1 2012 年度
修士論文
ブレビバチルスへのβ―アガラーゼ遺伝子導入
およびネオアガロオリゴ糖生産
Cloning of β-agarase gene to
Brevibacillus
and production of neoagarooligosaccharides
高知工科大学大学院 工学研究科
基盤工学専攻 物質・環境システム工学コース
1155003 小田 達
2 目次 緒言 1章 ブレビバチルスへのβ―アガラーゼ遺伝子導入および寒天オリゴ糖の生産 1.1 目的 1.2 実験方法 1.2.1 菌株の培養 1.2.2 Cellvibrio sp.株からの染色体 DNA の抽出 1.2.3 PCR による目的遺伝子の増幅 1.2.4 制限酵素による消化反応 1.2.5 PCR 産物とプラスミド消化物のライゲーション反応 1.2.6 形質転換 1.2.7 プラスミド抽出とインサート DNA の確認 1.2.8 組み換えBrevibacillusの培養 1.2.9 アガラーゼ生産組み換え菌による寒天オリゴ糖生産と HPLC 分析 1.3 結果と考察 1.3.1 導入プラスミドの電気泳動 1.3.2 Brevibacillus培養中の各培地のpH と濁度の変化 1.3.3 アガラーゼ生産組み換え菌による寒天オリゴ糖生産と HPLC 分析 2章 大腸菌へのα-アガラーゼ遺伝子導入および寒天オリゴ糖分解 2.1 背景と目的 2.2 実験方法 2.2.1 NCBI によるアミノ酸配列の相動性検索 2.2.2 primer 設計と PCR
3 2.2.3 形質転換 2.2.4 コロニーPCR 2.2.5 基質となるオリゴ糖の調製 2.2.6 組み換え大腸菌を用いた寒天オリゴ糖の分解 2.2.7 TLC 分析 2.3 結果と考察 2.3.1 NCBI によるアミノ酸配列の相同性検索 2.3.2 primer の設計と PCR 2.3.3 ネオアガロビオースの分解と HPLC 分析 2.3.4 ネオアガロビオース分解産物の TLC 分析 結言 謝辞 参考文献 補足
4 緒言 近年、微生物を用いた酵素生産の検討が積極的に行われている。その中で、遺伝子組み 換え技術は必要不可欠なものである。この技術によって、目的の酵素遺伝子を適当なプラ スミドベクターを用いて宿主にクローニングすることで酵素の大量生産が可能となった。 一方、我々の身近な食材の1つとして寒天がある。テングサなどの紅藻類の細胞壁中に存 在する多糖である寒天には過熱後の水溶液を室温に置くことで固まる性質がある。その性 質を利用して古くからゼリー菓子などに用いられてきた。また、寒天を精製して得られる アガロースは、現在、免疫学や遺伝子工学の分野でタンパク質やDNA 断片などの電気泳動 用の支持体として重要な役割も果たしている。さらに、寒天から生産されるヘテロオリゴ 糖に抗酸化作用、発がん予防作用、皮膚への保湿・美白効果などの生理活性があると報告 されている。 寒天の主成分であるアガロースは、D―ガラクトースと3,6-アンヒドロ-L-ガラク トースが交互にα-1,3 結合、β-1,4 結合した構造をとる(図 1)。この結合を加水分解する ことにより、アガロースから種々のヘテロオリゴ糖が生産される。これらは全て寒天オリ ゴ糖とよばれる。これまで、寒天オリゴ糖の生産は酸などの化学試薬を用いる方法で行わ れてきた。しかし、従来の方法ではアガロースの選択的な分解が行えず、特定の長さのヘ テロオリゴ糖を得ることが困難だった。これに対して、微生物が生産するアガロース加水 分解酵素(アガラーゼ)を用いた方法では、酵素の持つ基質特異性によりアガロースの選択 的な分解が可能であり、最終生産物の分離精製も通常の化学的方法で得られたオリゴ糖に 比べて容易であるなどの利点がある。 アガラーゼはその加水分解様式によって2 種類に分類され、α‐1,3 結合を切断するもの をα―アガラーゼ、β‐1,4 結合を切断するものをβ―アガラーゼとよぶ。アガラーゼを生 産する細菌として、主にAlteromonas、Cytophaga、Psedomonas、Pseudoalteromonas、 Streptomyces、Vibrioなどが報告されてきたが、それらは全てβ―アガラーゼに関する報
5
告であり、α―アガラーゼについてはほとんど報告されてこなかった。
これまで本研究室では、中村が高知県内の活性汚泥中から寒天分解菌Cellvibrio sp.を単 離し、井上がβ―アガラーゼ遺伝子agaA および agaB をEscherichia coliにクローニング することに成功するなど、寒天オリゴ糖生産に関する研究を行ってきた。しかしながら、 グラム陰性菌であるEscherichia coliは酵素をほとんど菌体外に分泌せず、培養後に超音波 破砕処理を行って酵素を回収する必要があった。そこで本研究では、β―アガラーゼの発 現に宿主としてグラム陽性菌であるBrevibacillusを用いることで酵素の菌体外大量生産を 試み、酵素生産と寒天オリゴ糖の同時生産を検討した。 また、α―アガラーゼ遺伝子の組み換えは報告が少ないため、今後の寒天オリゴ糖生産 に関する研究において重要である。最近、Cellvibrio sp.からα―アガラーゼを精製し、N-末端アミノ酸配列10 残基を決定した。そこで、本研究ではα―アガラーゼ遺伝子の大腸菌 へのクローニングを試みた。
図1 アガロースの構造
6 1 章Brevibacillusへのβ―アガラーゼ遺伝子導入および寒天オリゴ糖の生産 1.1 目的 β―アガラーゼの菌体外生産を行うため、アガラーゼ生産大腸菌E3 株にクローニングさ れたagaB 遺伝子のBrevibacillusへの導入を試みた。 1.2 実験方法 1.2.1 寒天分解菌の培養
本研究に用いられた寒天分解菌Cellvibrio sp.OA-2007( DDBJ Accession No.
