医薬品開発を促進する原薬物性の評価ならびに改善技術に関する研究
Physicochemical Evaluation and the Property Improvement Technologies of Drug Substance to Promote Drug Development
平成28年度
論文博士学位申請者
山本 克彦
(Yamamoto, Katsuhiko)
指導教員 深水 啓朗
本文中に用いた以下の用語、装置などは以下のように略記した。
DMSO: dimethyl sulfoxide
JP1: 1
stfluid of disintegration test in Japanese Pharmacopoeia JP": 2
ndfluid of disintegration test in Japanese Pharmacopoeia GCDC: glycochenodeoxycholic acid
MS: mass spectrometry
HPMC: hydroxy propyl methyl cellulose CC: cocrystal
API: active pharmaceutical ingredient CA: coamorphous
DSC: differential scanning calorimetry ITZ: itraconazole
FA: fumaric acid
TA:
L-tartaric acid
GRD: ground
LYO: lyophylized
RH: relative humidity
目次
第一章 序論と概要
... 1 - 3
第
1
節 医薬品の物性評価の必要性... 1
第
2
節 医薬品の物性改善手法... 2
第
3
節 研究概要、目的... 3
第二章 迅速かつ効果的な物性評価法の開発
... 4 - 19
第1
節 物性評価としての脂溶性および溶解度評価の必要性... 4
第
2
節 脂溶性評価法の開発... 5
第
3
節 高速溶解度測定法の開発... 9
第
4
節 本評価結果からの物理化学特性の考察... 14
第
5
節 物性改善・製剤設計戦略への繋がり... 17
第
6
節 小括... 19
第三章 効率的な共結晶探索法の構築
... 20 - 37
第1
節 共結晶の有用性と網羅的探索の必要性... 20
第
2
節 共結晶の結晶化と探索法... 21
第
3
節 効率的な共結晶探索法、Cocktail Cocrystal Grinding
法の開発... 24
第
4
節Cocktail Cocrystal Grinding
法の試行結果... 26
第
5
節Cocktail Cocrystal Grinding
法の特徴... 35
第
6
節 小括... 37
第四章 複合体非晶質
(
コアモルファス)
の物性評価と医薬品としての可能性... 38 - 55
第1
節 コアモルファスの定義と特徴... 38
第
2
節 イトラコナゾールのコアモルファスの物理化学的性質... 41
第
3
節 イトラコナゾールのコアモルファスの物理的安定性... 46
第
4
節 イトラコナゾールのコアモルファスの化学的安定性... 51
第
5
節 イトラコナゾールのコアモルファスの溶解性... 54
第
6
節 コアモルファスの医薬品としての実行可能性... 55
第
7
節 小括... 55
第五章 総括
... 56 - 57
謝辞... 58
実験の部
... 59
引用文献
... 67
1
第一章 序論と概要第
1
節 医薬品の物性評価の必要性医薬品において、有効性と安全性を担保するためにはその品質を保証するこ
とが必須であり、そのために安定性などの物理化学的性質
(
物性)
を調べる物性評 価は医薬品の開発において不可欠である1)。医薬品の主たる活性成分の多くは、化学合成によって創製・製造された低分 子化合物である。これら有機低分子化合物は、抗体等の生物製剤に比べ安定で コストが低く、製造性も高いという長所があるが、臨床試験・製造販売承認に 至るまでは、探索研究段階において膨大な数の化合物を新規に合成することが 必要である。同時に、それらの薬理活性や毒性、薬物動態特性をスクリーニン グし、開発化合物を選択する。近年、分子生物学的手法の発展により、優れた 薬理活性を持つ化合物を見出すことが可能となってきている。その一方、活性 向上を標的分子との結合の強さを上げることによって実現しているために、化 合物の分子量および脂溶性が増加し、医薬品の物性として適さない難水溶性化 合物が選択され、開発期間や費用が増大する場合や、開発自体の断念も余儀な くされることが見受けられている2)。こういった事態を避けるために、これまで は単なるキャラクタリゼーション、および製剤化研究の一環であった物性評価 を探索研究段階で実施し、良好な物性を持つ化合物を選択すること、加えて、
2
開発化合物の物性が完全ではない場合に、その物性改善を講じることが医薬品 開発において大変重要になってきている3, 4)。
第
2
節 医薬品の物性改善手法医薬品の物性評価を探索研究段階で早期に実施することによって、良好な物 性を持つ化合物を見定めることが可能となってきている。しかし、開発化合物 の選択は物性のみならず、薬理活性、安全性、薬物動態特性、特許性などとの 総合判断で行わる。また、開発の時系列、競合品の状況にも影響されるため、
必ずしも物性が完全ではない化合物が選択されることも起こりうる。したがっ て、開発品として選択された化合物について物性改善を行う必要が生じる5)。物 性改善については、原薬形態から行う方法と製剤技術から行う方法の二つのア プローチがある。原薬形態からのアプローチについては、水和物も含む結晶多 形検討、カウンターイオンによる化合物の塩化、シクロデキストリンなどによ る包接化などがある6, 7)。製剤技術からのアプローチは、非晶質固体分散体や自 己乳化型などの油系製剤を含む可溶化製剤8,9)、原薬の粉砕によるナノ化あるい は微細化、徐放化、小型化、遮光コーティングや防湿包装などの安定化、薬効 持続等のための投与経路の変更などがある。これらの物性改善は、化合物の試 料量や検討期間、投資予算など、医薬品の開発計画に応じて適切なタイミング
