が各 12.2%(15/123、 15 件)、AST 増加が 10.6%(13/123、13 件)、好酸球数増加が 8.1%(10/123、10 件)であっ た。2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
9.6.2 注射部位反応(副作用)発現率
注射部位反応の副作用発現率は、レボフロキサシン群で25.7%(35/136、104 件)セフトリ アキソン群で7.3%(9/123、19 件)であり、レボフロキサシン群で高かった。なお、注射部 位反応の副作用発現率の群間差は追加解析を行った結果、18.4%(95%CI:9.7~27.1)であっ た。 レボフロキサシン群での注射部位反応(副作用)は、注射部位紅斑が19.1%(26/136、50 件)、注射部位そう痒感が12.5%(17/136、38 件)、注射部位腫脹が 5.1%(7/136、7 件)、 注射部位疼痛が1.5%(2/136、2 件)、注射部位硬結、注射部位熱感、静脈炎、血管炎、注射 部位不快感、血管障害、穿刺部位疼痛が各0.7%(1/136、1 件)であった。 注射部位反応の重症度は95.2%(99 件/104 件)が軽度であり、104 件中 91 件は発現当日に 消失した。投与中止に至ったのは104 件中 3 件であり、処置が必要であったのは 104 件中 4 件と少数であった。 なお、注射部位反応を除いた副作用発現率は、レボフロキサシン群で38.2%(52/136、119 件)、セフトリアキソン群で54.5%(67/123、131 件)、群間差は−16.2%(95%CI:−28.2~ −4.2)であり、レボフロキサシン群で低かった。9.6.3 重症度別の有害事象発現率・副作用発現率
有害事象の重症度は、軽度がレボフロキサシン群で67.6%(92/136、289 件)、セフトリア キソン群で68.3%(84/123、237 件)、中等度がレボフロキサシン群で 21.3%(29/136、46 件)、 セフトリアキソン群で19.5%(24/123、34 件)、重度がレボフロキサシン群で 2.9%(4/136、5 件)、セフトリアキソン群で1.6%(2/123、2 件)であり、重症度の分布は両群で同様であっ た。 副作用の重症度は、軽度がレボフロキサシン群で48.5%(66/136、201 件)、セフトリアキ ソン群で54.5%(67/123、141 件)、中等度の副作用はレボフロキサシン群で 12.5%(17/136、 22 件)、セフトリアキソン群で 7.3%(9/123、9 件)であり、重度は両群で認められず、重症 度の分布は両群で同様であった。9.6.4 主要背景因子別の有害事象発現率・副作用発現率
レボフロキサシン群での主要背景因子別の有害事象発現率は、基礎疾患・合併症の有無を 除き、差は認められなかった。また、主要背景因子別の有害事象発現率は、レボフロキサシ ン群とセフトリアキソン群で同様であった。この傾向は副作用発現率でもほぼ同様であった。9.6.5 重篤な有害事象及び他の重要な有害事象
死亡に至った重篤な有害事象は、レボフロキサシン群は136 名中 2 名(肺炎が 2 名に 2 件)、 セフトリアキソン群は123 名中 2 名(咽頭癌・病期不明、及び肺の悪性新生物が各 1 名に 1 件)に認められた。転帰が「死亡」以外の重篤な有害事象は、レボフロキサシン群は136 名2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 害事象は肺小細胞癌・病期不明、肺炎、肺扁平上皮癌・病期不明、声門癌、肺腺癌、うつ病 が各1 名に 1 件(肺扁平上皮癌・病期不明と声門癌は同一被験者)認められた。セフトリア キソン群は肺非小細胞癌、胸腺腫が各1 名に 1 件認められた。両群でのすべての重篤な有害 事象の治験薬との因果関係は否定された。 本試験では、重篤なものを除く「担当医が重度と判定した有害事象」又は「治験薬の投与 中止に至った有害事象」を重要な有害事象とした。本試験の重要な有害事象は、いずれも治 験薬の投与中止に至った有害事象であり、レボフロキサシン群で7 名に 11 件、セフトリアキ ソン群で2 名に 2 件発現した。レボフロキサシン群で発現し投与中止に至った有害事象は、 譫妄、幻覚、頭痛、紅斑、そう痒症、背部痛、注射部位紅斑、口渇、ALT 増加、AST 増加、 血中ビリルビン増加が各1 名に 1 件であった。セフトリアキソン群では、薬疹、発熱が各 1 名に1 件認められた。両群でのすべての投与中止に至った有害事象の治験薬との因果関係は 否定されなかった。