AB332414 )は、中村光輝らによって 2004 年に高知県内の活性汚泥中より単離され、冷凍 保存されたものを使用した。 冷凍保存されたCellvibrio sp.を液体寒天培地(NaNO3 0.1w/v%,NaHPO4・12H2O 0.157w/v%,KH2PO4 0.09w/v%, MgSO4・7H2O 0.05w/v%, KCl 0.05w/v%, Agar, powder 0.1w/v%)において、25℃で 24 時間振とう培養し、液体寒天培地 1000ml に培地体積あたり 1%の濃度で植菌し、25℃で 2 日間、振とう培養した。 1.2.2 Cellvibrio sp.株からの染色体 DNA の抽出 Cellvibrio sp.培養液 1000ml を遠心分離(8000rpm, 10 分間, 4℃)し、菌体を集菌した。 菌体を20mM リン酸緩衝液に懸濁し、遠心分離により 2 回洗浄した。その後、20mM リン 酸緩衝液20ml に再び懸濁し、1.5ml チューブに分注し、DNA 抽出用Cellvibrio sp.溶液と して冷凍保存した。 冷凍保存した DNA 抽出用Cellvibrio sp.溶液を溶解し、遠心分離によ り菌体を集菌し、上清液を取り除いた。菌体にTE 溶液(補足 1)567μl を加え、穏やかに懸 濁した後、10%SDS 溶液 30μl と 5mg/ml プロテナーゼ K (Wako)3μl を加えた。37℃
7 で4 時間加温した後、5M NaCl 溶液 200μl を加え、穏やかに懸濁し、多糖の沈殿を行うた めに2%CTAB(補足2)溶液 80μl を加えた混合液を穏やかに懸濁し、65℃で 2 時間加温し、 RNaseA 溶液(補足3)10μl を加え、37℃で 1 時間加温した。 【精製】不要なタンパク質を取り除くため、懸濁液と等量のフェノール・クロロホルム溶 液(補足4)を加え、撹拌し、遠心分離(15,000rpm, 5 分間)した。中間層としてタンパク質 が出なくなるまで、フェノール・クロロホルムによるタンパク質除去を繰り返し行った。 その後、上清液(水層)を採取し、等量のクロロホルム・イソアミルアルコール溶液(補足 5)を加えた後、撹拌し、遠心分離(15,000rpm, 5 分間)し、再び上清液(水層)を採取し た。0.1 倍量の 3M 酢酸ナトリウム溶液と 2.5 倍量の 100%エタノールを加え、氷上にて 10 分間静置し、遠心分離(15,000rpm, 30 分間, 4℃)した。沈殿した DNA ペレットを確認し た後、上清液を取り除き、冷70%エタノールで洗浄・風乾し、DNA ペレットを TE 溶液中 に溶かし、染色体DNA として以後の実験に使用した。 1.2.3 PCR による目的遺伝子の増幅 Cellvibrio sp.の agaB 遺伝子の開始コドン直後から終止コドンの下流 91 塩基までを PCR によって増幅し、ベクタープラスミドpNCMO2(TaKaRa Bio)へクローニングするため、 Forward primer と Reverse primer を以下のように設計した。
Forward primer 5’ - GTCTCTAGAAAAAAAATCACTTCATGCATTGC - 3’ Reverse primer 5’ - ATTAAGCTTTCTGGCAAGCCACCAC - 3’
Forward primer の下線部は XbaⅠの制限酵素サイト、Reverse primer の下線部は Hind Ⅲの制限酵素サイトを示し、二重下線部は鋳型DNA と相補的な配列部を示す。
Cellvibrio sp.株の染色体 DNA を鋳型とし、TaKaRa EX Taq を用いて PCR により、agaB 領域を増幅した。 PCR 条件は以下の通りである。反応液組成:EX Taq 0.5μl, 10×EX Taq Buffer 5μl, dNTP mixture 4μl, Forward primer 1.5μl, Reverse primer 1.5μl, 鋳型
8
DNA 100ng, 滅菌蒸留水で全量 50μl に調製した(EX Taq を最後に添加)。PCR 条件:変 性温度94℃、アニーリング温度 59℃、伸長反応温度 72℃、伸長反応回数 25 回。PCR 産 物およびDNA の精製は、NucleoSpin Gel and PCR Clean-up(TaKaRa Bio)を用いて行っ た。
1.2.4 制限酵素による消化反応
