博
士
学
位
論
文
脂質・糖代謝制御を指向した MGAT2 および GPR119
リガンドの分子設計と合成ならびに生物学的評価
博
士
学
位
論
文
脂質・糖代謝制御を指向した MGAT2 および GPR119
リガンドの分子設計と合成ならびに生物学的評価
平成 27 年 10 月 14 日
目 次
序論 1 第一章 肥満および糖尿病の現状と課題 1 第二章 MGAT2 に関して 5 第三章 GPR119 に関して 7 本論 9 第一章 N-フェニルインドリン-5-スルホンアミド系 MGAT2 阻害剤の分子設計、合成と 生物活性 9 第一節 N-フェニルインドリン-5-スルホンアミド系リード化合物の創出 9 第一項 研究方針と分子設計 9 第二項 合成 10 第三項 生物活性と考察 14 第二節 経口吸収性改善に向けた構造最適化 18 第一項 研究方針と分子設計 18 第二項 合成 19 第三項 生物活性と考察 20 第三節 CYP3A4 に対する時間依存性阻害活性回避に向けた構造最適化 24 第一項 研究方針と分子設計 24 第二項 合成 25 第三項 生物活性と考察 27 第四節 活性増強と光毒性回避に向けた構造最適化 29 第一項 研究方針と分子設計 29 第二項 合成 30 第三項 生物活性と考察 32第五節 ピラゾリルピリミジン誘導体のプロファイルおよび経口脂質負荷試験 36 第六節 本章まとめ 39 第二章 インドリンカルバマートおよびインドリニルピリミジン系 GPR119 作動性 リガンドの分子設計、合成と生物活性 40 第一節 インドリンカルバマート系 GPR119 作動性リガンドの分子設計、合成 と生物活性 40 第一項 研究方針と分子設計 40 第二項 合成 40 第三項 生物活性と考察 43 第二節 インドリニルピリミジン系 GPR119 作動性リガンドの分子設計、合成 と生物活性 46 第一項 研究方針と分子設計 46 第二項 合成 47 第三項 生物活性と考察 49 第三節 化合物 104 および 121 の経口糖負荷試験 52 第四節 本章まとめ 54 第三章 総括 55 実験の部 57 引用文献 109 謝辞 発表論文一覧
序論
第一章 肥満および糖尿病の現状と課題
肥満は世界中で増加の一途をたどっており、先進国のみならず新興国や開発途上国でも深刻な 社会問題となっている。世界保健機関(World Health Organization、WHO)の報告によると、2014 年時点で 19 億人の成人が過体重状態にあり、6 億人の成人が肥満状態にあると推定されている 1。また、4000 万人を超える 5 歳以下の小児も過体重もしくは肥満状態にあると報告されている。 日本国内では、特に成人男性における肥満者の増加が顕著であり、成人男性の約 30%が肥満で あると推定されている2。 肥満とは体内の脂肪組織が過剰に蓄積された状態であり、エネルギーの過剰摂取や消費エネル ギーの減少により、相対的なエネルギー過剰状態となって引き起こされる。肥満度の分類には
body mass index(BMI)が広く使われており、その計算式と日本肥満学会3および WHO4による
肥満度の分類を Table 1 に示す。 肥満は、糖尿病や脂質異常症などの生活習慣病 5、心疾患や脳卒中などの循環器疾患 6、変形 性 関 節 症 な ど の 筋 骨 格 疾 患 7、 大 腸 癌 や 乳 癌 な ど の 癌 疾 患 8-10 と い っ た 非 感 染 性 疾 患 (Non-Communicable Diseases, NCDs)のリスクファクターとなることが報告されている。日本肥 満学会では、肥満に起因ないし関連する 11 の疾患(耐糖能障害、脂質異常症、高血圧、高尿酸 血症・痛風、冠動脈疾患、脳梗塞、脂肪肝、月経異常・妊娠合併症、睡眠時無呼吸症候群・肥満 低換気症候群、整形外科的疾患、肥満関連腎臓病)のうち 1 つ以上の健康障害を合併するか、そ の合併が予測される場合に肥満症として治療が必要な疾患と診断している3。また、肥満(特に 内臓脂肪型肥満)を背景基盤として、耐糖能異常、脂質代謝異常、高血圧といった動脈硬化危険 因子が集積する状態はメタボリックシンドロームという概念で広く受け入れられており11、これ らの危険因子が複数重なり合うことで冠動脈疾患の発症率が急激に上昇していくことがわかっ ている12, 13。すなわち、肥満状態の是正は、関連する様々な疾患の予防と治療の観点から極めて 重要である。 肥満症の治療は、摂取エネルギーと消費エネルギーのバランス調整を基本コンセプトとする。 初期には食事療法や運動療法などのライフスタイル改善が基礎となるが、これら非薬物療法のみ Table 1. BMI による肥満度の分類
Table 2. 日本および米国で承認されている代表的な肥満症治療薬 の効果は限定的であり、薬物療法に対する需要と期待度は高い14, 15。肥満症治療薬には中枢性と 末梢性のものがあり、期待できる主な作用機序として下記の 3 つが挙げられる16。摂取エネルギ ーを低下させる機序として、1)中枢神経系に対する作用を介した食欲の抑制(中枢性食欲抑制 薬)、2)末梢性作用を介した体内への栄養素吸収阻害(消化吸収阻害薬)、また消費エネルギー を増加させる機序として、3)末梢で摂取したエネルギーを熱に変えて放出させるエネルギー消 費促進(熱産生促進薬)、である。これらのうち、中枢性食欲抑制薬に関しては、これまでにい くつかの治療薬が承認に至っているが、末梢性の治療薬で承認されているのは膵リパーゼ阻害薬 (消化吸収阻害薬)のみである。現在、日本および米国で承認されている代表的な肥満症治療薬 を Table 2 に示す。日本で臨床利用されている肥満症治療薬は中枢性食欲抑制薬であるマジンド ールのみである。しかし、薬理学的特性がアンフェタミンと類似していることから BMI35 以上 の高度肥満患者のみの適応で、投与期間も 3 ヶ月に限定されている。2013 年に膵リパーゼ阻害 薬のセチリスタットが二型糖尿病および脂質異常症を合併した肥満症に対する治療薬として日 本で承認されたが、保険収載には至っていない。また最近、米国で複数の新規な抗肥満薬が承認 されたが17, 18、いずれも中枢性食欲抑制薬である。中枢性摂食抑制薬では中枢性の副作用が問題 となる場合が多く、過去に開発された中枢性食欲抑制薬の一つであるリモナバン(カンナビノイ ド受容体(CB1)拮抗薬)は、臨床試験において有意な抗肥満作用を示した一方、うつや自殺企 図などのリスクが高まることが判明したため、開発が中止されている19。このような肥満症治療 薬の開発状況の中で、高い安全性と有効性が期待できる末梢性の作用機序を有する新規な肥満症 治療薬の開発が切望されている。 糖尿病は糖代謝の異常によって高血糖を呈する進行性の疾患であり、その 90%を占める二型 糖尿病はインスリンの分泌不足もしくはインスリン抵抗性を特徴とする。世界の糖尿病患者数は 2013 年時点で 3 億 8 千万人と推定され、その数は 2035 年までに 5 億 9 千万人に増えることが見 込まれており20、糖尿病は肥満と並んで世界規模の重大なヘルスケア問題となっている。日本に おいても成人男性の 16%、また成人女性の 9%は糖尿病が強く疑われる状態にある2。前述した ように、肥満は糖尿病のリスクファクターと考えられており、二型糖尿病患者の 50–90%が肥満
Table 3. 代表的な経口糖尿病治療薬 状態にあると報告されている21。肥満と糖尿病の関連性の高さから、diabesity という概念も導入 されている5, 22。肥満における内臓脂肪の過剰な蓄積は、遊離脂肪酸による脂肪毒性(lipotoxicity) に加えて、脂肪細胞のアディポサイトカイン分泌異常による脂肪細胞毒性(adipotoxicity)を引 き起こし、これらがインスリン抵抗性惹起や二型糖尿病発症に関与していると考えられている 23。 慢性的な高血糖状態は、網膜症、腎症、神経障害等の微小血管障害による合併症を引き起こす。 さらに、虚血性心疾患、脳梗塞、動脈硬化等の大血管障害による合併症を誘発し、QOL の低下 のみならず、患者の高い死亡率につながっている24。したがって、これらの重篤な合併症リスク を低減させるために、適切な血糖コントロールは極めて重要である。糖尿病の治療には、運動療 法・食事療法に加えて、ビグアナイド薬、スルホニル尿素薬、グリニド薬、チアゾリジン薬等の メカニズムの異なる複数の血糖低下薬が使用されている(Table 3)。しかし、目標とする血糖値 に到達できていない患者の割合は多く25-28、長期間にわたる持続的な血糖コントロールという面 において高いアンメットニーズが存在する29, 30。このような観点から、新たなメカニズムの血糖 低下薬としては、低血糖や体重増加等の副作用が無いことに加えて、糖尿病の発症や進展に関与 する肥満状態を是正する作用や、持続的な血糖低下作用に寄与すると考えられる膵細胞の保護 作用を併せ持つことが重要である。 近年、新規な糖尿病治療薬としてインクレチン関連薬が注目されており、DPP-IV 阻害薬(経 口剤)および GLP-1 作動薬(注射剤)が実際に臨床利用されている31-33。インクレチンは膵細 胞からのインスリン分泌を促進する消化管ホルモンの総称であり、これまでに GLP-1 と GIP の 二種類が確認されている34, 35。GLP-1 は小腸下部に存在する L 細胞から、GIP は小腸上部に存在 する K 細胞から分泌され、インスリン分泌促進作用の他にも様々な作用を有する(Table 4)。イ ンクレチンによる膵細胞からのインスリン分泌作用はグルコース濃度依存的であるため、イン クレチン作用による低血糖誘導リスクは低いと考えられ、また膵細胞の保護作用を示す実験結
