博
士
学
位
論
文
p38 mitogen activated protein kinase 阻害剤の
合成研究
目 次 序論 1 第1 章 関節リウマチ 1 第2 章 炎症性サイトカインと p38 MAP キナーゼ 3 第3 章 代表的な p38 MAP キナーゼ阻害剤 4 本論 6 第 1 章 イミダゾ[1,2-b]ピリダジン誘導体に関する合成研究 6 第 1 節 イミダゾ[1,2-b]ピリダジン誘導体リード化合物の創出 6 第1 項 研究方針と分子設計 6 第 2 項 合成 8 第 3 項 生物活性と考察 9 第2 節 p38 MAP キナーゼ阻害活性向上に向けた構造最適化 10 第1 項 研究方針と分子設計 10 第 2 項 合成 11 第3 項 生物活性と考察 14 第3 節 CYP3A4 阻害活性改善に向けた構造最適化 16 第1 項 研究方針と分子設計 16 第2 項 合成 18 第3 項 生物活性と考察 19 第 4 節 代謝安定性改善に向けた構造最適化 21 第1 項 研究方針と分子設計 21 第2 項 合成 23 第3 項 生物活性と考察 25
第5 節 イミダゾ[1,2-b]ピリダジン誘導体の in vivo 評価 28 第 6 節 本章まとめ 30 第2 章 イミダゾ[4,5-b]ピリジン-2-オン誘導体に関する合成研究 31 第1 節 イミダゾ[4,5-b]ピリジン-2-オン誘導体の創出 31 第1 項 研究方針と分子設計 31 第2 項 合成 33 第3 項 生物活性と考察 34 第2 節 イミダゾ[4,5-b]ピリジン-2-オン誘導体リード化合物の創出 36 第1 項 研究方針と分子設計 36 第2 項 合成 37 第3 項 生物活性と考察 39 第3 節 ヒト全血アッセイにおける活性向上に向けた構造変換 45 第1 項 研究方針と分子設計 45 第2 項 合成 47 第3 項 生物活性と考察 50 第4 節 p38 MAP キナーゼ阻害活性向上に向けた構造最適化 51 第1 項 研究方針と分子設計 51 第2 項 合成 52 第3 項 生物活性と考察 57 第5 節 イミダゾ[4,5-b]ピリジン-2-オン誘導体のプロファイルおよび in vivo 評価 61 第6 節 本章まとめ 66 第3 章 総括 67
実験の部 68 引用文献 115 謝辞
序論
第1 章 関節リウマチ
関節リウマチ(RA)は古くから知られている疾患であり、紀元前 1500 年に Ebers Papyruralies により類似した疾患が明示されているが、1676 年にオランダの Masters と Sydenham により最初 の症例報告がなされた疾患である。その後、1859 年に Garrod によりリウマチ熱や通風と区別す る形で、1957 年に Charles Shortin により変形性関節症、脊椎関節症、全身性エリテマトーデスお よび結晶誘発性疾患と明確に区別する形で定義され、現在のRA として明確化された1。 RA は、地域、民族により発症頻度に差が認められるが、世界中でその発症が確認されている疾 患であり、世界人口の約1%が罹患していることが報告されている(Table 1)2-8。また、女性が 男性よりも発症リスクが高いのは、女性が男性よりも強い免疫力を有するためと考えられている 9-11。成人期に発症する傾向があり、発症から10年以内に RA 患者の多くが就業困難となるこ とが知られており、社会的経済的な影響が大きな疾患である12。
Table 1. Distribution and incidence of RA
Sociodemographic epidemiology Trends References2-8
Women vs men 2:1 to 3:1 2 Caucasian from North America 100/100,000 3 Rural and urban Africans 20–90/100,000 4 Native Americans 500/100,000 3 Asians 20–45/100,000 5 Caucasian from Europe 5–89/100,000 6 Latin America 10–50/100,000 7 Middle east countries 10–50/100,000 8
関節リウマチ(RA)は、関節、結合組織、筋肉、腱および線維組織に影響を及ぼす慢性の全 身性疾患であり、炎症性自己免疫疾患として知られている。RA には関節炎前期、関節炎移行期、 関節炎臨床期の3 つの異なる進行段階があると考えられている。関節炎前期は、RA 発症前であ り、RA の病因を蓄積していく段階と考えられている。その病因としては、遺伝的要因13-15、環 境的および後天的要因16-17が挙げられる。これらの要因が飽和状態になり、体内で耐性が失われ ると、関節炎移行期を経て、RA の臨床期に移行する。移行期は、軽度の外傷、感染、ホルモン、 心理的要因によって引き起こされる可能性がある。 RA の臨床期は、獲得免疫系および自然免疫系の活性化から始まり、白血球の滑膜組織への浸 潤により滑膜炎を引き起こされ、炎症性滑膜が構築される18-19。滑膜炎の原因の1 つとして、 腫瘍壊死因子-α(TNF-α)やインターロイキン(IL-1、IL-6、IL-8 など)などのサイトカイ
ンを放出するマクロファージが重要と考えられている20。 これらのサイトカインに対する選択的生物学的薬剤21,22として、TNF-α モノクローナル抗体 (インフリキシマブ23-25、アダリムムブ26,27)およびTNF-α 受容体融合タンパク質(エタネ ルセプト28)がRA 治療薬として使用され、有効性を示している。しかし、これら生物学的 薬剤は、自己抗体ができることにより治療効果が低減する懸念があること、注射剤である ためその使用に制限があること、などの欠点が存在する。従って、低分子 TNF-α 阻害剤を 開発できれば、生物学的薬剤に代わる治療オプションを提供することができる。
第2 章 炎症性サイトカインと p38 MAP キナーゼ
p38 mitogen activated protein(MAP)キナーゼは、セリン/スレオニンプロテインキナーゼファ ミリーのメンバーであり、内皮細胞、免疫細胞、および炎症細胞で広く発現しているリン酸化酵 素である。また、TNF-α、IL-1、IL-6、IL-8 などの炎症性サイトカインの産生および活性化の調 節に重要であり29-34、炎症反応刺激や熱などの細胞外ストレスによって活性化されることが知ら れている35(Figure 1)。実際に RA 患者から採取された滑膜組織においても、p38 MAP キナーゼ が過剰に発現していることが報告されたことから、RA 病態への関与が示唆されている36-42。こ れらのことから、TNF-α などのサイトカインカスケードの重要な要素である p38 MAP キナーゼ は、炎症組織において、炎症性サイトカインを増幅することにより、RA 病態に深く関与してい ることが示唆される。従って、その阻害剤は炎症性サイトカインの働きを抑制することが可能と 考えられ、有望なRA 治療薬として期待される43,44。
第3 章 代表的な p38 MAP キナーゼ阻害剤
代表的なp38 MAP キナーゼ阻害剤を Figure 2 および 3 に示した。これらは全て臨床試験に進 んだ化合物である45-50。Figure 1 に示した BIRB-79651,52、Scio-46951,53、BMS-58294951,54、VX-70251,55,56、
Ro-440225751,57、および TAK-71558-61は、臨床試験を中断した化合物であるが、その理由は、薬
効不足や毒性など化合物により様々であった。現在もFigure 2 に示した VX-74551,62、BCT-197、
LY-2228820、GW85655351,63,64、PH-79780465,66、AZD-7624、CHF-6297、FX-005、および ARRY-797
が臨床試験第II 相において、その有効性が検証されている67。従って、p38 MAP キナーゼ阻害
により生じる避けられない毒性が存在するわけではないと考えられ、p38 MAP キナーゼ阻害剤 として、新規な化学構造を有する化合物の開発が望まれる68-72。
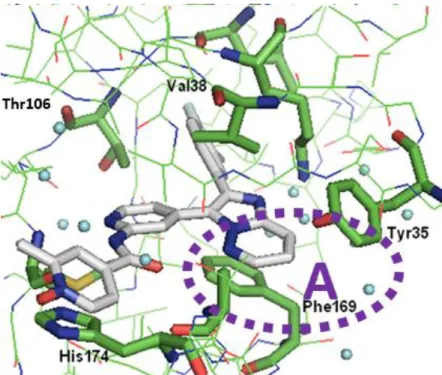
本論 第1 章 イミダゾ[1,2-b]ピリダジン誘導体に関する合成研究 第1 節 イミダゾ[1,2-b]ピリダジン誘導体リード化合物の創出 第1 項 研究方針と分子設計 武田薬品で開発された化合物1 は、臨床試験第 2 相試験において、明確な有効性を示すことが できず、薬剤としての開発を中断した60。化合物1 の p38 MAP キナーゼ阻害活性は IC 50 = 240 nM であり、筆者は、その酵素阻害活性を向上することにより、薬効増強が期待でき、有効性を示す ことが可能となると考えた。化合物1 と p38 MAP キナーゼの X 線共結晶構造解析(Figure 4)の 結果60、化合物1 は、p38 MAP キナーゼの ATP 結合サイトに結合し、複数の水素結合を形成し ていることが確認された。すなわち、化合物1 のアミノピリジン部位のピリジン窒素原子、およ びアミン水素原子は、p38 MAP キナーゼのヒンジ領域の Met109 主鎖の NH 水素原子およびカル ボニル酸素原子と水素結合を形成している。また、化合物1 のアミド酸素原子は p38 MAP キナ ーゼのAla172 主鎖の NH 水素原子と、チアゾール窒素原子は Lys53 側鎖のε-NH2の水素原子と 水素結合を形成している。一方で、リガンド近傍に存在するPhe169 の側鎖が 2 つのコンフォメ ーションA および B をとることが確認された。Phe169 の側鎖ベンゼン環と化合物 1 の距離から、 コンフォメーションA の場合、Phe169 の側鎖ベンゼン環は化合物 1 のエチルチアゾールとの疎 水性相互作用を形成しており、コンフォメーションB の場合、Phe169 の側鎖ベンゼン環は化合 物1 との相互作用を形成していないと解析された。したがって、2 つのコンフォメーション A お よびB が観察されたのは、化合物 1 と Phe169 側鎖ベンゼン環の疎水性相互作用が弱いためと考 えられる。この解析に基づき、2 つのコンフォメーションを、コンフォメーション A に収束させ ることにより、すなわち、Phe169 側鎖ベンゼン環との疎水性相互作用を増強することにより、 p38 MAP キナーゼ阻害活性を向上できると考えた。 上記の仮説に基づいて、p38 MAP キナーゼとの水素結合に重要な原子を保持する形で、化合 物1 のエチルチアゾール部位を縮合環へ変換した化合物をデザインした(Figure 5)。この化合物 設計により、活性発現に重要な水素結合に加え、Phe169 側鎖ベンゼン環とのπ-πスタッキング による疎水性相互作用が増強され、p38 MAP キナーゼ阻害活性が向上すると期待した。この化 合物設計に基づいて、エチルチアゾール部位を縮合環に変換した複数の化合物を合成した。これ ら化合物のp38 MAP キナーゼ阻害活性を評価したところ、イミダゾ[1,2-b]ピリダジン誘導体が 最も強力なp38 MAP キナーゼ阻害活性を示した。
(A) (B)
Figure 4. (A) Structure of inhibitor 1. The atoms involved in the hydrogen-bond interactions with p38
MAP kinase are shown in blue. (B) X-ray co-crystal structure of 1 bound with p38 MAP kinase (PDB code 6ANL). The blue dashed lines show hydrogen-bond interactions. The two conformation (A and B) of side chain of Phe169 is observed.
