The authors declare no con‰ict of interest. 九州大学先導物質化学研究所 e-mail: kazunari@ms.ifoc.kyushu-u.ac.jp ―Regular Article―
酵素反応の計算ミューテーション解析
土井富一城,蒲 池 高 志,吉 澤 一 成Computational Mutation Analysis of Enzymatic Reaction
Kazuki Doitomi, Takashi Kamachi, and Kazunari Yoshizawa Institute for Materials Chemistry and Engneering, Kyushu University;
744 Motooka, Nishi-ku, Fukuoka 8190395, Japan.
(Received June 18, 2012; Accepted August 1, 2012)
Density functional theory(DFT) calculations are established as a useful research tool to investigate the structures and reactivity of biological systems; however, their high computational costs still restrict their applicability to systems of several tens up to a few hundred atoms. Recently, a combined quantum mechanical/molecular mechanical (QM/MM) approach has become an important method to study enzymatic reactions. In the past several years, we have investigated B12-dependent diol dehydratase using QM/MM calculations. The enzyme catalyzes chemically di‹cult reactions by utilizing the high reactivity of free radicals. In this paper, we explain our QM/MM calculations for the structure and reactivity of diol dehydratase and report key ˆndings with respect to the catalytic roles of the active-site amino acid residues, computational mutational analysis of the active-site amino acid residues, assignment of the central metal ion, and function of the central metal ion. Our QM/MM calculations can correctly describe the structures and activation bar-riers of intermediate and transition states in the protein environment. Moreover, predicted relative activities of mutants are consistent with experimentally observed reactivity. These results will encourage the application of QM/MM research to the mechanistic study of enzymatic reactions, functional analysis of active-site residues, and rational design of en-zymes with new catalytic functions.
Key words―reaction mechanism; computational mutation; enzymatic reaction; quantum mechanical/molecular mechanical(QM/MM) method は じ め に 近年の急速なコンピュータの発達と優れたプログ ラムの開発により,量子化学計算,分子動力学計算 は理論計算の専門家でない実験化学者にとっても身 近になりつつある.また,2012 年秋に完成予定で ある次世代スーパーコンピュータ「京」の重要応用 分野に in silico 創薬の加速が含まれるなど計算化 学,計算創薬への期待は非常に大きくなっている. 筆者らはこれまで量子化学計算と分子力学計算を 組 み 合 わ せ た quantum mechanical / molecular