Table 4. インクレチンの代表的な作用 果が報告されている36。加えて、GLP-1 は食欲抑制や胃排出抑制作用を有することから34, 35、イ ンクレチン作用の増強は体重低下作用につながることが期待できる。前述したように、肥満は糖 尿病のリスクファクターとなるため、体重低下作用を有する糖尿病治療薬の創出は非常に意義深 い。したがって、生体内でインクレチンの分泌を促進し、その作用を増強させる経口低分子治療 薬は、次世代の優れた糖尿病治療薬となる可能性がある。
第二章 MGAT2 に関して トリアシルグリセロール(TG)は食事性脂肪の主成分であり、その消化管における吸収は多 段階を経由して効率的に行われている。具体的には、膵リパーゼによる TG のモノアシルグリセ ロール(MG)と脂肪酸への分解、分解産物の腸細胞への取り込み、腸細胞内での TG 再合成、 およびカイロミクロン構築を経て、TG は全身へと運ばれる(Figure 1-A)37-39。TG はヒトの重 要なエネルギー源の一つである一方、過剰な TG 摂取は脂肪細胞の肥大化・増殖を引き起こし、 肥満を誘導する40。したがって、TG の吸収過程の阻害は、肥満症治療の有効な手段になると期 待できる。 モノアシルグリセロールアシルトランスフェラーゼ(MGAT)は、MG とアシル CoA からジ アシルグリセロール(DG)を合成する酵素であり、これまでに三種のサブタイプ(MGAT1–3) が報告されている41, 42。その中で、MGAT2 は小腸に高発現している酵素であり、腸管内で TG 再合成の重要な役割を担っていると考えられている。実際に、最近報告された MGAT2 のノック アウトマウスでは、ワイルドタイプマウスと比べて脂肪負荷後に血中へ流入する TG 量が有意に 低下した43。さらに MGAT2 ノックアウトマウスは、高脂肪食負荷によって誘導されるる肥満、 耐糖能異常、高コレステロ-ル血症、脂肪肝等に対する保護作用を示した43-45。興味深いことに、 MGAT2 ノックアウトマウスではエネルギー消費の亢進作用が見られ、TG 吸収阻害作用に加え て、エネルギー消費促進作用が抗肥満効果に寄与していると考えられる。また、膵リパーゼ阻害 による脂肪吸収抑制作用を有するオルリスタットに関しては、副作用として脂肪便が報告されて いるが46、MGAT2 ノックアウトマウスではこのような脂肪便は観察されなかった。 著者は、上記の知見に基づいて、MGAT2 の阻害が末梢における脂質代謝を制御することによ って肥満症の新たな治療法になると考え(Figure 1-B)、経口吸収性を有する低分子 MGAT2 阻害 剤の創出に取り組んだ。研究開始当時、低分子 MGAT2 阻害剤の論文報告は無く47, 48、またこれ までに MGAT2 阻害剤を用いた臨床試験も報告されていない。著者はハイスループットスクリー ニングによって見出したスルホンアミド誘導体 1 を基点として合成展開を実施し、強力な
Figure 1. A) Schematic illustration of TG absorption in the small intestine. B) Postulated mechanism of anti-metabolic disease effects by MGAT2 inhibition.
Figure 2. Generation of N-phenylindoline-5-sulfonamide-based MGAT2 inhibitors.
MGAT2 阻害活性と優れた経口吸収性、および良好な化合物プロファイルを有する N-フェニルイ ンドリン-5-スルホンアミド誘導体 77c を見出した(Figure 2)。本化合物は、マウスを用いた経口 脂質負荷試験において、ノックアウトマウスで観察されたものと同様の有意な血中 TG 増加抑制 作用を示した。本論第一章にて、その分子設計と合成、および生物活性評価に関して詳述する。
第三章 GPR119 に関して
GPR119 は膵細胞および消化管に発現しているロドプシン-グループに属する G 蛋白質共役
型受容体(G-protein coupled receptor、GPCR)である49-53。GPR119 は Gs 蛋白質と共役しており、
その活性化は細胞内 cAMP の増加を引き起こす。興味深いことに、膵細胞および消化管に発現 している GPR119 は、活性化された後にそれぞれ異なる作用を発現する。膵細胞の GPR119 の 活性化はグルコース濃度依存的なインスリン分泌を促進する一方、消化管に発現している GPR119 の活性化はインクレチンを含む数種の消化管ホルモンの分泌を誘導する。小腸下部の L 細胞からは GLP-1 や PYY が、また小腸上部の K 細胞からは GIP が GPR119 の活性化によって分 泌される。分泌されたインクレチンもインスリン分泌作用を有しているため34, 35、GPR119 の作 動性リガンドは膵細胞に対する直接的な作用とインクレチンを介した間接的な作用の両方で 血糖低下作用に寄与できる。さらに、分泌された GLP-1 や GIP による膵細胞の保護作用や36、 GLP-1 や PYY による摂食抑制や胃排出遅延作用を介した抗肥満効果も期待できる34, 35。このよ うに、GPR119 の活性化は多様なメカニズムを介して糖代謝改善につながると考えられる(Figure 3)。 当初、GPR119 はオーファン GPCR として見出されたが、その後 oleoylethanolamide や MG を 始めとする複数の脂肪酸誘導体が内因性リガンドとして報告され54、fat sensor としての機能と役 割が議論されている 55。実際に、GPR119 は食事による GLP-1 分泌に関与しており56、MG が GPR119 の内因性リガンドとして重要な役割を果たしていると考えられている。しかし、報告さ れている内因性リガンドはいずれも GPR119 の作動活性が弱く55、GPR119 の活性化には高濃度 の内因性リガンドを要する。現在までに、より強力な活性を有する非内因性の GPR119 作動性リ ガンドが報告されており51, 52, 57, 58、臨床試験の成績から GPR119 作動性リガンドのヒトでの高い 安全性と認容性が見込まれている59-62。 筆者は、強力な GPR119 作動活性を有する低分子リガンドが、低血糖リスクの無い強力な血糖 低下作用に加え、膵細胞の保護作用や抗肥満効果を併せ持つ新たな糖尿病治療薬になると考え、
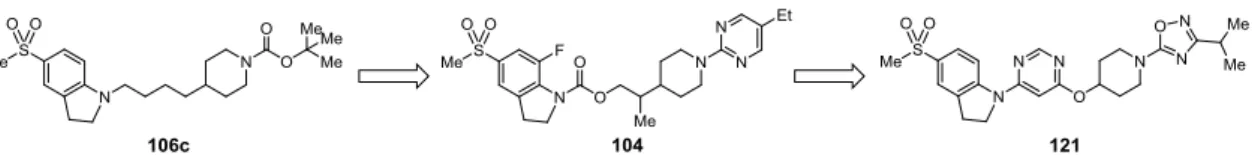
Figure 4. Generation of indoline carbamate- and indolinylpyrimidine-based GPR119 agonists. 新規リガンドの探索研究を実施した。初期リード化合物として見出したインドリン誘導体 106c からの合成展開を実施し、強力な活性を有するインドリンカルバマート誘導体 104 およびインド リニルピリミジン誘導体 121 を見出した(Figure 4)。これら二化合物は、二型糖尿病モデルラッ トを用いた経口糖負荷試験において、グルコース濃度依存的なインスリン分泌作用と血糖低下作 用を示した。本論第二章にて、その分子設計と合成、および生物活性評価に関して詳述する。
本論
第一章 N-フェニルインドリン-5-スルホンアミド系 MGAT2 阻害剤の分子設計、合成と生物活性
第一節 N-フェニルインドリン-5-スルホンアミド系リード化合物の創出
第一項 研究方針と分子設計