精製したPCR 産物(1.8kbp)とBrevibacillus用ベクタープラスミドpNCMO2(5.2kbp)を、 制限酵素HindⅢ、XbaⅠ(TaKaRa Bio)を用いて、37℃でそれぞれ消化した。滅菌蒸留 水 39μl、基質 DNA 4μl、市販制限酵素付属 Universal Buffer 5μl の順で混合し、最後 に制限酵素 1μl を加え、加温した。ベクタープラスミドおよび PCR 産物の消化反応時間 を3時間とした。所定時間後、前述した方法(1.2.2)に従い精製を行った。
1.2.5 PCR 産物とプラスミド消化物のライゲーション反応
DNA Ligation Kit <Mighty Mix> (TaKaRa Bio)を用いて説明書に従い、結合させ た。
1.2.6 形質転換
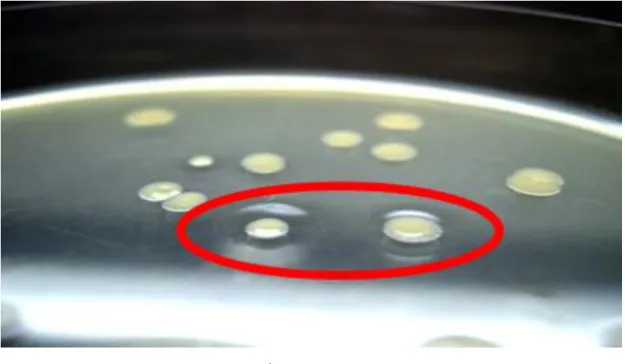
Brevibacillus Competent Cells(TaKaRa Bio)を用いて説明書に従い、NTP 法を用いて 形質転換を行った。その後、アガロース0.8%を含む LB 培地プレート上に塗布し、37℃で 16 時間培養後、周囲に凹みを生じたコロニー(図 2)を単離した。
1.2.7 プラスミド抽出とインサート DNA の確認
NucleoSpin Plasmid(TaKaRa Bio)を用いて、単離した菌体からのプラスミド DNA の抽 出を行った。その後、1.3kbp の産物が得られるように agaB の内部配列を基に、Forward
9
primer と Reverse primer を以下のように設計し、プラスミド DNA を鋳型として PCR を 行った。
Forward primer 5’ - CCGGCTGAGTTTAACTCGC - 3’ Reverse primer 5’ - GCCATCGTAAACACCGCC - 3’
また、抽出後のプラスミドDNA に対し、制限酵素 HindⅢおよび HindⅢ、XbaⅠの 2 重 消化を行った。
その後、少量の臭化エチジウム(補足6)を含む 0.8%アガロースゲル (TAKARA)を用いた 電気泳動によりインサートDNA の確認を行った。
1.2.8 組み換えBrevibacillusの培養
組換えBrevibacillusを、ネオマイシン(100μg/ml)(補足7)を含む LB 培地(Tryptone 1w/v%, Yeast Extract 0.5w/v%, NaCl 1w/v%)および 2SY 培地(グルコース 2w/v%,Soytone 4w/v%, Yeast Extract 0.5w/v%, CaCl2 2H2O 0.015w/v%)に植菌し、30℃で 16 時間前培養 した。培養液100μl を LB 培地、2SY 培地、100ml に植菌し 30℃で振とう培養を行った。 適宜、培養液のpH および濁度を測定した。 1.2.9 アガラーゼ生産組み換え菌による寒天オリゴ糖生産と HPLC 分析 アガラーゼ生産大腸菌E3 株および組み換えBrevibacillus を前培養後、LB 培地および 2SY 培地に植菌し、同時に 20g/l のアガロース粉末を加え、酵素と寒天オリゴ糖の同時生産 を開始した。適宜、培養液をサンプリングし、遠心分離(15,000rpm, 室温, 5 分間)で未 反応の寒天を取り除き、上清液を5分間煮沸し酵素反応を止めた。遠心分離(15,000rpm, 室 温, 5 分間)により、蛋白の沈殿を取り除き、上清液をフィルターろ過し、HPLC により分 析した。HPLC 分析は、展開溶液として脱気超純水を用い、流速 0.8 ml/min、カラム温度 50℃の条件で行った。
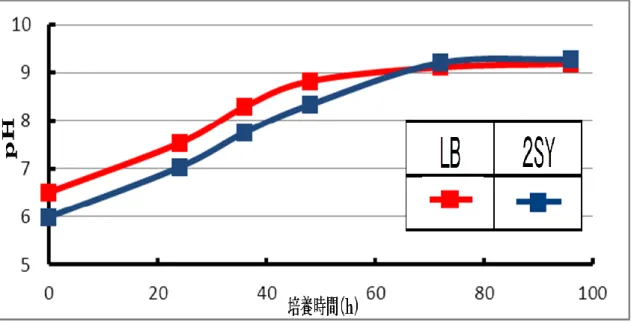
10 1.3 結果と考察 1.3.1 導入プラスミドの電気泳動 組み換えBrevibacillusから抽出したプラスミドDNA 鋳型に PCR をおこない、0.8%ア ガロースゲルによる電気泳動で1.3kbp の単一のバンドを得た。また、制限酵素 HindⅢに よる消化によって7.0kbp、HindⅢおよび XbaⅠによる 2 重消化によって 5.2kbp、1.8kbp の断片を得た(図 3)。 これによって、ブレビバチルスにagaB 遺伝子が導入されたことが示唆された。 1.3.2 Brevibacillus培養中の各培地のpH と濁度の変化 LB 培地、2SY 培地の 2 種の培地で組み換えBrevibacillusを培養した場合、両培地のpH の上昇速度や最大値に大きな差は見られなかった(図 4)。菌体濁度は両培地共に培養開始か ら48 時間時点で最大となった。菌体濁度の最大値は LB 培地で 11.2、2SY 培地で 16.7 と 大きな差がついた(図 5)。 