me-chanical(QM/MM)法により酵素の全原子を含む 「全酵素モデル」の計算を行い,酵素反応の機構を 明らかにしてきた.さらに,活性中心近傍に存在す るアミノ酸残基を置換して計算を行うことで特定残 基の機能を解明した.本稿では筆者らが行ってきた 計算ミューテーションによる酵素反応の解析につい て紹介したい. 計 算 方 法 1. QM/MM 法の概要 タンパク質などの生 体分子は数万原子からなる非常に大きな分子であ り,全原子に量子化学計算を適用することは非常に 高コストであり実用的ではない.そのため,酵素な どの巨大な分子を計算する上で量子化学計算と分子 力学計算を組み合わせた QM/MM 法が広く用いら れている.QM/MM 法は計算領域を QM 領域と MM 領域の 2 つの領域に分割して計算を行う方法 である.活性中心など反応に重要な領域(QM 領域) は電子状態や結合の生成,解離を記述できる ab in-itio や density functional theory (DFT)を用いて高 精度な計算を行い,それ以外の領域(MM 領域) はパラメータ依存の分子力学による安価で高速な計
Fig. 1. Image for QM and MM Region Partitioning 算を適用する(Fig. 1). QM/MM 法による全系のハミルトニアンは HQM/MM=HQM+HMM+HQM-MM (1) と記述できる.ここで HQMは QM 領域,HMMは MM領域,HQM-MMは QM 領域と MM 領域の相互 作用を表すハミルトニアンである.この方法におけ る一番の特徴は 2 つの領域間の相互作用を記述する HQMMMである.このハミルトニアンは HQM-MM=HelQM-MM+HvdWQM-MM+HbQM-MM (2) という 3 個の項に分割することができる.Hel QM-MM は QM 領域と MM 領域間の静電相互作用,HvdW QM-MM
は van der Waals 相互作用,Hb
QM-MMは結合性相互 作用を表すハミルトニアンである.Hel QM-MMの計 算では MM 領域の原子の電荷を QM 計算のハミル トニアンの中に含める(Eq. 3). Hel QM-MM=- electron
∑
i a∈MM∑
qa |ri-Ra| +∑
b∈QMa∈MM∑
qaZb |Rb-Ra| (3) ただし,qaは MM の点電荷,Zbは QM 原子の 電荷,Ra,Rb,riはそれぞれ MM 領域の原子,QM 領域の原子核,QM 領域の電子の位置を表す.また, a, b, i はそれぞれ MM 領域の原子,QM 領域の原 子核,QM 領域の電子の番号である.この方法を用 いることで QM 領域の電子状態を現実系に近い形 で取り扱うことが可能になる. また,QM/MM 法で取り扱いに注意しなければ ならないのは QM 領域と MM 領域の境界である. 酵素の計算を行う場合,共有結合が領域の境界に存 在することが多々ある.そのため,領域を分割する ためには結合を切断しなければならないが,切断し ただけでは不対電子が生成してしまう.そこで,切 断した結合に水素原子やメチル基をつけるリンク原 子法が広く用いられている. 2. QM/MM 計算のモデル作成 酵素の QM/ MM計算を行う場合,X 線結晶構造を基に計算モ デルを構築することが多い.しかし,X 線結晶構 造は水素の情報を持っておらず,アミノ酸残基のプ ロトン状態がわからない.また,QM/MM 計算で は溶媒の影響を取り込んだ計算を行うことが多く, X線結晶構造に溶媒分子を加える必要がある.そ のため,分子動力学(molecular dynamics; MD)計 算などを行い,QM/MM 計算で使用する計算モデ ルを構築する.筆者らは以下に述べるような手順で モデルを作成している. Protein Data Bankに登録された X 線結晶構造
に水素を付加し,PROPKA プログラム1,2)と Generalized Born法に基づく pKa予測プログラ ム35)により各アミノ酸残基のプロトン状態を 予測する. 溶媒分子の影響を計算に取り込むため半径 30 Å の球を形成するように水分子を発生させ, 酵素をその球の中に配置し,初期構造とする. 初期構造を作成した後はエネルギーの最小化を 行う.この操作は初期構造が持つ不自然な歪み を取り除き,MD 計算におけるエネルギーの発 散を避けるために実行される. 15 psかけて温度を 0 K から 300 K まで上昇さ せ,次に 300 K で 400 ps の平衡化を行う.こ の過程において水素原子には SHAKE 法によ る拘束を適用し,time step を 1.0 fs として計 算を行う. 400 ps平衡化した構造を用い,adapted basis
Newton-Raphson (ABNR)法6)による 5000 step
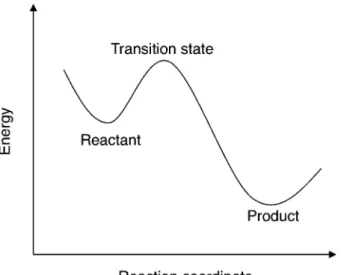
のエネルギーの最小化を行い,得られた構造を QM/MM 計算の計算モデルとする. モデル作成にあたり,QM 領域の原子とパラメー タの精度が不確かな補酵素など(後述の計算ではア デノシルコバラミン)は X 線結晶構造のままで固 定する.これらの計算は Discovery Studio7)を用い て行っている. 3. 理論計算に基づく反応経路の探索 ある化 学反応について理論計算を用いて検討を行うとき, その反応の 2 つの安定構造(Reactant, Product) とそれらを結ぶ 1 つの遷移状態(transition state; TS)を得ることが必要である.Figure 2 に示すよ うに,Transition state はエネルギー曲面の 1 次の