新規な MGAT2 阻害剤を創出する目的で、自社化合物ライブラリーのハイスループットスクリ
ーニングを行い、IC50 = 130 nM の MGAT2 阻害活性を示す化合物 1 を見出した(Figure 5-A)。初
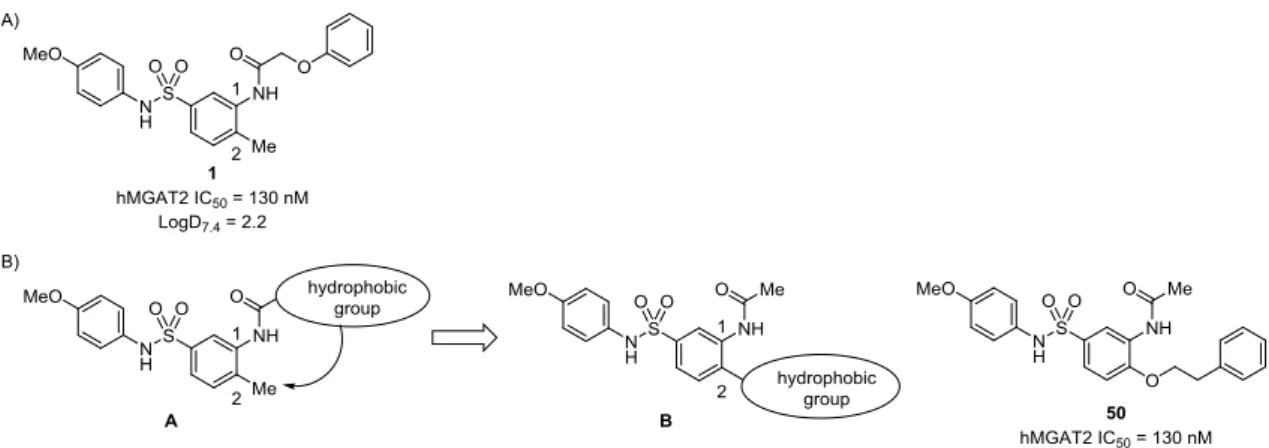
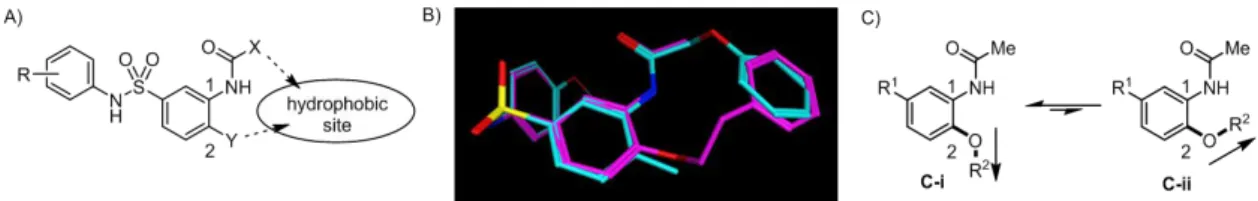
期の構造活性相関研究で、N-アリールベンゼンスルホンアミド構造(ArNHSO2Ar)は活性発現 に必須であり、このモチーフに対する構造変換は活性消失または大きな活性減弱につながること を明らかにした。一方、中央ベンゼン環 1 位の脂溶性置換基は活性発現に重要であるものの、構 造修飾に許容性を示した。MGAT2 が MG およびアシル CoA といった長鎖脂肪酸誘導体を基質 とする酵素であることから、リガンドの脂溶性部位が MGAT2 との親和性獲得に重要であり、脂 溶性置換基を適切な位置に配置することで MGAT2 阻害活性を増強できると考えた。 上記の仮説に基づいて脂溶性置換基の再配置を検討した結果、中央ベンゼン環 2 位へ脂溶性置 換基をシフトした化合物 50 が化合物 1 と同等の活性を示すことを見出した(Figure 5-B)。筆者 は、これら二化合物の分子右側のベンゼン環は MGAT2 との結合時に同一の脂溶性サイトを占有 していると考え、中央ベンゼン環 1 位と 2 位の間に MGAT2 の脂溶性サイトが存在するものと推 定した(Figure 6-A)。Figure 6-B に示した化合物 1 および 50 の重ね合わせ(Molecular Operating
Environment software(MOE)63の flexible alignment 機能を用いて計算)から、両化合物の分子右
側のベンゼン環が良い重なりを示すことを確認した。しかし、化合物 50 のフェネチルオキシ部
は、隣接位置換基の影響のためにコンフォメーション C-ii より C-i を好むと考えられ(Figure 6-C)、
Figure 6-B に示した化合物 50 のコンフォメーションはエネルギー的に不利な状態にある。そこ で、脂溶性置換基のコンフォメーションを制御する目的で、分子中央部に二環性骨格を導入した
Figure 5. A) Structure of an initial lead compound 1. B) Repositioning of a hydrophobic group from the 1-position to the 2-position.
Figure 6. A) Presumed hydrophobic space between the 1- and 2-positions. B) Flexible alignment of compounds 1 (cyan) and 50 (magenta) using MOE. C) Conformational preference of the alkoxy (OR2) moiety.
Figure 7. Design of orienting a substituent to a presumed hydrophobic site by introduction of bicyclic core systems. D の一般式で表される化合物をデザインした(Figure 7)。この分子設計により、活性発現に重要 な脂溶性置換基が適切な位置に配置され、MGAT2 と相互作用する際のエントロピー損失が減少 して、阻害活性が増強すると期待した。 第二項 合成 インドリン-5-スルホンアミド誘導体 10a–f は、Scheme 1 に示す方法を用いて合成した。5-ブロ モ-7-ニトロインドリン(2)を出発原料とし、インドリン窒素原子を Cbz 基で保護した後、ベン ジルメルカプタンとのパラジウムカップリング反応を行って、ベンジルスルフィド 4 を合成した。 スルホニルクロリド 5 の調製には、含水酢酸溶媒と NCS を用いた酸化的クロロ化反応を適用し 64、高収率で目的物を得ることができた。得られた 5 とアニリンとのスルホンアミド化反応は、 DMA を溶媒として用いることで、塩基の添加を必要とすることなく円滑に反応が進行し、化合 物 6a および 6b を合成した。続いて、ニトロ基の還元と生じたアミノ基のアセチル化を行って 化合物 8a、8b へと誘導した後、Cbz 基を除去して化合物 9a および 9b を得た。最後に、各種ア ルデヒドとの還元アルキル化反応を行ってインドリン環 1 位へ置換基を導入し、目的とする 10a– f を合成した。 化合物 10b からの誘導化を Scheme 2 に示す。化合物 10b のインドリン環は DDQ を用いるこ とで効率的に酸化することができ、対応するインドール誘導体 11 を合成した。一方、酸性条件 下で N-アセチル基を除去した後、ホルムアルデヒドとの還元アルキル化反応を行って、N-メチ ル体 13 へと誘導した。 インドリン環 7 位に置換基を有さない化合物は、Scheme 3 に示す方法で合成した。インドリ ン窒素原子を Boc 基で保護した 15 を調製した後、Scheme 1 に示した方法と同様の方法を用いて 目的とする化合物 20 を合成した。
Scheme 1. Synthesis of indoline-5-sulfonamide derivatives 10a–fa
aReagents and conditions: (a) NaH, CbzCl, THF, 0 °C to rt, 84%; (b) BnSH, Pd
2(dba)3, Xantphos,
DIEA, toluene, reflux, 73%; (c) NCS, AcOH, H2O, 0 °C to rt, 97%; (d) 4-methoxyaniline or
2,4-difluoroaniline, DMA, rt, 86–96%; (e) Fe, CaCl2, EtOH, H2O, reflux, 66% (for 7a); (f) Zn, AcOH, rt,
94% (for 7b); (g) AcCl, DMA, 0 °C to rt, 89–94%; (h) H2 (1 atm), Pd on carbon, THF, MeOH, rt, 92–
99%; (i) aldehyde, NaBH(OAc)3, AcOH, CH3CN, rt, 27–68% (for 10a–e); (j) PhCH2CH2CHO,
2-picoline-borane complex, MeOH, AcOH, rt, 93% (for 10f).
Scheme 2. Synthesis of indoline-5-sulfonamide derivatives 11 and 13a
a
Reagents and conditions: (a) DDQ, THF, rt, 54%; (b) HCl, H2O, MeOH, 80 °C, 58%; (c)
formaldehyde, NaBH(OAc)3, AcOH, THF, rt, 29%.
インダゾール誘導体 28 は、Scheme 4 に示す方法で合成した。鍵中間体となる 5-ブロモ-7-ニト ロインダゾール(22)は、o-トルイジン 21 のジアゾ化と続く自己環化反応によって合成した65。 化合物 22 と 2-(4-フルオロフェニル)エチルブロミドとの N-アルキル化反応では、位置選択性が 見られず、目的とする 1 位置換体 23 を 42%の収率で得た。生成物の構造は NMR 実験によって 決定し、インダゾール 3 位水素原子とフェネチル部 1'位水素原子の間で NOE クロスピークが観 察された異性体を 2 位置換体 23'とした(Figure 8)。得られた化合物 23 のインダゾール 5 位およ
Scheme 3. Synthesis of indoline-5-sulfonamide derivative 20a
aReagents and conditions: (a) Boc
2O, THF, rt, 95%; (b) BnSH, Pd2(dba)3, Xantphos, DIEA, toluene,
reflux, 100%; (c) NCS, AcOH, H2O, 0 °C to rt, 64%; (d) 2,4-difluoroaniline, pyridine, 0 °C to rt, 87%; (e)
HCl, AcOEt, rt, 86%; (f) PhCH2CHO, NaBH(OAc)3, AcOH, CH3CN, rt, 70%.