3
で実施されるべきであるが、技術革新により、それらの効果や実施の容易さ、
投資額は劇的に変わるものであり、新たな技術の開発とその有用性評価、タイ ムリーな開発化合物への適用は継続的に検討される必要がある。
第
3
節 研究概要、目的本研究では、医薬品開発における探索研究段階において適用が可能な迅速物 性評価法として、溶解度および脂溶性測定法を確立した。これらの評価法によ り、良好な物性をもつ開発化合物を早期から見定めることに寄与し、その判断 基準を与えることが可能となる。さらに選択した化合物についての物性改善や 適する製剤技術の指針を早期に提案することができる。続いて、物性改善技術 の開発として、近年、物性改善技術として注目されている共結晶について、そ
の効率的な探索法である
cocktail cocrystal grinding
法を新たに開発した。さら に、新規物性改善技術として研究され始めている複合体非晶質(コアモルファ ス)について物性評価を行い、医薬品としての実現可能性について判断した。4
第二章 迅速かつ効果的な物性評価法の開発第
1
節 物性評価としての脂溶性および溶解度評価の必要性医薬品化合物の水系溶媒に対する溶解度は、経口剤においては吸収性、注射 剤においては、製剤化の難易度を左右する重要な物理化学特性である10)。医薬 品として相応な溶解度の値は、化合物の薬理活性にも影響され、薬理活性が高 いほど溶解度の値は低くても許容されうる11)。しかし、経口医薬品の場合、腸 管膜への透過性の高いことが必要条件となり、かつ難水溶性化合物の場合は生 物学的利用率自体が低いため、服薬後の化合物血中濃度のバラつきも懸念され る。このような化合物の臨床試験においては、薬効や副作用の見極めが難しく なるなどの問題が生じる場合があり、その以前の段階でも、臨床試験申請のた めの毒性試験が成立しないことも起こり得る。したがって、難水溶性化合物の 開発には期間や費用がかかり、さらには開発自体困難となる場合がある。
脂溶性は薬理活性や毒性とも相関する指標であり、かつ溶解度や膜透過性と も密接に関わる12, 13)。脂溶性は、物理化学的パラメータとしては炭化水素系の 油系溶媒と水との間における化合物の分配係数を測定することで表される。脂 溶性は、文字通り脂質など疎水性部へのなじみやすさ、溶けやすさである。生 体は生体膜、タンパク質の内部など疎水的環境を持つため、化合物の脂溶性を 上げることで、薬理活性及び膜透過性を向上することが可能となる。しかし、
5
水系溶媒への溶解度は脂溶性と逆比例する性質があり、脂溶性向上によって活 性を高めた化合物の水に対する溶解性は低くなる傾向にある14)。したがって、
脂溶性の高い化合物は、医薬品としての性質の相応しさを示す
drug-likeness
が 不良となり、開発に問題が生じる場合が見受けられている。以上のことより、脂溶性は経口吸収性について広範に関与するパラメータであり、実際にその至 適範囲も論じられている15)。
したがって、両特性を探索研究段階において並行して評価することは、優れ た医薬品を創出するために必要である。本研究では、探索研究の時系列に見合 うように、溶解度および脂溶性を迅速に評価する系を開発した16)。
第
2
節 脂溶性評価法の開発脂溶性評価については、血液や組織の
pH
であるpH7.4
での分配係数、LogD
pH7.4を測定した。本研究では、LogD
pH7.4をLogD
と定義する。LogD
は、化合物を炭化水素系の有機溶媒である
1-
オクタノールと水溶液を接触させて分 配させ、(1)
式に示すようにそれぞれの化合物濃度C
orgとC
aqから、C
orgをC
aqで割ったものの対数として求められる。
LogD = Log
10(C
org/ C
aq) … (1)
6
LogD
の測定法として直接的に行う方法としては、C
orgとC
aqを測定することであるが、
LogD = 4
の場合、C
orgはC
aqの10,000
倍となり、測定に際し希釈等 の実験操作、定量についても特に濃度が低い水相の濃度についてLC-MS
などの 微量測定法が必要であり、複雑な操作およびその実験工数が要求されることから多検体の測定には適さない場合がある。したがって、
LogD
が様々な物理化学 理論と相関することを利用し、間接的にLogD
を測定することが行われている。例えば、キャピラリー電気泳動や電量滴定、クロマトグラフィーなどが報告さ
れている17, 18)。
本研究ではクロマトグラフィーを用い、逆相液体クロマトグラフィーにおい
て化合物の保持時間と
LogD
が相関することを利用した。(2)
式のように、保持 時間から導かれるLogk'
は、LogD
と一次相関する。LogD = alogk' + b … (2)
k' = (tR - t0)/t0, tR:
保持時間, t0:
カラム通過時間実際の測定としては、
LogD
既知化合物についてクロマトグラフを測定し、(2)
式を検量線として作成する。続いて、LogD
未知である対象化合物の測定を行い、保持時間から
(2)
式によりLogD
を算出する。本研究では、既報での条件を参考7
に、測定法のダウンサイズおよび高速化を実施した19)。カラムを内径
2.0 mm
、長さ
75 mm
のサイズとし、オクタデシル基を充填剤として内包したセミミクロカラムを用いることで、流速
0.2 mL/
分と移動相消費量の少ない条件を設定した。本条件はアイソクラティックであるため、カラムの平衡化に伴う測定のインタ
ーバルが無く、連続で測定を行うことが可能である。
LogD
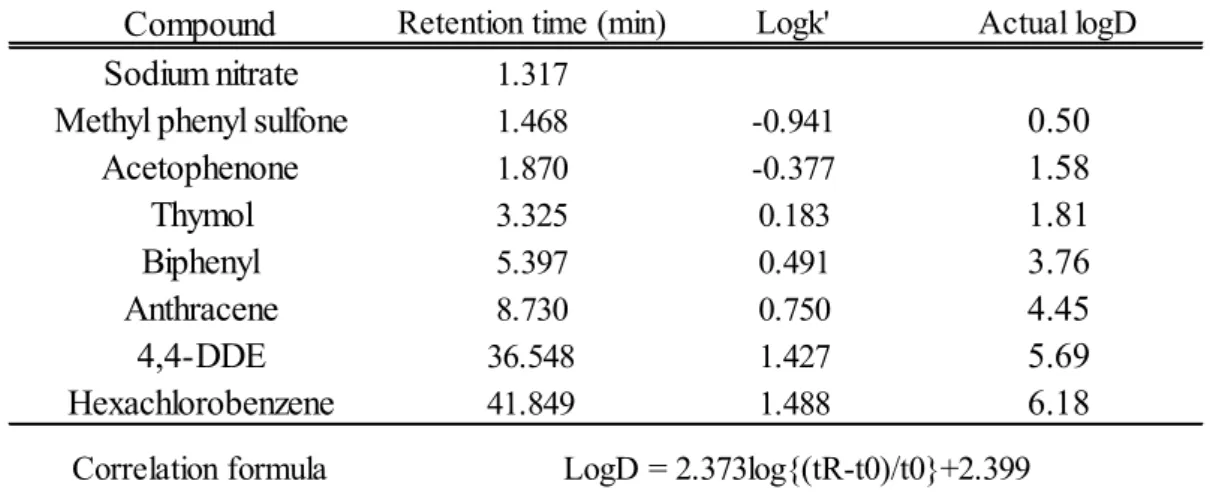
既知の化合物を用い て、条件の最適化を行った結果について、検量線、相関係数を表1
に、HPLC
条件を表2
に示す。実測値と検量線との相関係数は0.976
と相関の高い試験法 と設定した。本法では、
LogD = 4.45
のanthracene
が8.73
分に溶出しているように、LogD