Fig. 2. Potential Energy Surface along the Reaction Coor-dinate
鞍点つまり反応経路上におけるエネルギーの極大点, Reactant と Product は極小点に位置する.反応の 活性化エネルギーは Reactant と Transition state の エネルギー差となる.両者に共通していることはエ ネルギーの座標に対する 1 次微分(Gradient)は 0 ということである.しかし,Reactant と Product ではエネルギーの座標に対する 2 次微分(Hessian) はすべて正の固有値を持つが,Transition state で は Hessian は負の固有値を1つだけ持ち,残りは正 の値 とな る .安 定構 造 の探 索で は エネ ルギ ー と Gradientが小さくなる方向に最適化を繰り返す. 一方,遷移状態の探索では Hessian が負の固有値を 1つ持ち,Gradient が 0 になるように最適化が行わ れる.しかし,QM/MM 法においては数千,数万 原子 の計 算 を行 うの で ,す べて の 原子 につ い て Hessianを計算することは現実的ではない.そのた め,系を遷移状態に直接関与する数原子(Reaction core)と残りの原子(Environment)に分割して計 算する Microiterative scheme8)が用いられる.この 方法では Reaction core において遷移状態の探索を 行い,Environment において極小点の探索を行う. Reaction core の計算を行うとき,Environment の 原子は固定され,Environment の最適化を行うとき は,Reaction core の原子は固定される.Reaction core と Environment の計算を Gradient が収束する まで交互に繰り返す.また,QM/MM 計算では活 性中心から 610 Å 以内の 1000 原子程度に対して 構造最適化を行うことが一般的である.9)酵素反応 では各安定構造,遷移状態が連続的につながってい ることが必要であり,タンパク質のコンフォメーシ ョンが各安定構造,遷移状態において大きく変化し ないことが求められる.原子を固定することで構造 の変化を抑制する方法は計算時間を減らす上で有効 な手段である. 結 果・考 察 1. 全 酵 素 モ デ ル 計 算 に よ る 反 応 機 構 の 検 討 ビタミン B12 は生体内に取り込まれると,アデ ノシルコバラミンに変換され,十数種の酵素の補酵 素として働くことが知られている.このうちジオー ルデヒドラターゼは,1,2-プロパンジオール(pro-panediol; PDO),1,2-エタンジオール,グリセロー ル等のジオールの脱水反応を触媒する酵素である. こ の 酵 素 に よ る 反 応 は ア デ ノ シ ル コ バ ラ ミ ン (adenosylcobalamin; AdoCbl)中の CoC 結合が均
等に開裂することにより始まる[Fig. 3(A)].10)こ れにより生じたアデノシルラジカルが基質の 1 位の 炭素から水素を引き抜き,1,2-ジオールラジカルが 生成する.次に水酸基の転移が起き 1,1-ジオールラ ジカルを経て 1,1-ジオールを生成する.この 1,1-ジ オールから水分子が脱離することで,アルデヒドが 得られる[Fig. 3(B)].ジオールデヒドラターゼの 構造は虎谷らによる X 線結晶解析により明らかに され,1114)活性中心には K+又はこれと同程度のイ オン半径を持つカチオンが存在し,基質が直接カチ オンに配位していることが示された(Fig. 4). 筆者らは PDO,エチルラジカル(アデノシルラ ジカルのモデル),K+からなるモデル計算を行 い,反応機構の解析を行った.15)Figure 5に示すの は計算により求められた最適な反応経路とそのエネ ルギーダイアグラムである.遷移状態 TS2 を経由 する水酸基転移反応過程は協奏的に進行し,その活 性化エネルギーは 18.7 kcal/mol であることが分か った.また,K+イオンは基質を適切な位置に配位 させるために必要であることが提案された.TS1 か ら TS3 を経る 3 つの素過程のうち,TS2 の活性化 エネルギーが最大であり,水酸基転移反応過程が律 速段階であると予測された.しかし,Abeles らによ る速度論的同位体効果(kinetic isotope eŠect; KIE =10)の測定から,遷移状態 TS1 を経由する水素
Fig. 3. Minimal Mechanism of Diol Dehydratase Reaction
(A) Homolytic cleavage of the Co-C bond in AdoCbl. (B) Adenosyl radical-catalyzed rearrangement.