Figure 5. Design concept of p38 MAP kinase inhibitors. The atoms involving hydrogen-bond interactions
第2 項 合成 イミダゾ[1,2-b]ピリダジン誘導体 8a は、Scheme 1 に示した方法を用いて合成した。化合物 2a をブチルリチウムと処理することによりリチウム塩を反応系中で調製し、プロピレンイミン 3a と反応し、対応するケトン4a を収率 81%で合成した58,59。酢酸溶媒中4a の臭素化を行い、α-ブ ロモケトン5a へと誘導した。次に、3-アミノピリダジンとの反応を試みたが、目的とするイミ ダゾ[1,2-b]ピリダジン誘導体は得られず、3-アミノピリダジンの 1 位窒素がアルキル化された副 生成物を得た。これは、3-アミノピリダジンの 1 位窒素は 2 位窒素よりも相対的に求核反応性が 高いことが原因と考えられる。そこで、1 位窒素の求核性を低減した 3-アミノ-6-クロロピリダジ ンを用い、α-ブロモケトン 5a との反応を試みたところ、イミダゾ[1,2-b]ピリダジン環構築に成 功し、イミダゾ[1,2-b]ピリダジン誘導体 6a を収率 60%で得た。接触還元条件下脱塩素化を行い、 7a へと誘導し、ベンゾイルクロリドと反応後、アンモニアで処理することにより、目的とする 8a を収率 72%で合成した。
Scheme 1. Synthesis of compounds 8aa
aReagents and conditions: (a) i) nBuLi, THF, −78ºC then at 0ºC; ii) 3a, −78ºC, 81%; (b) Br
2, AcOH,
80ºC, 78%; (c) 3-aminopyridazine, EtOH, 80ºC; (d) 3-amino-6-chloropyridazine, EtOH, 80ºC, 60%; (e) H2 (0.5 MPa), Pd/C, AcOH, 50ºC, 54%; (f) i) benzoylchloride, Et3N, DCM, rt; ii) NH3/EtOH, rt, 72%.
第3 項 生物活性と考察 合成した化合物の生物学的評価は、p38 MAP キナーゼによる ATF2 リン酸化反応に対する阻害 活性、およびヒト単球THP-1 細胞における LPS 誘発 TNF-α産生に対する抑制活性を指標とした。 Table 2 に、本節第 1 項の化合物設計に基づいて合成したイミダゾ[1,2-b]ピリダジン誘導体 8a の 生物学的評価結果を示した。化合物8a は期待した通り p38 MAP キナーゼ阻害活性向上を達成し、 IC50 = 73 nM の強力な活性を示した。この阻害活性向上は、化合物 8a のイミダゾ[1,2-b]ピリダジ ン環とPhe169 側鎖ベンゼン環が効果的にπ-πスタッキングを形成し、脂溶性相互作用を増強し た結果であると考えられる。また、THP-1 細胞における LPS 誘発 TNF-α産生抑制活性は、化合 物1 と比較して 4 倍の活性向上を示した。p38 MAP キナーゼ阻害活性向上に伴い、THP-1 細胞 におけるTNF-α産生抑制活性も同様に向上したことから、細胞内を標的とする薬剤として必要 とされる細胞膜透過性については問題がないと考えた。 本節で記述した通り、化合物1 を基にリード化合物 8a を創出した。すなわち、化合物 1 の疎 水性相互作用増強を目的とした化合物デザインに基づき、イミダゾ[1,2-b]ピリダジン誘導体 8a を、合成、評価した結果、化合物1 と比較して、強力な p38 MAP キナーゼ阻害活性および THP-1 細胞におけるTNF-α産生抑制活性を示した。従って筆者は、更なる最適化研究のための有望な リード化合物として、イミダゾ[1,2-b]ピリダジン誘導体 8a を選定した。
Table 2. Inhibitory activities of imidazo[1,2-b]pyridazine derivative 8a against p38 MAP kinase and
production of TNF-α in human THP-1 cells.
Cmpd. p38 MAPK IC 50 (nM) a Production of TNF-α IC50 (nM) a 1 240 (170–350) 240 (110–540) 8a 73 (36–150) 58 (35–94) a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
95% confidence intervals.
第2 節 p38 MAP キナーゼ阻害活性向上に向けた構造最適化 第1 項 研究方針と分子設計 前節で記述した通り、強力なp38 MAP キナーゼ阻害活性および THP-1 細胞における TNF-α 産生抑制活性を示した有望なリード化合物として、イミダゾ[1,2-b]ピリダジン誘導体 8a を見出 した。続いて、イミダゾ[1,2-b]ピリダジン誘導体の更なる p38 MAP キナーゼ阻害活性向上を目 的として、構造活性相関情報の集積、解析を行い、構造最適化を行った。化合物8a は化合物 1 を基に設計された化合物であり、化合物1 と p38 MAP キナーゼの X 線共結晶構造解析情報(Figure 3)が活用可能と考えた。すなわち、Figure 6 に示した通り、化合物 8a が、化合物 1 と同様の結 合様式でp38 MAP キナーゼと結合していると仮定し、イミダゾ[1,2-b]ピリダジン誘導体の構造 活性相関情報の集積を行った。イミダゾ[1,2-b]ピリダジン環 2 位ベンゼン環上置換基は、Thr106 およびLeu104 などの疎水性アミノ酸残基により形成される空間的に小さな疎水性ポケットを占 有しているため、ベンゼン環上置換基は立体的に小さな置換基の探索を実施した。また、ピリジ ン環上アミド側鎖は溶媒領域に向くため、広範な変換が可能であると考え、アミド側鎖探索を実 施した。 上記の仮説に基づいて合成展開を行うことにより、化合物の変換可能な部位を限定することが でき、構造最適化を効率的に実施することが可能となり、p38 MAP キナーゼ阻害活性が向上す ると期待した。
第2 項 合成 イミダゾ[1,2-b]ピリダジン誘導体 8b–n は、Scheme 2 に示した方法を用いて合成した。化合物 2a–b のリチウム塩と、各種プロピレンイミン 3a–d あるいはワインレブアミド 3e を反応し、対 応するケトン 4b–f を合成した。酢酸溶媒中 4b-f の臭素化を行い、α-ブロモケトン 5b-f へと誘 導し、3-アミノ-6-クロロピリダジンと反応し、イミダゾ[1,2-b]ピリダジン誘導体 6b-f を得た。第 1 節で示したイミダゾ[1,2-b]ピリダジン誘導体 6a の酢酸溶媒中における接触還元条件下脱塩素 化反応は、用いる基質により、未反応の基質が残り、反応が完結しないことが確認された。これ は、不均一系での反応のため、撹拌効率など様々な原因が考えられるが、脱塩素化反応において 生じた塩化水素が未反応の基質と塩酸塩を形成し、反応系外へ析出する可能性も原因となる可能 性が考えられた。そこで、反応系外への析出を抑えるため、酢酸溶媒をDMF 溶媒に変更し、さ らにトリエチルアミンを添加することで反応が完結することが確認された。この条件を用い6b-f の接触還元条件下脱塩素化反応を行ったところ、収率が 44%と中程度から 95%までの高収率で アミン 7b-f を得た。なお、各基質に対する条件の最適化は実施しなかった。次に、合成したア ミン 7b-f を各種カルボン酸クロリドと反応後、アンモニアで処理することにより、目的とする 8b-n を合成した。 イミダゾ[1,2-b]ピリダジン誘導体 10a は、Scheme 3 に示した方法を用いて合成した。すなわち、 化合物7d を 2-クロロピリジン-3-カルボン酸クロリドと反応後、アンモニアで処理することによ り、収率92%で化合物 9a を得、接触還元条件下脱塩素化を行い、目的とする化合物 10a を収率 52%で合成した。
Scheme 2. Synthesis of compounds 8b–na
aReagents and conditions: (a) i) nBuLi, THF, −78ºC then at 0ºC; ii) 3a–e, −78ºC for 4b–f, 80–85% for
4c–e; (b) Br2, AcOH, 80ºC for 5b–e, 87–90% for 5c–e; (c) Br2, AcONa, AcOH, rt for 5f; (d)
3-amino-6-chloropyridazine, EtOH, 80ºC, 50–71% for 6c–e, 22–43% for 3 steps for 6b and 6f; (e) H2 (0.1
MPa), Pd/C, Et3N, DMF, rt, 44–95% for 7b–f; (f) i) acyl chloride derivative, Et3N, DCM, rt; ii)
NH3/EtOH, rt, 45–83% for 8b–j, 8m; (g) i) acyl chloride derivative, pyridine, DMA, rt; ii) NH3/EtOH,
THF, rt, 69–86% for 8k–l; (h) i) acyl chloride derivative, Et3N, THF, rt; ii) NH3/EtOH, THF, rt, 54% for 8n.