が5
以下であれば、1
化合物につき10
分以内で測定が可能である。Lipinski
の 報告から12)、また実際にも多くの医薬品化合物のLogD
は5
以下であることか ら、本法は10
分以内でほとんどの化合物のLogD
測定を行うことができる。本 法の試料溶液は、医薬品候補化合物ライブラリの保存溶液として用いられている
10 mM DMSO
溶液を200
倍希釈したものを使用するため、使用試料量はわずか
1-3 µL
である。8
表
1 LogD
既知の化合物の保持時間と相関式、相関英数表
2 LogD
測定のHPLC
条件Compound Retention time (min) Logk' Actual logD
Sodium nitrate 1.317
Methyl phenyl sulfone 1.468 -0.941 0.50
Acetophenone 1.870 -0.377 1.58
Thymol 3.325 0.183 1.81
Biphenyl 5.397 0.491 3.76
Anthracene 8.730 0.750 4.45
4,4-DDE 36.548 1.427 5.69
Hexachlorobenzene 41.849 1.488 6.18
Correlation factor
Correlation formula LogD = 2.373log{(tR-t0)/t0}+2.399 0.976
Column :
Column temp. : 40
oC Detection wavelength : 230 nm
Mobile phase : Britton-Robinson buffer / methanol (2:5, v/v), pH7.4
Flow rate : 0.2 mL/min
Run time :
Injection vol. : 2 μL
Concentration : 0.05 - 0.1 mg/mL
YMC-Pack pro C18 5 µ m, 2.0 x 75 mm (YMC Co., Ltd.)
10 min, 50 min for high lipophilic compounds
9
第3
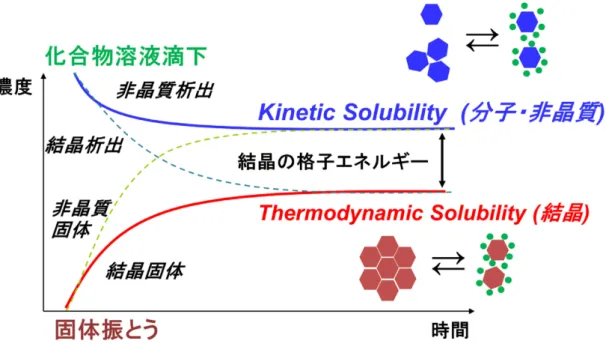
節 高速溶解度測定法の開発本研究では、化合物溶液を水溶液に滴下して析出させ、一定時間後にろ過し たろ液の濃度を定量することで求められる、析出法溶解度を測定法開発対象と
した。図
1
に示すように、多くの場合、析出法溶解度は分子および非晶質の溶 解度であるkinetic solubility
となる20)。溶解度にはもう一種類、結晶からの溶 解度であるthermodynamic solubility
がある。こちらは粉末試料を水溶液に添 加した後に一定時間振とうし、その飽和濃度を測定する振とう法で求められる21)。析出法でも結晶が析出した場合には
thermodynamic solubility
が、逆に非 晶質固体を振とうした場合には振とう法でもkinetic solubility
が測定される。Kinetic solubility
はthermodynamic solubility
より高い溶解度を示し、その差 は結晶の格子エネルギーに依存する。したがって、異なる結晶形はその格子エ ネルギーに応じたthermodynamic solubility
を有する。10
図
1 Kinetic solubility
とthermodynamic solubility
の関係本研究では
LogD
測定と同様、試料として化合物の10 mM DMSO
溶液を用 い、測定媒体に2.5 % (v/v)
で滴下して化合物濃度を測定した。測定媒体として は、人工胃液である日本薬局方崩壊試験第一液JP1 (pH1.2)
、および人工腸液で ある第二液JP2 (pH6.8)
を使用した。さらに摂食後の腸液のモデルとして、ヒト の腸内で最も濃度の高い胆汁酸であるglycochenodeoxycholic acid (GCDC)
を20 mM
、JP2
に加えたGCDC/JP2
も用いた。本研究では200 µL
のスケールで試験を実施したため、
1
測定条件あたり5 µL
の試料液量である。析出法では溶 液試料のみを用いるため、調液操作は96
穴、384
穴などのマルチウェルプレー トを用いて、分注ロボットによって高速化、自動化、多検体化が可能だが22)、11
ろ液の濃度定量については簡便かつ高速に行うのが難しいという問題があった。
したがって、本研究では溶解度測定のための高速定量法を検討した。
高速定量法としては、マルチウェルプレートごとプレートリーダーを用いて、
紫外可視吸収
(UV-vis)
スペクトルを測定することが最も多検体の測定に適して おり、高速化が可能である。しかし、その測定できる濃度範囲は限られており、さらに解離基を有し
pH
によってUV-vis
スペクトルが変化する化合物について は、そのpH
ごとに濃度標準溶液を用意する必要がある。HPLC-MS
も高速化 が可能な定量法であるが23)、溶解度が高い試料の場合には希釈操作が必要であ ること、濃度定量には化合物ごとに複数の濃度標準溶液の測定によって作成した検量線が必要なことから、測定が複雑かつ難解である。加えて
MS
検出器の 場合、導入費用や維持費用・工数が多くかかることも課題である。したがって、本研究では
HPLC-UV
を測定装置として適用して高速定量法を検討した。HPLC-UV
は安価で堅牢であり、測定濃度範囲が広く、一般的な溶解度の濃度範囲である
0.1 – 100 µg/mL
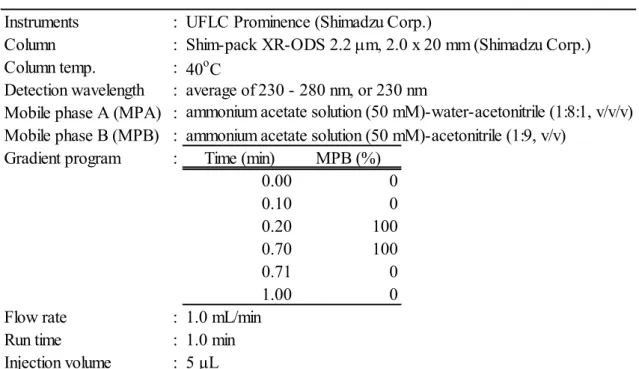
の間の測定に適している、濃度と感度の直線性も優 れるため溶解度測定に最適な装置である。本研究では、高速タイプの
HPLC
を用い、逆相測定での溶出の強溶媒である アセトニトリルの濃度が異なる二つの移動相を用いた高速グラジエントにより、化合物を高速で保持・溶出させてクロマトグラムを得る試験法を作成した。最
12
適化した条件を表
3
に示す。高速グラジエントはmobile phase B