Fig. 4. Crystal Structure of Diol Dehydratase (PDB ID 1EEX) 引き抜き反応過程又は遷移状態 TS3 を経由する水 素再結合反応過程が反応の律速段階であると考えら れ,16)この計算結果は実験結果と矛盾している.こ の原因は活性中心近傍の残基を計算に含めなかった ためと考えられる. 実際,Golding と Radom らによるモデル計算は 転移する水酸基にプロトンを与えること(push 効 果)で TS2 の活性化エネルギーが減少することを 示している.17)例えば,NH+ 4 や CH3OH+2 が転移す る水酸基にプロトンを与えることで活性化エネル ギーがそれぞれ 15.2, 26.4 kcal/mol 減少する.ま た,基質の 1 位の水酸基からのプロトン引き抜き (pull 効果)も TS2 の活性化エネルギーを低下させ る. こ れら の モデ ル計 算 は活 性中 心 に存 在す る His143と Glu170 が TS2 の活性化エネルギーを減 少させる重要な残基であることを示唆している. そこでわれわれは酵素の全原子からなる「全酵素 モデル」による計算(QM/MM 計算)を行い,周 辺アミノ酸残基の影響を検討した.18)QM 領域には Fig. 4 に示す 6 個のアミノ酸残基,中心金属 K,基 質(PDO),アデノシルのリボース部位を選択した. Figure 6 に示すのは QM/MM 計算による最適化構 造とエネルギーである.QM/MM 計算でも水酸基 転移反応はモデル計算と同様に協奏的に進行するこ とが予測された.そして TS2 の活性化エネルギー がモデル計算よりも 7.2 kcal/mol 低い 11.5 kcal/ mol となった.さらに,His143 が基質の 2 位の水 酸基と水素結合を形成している HIE モデルと水素 結合を形成していない HID モデルのエネルギーを
Fig. 5. Energy Diagram of the Model with K+Ion along the Reaction Path, from 1,2-Propanediol to 1,1-Propandiol Relative energies are in kcal/mol. Bond distances in Å.
Table 1. Calculated Barrier Heights (kcal/mol) in the Catal-ysis of Diol Dehydratase and Its Mutants
TS1 TS2 TS3 Relative activity (expb) Wild type 13.6 11.5 17.9a 1.0(1.0) His143Ala 11.7 16.4 19.6a 6.3×10-2(1.6×10-2) Glu170Gln 12.5 17.4 21.6a 2.5×10-3(2×10-4) Glu170Ala 9.1 13.6 22.6a 4.9×10-4(<1×10-4) Glu170Ala/ Glu221Ala 14.9 24.4a 19.3 6.4×10-18(N.A.)
aWe estimate the relative activities of the mutants using these values. bExperimental data from ref. 13.
比較して,His143 の水素結合(push 効果)により TS2の活性化エネルギーは 1.6 kcal/mol 低下する と見積もられ,残りの 5.6 kcal/mol の安定化は主 に Glu170 により引き起こされると予測された.ま た,QM/MM 計算では 3 つの素過程のうち TS3 の 活性化エネルギーが最大なので,水素再結合反応過 程が律速段階と予想された.水素の移動が反応の律 速段階という実験結果を再現しており,酵素反応を より正確に記述することができたと考えられる. 2. 計算ミューテーションによる変異型酵素の活 性予測 筆者らはこの反応において重要なアミノ 酸残基と考えられてきた His143 をアラニンに置換 した His143Ala 変異型,及び Glu170 を置換した Glu170Gln, Glu170Ala 変 異 型 ,さ ら に Glu170 と Glu221の 2 つを置換した Glu170Ala/Glu221Ala 変 異型(Fig. 7)についても QM/MM 計算を行い, 反応機構を検討した.19) これら変異型の反応も野生型と同様に 3 段階で進 行した.Table 1 に野生型と 4 個の変異型酵素の反 応活性及び各遷移状態の活性化エネルギーをまとめ た.His143Ala 変異型においてはヒスチジンの水素 結合による転移する水酸基の安定化の効果がなくな り,TS2 の活性化エネルギーが上昇した.さらに, 基質を最適な位置に固定することができないので TS3の活性化エネルギーが大きくなった.また,野 生型よりも TS2 の活性化エネルギーと TS3 の活性 化エネルギーの差が 3.2 kcal/mol 小さくなり,KIE の値が 4.0 と野生型の場合の 10 よりも小さくな り,水素引き抜きが部分律速になるという実験結 果20)に一致した. Glu170Gln 変異型では Gln170 と基質が水素結合 を形成するが,Glu170Ala 変異型では Glu221 と水 素結合を形成して構造が大きく変化した.これら変 異型酵素における TS2 の活性化エネルギーが野生 型のそれに比べ,Glu170Gln 変異型と Glu170Ala 変異型ではそれぞれ 5.9, 2.1 kcal/mol 高くなった. Glu170Ala 変異型が Glu170Gln 変異型に比べて低い 活性化エネルギーを示すのは Glu221 が Gln170 より も基質との間に強い水素結合を形成して,強い pull 効果を示すためだと考えられた.実際,Glu170Ala /Glu221Ala 変異型においては pull 効果を持つ残基
Fig. 6. Optimized Geometries of the Intermediates and Transition States for the Hydrogen Abstraction and the OH Group Migration
Bond distances in Å. Relative energies are measured from the initial complex.