Scheme 4. Synthesis of 1H-indazole-5-sulfonamide derivative 28a
a
Reagents and conditions: (a) NaNO2, AcOH, H2O, rt, 56%; (b) 2-(4-fluorophenyl)ethyl bromide,
Cs2CO3, DMF, rt, 42%; (c) BnSH, Pd2(dba)3, Xantphos, DIEA, toluene, reflux, 92%; (d) NCS, AcOH,
H2O, 0 °C to rt, 92%; (e) 2,4-difluoroaniline, DMA, rt, 87%; (f) H2 (1 atm), Pd on carbon, THF, MeOH,
rt, 95%; (g) AcCl, DMA, rt, 46%.
Figure 8. The isomer for which NOE cross peaks were observed between H-3 on the indazole ring and the H-1' of the phenethyl moiety was determined as 23'.
Scheme 5. Synthesis of 1H-benzimidazole-5-sulfonamide derivatives 35 and 36a
aReagents and conditions: (a) (1) ClSO
3H, 100 °C, (2) 2,4-difluoroaniline, DMA, rt, 72%; (b) (1)
NaNO2, CuCl2, H2SO4, HCl, AcOH, rt to 70 °C, (2) 4-fluorophenethylamine, THF, rt, 60%; (c) H2 (1 atm),
Pd on carbon, THF, rt, 76%; (d) formic acid, reflux, 96%; (e) HCl, MeOH, H2O, rt to 60 °C, 97%; (f)
AcCl, DMA, rt, 91%; (g) (1) AcOH, reflux, (2) AcCl, DMA, rt, 70%.
び 7 位置換基をそれぞれ Scheme 1 と同様の方法を用いて変換し、目的とする化合物 28 へ誘導し た。 ベンズイミダゾール誘導体 35 および 36 は、Scheme 5 に示す方法で合成した。まず、2,6-ジニ トロアニリンをクロロ硫酸で処理して直接的にクロロスルホニル化し66、生成したスルホニルク ロリドと 2,4-ジフルオロアニリンとのスルホンアミド化反応を行って化合物 30 を得た。続いて、 化合物 30 を Sandmeyer 反応に付し、4-フルオロフェネチルアミンとの求核置換反応を行って化 合物 31 を合成した。ニトロ基の還元を行ってトリアミノベンゼンスルホンアミド 32 へ誘導した 後、蟻酸中で加熱することでベンズイミダゾール環構築と N-ホルミル化が同時に進行し、化合 物 33 を高収率で得ることができた。最後に、化合物 33 の N-ホルミル基を N-アセチル基に変換 して、目的とする 35 を合成した。一方、化合物 32 を酢酸中で加熱した場合は、ベンズイミダゾ ール環形成のみが進行し、最後に未反応のアミノ基を N-アセチル化することで化合物 36 を合成 した。 1,3-ベンズオキサゾロン誘導体 45 の合成は、Scheme 6 に示す方法で合成した。2-アミノフェ ノール 3767を出発原料とし、N-アシル化とボラン還元の二工程を経てフェネチル基を導入した 化合物 39 を合成した。CDI を用いて 1,3-ベンズオキサゾロン骨格を構築した後、Scheme 1 と同 様の方法を用いて、化合物 40 から目的とする化合物 45 へ誘導した。 4-(2-フェニルエトキシ)ベンゼンスルホンアミド 50 は、Scheme 7 に示す方法で合成した。市販 のスルホニルクロリド 46 と 4-メトキシアニリンのスルホンアミド化反応により化合物 47 を合 成し、続いて 2-フェニルエタノールとの光延反応を行うことで化合物 48 へ誘導した。ニトロ基 の還元後、N-アセチル化を行って目的とする化合物 50 を合成した。
Scheme 6. Synthesis of 2-oxo-2,3-dihydro-1,3-benzoxazole-6-sulfonamide derivative 45a
aReagents and conditions: (a) phenylacetyl chloride, NaH, THF, 0 °C to rt, 42%; (b) borane-THF complex, THF, rt to 50 °C, 72%; (c) CDI, THF, rt, 81%; (d) BnSH, Pd2(dba)3, Xantphos, DIEA, toluene,
reflux, 91%; (e) NCS, AcOH, H2O, 0 °C to rt, 74%; (f) 2,4-difluoroaniline, DMA, rt, 73%; (g) Zn, AcOH,
0 °C to rt; (h) AcCl, DMA, rt, 35% over 2 steps.
Scheme 7. Synthesis of 4-(2-phenylethoxy)benzenesulfonamide derivative 50a
a
Reagents and conditions: (a) 4-methoxyaniline, DMA, rt, 89%; (b) 2-phenylethanol, DIAD, PPh3,
THF, toluene, 0 °C, 71%; (c) H2 (1 atm), Pd on carbon, THF, MeOH, rt, 91%; (d) Ac2O, pyridine, rt,
37%.
第三項 生物活性と考察
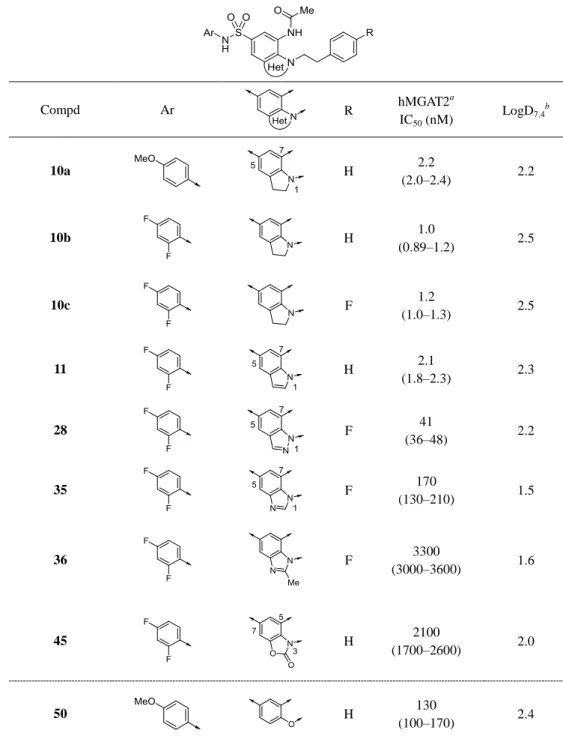
合成した化合物の in vitro MGAT2 阻害活性は、次の方法で評価した。FreeStyle293 細胞を用い て MGAT2 蛋白質を発現させ、その MGAT2 を含む膜画分を使用した DG 合成反応に対する化合 物の阻害率を求めた。Table 5 に、本節第一項の分子設計に基づいて合成した二環性化合物の MGAT2 阻害活性を示す。中央母核としてインドリン骨格を導入した化合物 10a は、期待した通
りに大幅な活性向上を達成し、IC50 = 2.2 nM の強力な MGAT2 阻害活性を示した。この活性向上
Table 5. MGAT2 inhibitory activity: SAR of central bicyclic systems Compd Ar R hMGAT2 a IC50 (nM) LogD7.4 b 10a H 2.2 (2.0–2.4) 2.2 10b H 1.0 (0.89–1.2) 2.5 10c F 1.2 (1.0–1.3) 2.5 11 H 2.1 (1.8–2.3) 2.3 28 F 41 (36–48) 2.2 35 F 170 (130–210) 1.5 36 F 3300 (3000–3600) 1.6 45 H 2100 (1700–2600) 2.0 50 H 130 (100–170) 2.4
aInhibitory activity against human MGAT2. IC
50 values are presented as means of duplicate
experiments with their 95% confidence intervals in parentheses. bLogD value at pH 7.4.68
結果であると考える。インドリン環 5 位アニリン部のベンゼン環上の電子密度は MGAT2 阻害活 性に大きな影響を与えず、2,4-ジフルオロアニリン誘導体 10b も化合物 10a と同等の強力な活性 を示した。また、インドリン環 1 位フェネチル基のベンゼン環上にフッ素原子を導入した 10c も同等の活性を示した。新たに見出したインドリン誘導体に続き、他の中央二環性骨格の導入を 検討した。インドール誘導体 11 はインドリン誘導体と同様に強力な活性を示したが、インダゾ
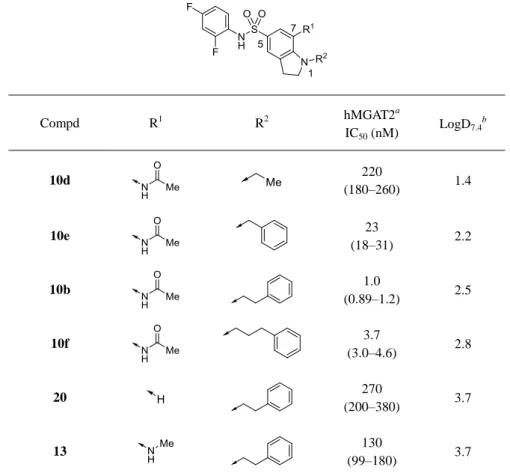
Table 6. MGAT2 inhibitory activity: SAR at the 1- and 7-positions of the indoline ring Compd R1 R2 hMGAT2 a IC50 (nM) LogD7.4 b 10d 220 (180–260) 1.4 10e 23 (18–31) 2.2 10b 1.0 (0.89–1.2) 2.5 10f 3.7 (3.0–4.6) 2.8 20 270 (200–380) 3.7 13 130 (99–180) 3.7
aInhibitory activity against human MGAT2. IC
50 values are presented as means of duplicate
experiments with their 95% confidence intervals in parentheses. bLogD value at pH 7.4.68
ール誘導体 28 やベンズイミダゾール誘導体 35 はインドリン誘導体と比較して 10 倍以上活性が 減弱した。さらに、ベンズイミダゾール環 2 位へのメチル基の導入(36)やベンズイミダゾロン 骨格の導入(45)は、より顕著な活性減弱を引き起こした。本系統誘導体の中央二環性部分は、 MGAT2 と結合する際、空間的に限られた脂溶性サイトに位置しており、中央母核へのヘテロ原 子の導入や置換基の導入が MGAT2 に対する親和性の低下を招いたものと考える。以上の結果か ら、インドリン骨格およびインドール骨格が脂溶性置換基の配向を適切に制御しながら、強力な MGAT2 阻害活性を発現させる有用なスキャホールドとなることを見出した。化合物の物性、お よび合成展開の許容性の観点からインドリン骨格を選択し、インドリン環 1 位および 7 位に関す る構造活性相関情報の取得を実施した。 インドリン誘導体 1 位および 7 位の変換体の MGAT2 阻害活性を Table 6 に示す。化合物 10b の 1 位フェネチル部のベンゼン環を除去した化合物 10d は大幅に活性が減弱し、末端脂溶性置 換基の活性発現に対する重要性が確認された。1-フェニルプロピル体 10f はフェネチル体 10b と 同等の活性を示したことから、想定している MGAT2 の脂溶性サイトはより大きな脂溶性置換基 を許容できる可能性がある。一方、ベンジル体 10e では約 10 倍の活性低下が見られ、MGAT2
Table 7. Selectivity of 10b over other acyltransferases Compd hMGAT2 IC50 (nM) hMGAT3a IC50 (nM) hDGAT1a IC50 (nM) hDGAT2a IC50 (nM) hACAT1a IC50 (nM) 10b 1.0 > 30000 > 30000 > 30000 35000 a
IC50 values are presented as means of duplicate experiments.