濃度を0.1
分 から0.2
分まで、一度に0%
から100%
へと変えることで行った。さらに体積の 小さい移動相ミキサーを装着することにより、円滑なグラジエントを実現した。両移動相の酢酸アンモニウム濃度は
5 mM
に一致させることで、グラジエント の際もクロマトグラムのベースライン変動を抑えて安定させた。カラムに関しては、分離理論段数の高い粒子径
2 µm
の充填剤を含有したカラムを複数検討し、最も背圧が低く、耐久性の高い
Shim-Pack XR-ODS
を採用した。流速について は、高速HPLC
の標準流速である0.5 mL/
分の2
倍である1 mL/
分として、線 流速を上げることで分離を高め、またグラジエントプログラムへの追随性を安定させた。以上の検討を実施したことにより、わずか
1
分にて濃度測定が可能 なHPLC
条件を構築した。試料注入に必要な装置の稼働時間は0.5
分であるた め、合計で1
試料につき1.5
分の測定時間となる。したがって、200
化合物の2
溶媒に対する溶解度を測定する場合、濃度標準溶液の1
測定と合わせて、1
化合 物当たり4.5
分の測定時間となる。200
化合物では900
分、15
時間で溶解度測 定を行うことが可能となった。13
表
3
溶解度測定用のHPLC
条件Instruments : UFLC Prominence (Shimadzu Corp.)
Column :
Column temp. : 40
oC
Detection wavelength : average of 230 - 280 nm, or 230 nm Mobile phase A (MPA) :
Mobile phase B (MPB) : ammonium acetate solution (50 mM)-acetonitrile (1:9, v/v) Gradient program : Time (min) MPB (%)
0.00 0
0.10 0
0.20 100
0.70 100
0.71 0
1.00 0
Flow rate : 1.0 mL/min
Run time : 1.0 min
Injection volume : 5 μL
Shim-pack XR-ODS 2.2 µ m, 2.0 x 20 mm (Shimadzu Corp.)
ammonium acetate solution (50 mM)-water-acetonitrile (1:8:1, v/v/v)
14
第4
節 本評価結果からの物理化学特性の考察本評価系における、モデル化合物の溶解度および脂溶性の測定結果を表
4
に 示す。化合物の各溶液に対する溶解度から、脂溶性との相関および酸塩基特性も評価することが可能であった。各化合物の特徴を述べると、
lansoprazole
に ついては、JP1
について低い溶解度を示していたが、クロマトグラムには本体 ピーク以外に多数のピークが観察されており、酸性溶液中にて不安定で分解を 示していた。本定量法はクロマトグラフィーによって行うため、化合物の溶液中安定性も同時に評価することが可能であった。
Hydrochlorothiazide
は高い溶 解度と低いLogD
を示した。Griseofluvin
については、良好な溶解度と中程度 のLogD
を示した。Diclofenac
、indomethacin
については酸性で低溶解度であ ったが、中性では良好な溶解度であった。Tamoxifen
は酸性、中性共に低い溶 解度であったが、LogD
が高いためGCDC/JP2
にはミセル溶解を示したため、良好な溶解度であった。
Albendazole
、pioglitazone
については、酸性、GCDC/JP2
については良好な溶解度であったが、中性では低い溶解度を示した。LogD
は中 程度であった。Candesartan
については、酸性で低溶解度であり、LogD
が高いため
tamoxifen
と同様にGCDC/JP2
に対し良好な溶解度を示した。以上の結果から、本法により各化合物の物理化学特性を迅速に判断することが可能とな った。
15
中性である
JP2
に対する溶解度と脂溶性のプロットを図2
に示す。Kinetic
solubility
の対数と脂溶性は、既報どおりほぼ線形の相関を示したが14)、一部この相関から外れる化合物が確認された。
Hydrochlorothiazide
の溶解度(
図2
中2
番)
は相関よりも低い溶解度の値であったが、析出法溶解度は化合物溶液の添加 量が溶解度の測定上限であるため、本化合物はこの上限よりも実際の溶解度が高いことによると考えられた。図
2
中2'
番として、既報での実測溶解度をプロ ットしたところ24)、ほぼ相関と合致していた。Albendazole
およびpioglitazone
についてもこの相関よりも低い溶解度を示した。原因として両化合物共に溶液 中で容易に結晶化して析出したため、結晶の溶解度であるthermodynamic
solubility
を示し、相関より低い値を示したことが考えられた。16
表
4
モデル化合物の溶解度と脂溶性図
2
モデル化合物のLogD
とJP2
への溶解度のプロット 図中の数字は表4
中のNo.
と対応するJP1, pH1.2 JP2, pH6.8 20mMGCDC in JP2
1 Lansoprazole 4.0 58.0 >99 1.789
2 Hydrochlorothiazide 64.8 >74 >74 -1.169
3 Griseofulvin 39.4 49.0 >88 1.643
4 Diclofenac 1.6 68.2 >80 2.107
5 Indomethacin 1.8 74.9 >90 2.359
6 Tamoxifen 1.2 0.2 >93 4.823
7 Albendazole 45.6 0.2 56.7 2.541
8 Pioglitazone 45.3 0.3 47.9 2.189
9 Candesartan <0.2 12.6 >110 4.320
Solubility (µg/mL)
Compound LogD
No.