Scheme 3. Synthesis of compounds 10aa
aReagents and conditions: (a) i) 2-chloropyridine-3-carbonyl chloride, Et3N, DCM, rt; ii) NH3/EtOH, rt,
第3 項 生物活性と考察 合成した化合物の生物学的評価は、本章第1 節に記載した方法と同様にして、p38 MAP キ ナーゼによる ATF2 リン酸化反応に対する阻害活性、およびヒト単球 THP-1 細胞における LPS 誘発TNF-α産生に対する抑制活性を指標とした。Table 3 に、本節第 1 項の化合物設計に基づい て、合成したイミダゾ[1,2-b]ピリダジン誘導体 8b–n および 10a の生物学的評価結果を示した。 化合物8f は 8a と同等の p38 MAP キナーゼ阻害活性を示したが、TNF-α産生抑制活性が減弱し た。この結果から、酵素阻害活性と細胞における活性間の乖離が小さいピリジン誘導体に注力し た。3-メチル体 8a と比較して、3-クロロ体 8b は p38 MAP キナーゼ阻害活性が 2 倍向上したが、 4-クロロ体 8c は活性が大幅に減弱した58,59。また、4 位へフッ素を導入した化合物 8d は劇的に 活性を向上した。また、化合物8e と 8g の比較から、ベンゼン環上置換基 R2は8e が最適である ことを見出した。これらの結果から、イミダゾ[1,2-b]ピリダジン環 2 位のベンゼン環上の 3 位置 換基は、強力なp38 MAP キナーゼ阻害活性に必須であり、メチル基あるいはクロロ基が許容さ れることが分かった。また、4 位にフッ素を導入する場合は、3 位はメチル基が最適であること が分かった。 続いて、ピリジン2 位アミド側鎖の置換基効果について構造活性相関情報を取得した。ベンゾ イル部位に、電子供与基としてメチル基を導入したところ、化合物8d と比較して、オルト位(8h) は活性が減弱したが、メタ位(8i)、パラ位(8j)は活性を保持した。次にメタ位に電子吸引基 として、フルオロ基(8k)、クロロ基(8l)を導入したところ、ともにメチル体(8i)と同等の p38 MAP キナーゼ阻害活性を示した。次に、化合物の脂溶性を調節するためにフェニル基をピ リジル基へ変換したところ、2-ピリジル体 8m では活性が減弱したが、3-ピリジル体 10a および 4-ピリジル体 8n ではフェニル体 8d よりも強力な p38 MAP キナーゼ阻害活性を示した。本節第 1 項で記述した通り、アミド側鎖は溶媒側に向いているため、極性基の導入が p38 MAP キナー ゼ阻害活性向上に寄与したと考えられた。さらに、3-ピリジル体 10a および 4-ピリジル体 8n は、 THP-1 細胞における LPS 誘発 TNF-α産生を、フェニル体 8d よりも強力に抑制することが確認 された。 本節で記述した通り、化合物1 の X 線共結晶構造情報を基にした、化合物 8a の推定結合様式 に基づき構造活性相関情報を集積、解析しながら、緻密な構造最適化を実施し、強力なp38 MAP キナーゼ阻害活性およびTHP-1 細胞における TNF-α産生抑制活性を示した化合物 10a および 8n を見出した。
Table 3. Inhibitory activities of imidazo[1,2-b]pyridazine derivative 8a–n against p38 MAP kinase and
production of TNF-α in human THP-1 cells.
Cmpd. X R1 R2 p38 MAPK IC50 (nM) a Production of TNF-α IC50 (nM) a 8a CH Ph 3-CH3 73 (36–150) 58 (35–94) 8b CH Ph 3-Cl 34 (28–41) 88 (40–190) 8c CH Ph 4-Cl 290 (200–440) 130 (69–250) 8d CH Ph 3-CH3, 4-F 9.4 (8.4–10) 52 (34–81) 8e CH 3-CF3-Ph 3-CH3, 4-F 11 (10–13) 13 (8.5–19) 8f N Ph 3-CH3 56 (50–63) 260 (140–510) 8g CH 3-CF3-Ph 3-Cl, 4-F 210 (160–280) >10000 8h CH 2-CH3-Ph 3-CH3, 4-F 84 (74–95) 1300 (610–2800) 8i CH 3-CH3-Ph 3-CH3, 4-F 11 (9.0–13) 19 (13–29) 8j CH 4-CH3-Ph 3-CH3, 4-F 23 (20–27) 1000 (110–9900) 8k CH 3-F-Ph 3-CH3, 4-F 12 (10–13) 18 (14–24) 8l CH 3-Cl-Ph 3-CH3, 4-F 14 (8.5–22) 14 (7.8–24) 8m CH 2-Py 3-CH3, 4-F 23 (19–28) 360 (220–610) 8n CH 4-Py 3-CH3, 4-F 5.4 (3.1–9.3) 16 (9.0–29) 10a CH 3-Py 3-CH3, 4-F 2.2 (0.58–8.0) 3.5 (2.2–5.5) a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
第3 節 CYP3A4 阻害活性改善に向けた構造最適化 第1 項 研究方針と分子設計 前節で記述した通り、p38 MAP キナーゼ阻害活性の向上が達成されたイミダゾ[1,2-b]ピリダジ ン誘導体10a および 8n が見出され、これら化合物は THP-1 細胞における TNF-α産生に対して も非常に強力な抑制活性を示した。イミダゾ[1,2-b]ピリダジン誘導体が in vitro 試験において、 優れたサイトカイン産生抑制作用を示すことができたため、次に、薬剤として必須の精査試験の 1 つである cytochrome P450 3A4(CYP3A4)阻害活性を評価した。その結果、Table 4 に示した通 り、イミダゾ[1,2-b]ピリダジン誘導体 10a および 8n がともに CYP3A4 阻害活性を有することが 明らかとなった。CYP3A4 は主要な薬物代謝酵素の 1 つであり、疎水性の高い薬物を水酸化する ことが知られている。CYP3A4 は、様々な薬物の代謝反応に関与しているため、関節リウマチの ような長期にわたり投薬が必要とされる慢性疾患の治療薬としては、薬物間相互作用の観点から、 回避すべき作用である。そこで、化合物10a および 8n の CYP3A4 阻害活性改善に取り組んだ。 CYP3A4 は活性中心にヘムを有するタンパク質であり、ヘムに結合可能な 3-ピリジル基あるい は4-ピリジル基を有する化合物は CYP3A4 の代謝活性を阻害し得ることが知られている58,73,74。 そこで、化合物10a および 8n の CYP3A4 阻害作用も、ピリジル基のヘムへの結合により発現し ていると考えられる。従って、ピリジル基のヘムへの結合を弱めることにより、CYP 阻害活性 を改善できると考えた。前節の構造活性相関情報から、アミド側鎖ベンゼン環にはメタ位および パラ位への置換基導入が可能である。従って、Figure 7 に示したように、ピリジン窒素隣接位へ 置換基を導入すれば、p38 MAP キナーゼ阻害活性を保持しながら、CYP3A4 阻害活性を改善で きると考えた。 上記の仮説に基づいて、CYP 阻害活性改善を目的として、ピリジル基のヘムへの結合を弱め る化合物をデザインした。すなわち、ピリジル基窒素の隣接位に置換基を導入することにより、 CYP 阻害活性が軽減すると期待した。
Table 4. CYP3A4 inhibition profiles of imidazo[1,2-b]pyridazine derivatives 10a and 8n. Cmpd. p38 MAPK IC 50 (nM) a Production of TNF-α IC50 (nM) a CYP3A4 inhibition (% inhibition) b 8n 5.4 (3.1–9.3) 16 (9.0–29) 85 10a 2.2 (0.58–8.0) 3.5 (2.2–5.5) 61 a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
95% confidence intervals. b Compound concentration is 10 μM.