との相互作用において 10e の末端ベンゼン環が好ましい位置まで到達できていないものと考え られる。また、インドリン環 7 位アセチルアミノ基の全て、もしくはカルボニル基を除去した場 合には活性が大きく減弱した(20、13)。上記の結果から、適切に配置されたインドリン環 1 位 の脂溶性置換基および 7 位カルボニル基の存在が本系統化合物の MGAT2 阻害活性発現に大きく 寄与していることを明らかにした。 続いて、新たに見出した N-フェニルインドリン-5-スルホンアミド誘導体の他のアシルトラン スフェラーゼに対する選択性を評価した。代表化合物として化合物 10b を選択し、MGAT3、
DGAT1、DGAT2、および ACAT1 に対する阻害活性評価を行った(Table 7)。その結果、化合物
10b は上記いずれの酵素に対しても 1 M 以上の IC50値を示し、非常に高い MGAT2 選択性を示 すことがわかった。 上述したように、ハイスループットスクリーニングを通じて見出した化合物 1 を基にしたリー ド創出を試み、脂溶性置換基の再配置とコンフォメーション制御を目的とした二環性中央骨格の 導入を行った。その結果、MGAT2 に対して強力かつ選択的な阻害活性を示す有望なリード化合 物として、化合物 10b に代表される N-フェニルインドリン-5-スルホンアミド誘導体を見出した。
第二節 経口吸収性改善に向けた構造最適化 第一項 研究方針と分子設計 前節で述べたように、MGAT2 に対して強力かつ選択的な阻害活性を示す有望なリード化合物 として、化合物 10b に代表される N-フェニルインドリン-5-スルホンアミド誘導体を見出した。 続いて、本系統の化合物の代謝安定性および体内動態を評価した。その結果、化合物 10b は in vitro 試験において肝ミクロソームによる代謝を受けて短時間で消失することが判明し、マウスを用い た動態試験においてもクリアラス値が高く、経口吸収性は低いことが明らかになった(Table 8、 CLtotal = 4977 mL/h/kg、F = 4.6%)。そこで、in vivo での評価に適した MGAT2 阻害剤を創出する
ために、経口吸収性の改善を目的とした構造変換に取り組んだ。 前述した in vitro 代謝安定性試験の結果から、経口吸収性を改善するためには酸化的代謝に対 する安定性を獲得する必要があると考えた。まず最初に、インドリン環 1 位の末端ベンゼン環の 酸化代謝を抑制する目的で、ベンゼン環上への置換基導入を行ったが、代謝安定性の大幅な改善 は見られなかった。この結果を受けて、インドリン環 1 位のアルキレンスペーサー部が酸化代謝 を受けている可能性を考えた。また、関連誘導体の in vitro 代謝物解析の結果から、インドリン 環 7 位の N-アセチル基が生体内で容易に加水分解を受ける可能性が示唆され、このことが低い 経口吸収性の一因になっていると考えた。これらの推定主代謝メカニズムを抑制する目的で、E で示す誘導体をデザインした(Figure 9)。すなわち、アルキレンスペーサー部の酸化代謝を抑制 するためにインドリン環 1 位へアシル型スペーサーを導入し、加えて N-アセチル基の加水分解 を抑制するためにインドリン環 7 位へラクタム等の環状構造を導入することで経口吸収性の改 善を試みた。
Table 8. Microsomal clearances and pharmacokinetic profiles of compound 10b
Compd
in vitro CLinta
(μL/min/mg) Pharmacokinetic profiles in mice
b, c HLM MLM CLtotal b (mL/h/kg) AUC0–8hc (ng·h/mL) Fc (%) 10b 101 220 4977 9.2 4.6
aHuman/mouse liver microsomal clearance. b0.1 mg/kg, iv. c1 mg/kg, po.
第二項 合成 インドリン環 7 位にヘテロ環を有する化合物は、Scheme 8 に示す方法で合成した。ラクタム 環形成の前駆体として、化合物 7b を 4-クロロブチリルクロリドまたは 5-クロロバレリルクロリ ドでアシル化した化合物 51a と 51b を調製した。これらの化合物を水素化ナトリウムで処理す ることにより、分子内環化反応が進行したラクタム 52a および 52b を合成した。環状ウレア誘 導体に関しては、化合物 7b とイソシアン酸アルキルとの反応で環化前駆体 51c と 51d を調製し、 ラクタム誘導体と同様の条件で環化反応を行って 52c および 52d を得た。また、環状カルバマー ト誘導体 52e は、クロロ蟻酸エチルを用いて同様の方法で合成した。得られた 52a–c の Cbz 基を 除去した後、インドリン環 1 位へ置換基を導入した。化合物 53a または 53b とフェニルアセト アルデヒドとの還元アルキル化反応においては、N-アセチル体 9b(Scheme 1)と反応性が異な り、トリアセトキシ水素化ホウ素ナトリウムを還元剤として用いる条件ではほとんど還元反応が 進行しなかった。この反応性の低下は、インドリン環 7 位に導入したヘテロ環による立体障害に 起因すると考えられる。目的とする還元アルキル化反応は、酸化白金触媒による水素添加条件を 用いることで効率的に進行することを見出し、化合物 54a および 54b を得ることができた。ま た、トリホスゲンをもちいたアニリン類とのウレア形成反応を行うことで、化合物 54c–f を合成 した。
Scheme 8. Synthesis of indoline-5-sulfonamide derivatives 52a–e and 54a–fa
aReagents and conditions: (a) 4-chlorobutyryl chloride or 5-chlorovaleryl chloride, DMA, 0 °C to rt; (b) 2-chloroethyl isocyanate or 3-chloropropyl isocyanate, THF, 60–70 °C; (c) 2-chloroethyl chloroformate, TEA, THF, 0 °C to rt; (d) NaH, DMF, 0 °C to rt, 59–92% over 2 steps (for 52a and 52b), 64–74% over 2 steps (for 52c and 52d), 11% over 2 steps (for 52e); (e) H2 (1 atm), Pd on carbon, THF, MeOH, rt, 94–
99%; (f) PhCH2CHO, H2 (1 atm), PtO2, THF, EtOH, rt, 41-62% (for 54a and 54b); (g) 4-substituted
第三項 生物活性と考察 合成した化合物に関しては、本章第一節に記載した方法で MGAT2 阻害活性を評価した。また、 代表化合物に関しては、in vitro 代謝安定性およびマウスへ経口投与した際の血中濃度を測定し た。本節第一項で述べた分子設計に基づいて、まずインドリン環 7 位へのラクタム環導入を検討 した(Table 9)。その結果、5 員環ラクタムのピロリドン誘導体 54a および 6 員環ラクタムのピ ペリドン誘導体 54b のいずれも強力な MGAT2 阻害活性を維持した。前節で述べた結果と合わせ ると、ラクタム環上のカルボニル基が活性発現において重要な役割を果たしていると考えられ、 MGAT2 との相互作用時に水素結合受容基(HBA)として機能している可能性がある。 インドリン環 7 位置換基上のカルボニル酸素原子の電子密度が活性に与える影響を考察する 目的で、ラクタム誘導体と関連する環状ウレアおよび環状カルバマート誘導体との比較を行った (Table 10)。これらの誘導体は同等の置換基サイズを持つ一方、カルボニル酸素原子上の電子密 度が異なるため、上記の目的に適していると考えた。Table 10 に示すカルボニル酸素原子上の電
子密度は、MOE を用いて半経験的 PM3 法(MOPAC)により計算した69。MGAT2 阻害活性に関
しては、環状ウレア誘導体 52c がラクタム誘導体 52a よりも強力な活性を示したのに対して、環 状カルバマート誘導体 52e は 52a と比べて活性が減弱した。カルボニル酸素原子の電子密度に関 しては、環状ウレア 52c>ラクタム 52a>環状カルバマート 52e の順で増加しており、阻害活性 と電子密度の間に良好な相関が見られた。一般に環状ウレアのカルボニル基は対応するラクタム のカルボニル基よりも水素結合受容能が高いことが報告されており70、このことは今回の計算結 果とも一致する。ラクタムから環状ウレアへの変換による活性向上は 6 員環誘導体 52b と 52d
Table 9. MGAT2 inhibitory activity: SAR at the 7-position of the indoline ring
Compd R hMGAT2 a IC50 (nM) LogD7.4b 10b 1.0 (0.89–1.2) 2.5 54a 6.7 (4.9–9.1) 2.9 54b 7.3 (6.5–8.2) 3.1
aInhibitory activity against human MGAT2. IC
50 values are presented as means of duplicate
Table 10. MGAT2 inhibitory activity: SAR at the 7-position of the indoline ring
Compd X n hMGAT2
a
IC50 (nM)
Electron charge on the
carbonyl oxygen of Fb LogD7.4c
52a CH2 1 14 (10–19) −0.353 2.8 52c NH 1 3.4 (2.9–3.9) −0.394 2.3 52e O 1 43 (33–56) −0.334 2.3 52b CH2 2 10 (8.3–13) −0.353 3.2 52d NH 2 2.2 (1.8–2.7) −0.399 2.6 a
Inhibitory activity against human MGAT2. IC50 values are presented as means of duplicate