Fig. 7. QM/MM-Optimized Structures of Mutants
(a) His143Ala (b) Glu170Gln (c) Glu170Ala (d) Glu170Ala/ Glu221Ala of diol dehydratase.
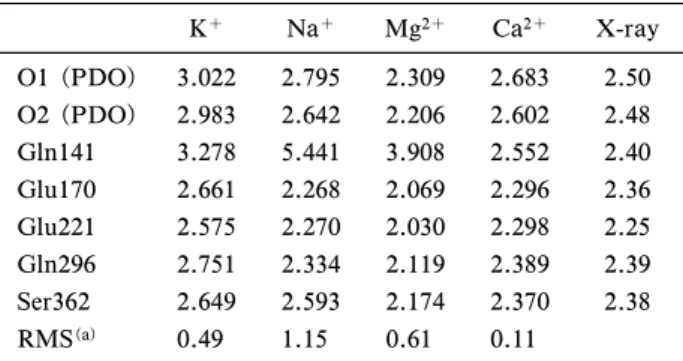
が存在しないため,TS2 の活性化エネルギーは著し く上昇した.また,Glu170Gln, Glu170Ala 変異型 では野生型の 1%以下の活性しか示さないことが知 られている.21)これは残基を置換することで基質を 水素再結合反応に最適な位置に固定することができ なくなり,立体的要因により TS3 の活性化エネル ギーが上昇するからだと予測された. このように 4 個の変異型酵素について計算ミュー テーションによる解析を適用して,それぞれの反応 活性を明らかにした.計算による予測値と実験値が よく一致しており,計算ミューテーションによる変 異型酵素の解析が有用であることが示された. 3. 理論計算による金属イオンの再同定及び反応 機構の再検討 これまで行った QM/MM 計算に よるジオールデヒドラターゼの活性中心における KO の平均結合距離は 2.8 Å であった.一方,X 線結晶構造におけるそれは 2.4 Å であった.Ryde らにより結晶構造と DFT により得られた安定構造
Table 2. QM/MM Optimized Metal-Oxygen Bond Lengths (Å) in the Substrate-binding Site at the B3LYP/SV(P) Lev-el of Theory K+ Na+ Mg2+ Ca2+ X-ray O1 (PDO) 3.022 2.795 2.309 2.683 2.50 O2 (PDO) 2.983 2.642 2.206 2.602 2.48 Gln141 3.278 5.441 3.908 2.552 2.40 Glu170 2.661 2.268 2.069 2.296 2.36 Glu221 2.575 2.270 2.030 2.298 2.25 Gln296 2.751 2.334 2.119 2.389 2.39 Ser362 2.649 2.593 2.174 2.370 2.38 RMS(a) 0.49 1.15 0.61 0.11
(a)RMS errors from experimental values.
Fig. 8. Computed Energy Diagram for the Hydrogen Ab-straction and the OH Group Migration in the Ca2+- and K+ -containing Models
Relative energies are in kcal/mol.