9.6.6 臨床検査値の評価
両群でのCRP、血小板数、白血球数及び白血球分画の変動は、感染症による炎症反応の治 癒過程を反映したものと考えられた。レボフロキサシン群で有害事象及び副作用として多く 認められたALT 増加、AST 増加、好酸球数増加の発現率は、セフトリアキソン群とほぼ同様 であった。9.7 結論
レボフロキサシン群の投与終了/中止時の臨床効果(有効率)は88.5%(92/104)であり、 セフトリアキソン群との群間差の95%信頼区間の下限値が−10%を上回ったため、レボフロキ サシンはセフトリアキソンに対する非劣性が検証された。また、レボフロキサシン注射剤の 安全性に重大な問題は認められなかった。よって、レボフロキサシン500 mg 1 日 1 回 7~14 日間静脈内投与は、S. pneumoniae を原因菌とする肺炎を含む、成人の市中肺炎(細菌性肺炎) に対して十分な治療効果が期待できると考えられる。2.7.6個々の試験のまとめ レボフロキサシン水和物 クラビット点滴静注バッグ500mg/100 mL クラビット点滴静注500ma/20 mL 10. 市中肺炎患者を対象としたレボフロキサシン 750mg 1日 1回 5日間投与と 500 mg 1日 1回 10日間投与の安全性及び有効性を比較する試験 …・…-添付資料番号5.3.5.1・2 (参考) 10.1 治験依頼者 10.2治験実施期間
20_年 I~
I 日 ~20_年 I~
I
日 10.3目的
軽症から重症の市中肺炎患者を対象にレポフロキサシン750mg 1日l回 5日間の静脈内又 は経口投与とレポフロキサシン500mg 1日l回 10日間の静脈内又は経口投与の有効性を比 較検討する。併せて、レポフロキサシンの安全性を検討する。 10.4治験方法 治験デザイン. 米国で実施した多施設共同、無作為化、 三重盲検、非劣性検証試験であった。 割付け . 被験者を、治験実施施設及びFineRisk Score(云70vs > 70 ~云 130) により無作為に 2つ の投与群(レボブロキサシン750mg 1日1回 5日間投与又はレポブロキサシン500mg 1日l 回 10日間投与)に1:1の割合で害I[付けた。 10.5 目標被験者数 臨床効果評価対象集団として各群 172 名(合計 344 名)を目標とし、 400~500 名の登録を 計画した。 登録 :530名 ITT解 析対象集団 :528名 (750mg : 256名、500mg : 272名) 安全性評価対象集団 :521名 (750mg : 256名、 500mg : 265名) 臨床効果評価対象集団 :390名 (750mg : 198名、 500mg : 192名) 微生物学的効果評価対象集団 :195名 (750mg:103名、 500mg : 92名) 10.6診断及び主な選択基準 年齢 18歳以上で性別は問わない 下気道感染の症状・ 所見及び治験薬投与開始前24時間以内の胸部X線写真上で肺炎2.7.6 個々の試験のまとめ
クラビット点滴静注バッグ500 mg/100 mL
レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
• 以下の項目を1 項目以上満たす者
発熱、低体温、白血球増加、桿状核球 > 10% • Fine Risk Score が 130 以下の者
10.7 被験薬、用量及び投与方法、バッチ番号
用量及び投与方法:レボフロキサシン750 mg 1 日 1 回静脈内又は経口投与(静脈内投与か ら経口投与への切り替え可) 被験薬:レボフロキサシンカプセル750 mg(GFI-25213-097-B-008)、市販の 20 mL バイア ル(レボフロキサシン500 mg 含有、NDC 0045-0069-51)10.8 対照薬、用量及び投与方法、バッチ番号