第2 項 合成 イミダゾ[1,2-b]ピリダジン誘導体 8o は、Scheme 4 に示した方法を用いて合成した。すなわち、 6-メチルピリジン-3-カルボン酸と塩化オキサリルの反応により調製した 6-メチルピリジン-3-カ ルボン酸クロリドと化合物7d を反応後、アンモニアで処理することにより、目的とする化合物 8o を低収率 12%ながら得ることに成功した。 イミダゾ[1,2-b]ピリダジン誘導体 10b は、Scheme 5 に示した方法を用いて合成した。すなわち、 2-クロロ-6-メチルピリジン-4-カルボン酸とオキシ塩化リンの反応により調製した 2-クロロ-6-メ チルピリジン-4-カルボン酸クロリドと化合物 7d を反応後、アンモニアで処理することにより、 化合物9b を得、接触還元条件下脱塩素化を行い、目的とする化合物 10b を合成した。
Scheme 4. Synthesis of compounds 8oa
aReagents and conditions: (a) i) 6-methylpyridine-3-carboxylic acid, (COCl)
2, Et3N, DMF, THF, rt; ii)
NH3/EtOH, THF, rt, 12%.
Scheme 5. Synthesis of compounds 10ba
aReagents and conditions: (a) i) 2-chloro-6-methylpyridine-4-carboxylic acid, POCl
3, 50ºC; ii) Et3N,
第3 項 生物活性と考察 合成した化合物の生物学的評価は、本章第1 節に記載した方法と同様にして、p38 MAP キナ ーゼ阻害活性、およびTHP-1 細胞における LPS 誘発 TNF-α産生に対する抑制活性を指標とした。 また合成した化合物のCYP3A4 阻害活性は、以下の方法で評価した。CYP3A4 発現ミクロソー ムを用い、テストステロンの代謝反応により生じる6β-ヒドロキシテストステロン量を測定し、 代謝反応の阻害率として算出することで、CYP3A4 阻害活性の指標とした。Table 5 に、本節第 1 項の化合物設計に基づいて合成したイミダゾ[1,2-b]ピリダジン誘導体 8o および 10b の生物学的 評価結果を示した。イミダゾ[1,2-b]ピリダジン誘導体 8o および 10b ともにメチル基の導入によ りp38 MAP キナーゼ阻害活性およびTNF-α産生抑制活性を保持することを示した。この結果は、 第2 節で記述したベンゼン環上へのメチル基導入による構造活性相関情報と一致し、ピリジン環 上置換基に対しても適用であることが示された。この化合物設計のCYPA4 阻害活性に対する効 果に関しては、興味深い結果を得た。すなわち、3-ピリジル基のヘムへの結合を減弱する目的で、 3-ピリジル環上 6 位へメチル基を導入した化合物 8o は、無置換 3-ピリジル体 10a と比較して、 CYP3A4 阻害活性が強くなることが明らかとなった。一方で、4-ピリジル基のヘムへの結合を減 弱する目的で、4-ピリジル環上 2 位へメチル基を導入した化合物 10b は、期待したとおり、無置 換4-ピリジル体 8n と比較して、CYP3A4 阻害活性を大幅に減弱することが確認できた。この結 果から、3-ピリジル体 10a と 4-ピリジル体 8n に対する CYP3A4 の阻害作用様式が異なると考え られる。すなわち、3-ピリジル体 10a は、ピリジン窒素とヘムの結合が主要な阻害様式ではなく、 4-ピリジル体 8n はピリジン窒素とヘムの結合が主要な阻害様式であると考えられる。また、ピ リジン窒素隣接位への置換基導入が、CYP 阻害回避の 1 つの手法となることを示し、化合物の CYP 阻害様式の違いにより、置換基導入の影響が異なることを明らかとした。 本節で記述した通り、ピリジン窒素隣接位への置換基導入により、強力なp38 MAP キナーゼ 阻害活性およびTHP-1 細胞における TNF-α産生抑制活性を保持しながら CYP3A4 阻害活性を改 善した化合物10b を見出すことに成功した。
Table 5. CYP3A4 inhibition profiles of imidazo[1,2-b]pyridazine derivatives 8o and 10b. Cmpd. R1 p38 MAPK IC50 (nM) a Production of TNF-α IC50 (nM) a CYP3A4 inhibition (% inhibition) b 8n 4-Py 5.4 (3.1–9.3) 16 (9.0–29) 85 10a 3-Py 2.2 (0.58–8.0) 3.5 (2.2–5.5) 61 8o 6-Me-3-Py 1.8 (0.63–5.1) 1.2 (0.93–1.6) 86 10b 2-Me-4-Py 4.4 (1.8–11) 15 (9.0–26) 42 a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
第4 節 代謝安定性改善に向けた構造最適化 第1 項 研究方針と分子設計 前節で記述した化合物10b は、強力な p38 MAP キナーゼ阻害活性および THP-1 細胞における TNF-α産生抑制活性を保持しながら CYP3A4 阻害活性を改善することを示した。続いて、経口 投与で使用可能な薬剤であるかどうかを評価する目的で、まずin vitro でのヒト肝ミクロソーム を用いた代謝安定性を評価した。その結果、イミダゾ[1,2-b]ピリダジン誘導体 10b が、短時間で 消失することが明らかとなった。この肝ミクロソームにおける代謝安定性試験は、酸化代謝を評 価する評価系であるため、化合物10b は酸化代謝を受け、消失していると考えられる。そこで、 化合物10b の酸化代謝に対する代謝安定性を改善する構造最適化に取り組んだ。 化合物10b が酸化的に代謝される部位を推定する目的で、化学構造が類似している化合物の 代謝安定性を評価した。Table 6 に示した通り、3-ピリジル体 10a と 4-ピリジル体 8n を比較して、 4-ピリジル体 8n が代謝的に不安定であることが確認された。また、6-メチル-3-ピリジル体 8o は、無置換3-ピリジル体 10a と比較して、代謝安定性に影響が認められなかった。一方で、2-メチル-4-ピリジル体 10b は、無置換 4-ピリジル体 8n と比較して、代謝安定性が改善する傾向を 示した。これらの解析から、筆者は、3-ピリジル誘導体および 4-ピリジル誘導体は異なるメカニ ズムで代謝を受けていると考え、4-ピリジル体 8n において確認されたメチル基導入による代謝 安定性の改善傾向に注目した。すなわち、2-メチル-4-ピリジル体 10b の代謝部位がピリジン窒 素であり、酸化代謝産物がピリジン-N-オキシド体であると推定した(Figure 8)。イミダゾ[1,2-b] ピリダジン誘導体の推定結合様式から、この部位は溶媒領域を向くため、置換基導入はp38 MAP キナーゼ阻害活性に対して許容されると考えられる。また、第2 節で記述した構造活性相関情報 に基づくと、この部位への極性基導入はp38 MAP キナーゼ阻害活性が向上する可能性があるこ とも考えられる。従って、ピリジン-N-オキシド体は、その代謝物自体が p38 MAP キナーゼ阻害 活性を有する活性代謝物である可能性が高いと考えられる。そこで、酸化代謝に対する代謝安定 性を改善するため、肝ミクロソームを用いた代謝物検索と検出された代謝物の同定を行うことを 考えた。 上記の仮説に基づいて、代謝安定性改善を目的として、ピリジン-N-オキシド体を合成するこ とにより、p38 MAP キナーゼ阻害活性を保持しながら、代謝安定性を改善できると考えた。
Table 6. Stability in human liver microsomes of imidazo[1,2-b]pyridazine derivatives 10a–b and 8n–o. Cmpd. R1 p38 MAPK IC50 (nM) a Production of TNF-α IC50 (nM) a Stability in human liver microsomes (μg/min/mg) b 8n 4-Py 5.4 (3.1–9.3) 16 (9.0–29) 177 8o 6-Me-3-Py 1.8 (0.63–5.1) 1.2 (0.93–1.6) 104 10a 3-Py 2.2 (0.58–8.0) 3.5 (2.2–5.5) 105 10b 2-Me-4-Py 4.4 (1.8–11) 15 (9.0–26) 132 a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
95% confidence intervals. b Compound concentration is 10 μM.