pH の上昇速度および最大値、菌体濁度が最大値に達する時間に大きな差がないことから、 組み換えBrevibacillusの培養において、LB 培地よりも 2SY 培地の方が適していると言え る。 1.3.3 アガラーゼ生産組み換え菌による寒天オリゴ糖生産と HPLC 分析 アガラーゼ生産大腸菌E3 株を LB 培地および 2SY 培地で培養し、同時にアガロースの 分解を行った結果、どちらの培地を用いた場合でも培養開始から80 時間経過後において 4 糖、6 糖共に殆ど生産されなかった(図 6)。一方で、組み換えBrevibacillusを用いて同様 の実験を行った結果、LB 培地を用いた場合では培養開始から 48 時間経過後に約 4.2g/l の 4 糖と 2g/l の 6 糖が生産された。さらに、2SY 培地を用いた場合では培養開始から 48 時間
11 経過後に約6g/l の 4 糖と 2g/l の 6 糖を生産した(図 7)。このことから、グラム陰性菌であ るE3 株はアガラーゼを菌体外に分泌せず、培養液中で直接寒天オリゴ糖を生産することが 困難であると考えられる。その一方で、グラム陽性菌である組み換えBrevibacillusは生産 したアガラーゼを菌体外に分泌し、培養液中での寒天オリゴ糖生産が容易であることが示 された。
12
図
2 組み換え
Brevibacillus
13
図
4 組み換え
Brevibacillus
培養における
培地
pH の経時変化
図
5 組み換え
Brevibacillus
培養における
14
図6
E3 株を用いた培地中の寒天オリゴ糖生産の経時変化
図7 組み換え
Brevibacillus
を用いた
15 2 章 大腸菌へのα―アガラーゼ遺伝子導入および寒天オリゴ糖分解 2.1 背景と目的 現在まで、β―アガラーゼの精製やクローニングについて数多く報告されてきたが、α ―アガラーゼについては精製の報告が極端に少なく、クローニングの報告は無い。そこで 本研究室ではα-アガラーゼ遺伝子のクローニングに取り組んできた。岡本はCellvibrio sp. 株からα―アガラーゼを精製し、アミノ酸のN 末端配列を以下のように決定した。 N 末端―GDLPEKKLSK この配列を基に大腸菌へのα-アガラーゼ遺伝子クローニングに取り組んだ。 2.2 実験方法 2.2.1 NCBI によるアミノ酸配列の相同性検索 上記のα―アガラーゼのN 末端アミノ酸配列および本研究室で保有する 2 種のβ―アガ ラーゼ(AgaA、AgaB )のアミノ酸配列を国立生物工学情報センター(NCBI)のデータベー スでBLAST プログラムを用いて相同性検索を行った。 2.2.2 primer 設計と PCR
前項の相同性検索で得られたアミノ酸配列を基にForward primer と Reverse primer を 設計し、前述した方法(1.2.3)で PCR および精製を行った。
2.2.3 組み換えプラスミドの調製
精製したPCR 産物(1.2kbp)と大腸菌用ベクタープラスミド pUC19(TaKaRa Bio)を制限 酵素HindⅢ、XbaⅠを用いて、37℃でそれぞれ消化した。滅菌蒸留水 39μl、基質 DNA 4
16
μl、市販制限酵素付属 Universal Buffer 5μl の順で混合し、最後に制限酵素 1μl を加え、 加温した。ベクタープラスミドおよびPCR 産物の消化反応時間を3時間とした。所定時間 後、前述した方法(1.2.2)に従い精製を行った。その後、DNA Ligation Kit <Mighty Mix >を用いて説明書に従い、結合させた。
2.2.4 形質転換
宿主大腸菌としてE.coli DH5αを選択した。前述の方法(2.2.3)で作成された組み換えプ ラスミド 1μl(10ng)を、E.coli DH5α competent Cells(TaKaRa) 100μl が入った 1.5ml チューブに加え、氷上で 10 分間静置した。その後、1.5ml チューブを 42℃で 45 秒 間加温し、組換えプラスミドをE.coli DH5α Competent Cells に導入し、形質転換を行っ た(Heat shock 法)。その後、SOC 培地(Trypton 2w/v%,Yeast Extract 0.5w/v%,NaCl 0.05w/v%,KCl 0.0186w/v%,pH7.0)1ml を 1.5ml チューブに加え、37℃で 90 分間振とうし、 pUC19 のアンピシリン耐性遺伝子の誘導を行った。誘導後、E.coli DH5αを 100μずつ、 アンピシリン(100μg/ml)(補足8)を添加した LB 寒天培地上に塗布し、37℃で 16 時間、 静置培養した。培養後、LB 寒天培地上でコロニーを得た。