用量及び投与方法:レボフロキサシン500 mg 1 日 1 回静脈内又は経口投与(静脈内投与か ら経口投与への切り替え可) 対照薬:レボフロキサシンカプセル500 mg(GFI-25213-097-B-006)、市販の 20 mL バイア ル(レボフロキサシン500 mg 含有、NDC 0045-0069-51) プラセボ:プラセボカプセル(FD 90000-000-EMX-31)、静脈内投与のプラセボが必要な場 合には、デキストロース又は塩化ナトリウム溶液を用いた。10.9 投与期間
750 mg 群はレボフロキサシンを 5 日間、その後プラセボを 5 日間、500 mg 群はレボフロ キサシンを10 日間投与した。10.10 評価項目
10.10.1 臨床効果
• 治療後1(Posttherapy Visit 3:Day 12~16)、治療後 2(Posttherapy Visit 4:Day 17~21) の臨床効果
症状・所見に基づき、「治癒」、「改善」、「無効」又は「評価不能」に分類した。 • 治療後に治癒又は改善したと考えられる被験者の試験後(Poststudy Visit 5:Day 31~
38)の臨床効果 症状・所見に基づき、「長期治癒」、「長期改善」、「再発」又は「評価不能」に分類した。 • 登録時より治療後までの症状・所見の変化 • 登録時より治療後及び試験後までのX 線所見の変化
10.10.2 細菌学的効果
• 治療後1、治療後 2 の細菌学的効果 陰性化率(「消失」、「存続」又は「不明」に分類)及び菌消失率(「消失」、「推定消失」、 「存続」、「推定存続」、「存続 + 耐性獲得」又は「不明」に分類)を評価した。2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL • 治療後に治癒又は改善したと考えられる被験者の試験後の細菌学的効果 陰性化率(「長期消失」、「再発」、「存続」又は「不明」に分類)及び菌消失率(「消失」、 「再発」、「推定再発」又は「不明」に分類)を評価した。
10.10.3 安全性
発現した有害事象(TEAE:treatment-emergent adverse event)、登録時から治療後までの臨 床検査値及びバイタルサインの変動を評価した。
10.11 統計手法
10.11.1 主要評価項目
レボフロキサシン投与終了後7~14 日(750 mg 群では治療後 1、500 mg では治療後 2)の 臨床効果を主要評価項目とした。臨床効果の群間差(500 mg 群−750 mg 群)の両側 95%信頼 区間を算出した。どちらかの群の有効率が90%以上の時、95%信頼区間の上限が 10%以下の 場合に有効性は同等であると判定した。10.11.2 副次評価項目
1) 治療後の陰性化率 2) 治療後の菌消失率 3) 登録時から治療後までの症状・所見の変化、登録時から治療後及び試験後までの X 線 所見の変化 4) 治療後に治癒又は改善と判定された被験者の試験後の臨床効果及び細菌学的効果 治療終了後の全体の菌消失率及び主要な菌の消失率について、群間差の両側95%信頼区間 を算出した。750 mg 群のレボフロキサシン投与 12~19 日後、500 mg 群のレボフロキサシン 投与7~14 日後の臨床効果(有効率)及び菌消失率についても解析した(以下、治療後 2 の 解析とする)。10.11.3 安全性
有害事象、登録時から治療後までの臨床検査値及びバイタルサインの変動については、記 述統計量(頻度、平均値、標準偏差)を用いて記載した。全体及び各器官分類別の有害事象 の発現率について、群間差の両側95%信頼区間を算出した。10.12 要約-結論
10.12.1 臨床効果
臨床効果評価対象集団のレボフロキサシン投与7~14 日後の有効率(治癒又は改善と判定 された被験者の割合)は、750 mg 群で 92.4%、500 mg 群で 91.1%、群間差の 95%信頼区間は −7.0~4.4 であった。治療後 2 の解析結果に基づき評価した場合でも同様の結果であった(有2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
10.12.2 細菌学的効果