experiments with their 95% confidence intervals in parentheses. bElectron density on the carbonyl oxygen of F was calculated by MOPAC using PM3 method. cLogD value at pH 7.4.68
の間でも同様に観察された。上記の結果から、インドリン環 7 位置換基上のカルボニル基は HBA として MGAT2 との相互作用に関与していることが示唆され、カルボニル酸素原子上の電子密度 を増加させることで活性増強につながることを見出した。 インドリン環 7 位へのラクタム環導入に続いて、インドリン環 1 位の置換基変換を検討した (Table 11)。インドリン環 1 位へウレア型スペーサーを導入した化合物 54c は、期待した通りに 代謝安定性の改善を示した。化合物 54c の MGAT2 阻害活性は、フェネチル体 54a に比べて減弱
したが、脂溶性効率(ligand lipophilicity efficiency(LLE):pIC50-LogD)71の観点で比較すると
両化合物は同等の値を示した(LLE = 5.3(54a)および 5.4(54c))。前節で述べたように、イン ドリン環 1 位置換基の脂溶性が MGAT2 との親和性獲得に寄与しているため、この部位の脂溶性 を高めることで活性増強につながると考えた。この仮説に基づいてベンゼン環上にトリフルオロ
メチル基を導入した化合物 54d は、54c と同等以上の代謝安定性を維持しながら、IC50 = 3.4 nM
(LLE = 5.4)の強力な阻害活性を示した。tert-ブチル基を導入した化合物 54e もトリフルオロメ チル体 54d と同等の強力な活性を示したが、代謝安定性に関しては 54d と同等の改善は見られ なかった。tert-ブチル体 54e は、トリフルオロメチル体 54d よりさらに脂溶性が増大しており、 このことが代謝安定性を低下させた原因として考えられる。また、脂溶性効率の観点から、トリ フルオロメチル基は tert-ブチル基よりも効果的に MGAT2 との相互作用を獲得できる有用な置換 基である。化合物 54d の MGAT2 阻害活性をさらに向上させるために、前述した知見に基づいて、 インドリン環 7 位に環状ウレア構造の導入を行った。その結果、化合物 54f は約 2 倍の活性向上
Table 11. MGAT2 inhibitory activity, microsomal clearances, and plasma concentration Compd R1 R2 hMGAT2 a IC50 (nM) LogD7.4b in vitro CLintc (μL/min/mg) AUC0–8hd (ng·h/mL) HLM MLM 54c 28 (26–31) 2.2 30 143 NT 54d 3.4 (3.0–3.9) 3.1 36 84 842 54e 3.1 (2.7–3.5) 3.6 66 146 15 54f 1.8 (1.3–2.4) 2.5 63 82 87 10b 1.0 (0.89–1.2) 2.5 101 220 9.2
aInhibitory activity against human MGAT2. IC
50 values are presented as means of duplicate
experiments with their 95% confidence intervals in parentheses. bLogD value at pH 7.4.68cHuman/mouse liver microsomal clearance. dMice. 1 mg/kg, po. NT = not tested.
を示し、脂溶性の低下と相まって大幅に LLE 値が向上した(LLE = 6.2)。また、化合物 54f は化 合物 54d と同等の代謝安定性改善を示した。
続いて、in vitro 代謝安定性の改善した化合物に関して、マウスにおける経口吸収性の評価を 行った。その結果、化合物 54d は期待した通りに良好な経口吸収性と高い血中濃度を示した(F
= 52%、AUC0-8h = 842 ng・h/mL)。一方、tert-ブチル体 54e に経口吸収性の改善は見られなかった。
代謝安定性の改善が不十分であったことに加え、脂溶性増大に伴う溶解度の低下(solubilitypH6.8 = 4.8 mg/mL(54e)、12 mg/mL(54d))が起因している可能性がある。また、インドリン環 7 位置 換基の環状ウレアへの変換も大幅な経口吸収性の低下を招いた。化合物 54f は 54d と同等の代 謝安定性を示しているにも関わらず、経口投与時の血中濃度は化合物 54d の約 1/10 であった。 化合物 54f は、水素結合供与基が増加したことで膜透過性が大幅に悪化しており(PAMPA 膜透 過性72(pH 7.4):396 nm/s(54d)、94 nm/s(54f))、このことが経口吸収性の低下に関与してい ると考えられる。上記の結果から、強力な MGAT2 阻害活性と良好な経口吸収性を併せ持つ化合
Figure 10. Effect of 54d on plasma TG elevation during oral fat tolerance test in C57BL/6J mice. 54d (3, 10, 30 mg/kg) was orally administered 6 h prior to oil challenge. Figure shows area under the curve (AUC) of plasma chylomicron/TG (CM/TG) levels during 4 h after oil challenge. Data are the mean + SD (n = 5). #, p ≤ 0.025 versus vehicle by William's test.
物 54d を in vivo 評価に用いる化合物として選択した。 MGAT2 阻害による in vivo 作用の評価には、C57BL/6J マウスを用いた経口脂質負荷試験を採 用した。本評価においては、リポプロテインリパーゼ(LPL)による血中 TG の加水分解を抑制 するために、LPL 阻害剤(Pluronic F127、Poloxamer 40773)をマウスに事前投与した条件で血中 の TG 蓄積量を定量した。化合物 54d をマウスに経口投与し、6 時間後にオリーブオイルを負荷 して、その後 4 時間にわたって血中 TG 濃度を測定した。その結果、化合物 54d はオリーブオイ ル負荷による血中 TG 増加を用量依存的かつ有意に抑制した(Figure 10)。MGAT2 ノックアウト マウスにおいて同様の血中 TG 増加抑制作用が見られており43、本結果は化合物 54d が MGAT2 阻害作用に基づいて TG 増加抑制作用を発揮したものと考える。 上述したように、リード化合物 10b に対して経口吸収性改善を目的とした最適化を実施し、 強力な活性と良好な経口吸収性を併せ持つ化合物 54d を見出した。本化合物は、C57BL/6J マウ スを用いた経口脂質負荷試験において、用量依存的かつ有意な血中 TG 低下作用を示し、低分子 MGAT2 阻害剤の in vivo における作用を実証した。
第三節 CYP3A4 に対する時間依存性阻害活性回避に向けた構造最適化 第一項 研究方針と分子設計 前節で述べた化合物 54d は、強力な MGAT2 阻害活性と良好な経口吸収性を示し、C57BL/6J マウスを用いた経口脂質負荷試験において有意な血中 TG 低下作用を示した。低分子 MGAT2 阻 害剤の in vivo における有用性を示すことができたため、化合物 54d のさらなる精査試験を行っ た。その結果、化合物 54d は薬物代謝酵素である cytochrome P450 3A4(CYP3A4)の時間依存性 阻害(time-dependent inhibition、TDI)活性を有することが判明した。CYP3A4 は最も主要な CYP アイソフォームの一つであり、多くの薬物の代謝反応に関与している。そのため、薬物間相互作
用の観点から、CYP3A4 の TDI は回避すべきプロファイルとして認識されている74。そこで、化
合物 54d の CYP3A4 に対する TDI 活性回避に向けた構造修飾に取り組んだ。
CYP3A4 の TDI 活性を誘発している構造を推定する目的で、化合物 54d の関連誘導体を評価 した結果、化合物 10b や 70c も同様に CYP3A4 に対して TDI 活性を示した(Table 12)。この結 果から、共通構造である 2,4-ジフルオロアニリン部が CYP3A4 に対する TDI 活性を誘発する原 因構造であるとの仮説を立てた。4-フルオロアニリン類は CYP による脱フッ素化を受けること が報告されており75, 76、この代謝は生体内活性化(bioactivation)を引き起こす可能性がある77, 78。 本系統の誘導体の TDI の推定メカニズムを Figure 11 に示す。2,4-ジフルオロアニリン部の酸化 代謝によってエポキシドやイミノキノンのような反応性代謝物が生成し、これらと CYP3A4 の アミノ酸残基の間に共有結合が形成されることで不可逆的な CYP3A4 の失活が誘導されると考 えた。この推定活性代謝物の生成を抑制する目的で、下記のコンセプトに基づいた 2,4-ジフルオ
Table 12. MGAT2 inhibitory activity and CYP3A4 TDI profiles
Compd hMGAT2 a IC50 (nM) CYP3A4 TDIb (% remaining) LogD7.4 c 54d 3.4 (3.0–3.9) 43 3.1 10b 1.0 (0.89–1.2) 13 2.5 70c 4.1 (3.4–4.9) 59 3.7
aInhibitory activity against human MGAT2. IC
50 values are presented as means of duplicate
experiments with their 95% confidence intervals in parentheses. bCYP3A4 time dependent inhibition assay (n = 2). The remaining activity of CYP3A4 after preincubation with a test compound was determined.