17
第5
節 物性改善・製剤設計戦略への繋がり前節のように、
kinetic solubility
とLogD
を並行して測定することにより、各化合物の物理化学特性を解析することが可能となった。さらにその特性を考 察することにより、各化合物への物性改善手段、および適用すべき製剤形態や 製剤技術などの対応策を提示することが可能である。各化合物についての対応
策を表
5
に示す。Lansoprazole
については、酸性溶液中で不安定であったが、中性では良好な溶解度を示したため、腸溶性製剤の適用が考えられた。実際、
lansoprasole
は腸溶コーティングを施した顆粒を含むカプセル剤として市販されている。
Hydrochlorothiazide
は良好な溶解度であったため、通常製剤で製剤 化可能と判断された。しかし、LogD
が-1.169
と低すぎるため、膜透過性が不良 である場合は用量の増加が必要であると推測された。Griseofluvin
についても 良好な溶解度を示したため通常製剤が適用可能と考えられたが、thermodynamic solubility
が低い場合や溶解速度が低い場合は、原薬の微細化やナノ化製剤が必要となると考えられた。
Diclofenac
、indomethacin
について は酸性で低溶解度、中性では良好な溶解度であったため、ナトリウムやカリウ ムなどのアルカリ性カウンターイオンとの塩化、塩基添加製剤の適用が考えら れた。実際、diclofenac
はナトリウム塩として市販されている。Tamoxifen
は 酸性、中性共に低い溶解度であったが、脂溶性が高くミセル溶解をする、また18
油系添加剤への溶解度が高いと考えられたため、自己乳化型製剤など油系製剤
の適用が示唆された。
Albendazole
、pioglitazone
については、酸性で良好な溶 解度を示したため、塩化または酸添加製剤の適用が考えられた。一方で結晶化 しやすく、その格子エネルギーの大きいことが中性での低溶解度の原因であると示唆されたため、非晶質固体分散体の適用も考えられた。
Candesartan
につ いては、酸性で低溶解度であるため、塩化または塩基添加製剤、LogD
が高いため
tamoxifen
と同様に油系製剤の適用が考えられた。表
5
モデル化合物の特性と対応策としての物性改善、製剤技術Model Compound Property Countermeasure
Lansoprazole acid-labile enteric-coating
Hydrochlorothiazide high solubility conventional formulation
Griseofulvin high solubility conventional formulation, nanocrystal
Diclofenac low acidic solubility salt formation, base loading
Indomethacin low acidic solubility salt formation, base loading
Tamoxifen low neutral solubility, high lipophilicity salt formation, acid loading, oily formulation Albendazole low neutral solubility, low lipophilicity salt formation, acid loading, solid dispersion Compound A low neutral solubility, low lipophilicity salt formation, acid loading, solid dispersion Compound B low acidic solubility, high lipophilicity salt formation, base loading, oily formulation
19
第6
節 小括本法は少量の溶液試料のみを使用することで
LogD
とkinetic solubility
を迅 速に測定することを可能とした。これらを並行して評価することによって、化 合物の物理化学的特性を考察することが可能となり、さらにその特性に合わせ た塩化などの物性改善手段、また油系製剤や固体分散体などの適切な製剤技術 を提案することも可能であると考えられた。20
第三章 効率的な共結晶探索法の構築第
1
節 共結晶の有用性と網羅的探索の必要性医薬品化合物の物性改善技術として、近年、共結晶
(cocrystal
、CC)
が活発 に研究および開発され、臨床化合物、製品の原薬形態としても適用されている。共結晶は、医薬品分子とカウンター分子
(cocrystal former, CCF)
間において塩 のようなプロトン移動を伴わずに、結晶格子内で水素結合やファンデルワール ス力などの非イオン性相互作用を有する、複成分からなる結晶である。共結晶 はフリー体結晶と異なる物理化学的性質を示すことから、溶解性・安定性など 物性改善の手段として多く用いられている25-26)。共結晶は解離基を持たない中性化合物へも適用が可能であり、さらに
CCF
としても中性の添加剤・甘味料な どの多種類の分子を用いることができるため、共結晶を形成する組み合わせは膨大となる。さらに、原則として塩については化合物と
CCF
の当量は解離可能 なプロトンの比率、例えば1:1
や2:1
のみであるが、共結晶については相互作用 可能な部位があれば当量は限定されず、様々な比率の共結晶が形成される可能性がある。したがって、共結晶を探索する際には、多種類の
CCF
、また複数の 比率について共結晶の形成の有無を調べる必要があり、多数の条件を行うこと が可能で、かつ効率的な探索法が必要とされている。21
第2
節 共結晶の結晶化と探索法共結晶は特徴的な複数の結晶化方法で形成させることが可能である。代表的 な
4
つの結晶化方法、それらの特徴を表6
に示す。表
6
代表的な共結晶の結晶化方法溶液からの晶析は27)、一般的な医薬品原薬の製造法として用いられている。
原薬の精製が可能であるため品質確保が容易であり、再現性及びスケールアッ プ性に優れるという利点がある。しかし、共結晶の結晶化においては、化合物
と
CCF
の溶解度の差が大きい場合、晶析溶媒に対して溶解度の低い方の成分が 溶液の冷却にしたがって先に単体として結晶化する。あるいは、溶媒との化合物との相互作用が化合物と
CCF
との相互作用より強い場合、同様に共結晶が結 晶化できず単体として結晶化することがある。そのため、晶析条件の設定には特定の溶媒中における化合物と
CCF
の比率、それらの濃度の三成分溶解度相図 を作成し、優先的に共結晶が結晶化する領域を見出す必要がある28)。探索法と して使用する際には、近年、塩・結晶多形スクリーニングにおいて行われてい調製法 長所 短所