微生物学的効果評価対象集団のレボフロキサシン投与7~14 日後の陰性化率は、750 mg 群 で93.2%、500 mg 群で 92.4%、群間差の 95%信頼区間は−8.6~7.0 であった。治療後 2 の解析 結果に基づき評価した場合でも同様の結果であった(菌陰性化率:750 mg 群 92.9%、500 mg 群92.4%)。 レボフロキサシン投与7~14 日後の菌消失率は、750 mg 群で 92.5%(136/147)、500 mg 群 で91.5%(118/129)であった。グラム陰性好気性菌の菌消失率は 750 mg 群で 500 mg 群より 高かった(菌消失率:750 mg 群 96.2%、500 mg 群 90.7%)。いずれかの投与群の 5 名以上で 分離されたその他の原因菌については、菌消失率は両群に違いは認められなかった。治療後 2 の解析結果でも同様の結果であった。10.12.3 安全性
本治験において、レボフロキサシンの安全性及び忍容性は良好であった。レボフロキサシ ン投与後14 日までの有害事象の発現率は、750 mg 群で 57.8%、500 mg 群で 59.6%であり、 群間差の95%信頼区間は−6.8~10.5 であった。全体の有害事象、器官分類別の有害事象、重 度の有害事象、重篤な有害事象、投与中止に至った有害事象及び顕著な臨床検査異常値の発 現率に群間差は認められなかった。750 mg 群で 5 名(1.9%)、500 mg 群で 9 名(3.4%)が死 亡したが、治験薬との因果関係はいずれも否定された。登録時から治療後までのバイタルサ インに有意な異常変動は見られなかった。本治験において、未知の有害事象は認められなか った。2.7.6個々の試験のまとめ レボフロキサシン水和物 クラビット点滴静注バッグ500mg/100 mL クラビット点滴静注500ma/20 mL 11. DR-3355
注射剤の市中肺炎または慢性呼吸器病変の二次感染を対象とした
一般臨床試験(後期第
11相/第
111相)
添付資料番号5.3.5.2・1 11 .1治験方法
表 2.7.6.11.1-1に治験方法の概略を示す。 治験の目的 対 象 表 2.7.6.11.1-1 治験方法の概略:日本一般臨床試験 成人の市中肺炎文は慢性呼吸器病変の二次感染に対するレボブロキサシン 500mg 1日 l回7 日から14日間点滴静脈内投与の有 効性と安全性を、オープンラベル試験にて確認する。併せて PopulationPharmacokinetics (PPK)法及びベイズ推定を用いて被験者ごとに薬物動態パラメー タを算出し、有効性及び有害事象発現との相闘を検討する。 また、少数例でE喜疾へ の移行性を 検討する。 l被験 者の組入れ基準 次の2項目を満たし、かつ1.1市中肺炎選択基準又は1.2慢性呼吸器病変の二次感染の選択 基 準を満たす愚者を選択した。 1)ステップ lでは同意取得時の年齢が20歳以上 79歳以下の男性及び女性。ステップ2では同 意取得時の年齢が80歳以上の愚者も登録可とした。 2)治験責任医師又は治験分 担医師が入院加療を必要と判断した患者。集中治療室管理もしく は 人工呼吸器装着を必要とする重篤な患者は除く。ただし、治療途中で退院し外来点滴治療を 行ってもよいこととした。 1.1市中肺炎の選択基準 1)病院外で日常生活をしていた人に発症し、市中肺炎と診断された患者。マイコプラス、マ肺炎、 クラミジア肺炎、及びレジオネラ肺炎を含む 2)治験薬投与開始日文はその前日に下記の症状・所見基準を満たす患者 (1)胸 部X線あるいは胸部CT検査で、急性に新たに出現した浸潤影が認められる。 (2)血液検査にて、 白血球数増多(施設上限値を超えるもの)又は CRP増加 (1.0mg/dL以上) のいずれかの急、性炎症所見を認める。 (3)上記 2項目を満たし、かつ下記の 4項目中 2項目以上を満足しなければならない。 i)三37.0C (版禽) ii)咳殿、 E客疾(膿性疾)、胸痛、呼吸困難などの呼吸器症状 iii)湿性ラ音 iv)略疾などの臨床検体から、原因菌と推定される微生物が確認されたものか、確認される 可能性の高い良質の検体が得られるもの 1.2慢性呼吸器病変の二次感染の選択基準 1)慢性呼吸器病変の二次感染と診断された患者 慢性気管支炎、ぴまん性汎級気管支炎、気管支拡張症、 肺気』重、肺線維症、気管支哨息、陳 │目性肺結核などのいずれかの二次感染 2)下記の症状・所見基準を満たす患者 (1)病歴や胸部X線などによって急性気管支炎や肺炎を除外し、慢性肺疾患の存在が確認され ている。 (2)咳歌・疾の新たな出現、あるいは等主主量の増加や膿性度の悪化を認める。 (3)CRPの増加 (0.7mg/dL以上、あるいは施設上限値を超えるもの)を認める。 (4)下記の条件を少なくとも lつ満たしていること。 i)原因菌が明確であること ii)三37.0C (膝禽) iii)白血球数増多 (:>-8000lmmに あ る い は 施 設上限値を超えるもの)2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 表2.7.6.11.1-1 治験方法の概略:日本一般臨床試験(続き) 被験者数 計画された被験者:200 名 同意取得:206 名 登録被験者:206 名 投与被験者:206 名(市中肺炎:165 名、慢性呼吸器病変の二次感染:41 名) 投与完了被験者:173 名(市中肺炎:138 名、慢性呼吸器病変の二次感染:35 名) 投与中止被験者:33 名(市中肺炎:27 名、慢性呼吸器病変の二次感染:6 名) 解析対象被験者 安全性解析対象集団: 206 名(市中肺炎:165 名、慢性呼吸器病変の二次感染:41 名) 最大の解析対象集団(FAS): 194 名(市中肺炎:155 名、慢性呼吸器病変の二次感染:39 名) 治験実施計画書に適合した集団(PPS): 181 名(市中肺炎:146 名、慢性呼吸器病変の二次感染:35 名) PK/PD 解析対象集団(安全性): 195 名(市中肺炎:154 名、慢性呼吸器病変の二次感染:41 名) PK/PD 解析対象集団(有効性): 77 名(市中肺炎:56 名、慢性呼吸器病変の二次感染:21 名) 治験デザイン 多施設共同一般臨床試験(中央登録方式、オープンラベル) 治験薬 DR-3355inj(ロット番号 050171) 用法・用量 レボフロキサシンを1 日 1 回 500 mg/100 mL を約 60 分間かけて点滴静脈内投与した。 投与期間 7~14 日間 評価項目 1 有効性評価項目 1.1 主要評価項目 投与終了/中止時の臨床効果(有効率) 日本化学療法学会「呼吸器感染症における新規抗微生物薬の臨床評価法(案)」の臨床効果判定 基準を参考に、投与終了/中止時の臨床効果を「有効」、「無効」、又は「判定不能」に分類し、 市中肺炎及び慢性呼吸器病変の二次感染ごとに有効率で評価した。 1.2 副次評価項目 1)投与開始 3 日後の臨床効果(有効率) 2)投与開始 7 日後の臨床効果(有効率) 3)最終観察時の臨床効果(有効率) 4)投与終了/中止時の微生物学的効果(陰性化率) 5)投与終了/中止時の微生物学的効果(菌消失率) 1.3 その他の評価項目 1)投与終了/中止時の主要背景因子別の臨床効果(有効率) 2)肺炎球菌による感染被験者での投与終了/中止時の臨床効果(有効率) 3)投与終了/中止時の症状・所見の改善率 4)直前抗菌化学療法無効被験者での投与終了/中止時の臨床効果(有効率) 5)投与終了/中止時の原因菌別の臨床効果(有効率) 6)投与終了/中止時の原因菌別の微生物学的効果(菌消失率) 7)投与終了/中止時の原因菌別 MIC 別の臨床効果(有効率) 8)投与終了/中止時の原因菌別 MIC 別の微生物学的効果(菌消失率) 9)臨床効果有効被験者での投与終了/中止後の抗菌薬治療実施率 10)投与終了/中止時の菌交代率 11)投与終了/中止時の原因菌種数別の臨床効果(有効率) 12)投与終了/中止時の原因菌種数別の微生物学的効果(陰性化率) 項目 計算式 臨床効果 (有効率) 有効率 = 「有効被験者数」/「解析対象被験者数」 (投与終了/中止時、投与開始3 日後、及び投与開始 7 日後では、PPS での判定不能は分母から除き、FAS の判定不能は含める。) (最終観察時では、PPS の判定不能及び観察中止は分母から除き、FAS の判定不能は含める。) 微生物学的効果 (陰性化率) 陰性化率 = 「菌消失又は推定菌消失被験者数」/「解析対象被験者数」 (判定不能は分母から除く) 微生物学的効果 (菌消失率) 菌消失率 = 「消失菌株数」/「解析対象菌株数」(判定不能は分母から 除く)