2.2.5 コロニーPCR
得たコロニーを滅菌した竹串で取り、2.2.2 で設計した Forward primer と Reverse primer を用いて前述した方法(1.2.3)で PCR を行った。
2.2.6 ネオアガロビオースの調製
基質となるネオアガロビオースの調製は山善株式会社の中圧分取液体クロマトグラフィ ーを用いて、展開溶液として超純水、流速25 ml/min の条件で行った。
17 2.2.7 組み換え大腸菌を用いた寒天オリゴ糖分解と HPLC 分析 組換え大腸菌を、アンピシリン(100μg/ml)を含む LB 培地に植菌し、25℃で 24 時間 前培養した。培養液100μl を、アンピシリン(100μg/ml)を含む LB 培地 500ml に植菌 し、25℃で 24 時間振とう培養した。菌体を遠心分離(8,000rpm,15 分間,4℃)により集菌 し、上清液を取り除いた。菌体を冷20mMリン酸緩衝液に懸濁し、遠心分離(8,000rpm、 15 分間、4℃)により2回洗浄した。その後、再び菌体を冷 20mM リン酸緩衝液 50ml に 懸濁し、氷冷しながら超音波で菌体を破砕した(60kHz、一秒周期で ON/OFF, 20 分間)。 遠心分離(8,000rpm、15 分間、4℃)により菌体残渣を取り除き、上清液を粗酵素溶液と して得た。その後、粗酵素溶液を加えた20mM リン酸緩衝液 5ml に、2.2.5 で調製したネ オアガロビオース(HPLC 面積値 100,000)を加え、25℃で加温しながら分解を行った。適宜、 反応液を5分間煮沸し酵素反応を止めた。遠心分離(10,000rpm, 室温, 5 分間)により、 蛋白の沈殿を取り除き、上清液をフィルターろ過し、HPLC により分析した。 2.2.8 TLC 分析 酵素反応生産物の分析のため、薄層クロマトグラフィー (TLC)を行った。2.2.7 でネオア ガロビオースの分解を行った試料を Silica gel 60 プレート (10×20cm, MERCK)にスポッ トした後、展開溶媒 (n-ブタノール:エタノール:水 = 3:3:1)で展開した。発色試薬としてナ フトレゾルシノール(SIGMA)0.06w/v%を加えた 20%硫酸を吹きつけた後、ホットプレート で加熱して可視化した。標準物質として1g/l の D-ガラクトース(Wako)、アンヒドロガラ クトース、ネオアガロビオースを用いた。
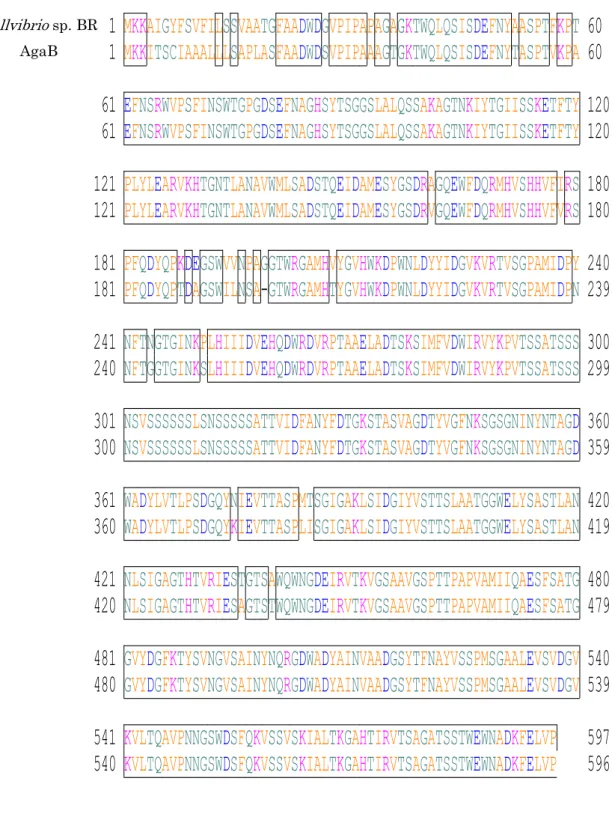
18 2.3 結果と考察 2.3.1 NCBI によるアミノ酸配列の相同性検索 国立生物工学情報センター(NCBI)のデータベースで BLAST プログラムを用いてα― アガラーゼのN 末端アミノ酸配列との相同性検索を行った結果、Cellvibrio sp. BR のゲノ ムDNA の中に、開始アミノ酸の1残基下流から10残基、完全に一致する部位を発見した (図 8)。さらに、本研究室で保有する 2 種のβ―アガラーゼのアミノ酸配列を用いて同様の 検索を行った結果、Cellvibrio sp. BR は非常に相同性の高いゲノム領域を持っていること が分かった(図 9、図 10)。これは、Cellvibrio sp. OA-2007 とCellvibrio sp. BR が非常に 近い遺伝子配列を持っていることを示唆している。
2.3.2 primer の設計と PCR
Cellvibrio sp. BR のゲノム DNA の中で、α―アガラーゼの N 末端アミノ酸配列と完全 に一致したタンパク質をコードする部位を基に以下のようなForward primer と Reverse primer を設計した。
Forward primer 5-AAGAAGCTTATGGGTGATCTTCCAGA-3 Reverse primer 5-AATTCTAGATCAGACGTTCTGAAAGGT-3
Forward primer の下線部は HindⅢ、Reverse primer の下線部は XbaⅠを示し、二重下 線部は鋳型DNA と相補的な配列部を示す。
このprimer を用いて、Cellvibrio sp. OA-2007 の染色体 DNA を鋳型に PCR を行った結 果、1.2kbp のシングルバンドを得た。この大きさが基のCellvibrio sp. BR の遺伝子部位と ほぼ一致したことから、得られた遺伝子はCellVibrio sp. OA-2007 由来のα―アガラーゼ 遺伝子の可能性があると考えた。