溶液からの晶析 精製、スケールアップ 溶媒の影響、化合物とCCFの溶解度差
溶媒懸濁(スラリー) スループット、安定形 終点が不明、溶媒の影響
融解冷却(コフラー法) 反応性高い 融解分解に適用不可、スケールアップ難しい 粉砕法(grinding法) 反応性高い、メカニカルに強い 試料量・工数が多い
22
るのと同様に、マルチウェルプレートを適用できる。
CCF
と溶媒種をマトリッ クス状に配置してウェルプレートごと冷却、または溶媒除去を行って晶析する ことによって、少量で網羅的に行うことが可能でなる。しかし、設定条件が上 記三成分溶解度相図における、共結晶晶出領域と一致している場合にのみ共結 晶が取得されるものであるため、条件設定・効率という面で工夫が必要である。溶媒懸濁
(
スラリー)
法は、化合物とCCF
をそれぞれの飽和溶解度を超える濃 度で溶媒に共に添加し、懸濁状態でオストワルト則により共結晶の結晶化を促 す方法である29)。実験操作も簡便で熱力学的安定形が得られやすいという特徴 がある。一方、共結晶の結晶化反応にかかる期間はその結晶の特性に依存するため、
in situ
モニタリング等で捕捉可能ではあるものの30)、反応終点が不明瞭であり、
1
週間、2
週間など比較的長期の試験期間を設定する必要がある。また、溶媒と化合物の相互作用により溶媒含有結晶が生じやすく、共結晶の結晶化に 必ずしも有利とならない場合があり、加えて共結晶の溶媒含有結晶も晶出され うる。探索法としては、溶液からの晶析の際と同様にマルチウェルプレートが
適用可能であり、化合物と
CCF
の粉体をウェルに入れて溶媒を分注し懸濁させ ることで評価できる。しかし、化合物が溶けやすい溶媒を用いる場合、懸濁状 態となるまでには多くの化合物量の添加が必要である。さらには、懸濁後に共 結晶が結晶化していても、化合物およびCCF
との混合物となっている場合、共23
結晶の検出および判別が難しく共結晶を見逃すことがある。
融解冷却法
(
コフラー法)
は化合物とCCF
が溶融状態で混合するため、両者の 相互作用において溶媒の影響がなく、共結晶の結晶化が促されやすい31)。短所 としては融解分解を起こす化合物には適用できない点、試料量が多い場合は、均一となりにくいためにスケールアップが難しいという点がある。近年、本原
理を利用した、加熱押し出し成形機
(hot melt extruder)
による共結晶の結晶化 が報告されており、実製造への適用も行われるものと予測される32)。探索法としては、本法は溶媒が不要であるため、溶媒種を検討する必要が無い。しかし、
CCF
として用いられる有機酸の一部には融解分解、あるいは昇華性で明確な融 点を持たないものがあるため適用が限られる。さらに、本法にはマルチウェルプレートを適用できないため、多検体の試料処理能力
(
スループット)
が低いこと が課題として挙げられる。しかし、熱分析装置を用いて、共結晶形成に伴う熱 挙動をモニターしながら本法を行うことで、より効率的に本原理により共結晶 を探索することが可能である。粉砕法は、化合物と
CCF
をボールミルなどで混合粉砕することにより共結晶 を得る方法である33 - 35)。粉砕時の応力により化合物およびCCF
に結晶格子の 欠陥もしくは非晶質化を生じさせ、その部位を利用して分子間相互作用を促す。本法も溶媒の影響が無く、反応性が高いため共結晶を結晶化させやすい。また
24
外部からの加熱を行わないため、融解冷却法を適用できない、融解時に分解を 起こす、あるいは熱に対して不安定な化合物にも適用可能である。加えて、粉 砕のせん断力に抗って結晶化させるため、物理的にも強固な結晶となることも 特徴である。しかし、探索法への適用を考える場合、本法粉砕操作の都合上、
試験系のスケールが数百
mg
となるなど大きく、試料のロスも生じるために多く の試料量が必要である。したがって、化合物量の限られる医薬品研究の探索研 究段階には用いにくい。さらに、必然的に粉体を取り扱う実験工数が多く、秤 量、静電気対策などが煩雑であるため、スループットという点で探索法として の実施が難しいということが課題である。第
3
節 効率的な共結晶探索法、Cocktail Cocrystal Grinding
法の開発 本研究では、粉砕法の長所を持ちつつも、スループットが高く効率的なcocktail cocrystal grinding
法を開発した36)。本法では、図3
のように、あらかじめ
4
つのCCF
を物理混合したCCF
混合物との粉砕を行い、1
次スクリーニ ングとして共結晶形成能を調べる。1
次スクリーニングにて共結晶の結晶化が認 められた場合、続いて2
次スクリーニングとしてヒットした混合物内の個々のCCF
との混合粉砕を行い、どのCCF
との共結晶が形成されたかを絞り込む。例えば
20
種類のCCF
の中から1
つの共結晶のみが見出される場合、1
対1
の25
従来法では
20
回の粉砕が必要となるのに対し、本法では混合物の5
回に加え、混合物内の個々の
4
つのCCF
との4
回、計9
回の粉砕実験で可能となる。した がって、試料量・実験数を従来法の半分以下に減らすことが可能である。図
3 Cocktail cocrystal grinding
法と従来法の比較CCF
混合物の設計については、どのような組み合わせでカウンター分子を混合しても、本法に適用することは可能であると推測された。さらに
CCF
混合物 として、同じ部分構造、官能基を持つCCF
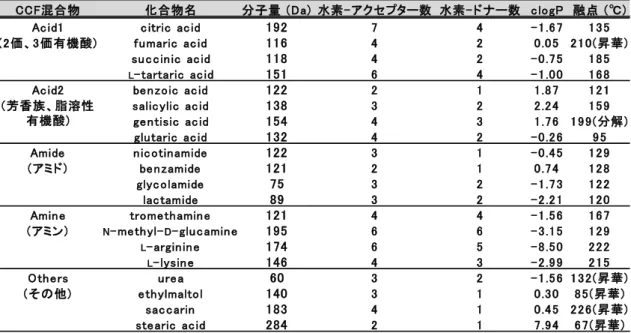
を混合することで、化合物との相互 作用を優位に行える部分構造を見出すことを可能とした。表7
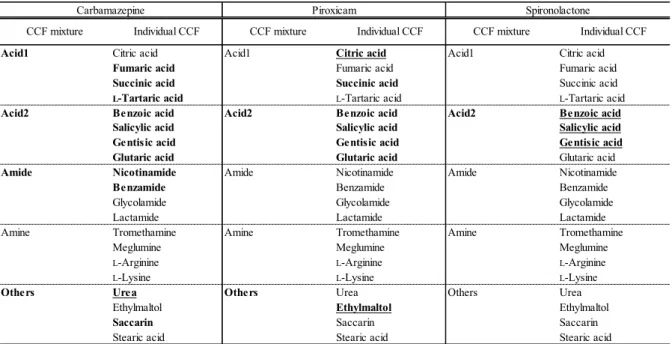
のように、添加 剤として使用実績のある20
種類のCCF
を選択し、同じ部分構造を持つ4
つのCCF
から成る混合物を4
種、Acid 1
及びAcid 2
、Amide
、Amine
として作成26
した。
Others
については異種CCF
間のモデルとして、異なる部分構造を持つ4
つの
CCF
からなる混合物とした。表
7 CCF
混合物一覧第
4
節Cocktail Cocrystal Grinding
法の試行結果本法を試行するにあたり、
CCF
混合物内のCCF
同士が粉砕によって共結晶 を形成する場合は、化合物との共結晶形成が優先的に行われないおそれがあるため、始めに
CCF
混合物のみでの粉砕を行った。共結晶の検出には、結晶構造 を反映し、その変化に鋭敏である粉末X
線回折を用いた。粉砕前後のCCF
混合 物の粉末X
線回折ピークを図3
に示す。Acid 1
およびAcid 2, Others