Figure 11. Postulated mechanism of CYP3A4 inactivation.
Figure 12. Compound design to mitigate CYP3A4 TDI activity.
ロアニリン部の構造修飾を計画した(Figure 12)。 1)4 位フッ素原子を代謝安定な置換基へ変換 2)酸化代謝に対する安定性を獲得するためにベンゼン環上へ電子吸引性置換基を導入 第二項 合成 4-クロロ-2,6-ジフルオロアニリン誘導体 60a および 60b は、Scheme 9 に示す方法で合成した。 本章第一節第二項で述べたように、スルホニルクロリド 5 と 2,4-ジフルオロアニリンとのスルホ ンアミド化反応は、DMA 溶媒中で塩基の添加を必要とすることなく円滑に進行した。しかし、 同様の条件で 4-クロロ-2,6-ジフルオロアニリンと反応を行った場合、低収率(10–20%)で目的 物を与えるのみであった。これは、4-クロロ-2,6-ジフルオロアニリンの塩基性/求核性が著しく 低いことに起因すると考えられる。反応条件を検討した結果、溶媒として 2,2,2-トリフルオロエ タノールを用いた加熱条件で目的とするスルホンアミド化反応が効率良く進行することを見出 した。この条件では、ピリジンや水素化ナトリウムなどの塩基を用いた反応条件よりも良好な結 果を与えた。溶媒としてエタノールや酢酸エチルを用いた場合には、ほとんど反応が進行しなか ったことから、2,2,2-トリフルオロエタノールの酸性度の高さ(pKa = 12.4)が重要であり、スル ホニルクロリドの活性化に関与していると考えられる。また、酸に不安定な基質を用いる際は、 反応の進行に従って生じる塩化水素の捕捉剤として、モレキュラーシーブス 4A や炭酸水素ナト リウムの添加が有効であることを見出した。本反応条件を用いてスルホンアミド 55 を合成した 後、亜鉛粉末を用いてニトロ基の還元を行い、アニリン 56 を良好な収率で得た。続いて、Scheme 8 と同様の方法を用いて、インドリン環 7 位にラクタム環を構築し、化合物 58 へ誘導した。イ ンドリン環 1 位の Cbz 基を除去した後、ウレア型もしくはカルバマート型の置換基導入を行っ
Scheme 9. Synthesis of indoline-5-sulfonamide derivatives 60a and 60ba
aReagents and conditions: (a) 4-chloro-2,6-difluoroaniline, 2,2,2-trifluoroethanol, MS4A, reflux; (b) Zn, AcOH, rt, 86% over 2 steps; (c) 4-chlorobutyryl chloride, DMA, 0 °C to rt; (d) NaH, DMF, 0 °C to rt, 91% over 2 steps; (e) NaOH, H2O, THF, MeOH, rt, 87%; (f) 4-(trifluoromethyl)aniline, triphosgene,
pyridine, THF, 0 °C; then 59, TEA, THF, 0 °C to rt, 83%; (g) 62, triphosgene, TEA, THF, 0 °C to rt; then 59, THF, 0 °C to rt, 87%; (h) 2,2,3,3,3-pentafluoropropyl trifluoromethanesulfonate, TEA, THF, 0 °C to rt, 88%.
Scheme 10. Synthesis of indoline-5-sulfonamide derivatives 70a–ca
a
Reagents and conditions: (a) Na2S2O4, EtOH, THF, H2O, 60 °C, 73%; (b) 4-chlorobutyryl chloride,
DMA, 0 °C to rt; (c) NaH, DMF, 0 °C to rt, 82% over 2 steps; (d) BnSH, Pd2(dba)3, Xantphos, DIEA,
toluene, reflux, 96%; (e) NCS, AcOH, H2O, rt, 81%; (f) 2-fluoro-4-(trifluoromethyl)aniline,
2,2,2-trifluoroethanol, 80 °C, 87%; (g) 2,4,6-trifluoroaniline, pyridine, rt, 70%; (h) H2 (1 atm), Pd on
て、目的とする 60a および 60b を合成した。化合物 60b の合成に必要なピペリジン誘導体 62 は、 4-ヒドロキシピペリジンとトリフルオロメタンスルホン酸 2,2,3,3,3-ペンタフルオロプロピルか ら調製した。 インドリン環 5 位に種々のアニリンを導入した化合物を効率的に合成する目的で、Scheme 9 に示した合成ルートとは別に、合成の後半でアニリンを導入する合成ルートの確立を行った (Scheme 10)。スルホンアミドの導入に先立ってインドリン環 7 位にラクタム環を構築するため に、亜ジチオン酸ナトリウムを用いて化合物 3 のニトロ基を選択的に還元し、アニリン 63 を合 成した。続いて、4-クロロブチリルクロリドによるアシル化と分子内環化反応によりインドリン 環 7 位にラクタム環を構築し、化合物 65 へ導いた。その後、ベンジルメルカプタンとのパラジ ウムカップリング反応によってベンジルスルフィド 66 へと誘導し、NCS を用いた酸化的クロロ 化反応を行って鍵中間体となるスルホニルクロリド 67 を良好な収率で得た。得られた 67 とアニ リンとのスルホンアミド化反応を行った後、Scheme 9 と同様の方法でインドリン環 1 位の構造 修飾を行い、目的とする 70a–c を合成した。 第三項 生物活性と考察
合成した化合物に関しては、MGAT2 阻害活性および CYP3A4 に対する TDI 活性を評価した。
CYP3A4 の TDI 活性に関しては、下記の方法で評価した。化合物とヒト肝ミクロソームを NADPH
の存在下でプレインキュベーションした後に CYP3A4 の酵素活性を評価し、プレインキュベー ションしない場合の酵素活性との比(% remaining)を算出して、TDI 活性の指標とした。Table 13 に合成した化合物の MGAT2 阻害活性と CYP3A4 に対する TDI 活性を示す。
化合物 70c の 2,4-ジフルオロアニリン部の 4 位フッ素原子をトリフルオロメチル基に変換した 化合物 70a は大幅に CYP3A4 の TDI 活性が減弱した。また、ベンゼン環上の電子密度低下を目 的としてさらにフッ素原子を導入した 2,4,6-トリフルオロアニリン体 70b においても、TDI 活性 が消失した。これらの結果は、化合物 10b、54d、および 70c の CYP3A4 に対する TDI 活性が仮 説通りに 2,4-ジフルオロアニリン部に起因することを示している。しかし、これらの構造変換は、 TDI 改善効果と同時に、対応する 2,4-ジフルオロアニリン誘導体と比べて 10–20 倍の MGAT2 阻 害活性の減弱をもたらした。さらなる置換アニリンの検討を行った結果、化合物 60b に代表さ れる 4-クロロ-2,6-ジフルオロアニリン構造が、MGAT2 阻害活性の減弱が少なく、CYP3A4 に対 する TDI リスクを改善するバランスのとれたモチーフであることを見出した。脂溶性効率71の 観点から評価した場合にも、4-クロロ-2,6-ジフルオロアニリン誘導体 60b が最も高い LLE 値を 示した。この 4-クロロ-2,6-ジフルオロアニリン構造の有用性は、インドリン環 1 位にフェニルウ レア構造を有する誘導体においても確認され、化合物 60a は化合物 54d と比較して CYP3A4 の TDI 活性が大幅に減弱した。 上述したように、2,4-ジフルオロアニリン誘導体の CYP3A4 に対する TDI 活性回避を目的とし て、推定 TDI 誘発メカニズムに基づいた構造変換を行い、MGAT2 阻害活性を保持しながら TDI
Table 13. MGAT2 inhibitory activity and CYP3A4 TDI profiles Compd R1 R2 hMGAT2 a IC50 (nM) CYP3A4 TDIb (% remaining) LogD7.4 c LLEd 70a 42 (32–55) 93 4.1 3.3 70b 75 (58–96) 95 3.4 3.7 60b 19 (15–24) 82 3.8 3.9 60a 30 (26–34) 89 3.3 4.2 70c 4.1 (3.4–4.9) 59 3.7 4.7 54d 3.4 (3.0–3.9) 43 3.1 5.4 a
Inhibitory activity against human MGAT2. IC50 values are presented as means of duplicate
experiments with their 95% confidence intervals in parentheses. bCYP3A4 time dependent inhibition assay (n = 2). The remaining activity of CYP3A4 after preincubation with a test compound was determined. cLogD value at pH 7.4.68dLLE = pIC50 − LogD7.4.