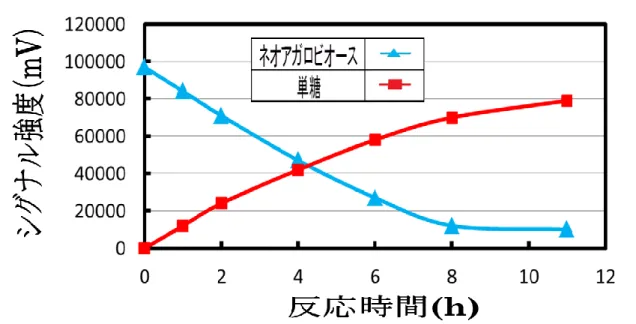
19 2.3.3 ネオアガロビオースの分解と HPLC 分析 組み換え大腸菌から得た粗酵素液を用いてネオアガロビオースの分解を行った結果、基 質となるネオアガロビオースは徐々に減少し、分解産物である単糖が生産された(図 11)。こ のことから、組み換え大腸菌にα―アガラーゼ遺伝子が導入されていることが示唆された。 2.3.4 ネオアガロビオース分解産物の TLC 分析 組み換え大腸菌の粗酵素液添加後、時間が経過するごとにネオアガロビオースのスポッ トが薄くなり、逆にガラクトースとアンヒドロガラクトースのスポットが徐々に濃くなっ た(図 12)。このことからも、組み換え大腸菌がα―アガラーゼを生産していることが示さ れた。
20
図
8 α―アガラーゼの N 末端アミノ酸配列と
相同性の高い
Cellvibrio
sp. BR のゲノム領域
21
図
9 AgaA と相同性の高い
Cellvibrio
sp. BR のゲノム領域
Cellvibrio sp. BR AgaA
22
図
10 AgaB と相同性の高い
Cellvibrio
sp. BR のゲノム領域
BR_E3AA.seq 1
M
KK
AI
G
YF
S
VFIL
SS
VAA
TG
FAA
D
W
D
G
VPIPAPA
G
A
G
K
TWQ
L
QS
I
S
DE
F
N
YAA
S
P
T
F
K
P
T
60
E3_AA.seq
1
M
KK
I
TSC
IAAALLL
S
APLA
S
FAA
D
W
D
S
VPIPAAA
GTG
K
TWQ
L
QS
I
S
DE
F
N
Y
T
A
S
P
T
V
K
PA
60
BR_E3AA.seq 61
E
F
NS
R
W
VP
S
FI
NSWTG
P
G
D
S
E
F
N
A
G
H
S
Y
TSGGS
LAL
QSS
A
K
A
GTN
K
IY
TG
II
SS
K
E
T
F
T
Y
120
E3_AA.seq
61
E
F
NS
R
W
VP
S
FI
NSWTG
P
G
D
S
E
F
N
A
G
H
S
Y
TSGGS
LAL
QSS
A
K
A
GTN
K
IY
TG
II
SS
K
E
T
F
T
Y
120
BR_E3AA.seq 121
PLYL
E
A
R
V
KH
TGNT
LA
N
AV
W
ML
S
A
D
STQ
E
I
D
AM
E
S
Y
GS
D
R
A
GQ
E
W
F
D
Q
R
M
H
V
S
HH
VFI
R
S
180
E3_AA.seq 121
PLYL
E
A
R
V
KH
TGNT
LA
N
AV
W
ML
S
A
D
STQ
E
I
D
AM
E
S
Y
GS
D
R
V
GQ
E
W
F
D
Q
R
M
H
V
S
HH
VFV
R
S
180
BR_E3AA.seq 181
PF
Q
D
Y
Q
P
K
DE
GSW
VV
N
PA
GGTW
R
G
AM
H
VY
G
V
H
W
K
D
P
WN
L
D
YYI
D
G
V
K
V
R
T
V
SG
PAMI
D
PY
240
E3_AA.seq 181
PF
Q
D
Y
Q
P
T
D
A
GSW
IL
NS
A
-
GTW
R
G
AM
H
T
Y
G
V
H
W
K
D
P
WN
L
D
YYI
D
G
V
K
V
R
T
V
SG
PAMI
D
P
N
239
BR_E3AA.seq 241
N
F
TNGTG
I
N
K
PL
H
III
D
V
E
H
Q
D
W
R
D
V
R
P
T
AA
E
LA
D
TS
K
S
IMFV
D
W
I
R
VY
K
PV
TSS
A
TSSS
300
E3_AA.seq 240
N
F
TGGTG
I
N
K
S
L
H
III
D
V
E
H
Q
D
W
R
D
V
R
P
T
AA
E
LA
D
TS
K
S
IMFV
D
W
I
R
VY
K
PV
TSS
A
TSSS
299
BR_E3AA.seq 301
NS
V
SSSSSS
L
SNSSSSS
A
TT
VI
D
FA
N
YF
D
TG
K
ST
A
S
VA
G
D
T
YV
G
F
N
K
SGSGN
I
N
Y
NT
A
G
D
360
E3_AA.seq 300
NS
V
SSSSSS
L
SNSSSSS
A
TT
VI
D
FA
N
YF
D
TG
K
ST
A
S
VA
G
D
T
YV
G
F
N
K
SGSGN
I
N
Y
NT
A
G
D
359
BR_E3AA.seq 361
W