第2 項 合成 イミダゾ[1,2-b]ピリダジン誘導体 15a およびその硫酸塩 16 は、Scheme 6 に示した方法を用い て合成した。すなわち、化合物11 を接触還元条件下脱塩素化後、酢酸溶媒中過酸化水素-尿素複 合体によりピリジン-N-オキシド 13a へと変換し、エステル基の加水分解を行い、カルボン酸 14a を得た。化合物7d と縮合し、目的とする化合物 15a を合成した。アニリン 7d とカルボン酸 14a のアミド化反応の縮合剤として HATU を使用することで、高収率 94%で目的物を得ることがで きた。続いて、化合物15a の酢酸溶液に当量の濃硫酸と適切な量のエタノールを加え加熱するこ とにより硫酸塩16 を 87%の高収率で調製した。 イミダゾ[1,2-b]ピリダジン誘導体 15b は、Scheme 7 に示した方法を用いて合成した。すなわち、 化合物 12b を、酢酸溶媒中過酸化水素-尿素複合体によりピリジン-N-オキシド 13b へと変換後、 エステル基の加水分解を行い、カルボン酸14b を得た。化合物 7d と縮合し、目的とする化合物 15b を合成した。
Scheme 6. Synthesis of compounds 15a and 16a
aReagents and conditions: (a) H2 (0.1 MPa), Pd/C, Et3N, DMF, rt, 61%; (b) H2O2 urea complex, AcOH,
rt; (c) 8 N NaOH (aq.), THF, MeOH, 70°C, 43% for 2steps; (d) 7d, HATU, iPr2EtN, pyridine, 70ºC, 94%;
Scheme 7. Synthesis of compounds 15ba
aReagents and conditions: (a) H
2O2 urea complex, AcOH, rt; (b) 8 N NaOH (aq.), THF, MeOH, 70°C;
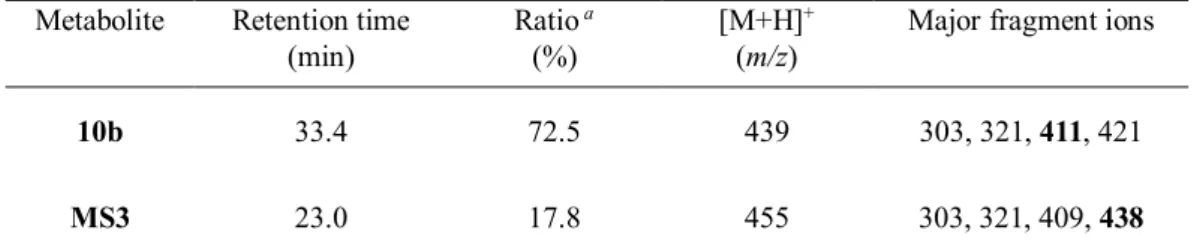
第3 項 生物活性と考察 本節第1 項で記述した通り、2-メチル-4-ピリジル体 10b の代謝物を同定するため、まず肝ミ クロソームを用いた代謝物検索を実施した。代謝物検索に関しては、in vitro で化合物を肝ミク ロソームと反応させる以下の方法で行った。化合物と肝ミクロソームの共存下、反応開始剤とし てのNADPH 再生試薬を添加後、30 分間インキュベーションした。反応後得られた上清を分析 することにより、代謝物の構造推定を実施した。Table 7 に示した通り、原体である化合物 10b が33.4 分に 72.5%の割合で、主代謝物 MS3 が 23.0 分に 17.8%の割合で検出された。化合物 10b は、m/z 439 でプロトン化分子[M+H]+を生成し、主代謝物MS3 は、m/z 455 でプロトン化分子 [M+H]+を生成したことから、主代謝物MS3 は化合物 10b から 16u だけ増加したことが確認され た。また、化合物10b および主代謝物 MS3 の LC/UV/MS 分析に基づく主要なフラグメントイオ ンを解析した結果、主代謝物MS3 には 438、303、321、409 が含まれており、特に m/z 438 はヒ ドロキシラジカルの開裂により生じたものと推定できる。また、化合物10b のフラグメントイ オンと類似していることから、アミド側鎖部位が酸化されたことが示唆された。この解析の結果、 主代謝物MS3 の化学構造は、期待した通り、ピリジン-N-オキシド体であることが強く示唆され た。実際に合成されたピリジン-N-オキシド 15a は、HPLC 分析により MS3 と一致することが確認 され、代謝安定性を劇的に改善した(Table 8)。さらに、化合物 15a は、化合物 10a と同等の 強力なp38 MAP キナーゼ阻害活性および THP-1 細胞における TNF-α産生抑制活性を示した。 一方、6-メチル-3-ピリジル体 8o に対応するピリジン-N-オキシド体 15b は強力な p38 MAP キナ ーゼ阻害活性およびTHP-1 細胞における TNF-α産生抑制活性を示したが、代謝安定性の改善は 軽微であった。このことから、本節第1 項で記述した通り、3-ピリジル誘導体および 4-ピリジル 誘導体は、異なるメカニズムで酸化的に代謝を受けていることが示唆される。 本節で記述した通り、化合物10b の代謝安定性改善を目的として、代謝物検索と代謝物の同 定を行うことにより、強力なp38 MAP キナーゼ阻害活性および TNF-α産生抑制活性を保持しな がら、代謝安定性を劇的に改善したピリジン-N-オキシド体 15a を見出すことに成功した。
Table 7. HPLC and LC/MSn data of 10b and its metabolites after incubation with human liver
microsomes.
Metabolite Retention time (min)
Ratio a
(%)
[M+H]+
(m/z)
Major fragment ions
10b 33.4 72.5 439 303, 321, 411, 421
MS3 23.0 17.8 455 303, 321, 409, 438
Table 8. Stability in human liver microsomes of imidazo[1,2-b]pyridazine derivatives 15a.
Cmpd. R1 p38 MAPK
IC50 (nM) a
Production of TNF-α IC50 (nM) a
Stability in human liver microsomes (μL/min/mg) b 8n 4-Py 5.4 (3.1–9.3) 16 (9.0–29) 177 8o 6-Me-3-Py 1.8 (0.63–5.1) 1.2 (0.93–1.6) 104 10a 3-Py 2.2 (0.58–8.0) 3.5 (2.2–5.5) 105 10b 2-Me-4-Py 4.4 (1.8–11) 15 (9.0–26) 132
15a 2-Me-4-Py N-oxide 5.4 (4.6–6.4) 18 (10–33) 32
15b 6-Me-3-Py N-oxide 2.7 (2.4–3.0) 16 (8.9–28) 89
a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
95% confidence intervals. b Compound concentration is 10 μM.
次に、p38 MAP キナーゼ阻害剤としての新規な化学構造であるイミダゾ[1,2-b]ピリダジン誘導 体の結合様式を理解するために、化合物15a と p38 MAP キナーゼの X 線共結晶複合体構造を取 得した。得られたX 線共結晶構造を解析することにより、化合物 15a が単一の Phe169 コンフォ メーションを採用するp38 MAP キナーゼのヒンジ領域に結合することが明らかとなった(Figure 9)。この複合体では、p38 MAP キナーゼと化合物 1 の X 線共結晶構造で観察された水素結合が 期待した通り保存されていることが確認された。さらに、ピリジン-N-オキシドの溶媒領域に曝 露された酸素原子は、His174 の側鎖イミダゾール NH と水素結合を形成することが新たに確認 できた。4-フルオロ-3-メチルフェニル環は、Thr106 および Leu104 により形成される空間的に小 さい内部の疎水性ポケットを占有していることも確認できた。最後に、イミダゾ[1,2-b]ピリダジ ン環部位は、Tyr35、Val38、および Phe169 の側鎖と疎水性相互作用を形成し、期待した通り、 Phe169 との π-π 相互作用を形成している。エチルチアゾール部位のイミダゾ[1,2-b]ピリダジン環 への変換によって増強されたこの疎水性相互作用は、これまで本章を通して構造活性相関研究で 示されたように、p38 MAP キナーゼ阻害活性の向上に重要である。
p38 MAP キナーゼ阻害剤としての新規な化学構造であるイミダゾ[1,2-b]ピリダジン誘導体の キナーゼ阻害の選択性を確認するために、代表化合物として、化合物15a を用いて、p38 MAP
キナーゼ以外の42 種類のキナーゼに対する阻害活性を評価した。化合物 15a の p38 MAP キナー ゼ阻害活性と比較して、42 種類のキナーゼのうち 40 種類のキナーゼに対して 100 倍以上弱い阻 害活性を、Her4 および CK1δ に対して数倍弱い阻害活性を示した(data not shown)。この結果から、
化合物15a は、p38 MAP キナーゼを選択的に阻害することが確認された。
第5 節 イミダゾ[1,2-b]ピリダジン誘導体のプロファイルおよび in vivo 評価
各種in vitro 試験系における構造最適化により見出した化合物 15a の in vivo 薬効試験を実施し、
その経口薬としてのポテンシャルを薬効面から評価した。前節までに記述した通り、化合物15a
はヒト代謝安定性が改善された。この過程において、ラット代謝安定性についても同様に改善さ れ、16 μL/min/mg であることが分かった(Table 9)。そこで、in vivo 薬効評価に先立って、化合
物15a のラットにおける経口吸収性に関する評価を実施した。Table 10 に化合物 15a およびその
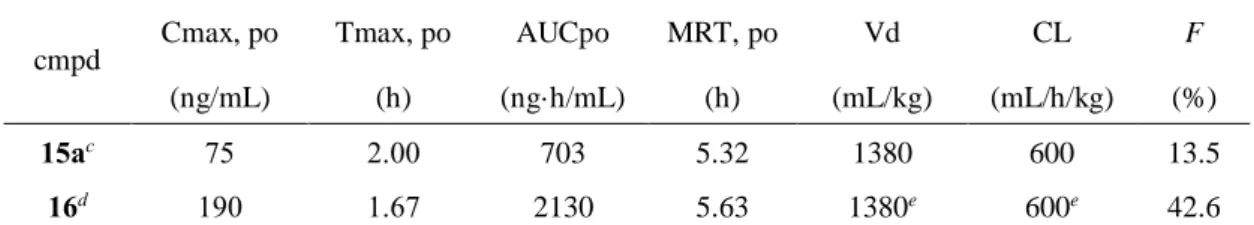
硫酸塩16 の薬物動態特性を示した。期待した通り、化合物 15a は、良好な経口吸収性と高い血
中濃度を示した(F = 13.5%、AUC = 703 ng·h/mL)。また硫酸塩 16 は、化合物 15a を上回る良好
な経口吸収性と高い血中濃度を示した(F = 42.6%、AUC = 2130 ng·h/mL)。上記の結果から、よ
り高い血中濃度を示した硫酸塩16 を in vivo 薬効評価に用いる化合物として選択した。
Table 9. Profile summary of 15a.