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 表2.7.6.11.1-1 治験方法の概略:日本一般臨床試験(続き) 評価項目(続き) 2 薬物動態評価項目 1)血漿中薬物濃度 2)母集団薬物動態パラメータ 3)ベイズ推定による被験者ごとの薬物動態パラメータ 4)PK/PD パラメータと有効性の相関、薬物動態パラメータと有害事象発現の相関 5)喀痰中薬物濃度 3 安全性評価項目 1)有害事象発現率及び副作用発現率 2)主要背景因子別の有害事象発現率及び副作用発現率 3)臨床検査値の推移 評価スケジュール 表2.7.6.11.1-2 参照 統計解析手法 1 有効性の解析 有効性解析の主たる対象集団はPPS とする。主要評価項目並びに一部の副次評価項目について は、FAS による解析も参考として行った。 各評価項目について点推定値を求め、その値の正規近似に基づく両側95%信頼区間(95%CI) を求めた。 2 薬物動態の解析 本治験のPK/PD 解析対象集団(安全性)と臨床薬理 3 試験の被験者のデータをもとに母集団薬 物動態解析を実施し、ベイズ推定により各被験者の薬物動態パラメータ(Cmax、C24h、及び AUC0−24h)を算出した。また、本治験のPK/PD 解析対象集団(有効性)の各被験者の薬物動態 パラメータと各原因菌のMIC から PK/PD パラメータ(AUC0−24h/MIC 及び Cmax/MIC)を算出し た。 PK/PD 解析対象集団(有効性)を対象として PK/PD パラメータと有効性評価項目(投与終了/ 中止時の菌消失率及び有効率)との関係を、またPK/PD 解析対象集団(安全性)を対象として 薬物動態パラメータと安全性評価項目(有害事象及び副作用)との関係を検討した。 3 安全性の解析 安全性解析対象集団を対象として、有害事象及び副作用を発現した被験者数、被験者の割合と その両側95%CI、件数を示し、器官別大分類・基本語別の集計も併せて行った。 臨床検査の血液一般検査と血液生化学検査については、投与前後の要約統計量を算出し、異常 値(高値・低値)の頻度を示すとともに、Wilcoxon の符号付順位検定により投与前後を比較し た。また、投与前と投与後の散布図を作成した。尿一般検査については、投与前後の分割表を 作成し、異常値の頻度を示すとともに、Wilcoxon の符号付順位検定により投与前後を比較した。 また、各検査項目の基準値からの逸脱を示すシフトテーブルを作成した。
2.7.6 個々の試験のまとめ
クラビット点滴静注バッグ500 mg/100 mL
レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL
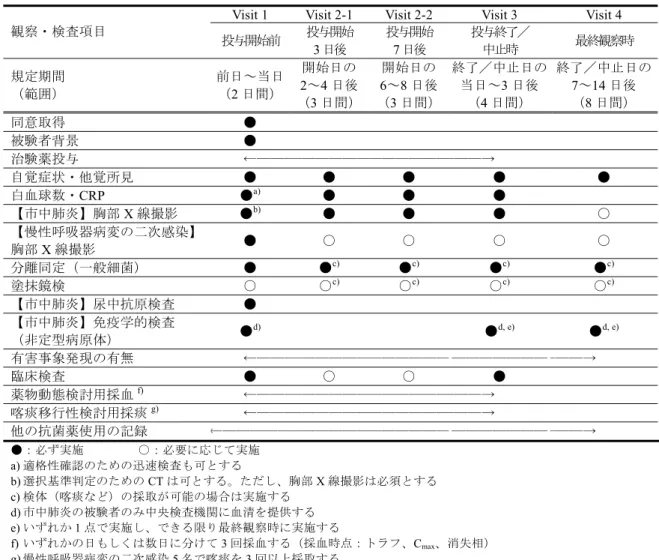
表2.7.6.11.1-2 調査、観察、検査項目、及び試料採取の実施時期
Visit 1 Visit 2-1 Visit 2-2 Visit 3 Visit 4 観察・検査項目 投与開始前 投与開始 3 日後 投与開始 7 日後 投与終了/ 中止時 最終観察時 規定期間 (範囲) 前日~当日 (2 日間) 開始日の 2~4 日後 (3 日間) 開始日の 6~8 日後 (3 日間) 終了/中止日の 当日~3 日後 (4 日間) 終了/中止日の 7~14 日後 (8 日間) 同意取得 ● 被験者背景 ● 治験薬投与 ←――――――――――――――――――→ 自覚症状・他覚所見 ● ● ● ● ● 白血球数・CRP ●a) ● ● ● 【市中肺炎】胸部X 線撮影 ●b) ● ● ● ○ 【慢性呼吸器病変の二次感染】 胸部X 線撮影 ● ○ ○ ○ ○ 分離同定(一般細菌) ● ●c) ●c) ●c) ●c) 塗抹鏡検 ○ ○c) ○c) ○c) ○c) 【市中肺炎】尿中抗原検査 ● 【市中肺炎】免疫学的検査 (非定型病原体) ● d) ●d, e) ●d, e) 有害事象発現の有無 ←――――――――――――――― ――――――――――→ 臨床検査 ● ○ ○ ● 薬物動態検討用採血f) ←――――――――――――――――――→ 喀痰移行性検討用採痰g) ←――――――――――――――――――→ 他の抗菌薬使用の記録 ←――――――――――――――――― ――――――――――→ ●:必ず実施 ○:必要に応じて実施 a) 適格性確認のための迅速検査も可とする b) 選択基準判定のための CT は可とする。