A
D
YLV
T
LP
S
D
GQ
Y
N
I
E
V
TT
A
S
PM
TSG
I
G
A
K
L
S
I
D
G
IYV
STTS
LAA
TGGW
E
LY
S
A
ST
LA
N
420
E3_AA.seq 360
W
A
D
YLV
T
LP
S
D
GQ
Y
K
I
E
V
TT
A
S
PLI
SG
I
G
A
K
L
S
I
D
G
IYV
STTS
LAA
TGGW
E
LY
S
A
ST
LA
N
419
BR_E3AA.seq 421
N
L
S
I
G
A
GT
H
T
V
R
I
E
STGTS
A
WQWNG
DE
I
R
V
T
K
V
GS
AAV
GS
P
TT
PAPVAMII
Q
A
E
S
F
S
A
TG
480
E3_AA.seq 420
N
L
S
I
G
A
GT
H
T
V
R
I
E
S
A
GTSTWQWNG
DE
I
R
V
T
K
V
GS
AAV
GS
P
TT
PAPVAMII
Q
A
E
S
F
S
A
TG
479
BR_E3AA.seq 481
G
VY
D
G
F
K
T
Y
S
V
NG
V
S
AI
N
Y
NQ
R
G
D
W
A
D
YAI
N
VAA
D
GS
Y
T
F
N
AYV
SS
PM
SG
AAL
E
V
S
V
D
G
V
540
E3_AA.seq 480
G
VY
D
G
F
K
T
Y
S
V
NG
V
S
AI
N
Y
NQ
R
G
D
W
A
D
YAI
N
VAA
D
GS
Y
T
F
N
AYV
SS
PM
SG
AAL
E
V
S
V
D
G
V
539
BR_E3AA.seq 541
K
VL
TQ
AVP
NNGSW
D
S
F
Q
K
V
SS
V
S
K
IAL
T
K
G
A
H
T
I
R
V
TS
A
G
A
TSSTW
E
WN
A
D
K
F
E
LVP
597
E3_AA.seq 540
K
VL
TQ
AVP
NNGSW
D
S
F
Q
K
V
SS
V
S
K
IAL
T
K
G
A
H
T
I
R
V
TS
A
G
A
TSSTW
E
WN
A
D
K
F
E
LVP
596
Cellvibrio sp. BR AgaB
23
図
11 組み換え大腸菌の粗酵素液による
ネオアガロビオース分解の経時変化
図
12 組み換え大腸菌の粗酵素液による加水分解産物の
24 結言
Cellvibrio sp. OA-2007 由来のβ―アガラーゼ遺伝子 agaB をBrevibacillusへ導入し、 酵素と寒天オリゴ糖の菌体外同時生産に成功した。Cellvibrio sp. BR のゲノム DNA 配列を 基に、Cellvibrio sp. OA-2007 由来のα―アガラーゼ遺伝子の大腸菌へのクローニングに成 功した。
25 謝辞 本研究を行うに当たり多大な御助力と厳しくも温かい御指導を賜りました高知工科大学 工学部 物質・環境システム工学科 有賀修 准教授に心からの深謝を申し上げます。本論文 の審査において御助言を賜りました、物質・環境システム工学科 榎本恵一 教授、堀澤栄 准 教授に深謝を申し上げます。本研究で用いた組み換え菌を作成し、遺伝子組み換え実験に ついて一から御教授賜り、御助力頂いた大濱武 教授に感謝を申し上げます。有益な御助言 を頂いた細川覚司氏に感謝を申し上げます。最後に日頃から御協力いただいた有賀研究室 の先輩方、後輩方、物質・環境システム工学科の先生方、友人方に感謝を申し上げます。
26 参考文献
【1】 Sugano,Y.,Kodama,H.,Terada,I.,Yamazaki,Y.,Noma,M,Purification and Characterization of a Novel Enzyme, α―Neoagarooligosaccharide Hydrolase,
from a Marine Bacterium, Vibrio sp. Strain JT0107(1994)
【2】 Suzuki,H.,Sawai,Y.,Suzuki,T.,Kawai,K.Purification and Characterization of an Extracellular α―Neoagarooligosaccharide Hydrolase from Bacillus sp. MK03(2002)
【3】 Araki,T.,Hayakawa,M.,Lu,Z.,Karita,S.,Morishita,T.Purification and
characterization of agarases from a marine bacterium,Vibrio sp. PO-303(1998)