Cmpd. p38 MAPK IC 50 (nM) a Production of TNF-α IC50 (nM) a Solubility in pH = 6.8 (μg/mL) 15a 5.4 (4.6–6.4) 18 (10–33) 0.91 Cmpd. CYP3A4 inhibition (% inhibition) b
Stability in human liver microsomes (μL/min/mg) b
Stability in rat liver microsomes (μL/min/mg) b
15a 38 32 16
a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
95% confidence intervals. b Compound concentration is 10 μM.
Table 10. Pharmacokinetic properties of 15a and 16a in ratsb.
cmpd Cmax, po Tmax, po AUCpo MRT, po Vd CL F (ng/mL) (h) (ng·h/mL) (h) (mL/kg) (mL/h/kg) (%)
15ac 75 2.00 703 5.32 1380 600 13.5
16d 190 1.67 2130 5.63 1380e 600e 42.6 a H
2SO4 salt of 15a. b Mean values of measurements conducted in three animals. c i.v. 0.2 mg/kg, p.o. 3.0
mg/kg in 0.5% methyl cellulose suspension. d p.o. 3.0 mg/kg in corn oil suspension. e Because the main
化合物16 の in vivo 薬効評価は、ラットのコラーゲン誘発関節炎モデルを用いて評価された (Figure 10)。このモデルラットは、以下の方法で構築された。すなわち、雌ルイスラットの背 中に、ウシ由来Ⅱ型コラーゲンを経皮投与後、その7 日後にウシ由来Ⅱ型コラーゲンを尾部より 注射することで炎症を惹起する。時間経過に伴い、後肢関節腫脹の増大が観察される。この後肢 関節腫脹の増大を、その関節体積を測定することにより評価し、抗炎症作用の指標とした。関節 炎を発症したラットに対し、化合物16 を 1 日 2 回経口投与で、5 用量(0.1、0.3、1、3、および 10 mg/kg)が 11 日間投与された。その結果、化合物 16 は、1 mg/kg から用量依存的に有意に後 肢関節腫脹の抑制作用が認められた。特に、10 mg/kg の用量においては、顕著な抑制作用を達 成した。この結果から、p38 MAP キナーゼ阻害剤としての新規な化学構造であるイミダゾ[1,2-b] ピリダジン誘導体のin vivo 薬効動物における抗炎症作用を確認できた。 本節で記述した通り、強力なp38 MAP キナーゼ阻害活性および TNF-α産生抑制活性と良好な 経口吸収性を併せ持つ化合物16 に関して、コラーゲン誘発関節炎モデルを用いて、in vivo 薬効 評価を実施し、1 mg/kg から用量依存的に有意に抗炎症作用を示すことが確認され、イミダゾ [1,2-b]ピリダジン誘導体の in vivo における有用性を実証することができた。すなわち、p38 MAP キナーゼ阻害剤としての新規な化学構造であるイミダゾ[1,2-b]ピリダジン誘導体の in vivo にお ける有用性を実証することに成功した。
Figure 10.Anti-inflammatory effects of 16 in the model of collagen-induced arthritis in the rat (po, 11 days treatment, n = 8 per group). **; p ≤ 0.01 vs. control by Dunnett's test.
0 0.1 0.2 0.3 0.4 0.5 0.6 Cont. 0.1 0.3 1 3 10 In c re a s e o f p a w v o lu m e fr o m d a y 0 (m L ) Compound 16 (mg/kg)
**
**
**
第6 節 本章まとめ
p38 MAPK 阻害剤 1(IC50 = 240 nM)と p38 MAP キナーゼの X 線共結晶構造解析情報から、
酵素阻害活性の向上を目的として、化合物1 の疎水性相互作用を増強する化合物設計を行った。 この化合物設計に基づくエチルチアゾール骨格の縮合環への変換により、強力なp38 MAP キナ ーゼ阻害活性(IC50 = 73 nM)を有するイミダゾ[1,2-b]ピリダジン誘導体 8a を見出した。リード 化合物8a のp38 MAPK 阻害活性の向上を目的とし、構造活性相関情報を集積、解析し、構造最 適化を実施した。その結果、酵素阻害活性としてIC50 = 5.4 nM を示す高活性なイミダゾ[1,2-b] ピリダジン誘導体8n を見出した。次に、CYP 阻害活性の低減を目的として、ピリジン窒素の隣 接位へのメチル基導入を行い、CYP 阻害活性を低減したイミダゾ[1,2-b]ピリダジン誘導体10b を見出した。続いて、ヒト肝細胞における酸化代謝に対する安定性の改善に取り組んだ。ピリジ ン窒素原子が代謝され、ピリジン-N-オキシド誘導体が生じることが要因であると考え、ピリジ ン-N-オキシド誘導体を合成したところ、p38 MAP キナーゼ阻害活性を保持しながら、ヒト代謝 安定性も劇的に改善する化合物15a (16: 硫酸塩)の発見に至った。化合物 15a は、42 種類のキナ ーゼパネル試験において優れた選択性を示した。硫酸塩16 は、良好な経口吸収性を示し、ラッ トのコラーゲン誘発関節炎モデルにおいて、後肢関節腫脹を用量依存的に有意に抑制した。以上 の結果から、イミダゾ[1,2-b]ピリダジン誘導体15aの動物モデルにおける有効性を実証すること ができ、p38 MAP キナーゼ阻害剤としての新規な化学構造であるイミダゾ[1,2-b]ピリダジン誘導 体が炎症性疾患の治療薬となる可能性を示した。筆者は、代表化合物である15a は、有望な開発 候補化合物となり得るのみならず、p38 MAPK の生理作用の解明に使用可能な生物学的ツールと しても有用であると考える。
第2 章 イミダゾ[4,5-b]ピリジン-2-オン誘導体に関する合成研究 第1 節 イミダゾ[4,5-b]ピリジン-2-オン誘導体の創出
第1 項 研究方針と分子設計
新規なp38 MAP キナーゼ阻害剤を創出する目的で、自社化合物のハイスループットスクリー ニングを行い、IC50 = 140 nM の p38 MAP キナーゼ阻害活性を示した化合物 17 を見出した(Figure
11(A))。化合物 17 と p38 MAP キナーゼの X 線共結晶構造解析(Figure 11(B))の結果、化合
物17 は、p38 MAP キナーゼの ATP 結合サイトに結合し、特徴的な水素結合を形成していること が確認された。すなわち、化合物17 のカルボニル基が p38 MAP キナーゼのヒンジ領域の Met109 およびGly110 の主鎖 NH 水素原子との 2 つの水素結合を介して酵素と相互作用している。Figure 11(C)に示した通り、p38 MAP キナーゼのヒンジ領域はアミド結合カルボニル基が交互に配列 されたコンフォメーションをとり、Met109 のカルボニル基がリガンド結合部位に向き、ATP な どのリガンドとの水素結合に利用される。しかし、化合物17 が結合することにより、Met109 と Gly110 の間のペプチド結合が反転することにより、水素結合アクセプターとドナーの配列分布 が切り替わったコンフォメーションが誘導されていることが確認された。このペプチドフリップ は、Gly110 のように側鎖サイズが小さなアミノ酸残基の場合にエネルギー的に許容される。一 方、より大きな側鎖を有するアミノ酸残基の場合は、エネルギー的に不利となるため、このペプ チドフリップは起こりにくい43,75,76。
p38 MAP キナーゼの Gly110 に対応する配列位置 X + 4(X はゲートキーパー残基)に Gly を 有するキナーゼは、すべてのヒトプロテインキナーゼの約9.2%のみであることが知られている ため、ペプチドフリップを誘導することは、高いキナーゼ選択性を達成するために有利に働くと 考えられる51,76。従って、化合物17 の水素結合アクセプターとして機能するカルボニル基を保 持しながら、立体的にかさ高いピペリジン部位を芳香環へと変換し、カルボニル基の方向を規定 できるよう、縮環する化合物設計を行った(Figure 12)。デザインされた化合物は、His107 のカ ルボニル酸素との水素結合も新たに狙うことが可能と考えられる77。 上記の仮説に基づいて、p38 MAP キナーゼとの水素結合に重要なカルボニル酸素原子を保持 する形で、p38 MAP キナーゼのペプチドフリップを誘導し、His107 との水素結合を新たに獲得 可能な骨格へ変換した化合物をデザインした(Figure 12)。この化合物設計により、強力かつ高 選択的なp38 MAP キナーゼ阻害活性を有する骨格に変換できると考えた。 この化合物設計に基づいて、複数の骨格変換を行い、p38 MAP キナーゼ阻害活性を評価した ところ、いずれの化合物も強力なp38 MAP キナーゼ阻害活性を示した。さらに物性評価の指標 として溶解度を評価したところ、イミダゾ[4,5-b]ピリジン-2-オン誘導体が、強力な活性と良好な 溶解性を併せ持つ均衡のとれた骨格であることを見出した。 なお、Figure 12 の構造から Thr106 および Leu104 などの疎水性アミノ酸残基により形成される 空間的に小さな疎水性ポケットを占有すると考えられるフェニル基上には、活性向上を目的とし て、小さな置換基を配置することとした。
(A) (B) (C)
Figure 11. (A) Structure of inhibitor 17. The atoms involved in the hydrogen-bond interactions with p38
MAP kinase are shown in blue. (B) X-ray co-crystal structure of 17 bound with p38 MAP kinase (PDB code 6M95). The gray dashed lines show hydrogen-bond interactions. The purple dashed circle
highlighted the peptide flipped hinge conformation. (C) Typical hinge conformation and binding mode of
17 with the peptide flipped hinge conformation.