Fig. 9. Optimized Structure of the Transition State for the OH Group Migration in the Simple Model Calculation
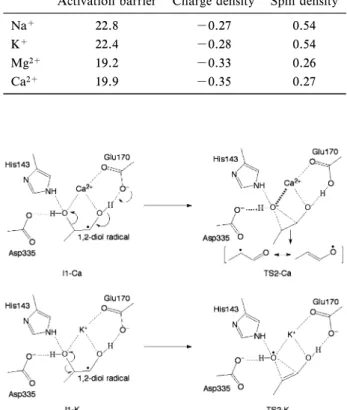
Units in Å. の 長 さ の 誤 差 は 平 均 0.1 Å 以 下 と 報 告 さ れ て お り,22)0.4 Åという違いは非常に大きい.さらに,7 配位の K+イオンを含む Staphylcoccus aureus 由来 のアルセナイトレクターゼの平均 KO 結合長は 2.81 Å であり,23)QM/MM 計算によって得られた 結果に近い長さになった.これらの結果から,筆者 らは活性中心の金属イオンは K+ではなく,別の金 属イオンのではないかと考え,QM/MM 計算を用 いて金属イオンの再同定を行った.金属イオンは K+ と 周 期 表 で K の 近 隣 に 位 置 す る Ca2+, Na+, Mg2+を考えた. Table 2に示すのは QM/MM 計算によって最適 化された活性中心の金属イオンと配位酸素の結合距 離と RMS(実験値と計算値の誤差)である.24)こ れまで中心金属と考えられてきた K+の場合におけ る RMS は 0.49 であったが,Ca2+ の場合の RMS が 0.11 と K+の場合よりも小さくなった.また, Na+と Mg2+の場合は Gln141 が金属イオンに配位 せず六配位構造をとったため,RMS が大きくなっ た.われわれはこの計算結果から中心金属は Ca2+ であると提案した.最近,虎谷らによる EDTA の 吸着実験によって金属イオンは Ca2+であることが 確認された.25) この結果に基づき筆者らは中心金属が Ca2+の場 合における反応機構の再計算を行った.26)また, QM/MM 計算では QM 領域の原子を適切に選択す る必要があるため,再計算では実験により反応に重 要な残基と提案されている Asp33521)を QM 領域に 加えた. Figure 8に示すのは K+と Ca2+それぞれの場合 における反応のエネルギーダイアグラムである.3 つの素過程のうち K+の場合は TS2, Ca2+の場合は TS3 が最も大きな活性化エネルギーを持つので,反 応の律速段階はそれぞれ水酸基転移反応過程,水素 再結合反応過程と予測された.前述した通り,KIE の値から反応の律速段階は水素引き抜き反応過程又 は水素再結合反応過程であるので,反応の律速段階 も中心金属が Ca2+の場合を支持している.また, 筆者らは水酸基転移反応において Ca2+の方が K+ よりも活性化エネルギーが 8.9 kcal/mol 低いこと に注目した.この違いを明らかにするために金属イ オン(Na+, K+, Mg2+, Ca2+)とプロパンジオール ラジ カル か らな る 簡易 モデ ル の計 算を 行 った . Figure 9はモデル計算の遷移状態の構造であり,そ の構造における活性化エネルギー,転移する水酸基 の電荷密度とスピン密度を Table 3 にまとめた.一 価のカチオンよりも二価のカチオンの活性化エネル
Table 3. Calculated Activation Barrier (kcal/mol) of the OH Group Migration and Mulliken Charge and Spin Densities on the Migrating OH Group for a Cluster Model Using a Dielectric Constant of 6.0 at the B3LYP/6-311GLevel of Theory
Activation barrier Charge density Spin density
Na+ 22.8 -0.27 0.54
K+ 22.4 -0.28 0.54
Mg2+ 19.2 -0.33 0.26
Ca2+ 19.9 -0.35 0.27
Fig. 10. A Cartoon Showing the Electronic Changes for the Transition State of the OH Group Migration in the Ca2+ -and K+-containing Active Site of Diol Dehydratase
ギーが数 kcal/mol 小さくなった.しかし,Ca2+の 効果だけで TS2 の活性化エネルギーの減少を十分 に説明することができなかった.それは QM/MM 計算において Glu170 が活性エネルギーの減少に大 きく寄与しているからである.Ca2+の場合,遷移 状態 TS2 において Glu170 が 1 位の水酸基の水素を 引き抜き,共鳴構造を取ることができるようにな り,活性化エネルギーが低下するが,K+の場合は 1位の水酸基の水素を Glu170 が引き抜くことがで きないので,共鳴構造を取ることができず,TS2 の 活性化エネルギーは大きくなると予測された(Fig. 10).また,Asp335 は負電荷を持つので,転移する 水酸基と静電的に反発し,TS2 の活性化エネルギー を上げることで,反応の進行を妨げることが示され た. 結 論 以上の研究が示すように QM/MM 計算により活 性部位アミノ酸残基の役割を明らかにすることがで きた.さらに,量子化学計算による酵素変異という 新しい試みに挑戦し,変異型酵素の活性予測を行っ た.その予測は変異型酵素の実際の測定結果とよい 一致を示した.また,理論計算により X 線結晶解 析の誤りを正したことにより,量子化学計算による 酵素の研究は実験結果を補完する有用な解析ツール であると示すことができた.これまで理論計算は実 験結果の説明が主な役割と見なされてきたが,近い 将来,計算化学的アプローチが酵素の設計と機能制 御に重要な役割を果すことを期待している. REFERENCES
1) Li H., Robertson A. D., Jensen J. H., Pro-teins, 61, 704721 (2005).