【4】 Sugano,Y.,Terada,I.,Arita,M.,Noma,M.,Matsumoto,T.Purification and
characterization of a new agarase from a marine bacterium,Vibrio sp. Strain JT0107(1993) 【5】 中村光輝, 寒天オリゴ糖の選択的生産方法の開発. 修士論文(2004). 【6】 井上貴由, Cellvibrio sp.からの新規β-アガラーゼ遺伝子のクローニングとネオア ガロオリゴ糖生産. 修士論文(2008). 【7】 久保元, Cellvibrio sp. OA-2007 由来の組み換えβ-アガラーゼの精製とキャラクタ ライゼーション.修士論文(2009) 【8】 Ariga,O.,Inoue.T.,Kubo,H.,Minami,K.,Nakamura,M.,Iwai,M.,Moriyama,H.,Yan agisawa,M.,Nakasaki,K. Cloning of agarase gene from non-marine agarolytic bacterium Cellvibrio sp.(2012)
27 補足 ・1M トリス塩酸緩衝液(密栓して、オートクレーブ滅菌) Tris(hydroxymethyl)aminomethane(SIGMA)60.55g を 400ml の蒸留水に溶かし、 塩酸を加えながらpH8.0 に合わせ、500ml にメスアップした。 ・トリス・フェノール(遮光して冷蔵保存) 結晶フェノール(SIGMA)500g をビンごと 70℃のウォーターバスに入れ、試薬を 溶かす。溶解したフェノールをフタのできる大きめのガラスビンに移し、トリス塩酸緩衝 液400ml とキノリノール(SIGMA)0.5g を加え、ビンのフタを閉めて 30 分間よく混合し た。その後、上層液をガラスピペットで取り除き、さらに0.5M トリス塩酸緩衝液 400ml を加えた。 ・0.5M EDTA EDTA2Na・2H2O(同仁化学研究所株式会社) 93.06g 固形NaOH(和光純薬工業株式会社:Wako) 10g 5N NaOH 適量 400ml の蒸留水に EDTA を懸濁し、撹拌しながら pH を測定した後、固形 NaOH を徐々に 加え5N NaOH で pH8.0 に調節した。蒸留水で 500ml に調整した後、オートクレーブ滅菌 した。 【1】TE 溶液(オートクレーブ滅菌) 1M トリス塩酸緩衝液 5ml 0.5M EDTA 1ml 蒸留水 全量 500ml に調整 【2】2% CTAB 溶液
28 1M トリス塩酸緩衝液 20ml 0.5M EDTA 8ml 5M NaCl(SIGMA) 56ml Polyvinylpyrrolidone 2g 上記の各試薬を混合し、蒸留水で全量200ml に調整した。 【3】10mg/ml RNaseA(密栓して 15 分間煮沸した。) 牛膵臓RNaseA(TaKaRa Bio) 0.1g 1M トリス塩酸緩衝液, pH7. 0.1ml 5M NaCl(SIGMA) 0.03ml 滅菌水 9.87ml 【4】フェノール・クロロホルム溶液(遮光して冷蔵保存) トリス・フェノールとCIA を等量ずつ入れて混合した。 【5】クロロホルム・イソアミルアルコール混合液(CIA)(室温で保存) クロロホルム(SIGMA)とイソアミルアルコールを 24:1 の比率で混合した。 【6】1% 臭化エチジウム溶液(遮光して室温保存) 臭化エチジウム(Wako) 2g 蒸留水 200ml スターラーバーで一晩撹拌した。 【7】ネオマイシン保存溶液(冷凍保存) ネオマイシン(SIGMA)1g を滅菌水 5ml に溶かし、保存溶液とした。使用濃度は 100μg/ml。 【8】アンピシリン保存溶液(冷凍保存) アンピシリンナトリウム(Wako)1g を滅菌水 10ml に溶かし、保存溶液とした。使
29 用濃度は100μg/ml。 使用機材 ・電気泳導装置 GelMate2000(東洋紡績株式会社) ・加温機 BLOCK INCUBATOR(株式会社アステック) CI-410(アドバンテック東洋株式会社) ・遠心分離機 himac CF15D2(日立工機株式会社:HITACHI) ・トランスイルミネーター TDM-15(UVP 社) ・PCR 装置 GeneAmp PCR system 9700(Applied Biosystems Japan Ltd.)
・遺伝子情報処理ソフトフェア GENETYX Ver.8・Windouws 版(株式会社ゼネティック ス)