第四節 活性増強と光毒性回避に向けた構造最適化 第一項 研究方針と分子設計 前節で、CYP3A4 に対する TDI リスクを回避した 4-クロロ-2,6-ジフルオロアニリン誘導体の 発見に関して述べた。前述したように、この構造修飾によって CYP3A4 の TDI プロファイルが 改善した一方、MGAT2 阻害活性の減弱を伴ったため、再び活性増強を目的とした分子設計を行 った。第一節および第二節で述べたように、本系統の誘導体ではインドリン環 1 位に導入した脂 溶性置換基が MGAT2 阻害活性発現に大きく寄与しており、特に末端のベンゼン環およびトリフ ルオロメチル基が MGAT2 との鍵相互作用を形成していると考えた。そこで、末端脂溶性モチー フのコンフォメーション固定化による活性増強の可能性を考え、Figure 13 に示すビアリール誘 導体をデザインした。このデザインに基づいて合成した化合物 73 は大幅に活性が向上し、IC50 = 0.25 nM の強力な MGAT2 阻害活性を示した。しかし、化合物 73 は in vitro 評価において新たに 光毒性ポテンシャルを有することが判明した。光毒性は薬剤誘発の副作用の一つであり、光照射 によって皮膚等に傷害が惹起されることを特徴とする79。生体における薬剤による光毒性発現に は、光照射に対する化合物の反応性に加えて、皮膚や目などの光照射に晒され易い組織への化合 物の分布の度合が影響する。しかし、安全性の高い薬物開発の観点からは、本質的に光毒性ポテ ンシャルの低い化合物を創出することが重要である。そこで、化合物 73 の光毒性リスク回避に 向けた構造最適化に取り組んだ。 光毒性発現の主要なメカニズムとして、光照射により化合物がエネルギー的に励起され、その 励起状態からエネルギーや電子が放出されることでスーパーオキシドアニオンラジカルや一重 項酸素などの活性酸素種が生成し、これらを介して組織が傷害を受ける機序が考えられる80, 81。 ビアリール誘導体 73 が光毒性リスクを示す一方、非ビアリール誘導体 60a は光毒性ポテンシャ ルを示さなかったことから、インドリン環 1 位のビアリール構造がクロモフォアとして光毒性誘 発に関与していると考えた。筆者はビアリール部位の光照射によるエネルギー励起に着目し、光 毒性ポテンシャルの低減を目的として、ビアリール部位の HOMO–LUMO ギャップを増大させる アプローチの適用を試みた81, 82。半経験的分子軌道法(AM1)を用いた計算から、分子を簡素化
したビアリールモチーフ 73-I の HOMO は主に A 環に局在しており、LUMO は B 環に広がって いると考えられた(Figure 14-A)。この軌道の局在性に基づいて、HOMO が局在している A 環の 電子密度の低下、または LUMO が局在している B 環の電子密度の増加によって、HOMO–LUMO ギャップを増大できると考え、J で示される化合物をデザインした(Figure 14-B)。すなわち、
Figure 14. A) Molecular orbital of HOMO and LUMO of compound 73-I calculated by MOE (MOPAC, AM1). B) Design for mitigation of phototoxicity based on HOMO–LUMO gap theory.
適切な環構造の組み合わせを用いることで、強力な活性を示すビアリール構造を保持したまま、 光毒性リスクの低減を試みた。
第二項 合成
1-フェニルインドリン誘導体 73 は、Scheme 11 に示す方法で合成した。インドリン環 1 位への フェニル基導入は、アリールハライドと二級アミンの C–N 結合形成に対する有用性が報告され
ている RuPhos precatalyst83, 84を用いた Buchwald–Hartwig アミノ化反応によって達成することが
できた。得られた化合物 71 に対して 1 当量の NBS を作用させることで選択的なブロモ化反応が 進行し、化合物 72 を得た。最後に、ボロン酸との鈴木–宮浦カップリング反応を行うことで、目 的とする化合物 73 を合成した。
1-(ピリミジン-2-イル)インドリン誘導体 77a–f は、Scheme 12 に示す方法で合成した。インド リン環1位へのピリミジン環導入反応は、化合物 59 と 2-クロロ-5-ニトロピリミジンを THF 中
Scheme 11. Synthesis of indoline-5-sulfonamide derivative 73a
aReagents and conditions: (a) PhBr, RuPhos precatalyst, RuPhos, NaOtBu, DME, reflux, 65%; (b) NBS, DMF, 0 °C, 99%; (c) 4-(trifluoromethyl)phenylboronic acid, Pd(PPh3)4, K2CO3, THF, reflux, 63%.
Scheme 12. Synthesis of indoline-5-sulfonamide derivatives 77a–fa
aReagents and conditions: (a) 2-chloro-5-nitropyrimidine, THF, reflux, 98%; (b) H
2 (1 atm), PtO2, THF,
EtOH, rt, 93%; (c) n-pentyl nitrite, CuBr2, CH3CN, 0 °C to 60 °C, 45%; (d) ArB(OH)2, Pd(PPh3)4,
Na2CO3, THF/H2O or DME/H2O, 110–130 °C (microwave), 50–87% (for 77a and 77b); (e) ArB(OH)2,
Pd(PPh3)4, K2CO3, THF, reflux, 38% (for 77c); (f) substituted pyrazole, CuI, 8-quinolinol, K2CO3,
DMSO, 130°C, 11–42% (for 77d–f).
Figure 15. ORTEP of 77e, thermal ellipsoids are drawn at 50% probability.
Figure 16. NOE cross peaks were observed between H-4 (or H-6) on the pyrimidine ring and the methyl proton on the pyrazole 1-position of compound 77f.
Scheme 13. Synthesis of indoline-5-sulfonamide derivative 84a
aReagents and conditions: (a) 2-chloroethyl isocyanate, cat. HCl, THF, 70 °C; (b) NaH, DMF, 0 °C to rt, 77% over 2 steps; (c) NaOH, H2O, THF, MeOH, reflux, 92%; (d) 2-chloro-5-nitropyrimidine, THF, reflux,
92%; (e) H2 (1 atm), PtO2, THF, EtOH, rt, 97%; (f) tert-butyl nitrite, CuBr2, CH3CN, rt, 39%; (g)
1-methyl-3-trifluoromethyl-1H-pyrazole-5-boronic acid pinacol ester, Pd(PPh3)4, K2CO3, THF, 100 °C
(microwave), 46%. で加熱還流することで速やかに進行し、化合物 74 を高収率で得ることができた。この SNAr 反 応は、ピリミジン環上にニトロ基やアルコキシカルボニル基等の電子吸引基が存在する場合にの み円滑に進行した。化合物 74 のニトロ基を Adams 触媒を用いて還元し、続いて Sandmeyer 反応 に付すことでブロモ体 76 へ誘導した。最後に、ボロン酸との鈴木-宮浦カップリング反応を行っ て目的とする 77a–c を合成した。また、銅触媒を用いたピラゾールとの N-アリール化反応を行 うことで化合物 77d–f を合成した。化合物 77e の構造は、単結晶 X 線構造解析によって確認し た(Figure 15)。一方、化合物 77f に関しては、NMR 実験においてピリミジン環 4 位(6 位)水 素原子とピラゾール環 1 位メチル基の間で NOE のクロスピークが確認されたことから、期待通 りにピラゾール環 5 位がアリール化された構造と決定した(Figure 16)。 インドリン環 7 位に環状ウレア構造を有する化合物 84 は、Scheme 13 に示す方法で合成した。 化合物 56 とイソシアン酸 2-クロロエチルによるウレア結合形成反応においては、触媒量の塩化 水素の添加が反応の進行を加速化させ、またスルホンアミド窒素原子上でウレア形成反応が進行 する副反応を抑制できることを見出した。得られた化合物 78 を水素化ナトリウムで処理するこ とで分子内環化反応が円滑に進行し、環状ウレア 79 へと誘導した。以降、Scheme 12 と同様の 方法で目的とする化合物 84 を合成した。 第三項 生物活性と考察 合成した化合物に関しては、MGAT2 阻害活性および in vitro における光毒性ポテンシャルの評 価を行った。光毒性ポテンシャルに関しては、BALB/c 3T3 細胞を用いて、UV 照射および非照