Figure 12. Design concept of p38 MAP kinase inhibitors. The atoms involving hydrogen-bond
interactions with p38 MAP kinase are shown in blue. The blue dashed arrow represents potential hydrogen-bond interaction.
第2 項 合成 イミダゾ[4,5-b]ピリジン-2-オン誘導体 23a-c は、Scheme 8 に示した方法で合成した。化合物 18 を出発物質として、フッ素を導入後、アニリンと加熱することで、ピリジン 2 位へ位置選択 的に反応が進行し、化合物 20a を収率 80%で得た。続くバックワルドハートウィッグアミノ化 反応により、対応するアミンとの反応を行い、ジアミン21a-c を収率 72–92%で得た。エステル 基の加水分解後、クルチウス転移反応に続く分子内環化反応によりイミダゾピリジン環を構築し、 目的とする化合物23a-c を中程度の収率で合成した。
Scheme 8. Synthesis of compounds 23a–ca
aReagents and conditions: (a) CsF, DMSO, 50°C, 87%; (b) PhNH2, DMSO, 100°C, 80%; (c) R1NH2,
XPhos, Pd2(dba)3, Cs2CO3, toluene, 100°C, 72–92% for 21a and 21c; (d) 1 N NaOH, THF-MeOH-H2O,
rt, 43% for 22a, 70% for 2 steps for 22b; (e) DPPA, Et3N, toluene, 100°C, 41–62% for 23a–b, 17% for
第3 項 生物活性と考察
合成した化合物の生物学的評価は、p38 MAP キナーゼによる ATF2 リン酸化反応に対する阻害 活性を指標とした。Table 11 に、本節第 1 項の化合物設計に基づいて合成したイミダゾ[4,5-b]ピ リジン-2-オン誘導体 23a–c の生物学的評価結果を示した。化合物 23a は、IC50 = 390 nM と化合
物17 (IC50 = 140 nM)とほぼ同等の p38 MAP キナーゼ阻害活性を示した。Figure 9 (B)に示し
たように、化合物17 のベンジル基は、Thr106 および Leu104 で構成される空間的に小さな疎水 性ポケットに向いており、化合物23a が同じ結合様式で p38 MAP キナーゼと相互作用している と仮定すると、第1 章第 2 節で記述した通り、R1置換基はp38 MAP キナーゼ阻害活性発現に重 要であり、立体的に小さな置換基のみ許容する。また、第1 章第 2 節で記述した通り、効率的に 構造最適化研究を進める上で、結合様式を知ることは有益な情報となる。従って、まずR1置換 基の探索を実施し、イミダゾ[4,5-b]ピリジン-2-オン誘導体が化合物 17 と同様の結合様式で相互 作用しているかどうかを確認した。その結果、3-メチル体 23b は活性が消失し、2,4-ジフルオロ 体23c は IC50 = 63 nM と活性が大きく向上することを確かめた。この結果より、イミダゾ[4,5-b] ピリジン-2-オン誘導体は、期待した通り、化合物 17 と同様の結合様式で p38 MAP キナーゼと 相互作用していることが示唆された。 また、経口投与で使用できる薬剤を考える上で物理化学的性質の優れた化合物が望まれるため、 物理化学的評価の指標として、溶解度を測定したところ、強力なp38 MAP キナーゼ阻害活性を 示した2,4-ジフルオロ体 23c が、4.7 μg/mL の良好な溶解性を有することが確認された。 本節で記述した通り、p38 MAP キナーゼ阻害剤の新規骨格として、化合物 17 を基にイミダゾ [4,5-b]ピリジン-2-オン誘導体を創出した。すなわち、化合物 17 のカルボニル基を保持しながら かさ高いピペリジン部位を芳香環へと変換することにより、イミダゾ[4,5-b]ピリジン-2-オン誘導 体を見出すことに成功した。続いて、R1置換基の構造活性相関情報からp38 MAP キナーゼ阻害 活性に対して鋭敏に影響することが確認されたことから、イミダゾ[4,5-b]ピリジン-2-オン誘導体 は化合物17 と同様の結合様式で p38 MAP キナーゼと相互作用していることが示唆された。また、 イミダゾ[4,5-b]ピリジン-2-オン誘導体が置換基変換によって、優れた溶解度を示すことも確認し た。従って、筆者は、有望なリード骨格として、イミダゾ[4,5-b]ピリジン-2-オン骨格を選定した。
Table 11. Inhibitory activities of imidazo[4,5-b]pyridin-2-one derivatives 23a–c against p38 MAP kinase
and corresponding solubility.
Cmpd. R1 p38 MAPK IC50 (nM)a Solubilityb (μg/mL) 23a 2-Me 390 (330–470) 0.46 23b 3-Me > 10000 0.34 23c 2,4-diF 63 (55–72) 4.7 a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent
95% confidence intervals. b Solubility in pH = 6.8.
第2 節 イミダゾ[4,5-b]ピリジン-2-オン誘導体リード化合物の創出 第1 項 研究方針と分子設計 前節で記述した通り、強力なp38 MAP キナーゼ阻害活性を示したイミダゾ[4,5-b]ピリジン-2-オン誘導体23c を見出し、初期の構造活性相関情報から推定した結合様式に基づき、更なる p38 MAP キナーゼ阻害活性向上を目的として、構造活性相関情報を集積、解析を行い、構造最適化 を行った。すなわち、Figure 13 に示した通り、2,4-ジフルオロフェニル基は、Thr106 および Leu104 などの疎水性アミノ酸残基により形成される空間的に小さな疎水性ポケットを占有している。イ ミダゾ[4,5-b]ピリジン-2-オン環 3 位置換基は溶媒領域へ向くため、広範な変換が可能であると考 えられる。 上記の仮説に基づいて合成展開を行うことにより、化合物の変換可能な部位を限定することが でき、構造最適化を効率的に実施することが可能となり、p38 MAP キナーゼ阻害活性が向上す ると期待した。
第2 項 合成 イミダゾ[4,5-b]ピリジン-2-オン誘導体 23d-s は、Scheme 9 に示した方法で合成した。化合物 19 を出発物質として、対応するアニリンと加熱することで、ピリジン 2 位へ位置選択的に反応 が進行し、化合物20b-o を得た。続くバックワルドハートウィッグアミノ化反応により、対応す るアミンとの反応を行い、ジアミン化合物20d-s 得た。エステル基の加水分解後、クルチウス転 移反応に続く分子内環化反応によりイミダゾピリジン環を構築し、目的とする化合物23d-s を合 成した。
Scheme 9. Synthesis of compounds 23d–sa
aReagents and conditions: (a) R1NH2, DMSO, 100°C; (b) R2NH2, XPhos, Pd2(dba)3, Cs2CO3, toluene,
100°C, 16%–quant for 21e–f and 21r; (c) 1 N NaOH, THF-MeOH-H2O, rt, 10–56% for 22e–f and 22r,
4–77% for 2 steps for 22k, 22o, and 22s; (d) DPPA, Et3N, toluene, 100°C, 10–23% for 3 steps for 23d, 23p, 25–60% for 23e–f, 23k, 23o, 23r, and 23s, 10–20% for 4 steps for 23g–j, 23l–n, and 23q.