混合物に おいて粉砕後にわずかな新規の回折ピークが確認されたが、粉砕前のパターンCCF混合物 化合物名 分子量 (Da) 水素- アクセプター数 水素- ドナー数 c logP 融点 (℃)
Ac id1 c itric ac id 192 7 4 - 1 .6 7 1 3 5
(2 価、3 価有機酸) fu maric ac id 116 4 2 0 .0 5 2 1 0 (昇華)
su c c in ic ac id 118 4 2 - 0 .7 5 1 8 5
L- tartaric ac id 151 6 4 - 1 .0 0 1 6 8
Ac id2 be n zoic ac id 122 2 1 1 .8 7 1 2 1
salic ylic ac id 138 3 2 2 .2 4 1 5 9
ge n tisic ac id 154 4 3 1 .7 6 1 9 9 (分解)
glu taric ac id 132 4 2 - 0 .2 6 9 5
Amide n ic otin amide 122 3 1 - 0 .4 5 1 2 9
(アミド) be n zamide 121 2 1 0 .7 4 1 2 8
glyc olamide 75 3 2 - 1 .7 3 1 2 2
lac tamide 89 3 2 - 2 .2 1 1 2 0
Amin e trome th amin e 121 4 4 - 1 .5 6 1 6 7
(アミン) N- me th yl-D- glu c amin e 195 6 6 - 3 .1 5 1 2 9
L- argin in e 174 6 5 - 8 .5 0 2 2 2
L- lysin e 146 4 3 - 2 .9 9 2 1 5
Oth e rs u re a 60 3 2 - 1 .5 6 1 3 2 (昇華)
(その他) e th ylmaltol 140 3 1 0 .3 0 8 5 (昇華)
sac c arin 183 4 1 0 .4 5 2 2 6 (昇華)
ste aric ac id 284 2 1 7 .9 4 6 7 (昇華)
(芳香族、脂溶性 有機酸)
27
はほぼ保存されていた。新規ピークの強度自体も非常に低いものであった。し
たがって、各
CCF
混合物内での共結晶形成または変化は微小なものであり、化 合物との混合粉砕に影響は軽微と考えられたため、CCF
混合物を用いる本法は 実施可能であると判断された。図
4 CCF
混合物のみでの粉砕前後の粉末X
線パターンa – I: Acid1
粉砕前, II: Acid1
粉砕後, III: Acid1
粉砕前, IV: Acid1
粉砕後b – I: Amide
粉砕前, II: Amide
粉砕後, III: Amine
粉砕前, IV: Amine
粉砕後c – I: Others
粉砕前, II: Others
粉砕後IV
III
II
I
IV
III
II
I
II
I
a b
c
28
CCF
混合物との粉砕後の共結晶の検出については、DSC
による融点の吸熱挙 動の変化による検出も試みた。しかし、混合による融点降下および共融解の影響があり、かつ昇華する
CCF
の分析も困難なことから、検出法として不適であ ると判断した。赤外吸収、ラマン散乱などの分光法による検出は微量試料での 測定が可能であるという観点で有利であるが、主に化学構造を反映する方法で あるため、結晶構造の検出感度が低いという問題から、共結晶形成によるスペ クトルの変化が顕著である場合にのみ適用できると考えられた。実際に本法を医薬品化合物へ適用した結果、新規のものも含め複数の共結晶
が確認された。まず、複数の共結晶が報告されている
carbamazepine (CBZ)
へ 本法を試行した37)。図5 a
に化学構造を示す。化合物との混合粉砕操作につい ては、化合物10 mg
とCCF
混合物10 mg
を物理混合したものについて、CCF
混合物のみを粉砕した際と同様に実施した。CBZ
とCCF
混合物との混合粉砕後、Amine
混合物以外のすべてのCCF
混合物において新規の回折ピークが認められ、共結晶の形成が示唆された。粉末
X
線回折パターンを図6
に示す。つづいて、
CCF
混合物との粉砕において示唆された共結晶形成は真であるか、具体的にどの
CCF
と共結晶を形成したかを検証するため、CCF
混合物内の個々 のCCF
との粉砕を行った。図7
に示すように、新規ピークが認められたCCF
混合物内の個々のCCF
と粉砕した後に、新規回折ピークが確認された。CCF
29
混合物と個々の
CCF
の試行において相反しない結果が得られた。以上の結果よ り、CBZ
へのcocktail cocrystal grinding
法の適用から、本法のコンセプトが 確認され、効率的に共結晶を見出すことが可能であると考えられた。図
5
モデル医薬品化合物の化学構造a – CBZ, b – PRX, c – SPI
30
図
6 CBZ
とCCF
混合物との粉砕後の粉末X
線回折パターンa – I: CBZ, II: Acid1, III: CBZ-Acid1, IV: Acid2, V: CBZ-Acid2
b – I: CBZ, II: Amide, III: CBZ-Amide, IV: Amine, V: CBZ-Amine c – I: CBZ, II: Others, III: CBZ-Others
図中矢印は新規回折ピークを示す
a b
c
V IV III II I
V IV III II I
III II
I
31
CBZ-
L-tartaric acid
L
-tartaric acid CBZ-succinic acid succinic acid CBZ-fumaric acid fumaric acid CBZ-citric acid citric acid CBZ
CBZ-glutaric acid glutaric acid CBZ-gentisic acid gentisic acid CBZ-salicylic acid salicylic acid CBZ-benzoic acid benzoic acid CBZ
CBZ-lactamide lactamide CBZ-glycoamide glycoamide CBZ-benzamide benzamide
CBZ-nicotinamide
nicotinamide
CBZ
32
図
7 CBZ
とCCF
混合物内の個々のCCF
との粉砕後の粉末X
線回折パターン 図中矢印は新規回折ピークを示す下線は共結晶のヒットを示す
CBZ-
L-lysine
L
-lysine
CBZ-
L-alginine
L
-arginine CBZ-meglumine meglumine
CBZ-tromethamine tromethamine CBZ
CBZ-steatic acid steatic acid CBZ-saccharin saccharin
CBZ-ethylmaltol