2) Bas D. C., Rogers D. M., Jensen J. H., Pro-teins, 73, 765783 (2008).
3) Bashford D., Karplus M.,J. Phys. Chem., 95, 95569561(1991).
4) Dominy B. N., Brooks C. L. III, J. Phys. Chem. B, 103, 37653773 (1999).
5) Spassov V. Z., Yan L.,Protein Sci., 17, 1955 1970 (2008).
6) Press W. H., Flannery B. P., Teukolshy S. A., Vetrrling W. T., ``Numerical Recipes: The Art of Scientiˆc Computing,'' University Press, Cambridge, 1987.
7) Discovery Studio 2.0; Accelrys software Inc.; San Diego, 2007.
8) Turner A. J., Moliner V., Williams I. H., Phys. Chem. Chem. Phys., 1, 13231331 (1999).
9) Thiel W., ``Multiscale Simulation Methods in Molecular Science, QM/MM methodology: fundamentals, scope, and limitations,'' eds. by Grotendorst J., Attig N., Bl äugel S., Marx D., Institute for Advanced Simulation, For-schungszentrum J äulich, 2009, pp. 203214. 10) Toraya T., Chem. Rev., 103, 20952127
(2003).
11) Shibata N., Masuda J., Tobimatsu T., Toraya T., Suto K., Morimoto Y., Yasuoka N., Struc-ture, 7, 9971008 (1999).
12) Masuda J., Shibata N., Morimoto Y., Toraya T., Yasuoka N.,Structure, 8, 775788 (2000). 13) Shibata N., Masuda J., Morimoto Y., Yasuo-ka N., Toraya T., Biochemistry, 41, 12607 12617(2002).
14) Shibata N., Nakanishi Y., Fukuoka M., Yamanishi M., Yasuoka N., Toraya T., J. Biol. Chem., 278, 2271722725 (2003). 15) Eda M., Kamachi T., Yoshizawa K., Toraya
T., Bull. Chem. Soc. Jpn., 75, 14691481 (2002).
16) Abeles R. H., Dolphin D.,Acc. Chem. Res., 9, 114120(1976).
17) Smith D. M., Golding B. T., Radom L., J. Am. Chem. Soc., 123, 16641675 (2001). 18) Kamachi T., Toraya T., Yoshizawa K.,J. Am.
Chem. Soc., 126, 1620716216 (2004). 19) Kamachi T., Toraya T., Yoshizawa K.,Chem.
Eur. J., 13, 78647873 (2007).
20) Kinoshi K., Kawata M., Ogura K., Yamasaki A., Watanabe T., Komoto N., Hieda N., Yamanishi M., Tobimatsu T., Toraya T., Biochemistry, 47, 31623173 (2008).
21) Kawata M., Kinoshi K., Takahashi S., Ogura K., Komoto N., Yamanishi M., Tobimatsu T., Toraya T.,J. Biol. Chem., 281, 1832718334 (2006).
22) Ryde U.,Dalton Trans., 6, 607625 (2006). 23) Lah N., Lah J., Zergers I., Wyns L., Messens
J., J. Biol. Chem., 278, 2467324679 (2003). 24) Kamachi T., Takahata M., Toraya T., Yoshizawa K., J. Phys. Chem. B, 113, 8435 8438 (2009).
25) Toraya T., Honda S., Mori K.,Biochemistry, 49, 72107217 (2010).
26) Kamachi T., Doitomi K., Takahata M., Toraya T., Yoshizawa K., Inorg. Chem., 50, 29442952 (2011).