続いて、イミダゾ[4,5-b]ピリジン-2-オン誘導体 26 は、Scheme 10 に示した方法で合成した。 すなわち、化合物19 と 2,4-ジフルオロアニリンをバックワルドハートウィッグアミノ化反応条 件下加熱することで、ピリジン5 位のみならず、2 位の反応も同時に進行したため、過剰量の 2,4-ジフルオロアニリンを追加し、化合物24 を 25%の低収率ながら得た。加水分解後、クルチウス 転移反応に続く分子内環化反応によりイミダゾピリジン環を構築し、目的とする化合物26 を収 率7%で合成した。また、イミダゾ[4,5-b]ピリジン-2-オン誘導体 30a-c は、Scheme 11 に示した方
法で合成した。すなわち、ピリジン2 位がクロロ基である化合物 18 をバックワルドハートウィ ッグアミノ化反応条件下、リガンドとしてXantphos を用いて 100 度で、対応するアニリンとの 反応を行い、ピリジン5 位選択的にアミノ基を導入することに成功し、化合物 27a-b を得た。続 いて、バックワルドハートウィッグアミノ化反応条件下、リガンドとして BINAP を用いて 150 度で、対応するアニリンとの反応を行い、ピリジン2 位へのアミノ基の導入を行い、化合物 28a-c を得た。これら合成法により、ピリジン2 位および 5 位に選択的にアミノ基を導入する方法が確 立できた。化合物28a-c を加水分解後、クルチウス転移反応に続く分子内環化反応によりイミダ ゾピリジン環を構築し、目的とする化合物30a-c を合成した。
Scheme 10. Synthesis of compounds 26a
aReagents and conditions: (a) 2,4-Difluoroaniline, Xphos, Pd
2(dba)3, Cs2CO3, 60°C, 25%; (b) 1 N
NaOH, THF-MeOH-H2O, rt, quant; (c) DPPA, Et3N, toluene, 100°C, 7%.
Scheme 11. Synthesis of compounds 30a–ca
aReagents and conditions: (a) R2NH
2, Xantphos, Pd2(dba)3, Cs2CO3, toluene, 100°C, 50–59%; (b)
R1NH
2, BINAP, Pd(OAc)2, Cs2CO3, toluene, MW, 150°C, 17–83%; (c) 1 N NaOH, THF-MeOH-H2O, rt,
第3 項 生物活性と考察 合成した化合物の生物学的評価は、本章第1 節に記載した方法と同様にして、p38 MAP キナ ーゼ阻害活性を指標とした。また、物理化学的性質の指標の1 つとして、溶解度も合わせて評価 した。Table 12-1 および Table 12-2 に、本節第 1 項の化合物設計に基づいて、合成したイミダゾ [4,5-b]ピリジン-2-オン誘導体 23d–s, 26,および 30a–c の生物学的評価結果を示した。化合物 23c と比較して、2-フルオロ体 23d は、より強力な p38 MAP キナーゼ阻害活性を示し、IC50値は27 nM であり、2-フルオロ体 23d の 2,4-ジフルオロフェニル基を 2,6-ジフルオロフェニル基(30a)に 変換したところ、p38 MAP キナーゼ阻害活性が低下した。したがって、4-フルオロ基は、浅いポ ケットを効果的に占有していると考えられる。従って、R2置換基は2,4-ジフルオロ基に固定して、 R1置換基の検討を行った。2,4-ジフルオロ体 26 の阻害活性は低下したが、2,6-ジフルオロ化合物 30b は 23d よりも強力な阻害活性を示した。さらに、R1フェニル基に対するオルト置換基の効果 を確認するために、2,6-ジクロロ体 30c が評価され、IC50 = 5.6 nM とより強力な阻害活性を示し た。しかしながら、化合物30c は溶解度を大きく低下させることも明らかとなった。これらの結 果は、フェニル基の置換基効果が電子吸引効果よりも立体的な効果の方が、強力な阻害活性に重 要であることを示唆している。この構造活性相関情報から R1フェニル基とイミダゾ[4,5-b]ピリ ジン-2-オン骨格は、空間的に相互に直交する平面上にあると考えられる。このことから、R1置 換基が空間的に大きなサイズでも許容されることが確認できたので、優れた活性および溶解度を 併せ持つ化合物を探索することを目的として、更なる置換基効果を検証した。ベンジル体 23e は、23c と同等の活性を示したが、炭素鎖を伸長したフェネチル化合物 23f は活性が低下した。 ベンジル体23e のメチレン部位にメチル基を導入したモノメチル体 23g は活性を保持したが、ジ メチル体23h は活性が減弱した。合成されたベンジル誘導体(23i-m)の中で化合物 23m が強力 な活性を示したが、溶解度の改善には至らなかった。次に、23g が中程度の活性を保持しながら、 非常に優れた溶解度を示したことに注目し、R1置換基として、分岐アルキル基の探索を行った。 イソプロピル体 23n は 23g よりも強力な阻害活性を示し、良好な溶解度を示した。このことに より、23g のフェニル基がメチル基で置換可能であると考えられる。すなわち、これは、リガン
ド効率(ligand efficiency: LE = pIC50/Heavy atom count)や脂溶性効率(ligand lipophilicity efficiency:
LLE = pIC50-LogD)の観点から非常に重要な知見であると考えられる。イソブチル体 23o では、
活性を保持し、s-ブチル体 23p および 3-ペンチル体 23q で活性が増強することを確認できた。以
上の結果、23o および 23p の比較から、窒素原子に隣接する位置のメチル基が p38 MAP キナー
ゼ阻害活性に重要であることが分かり、さらに、溶解度にも同様にこの分岐メチル基が重要であ ることが確認できた。
Table 12-1. Inhibitory activities of imidazo[1,2-b]pyridazine derivatives 23c–m, 26, and 30a–c against
p38 MAP kinase and corresponding solubility.
Cmpd. R1 R2 R3 p38 MAPK IC50 (nM) a Solubilityb (μg/mL) 23c Ph 2,4-diF NH 63 (55–72) 4.7 23d 2-F-Ph 2,4-diF NH 27 (24–30) 3.3 23e PhCH2 2,4-diF NH 29 (26–32) 1.8 23f PhCH2CH2 2,4-diF NH 110 (90–120) 0.26 23g PhCH(CH3) 2,4-diF NH 31 (26–36) 10 23h PhC(CH3)2 2,4-diF NH 150 (130–160) 3.0 23i 2-F-PhCH2 2,4-diF NH 15 (13–16) 0.49 23j 3-F-PhCH2 2,4-diF NH 37 (33–42) 1.8 23k 4-F-PhCH2 2,4-diF NH 52 (44–60) 0.81 23l 2-Cl-PhCH2 2,4-diF NH 100 (91–120) 0.14 23m 2,6-diF-PhCH2 2,4-diF NH 12 (11–13) 0.11 26 2,4-diF-Ph 2,4-diF NH 100 (90–110) 4.5 30a 2-F-Ph 2,6-diF NH 87 (76–99) 0.69 30b 2,6-diF-Ph 2,4-diF NH 9.9 (8.9–11) 1.7 30c 2,4-diCl-Ph 2,4-diF NH 5.6 (4.9–6.4) < 0.12 a IC
50 values shown are the mean values of quadruple measurements. Numbers in parentheses represent


![Table 2. Inhibitory activities of imidazo[1,2-b]pyridazine derivative 8a against p38 MAP kinase and production of TNF-α in human THP-1 cells](https://thumb-ap.123doks.com/thumbv2/123deta/9938870.1390611/13.892.215.682.705.968/table-inhibitory-activities-imidazo-pyridazine-derivative-kinase-production.webp)
![Table 3. Inhibitory activities of imidazo[1,2-b]pyridazine derivative 8a–n against p38 MAP kinase and production of TNF-α in human THP-1 cells](https://thumb-ap.123doks.com/thumbv2/123deta/9938870.1390611/19.892.123.770.227.1036/table-inhibitory-activities-imidazo-pyridazine-derivative-kinase-production.webp)

![Table 6. Stability in human liver microsomes of imidazo[1,2-b]pyridazine derivatives 10a–b and 8n–o](https://thumb-ap.123doks.com/thumbv2/123deta/9938870.1390611/26.892.145.745.199.565/table-stability-human-liver-microsomes-imidazo-pyridazine-derivatives.webp)
![Table 8. Stability in human liver microsomes of imidazo[1,2-b]pyridazine derivatives 15a](https://thumb-ap.123doks.com/thumbv2/123deta/9938870.1390611/30.892.135.752.202.651/table-stability-human-liver-microsomes-imidazo-pyridazine-derivatives.webp)