ethylmaltol
CBZ-urea
urea
CBZ
33
CBZ
と同様に共結晶が報告されている医薬品化合物、piroxicam (PRX)
38)および
spironolactone (SPI)
39)についても本法を試行した。PRX
およびSPI
の化 学構造をそれぞれ図5 b
、c
に示す。いずれの化合物についても、CCF
混合物で 新規回折ピークが認められたものは、全て個々のCCF
とも共結晶が確認された。したがって、
CCF
混合物ではヒットしたものの、個々のCCF
ではヒットしないという
false positive
は確認されず、本法のスクリーニング法としての高い整合性が示唆された。
CCF
混合物、また個々の各CCF
との混合粉砕後の共結晶 形成結果の一覧を表8
に示す。文献などで既知の共結晶については、本法によ りほぼ見落とすことなく確認されたことから、本法の検出性能に問題はないと示された。また、新規の共結晶である、
CBZ – Urea
およびPRX - citric acid
、PRX – ethyl maltol
、SPI – benzoic acid
、SPI – salicylic acid
、SPI – gentisic acid
も見出された。しかし、PRX
については、CCF
混合物のAcid 1
グループ においてヒットが観察されなかったものの、Acid 1
内の個々のCCF
であるcitric
acid
とsuccinic acid
とで、PRX
との共結晶形成が確認された。この結果は、本試行結果の中で唯一、
CCF
混合物ではヒットしないにもかかわらず、個々のCCF
では実はヒットするというfalse negative
であった。34
表
8
モデル化合物とのcocktail cocrystal grinding
法試行結果Acid1 Citric acid Acid1 Citric acid Acid1 Citric acid
Fumaric acid Fumaric acid Fumaric acid
Succinic acid Succinic acid Succinic acid
L-Tartaric acid L-Tartaric acid L-Tartaric acid
Acid2 Benzoic acid Acid2 Benzoic acid Acid2 Benzoic acid
Salicylic acid Salicylic acid Salicylic acid
Gentisic acid Gentisic acid Gentisic acid
Glutaric acid Glutaric acid Glutaric acid
Amide Nicotinamide Amide Nicotinamide Amide Nicotinamide
Benzamide Benzamide Benzamide
Glycolamide Glycolamide Glycolamide
Lactamide Lactamide Lactamide
Amine Tromethamine Amine Tromethamine Amine Tromethamine
Meglumine Meglumine Meglumine
L-Arginine L-Arginine L-Arginine
L-Lysine L-Lysine L-Lysine
Others Urea Others Urea Others Urea
Ethylmaltol Ethylmaltol Ethylmaltol
Saccarin Saccarin Saccarin
Stearic acid Stearic acid Stearic acid
太字: 見出された既知の共結晶, 太字下線: 新規共結晶, 修飾なし: 共結晶が確認されなかったもの
Carbamazepine Piroxicam Spironolactone
CCF mixture Individual CCF CCF mixture Individual CCF CCF mixture Individual CCF
35
第5
節Cocktail Cocrystal Grinding
法の特徴本法の利点について考察すると、本法は粉砕法の短所の一つである、多くの 試料量・実験工数を要する点について、それらの削減を可能としたことが最も
大きなものである。さらに、本法では
CCF
混合物との粉砕において、複数のCCF
が化合物との相互作用の際に競合し、打ち勝ったCCF
が共結晶を形成す ると考えられる。したがって、本法で見出された共結晶は、より強い相互作用 を有する、物理的に安定な共結晶であると推察された。粉砕法は反応性の高い 結晶化法であるため、一対一での従来法では、相互作用の弱い共結晶も見出してしまう可能性がある。この点において、本法では
CCF
間での相互作用の競合 が生じるため、このリスクは低減されると考えられる。他の利点としては、本法で用いた
CCF
混合物は同じ部分構造を持つCCF
を含有するように設計した ことから、相互作用を引き起こす構造をベースとした探索法となっていることが挙げられる。例えば、ある化合物が本法において
fumaric acid
とsuccinic acid
とのみ共結晶形成が確認されたものの、両共結晶の物性改善効果が期待された レベルではなかった場合を想定する。その際は、本法で見出された化合物とカルボン酸との相互作用を利用して、他のカルボン酸
CCF
との共結晶探索に展開 し、望まれる物性の共結晶を得ることが期待される。一方、欠点としては、
CCF
同士が競合することによって、化合物との相互36
作用を互いに妨害し、
CCF
混合物との粉砕において共結晶が形成されないとい う可能性がある。もしくは、複数のCCF
が同時に化合物に相互作用した結果、例えば相互作用が三すくみとなる、あるいは複合体を形成するものの結晶化は
しなかった場合、本法では共結晶を検出することは出来ない。これらの場合、
CCF
混合物との粉砕ではなく、一対一の粉砕でのみ共結晶が確認されるというfalse negative
を生じる。実際に本研究での試行において、PRX
とAcid 1
混合 物との粉砕の際にこの現象が確認された。今後、本法におけるこれらの欠点に ついては、テラヘルツ分光法など直接的に相互作用を検出できる測定技術の発 展により解決できるものと見込まれる40)。今後の共結晶探索の展開としては、単に共結晶を得るのみでなく、その溶解 性・安定性といった機能性評価も合わせた評価系の構築が理想的である。その
際、化合物と
CCF
の相互作用に関する情報を有効に利用することで、求められ る物性改善に繋げられると推測される。また、このような分子間相互作用の知 見は化合物と賦形剤、高分子基剤などとの相互作用にも展開が可能であり、製 剤の更なる高機能化に繋がることが期待される。37
第6
節 小括本研究で開発した