ただし、胸部 X 線撮影は必須とする c) 検体(喀痰など)の採取が可能の場合は実施する d) 市中肺炎の被験者のみ中央検査機関に血清を提供する e) いずれか 1 点で実施し、できる限り最終観察時に実施する f) いずれかの日もしくは数日に分けて 3 回採血する(採血時点:トラフ、Cmax、消失相) g) 慢性呼吸器病変の二次感染 5 名で喀痰を 3 回以上採取する
11.1.1 被験者数の設定根拠
本剤の申請データパッケージでは、患者を対象として、本治験及び第III 相比較試験を予定 していた。安全性評価の集積被験者数として、発現率1%の有害事象を 95%の確率で検出可 能なように300 名以上を集積することとした。第 III 相比較試験では本薬群 100~150 名程度 の被験者数を予定していたことから、本治験では200 名を集積することとした。中間評価は 80 名~100 名を目標被験者数とした。11.1.2 用法・用量の設定根拠
キノロン系抗菌薬の治療効果に相関する主要なPK/PD パラメータは、一般に血中 24 時間 AUC と MIC の比(AUC/MIC)とされている。レボフロキサシンが肺炎の代表的な原因菌で ある肺炎球菌に対して効果を示す条件はAUC/MIC 30 以上とされている。レボフロキサシン を250~1000 mg 静脈内単回投与したときの AUC0-infは表2.7.6.11.1-3 に示すとおりである。肺炎球菌の臨床分離株に対するレボフロキサシンのMIC90は1 µg/mL であることから、臨床
2.7.6 個々の試験のまとめ クラビット点滴静注バッグ500 mg/100 mL レボフロキサシン水和物 クラビット点滴静注500 mg/20 mL 表2.7.6.11.1-3 レボフロキサシンの PK/PD からの臨床推奨用量の検討 レボフロキサシンの静脈内投与量(単回投与) PK パラメータ 250 mg 500 mg 750 mg 1000 mg AUC0-inf(µg・h/mL) 23.45 52.09 91.12 127.85 下線はAUC/MIC ≥ 30 であり効果を示す AUC AUC0-infは健康成人での第I 相単回投与試験より引用 一方、キノロン系抗菌薬への肺炎球菌の耐性化を防止するためには、CmaxとMIC の比 (Cmax/MIC)を 5~10 以上とすることが必要とされている。表 2.7.6.11.1-4 にレボフロキサシ
ンに対する肺炎球菌のMIC と各用法・用量における Cmax/MIC との関係を示した。国内にお
けるレボフロキサシンの経口剤の標準用量である100 mg 1 日 3 回投与では、MIC90が1 μg/mL
の菌ではCmax/MIC が 2.62 であり、耐性化抑制に必要とされる Cmax/MIC は達成できていない。
一方、レボフロキサシン注射剤の500 mg 1 日 1 回投与では Cmax/MIC が 9.79 となり、耐性化 抑制に必要とされるCmax/MIC を達成できる。 表2.7.6.11.1-4 LVFX の PK/PD パラメータ LVFX 100 mg 1 日 3 回経口投与 LVFX 500 mg 1 日 1 回静脈内投与 LVFX MIC (µg/mL)
Cmax/MIC Cmax/MIC
8 0.33 1.23 4 0.66 2.45 2 1.31 4.90 1 2.62 9.79 0.5 5.24 19.58 耐性化抑制が期待されるCmax/MIC 上記PK/PD の考え方とモンテカルロ・シミュレーションの手法を用い、日本人の肺炎球菌 性肺炎患者の臨床効果を予測したところ、500 mg 1 日 1 回投与の有効率は 95%以上であるこ とが予想された。 以上より、レボフロキサシンの用量・用法を500 mg 1 日 1 回静脈内投与とした。