2014 年度学術発表会 特別講演
京都大学大学院 医学研究科 腫瘍生物学講座骨髄異形成症候群の分子プロファイリング
小川誠司
( これは,2014 年 6 月 5 日天理よろづ相談所学術発表会で, 主として非専門家を対象に行われた小川教授の特別講演を 当医学研究所で編集したものである. ) 図 1. 骨髄異形成症候群MDS, myelodysplastic syndrome; sAML, secondary acute myeloid leukemia はじめに 私は,昨年 (2013年) 9 月に京都大学に赴任した. 東京に30 年ほど居たので,初めて住む京都には まだなかなか慣れない.しかし,歴史のある京都 の町ではさまざまな文化の香りが漂い,四季の移 り変わりも身近に感じることができる.私が間借 りをしている大徳寺の聚光院は,狩野松栄・永徳 父子が描いた国宝の方丈障壁画で有名な禅寺であ る.窓から外を眺めると400 年前の風情を感じさ せる景色で,空気が外界とは異なり朝晩の心の切 り替えが上手くできる.このように落ち着いて研 究に専念できる環境を与えていただいた聚光院住 職のご支援に感謝している.さて,本日は,骨髄 異形成症候群 (myelodysplastic syndrome; MDS) の 概要と,近年の知見や今後の展望を概説したい. 骨髄異形成症候群 (MDS) の病態と現状 MDS は,骨髄中の造血幹細胞の異常が原因で発 症する腫瘍性疾患である.赤血球や白血球,血小 板が減少し,貧血や感染症,場合によっては重篤 な出血をきたす難治性の疾患である.また,MDS の 約30% が最終的には急性骨髄性白血病 (acute
myeloid leukemia; AML) に進行する.MDS の特徴 の一つは血球形態の異常であり,この疾患が認識 される契機となった ( 図1).つまり,骨髄異形成 の“異形成”にはさまざまな意味があるが,骨髄 で異形血球がつくられるという意味が含まれてい る. 年間のMDS 新規患者は,米国では約 1 万人~ 1 万5 千人,日本では約 5 千人と推定されている. 患者年齢は60 歳以上が 90% を超え,MDS の患者 数は近年ますます増加傾向にある.MDS は通常の 化学療法では根治は難しく,同種造血幹細胞移植 が唯一の根治的治療法である.しかし,造血幹細 胞移植は高齢者には実施することができないので, 高齢者が大半を占めるMDS には,残念ながら根治 的治療法がない.近年,米国で開発された新薬で あるアザシチジンが導入された.この治療薬で余 命が少し延長する効果はあるが,これも最終的に は根治に至らしめることはできない.今後わが国 の人口が高齢化するに従って,MDS はますます増 加していく難病である. 我々の研究の目的の一つは,MDS の発症原因 を明らかにして治療成績の向上をめざすことであ MDS sAML
る.今のところ優れた治療薬の開発に結びついて はいないが,難治性疾患を地道に研究し続けるこ とによって最終的に成果があった事例が幾つかあ る.たとえば,慢性骨髄性白血病(chronic myeloid leukemia; CML)は,私が研修医であった頃には造 血幹細胞移植をしなければ治る見込みのない疾患 であったが,イマチニブという分子標的薬が開発 されたことによって,今日の若い医師にとっては, かつてCML が不治の病であったことは想像もでき ないであろう.また,急性前骨髄球性白血病(acute
promyelocytic leukemia; APL)も以前は死亡するこ とが多かったが,オールトランス型レチノイン酸 (all-trans retinoic acid; ATRA)による分化誘導療法 の導入以来,優れた治療効果が得られ生存率が高 くなった.このように治療成績が劇的に改善する ことがある.それは研究の積み重ねの成果であり, MDS も将来克服できる時が来るかもしれない. 癌の発症 MDS は,最終的には AML という血液癌に進行 する,いわゆる前癌状態ということができる.では, 一般的に癌はどのようにして発症するのか.ヒト の細胞は,分裂する際に等しく受け継がれる遺伝 情報を持つ.遺伝情報はDNA 分子によって担われ る.DNA 上には,アデニン (A),チミン (T),グ アニン (G),シトシン (C) という 4 種類の塩基が 全部で約30 億個も並んでいる.30 億個というと, 1 秒間に 1 個ずつ読んでも全遺伝情報を解読する のは何十年もかかるほどの膨大な情報量である. たった4 種類の塩基でゲノムに含まれる膨大な遺 伝情報が書かれ,細胞が分裂する度にほぼ正確に 受け継がれて必要な情報が記載されていく.しか し,膨大な情報を正確に複製するのは難しく,時々 間違ってはいけない場所,いわゆる癌遺伝子や癌 抑制遺伝子などの,細胞の機能に重要な役割を果 たしている遺伝子にエラーが起こる.ゲノム複製 時のエラーによって遺伝情報が書き換えられ,細 胞が本来の正確な情報と異なった振る舞いをする ようになる.このように理論的には,どのような 癌も或る種の幹細胞のような前駆細胞に,遺伝子 の変異が蓄積して発症すると理解できる.そこで, まず,どのような遺伝子が変異しているのか,そ してそれらの遺伝子変異が病態とどのように関連 しているのかを明らかにすることが,MDS を含む 癌を理解する上で大変重要となる. MDS における遺伝子変異の同定 ( ~ 1993 年 ) MDS は造血幹細胞に遺伝子の変異が蓄積して発 症する.では,どういう遺伝子が変異しているの か.MDS で最初に報告された変異遺伝子は N-ras (NRAS) である.これは,恩師の平井久丸教授(前 東京大学大学院 血液・腫瘍内科 ) が,1987 年に世 界に先駆けてNature 誌に報告した .1 つまり,MDS ではNRAS の変異によって或る特定のアミノ酸が 変わってしまうことが分かった.次いで,1993 年 に平井教授らのグループは p53 (TP53) が MDS で も変異していることを報告した.2 TP53 は癌の発 生にとっては極めて重要な遺伝子の一つで,ヒト の癌の約半分に変異が認められる.TP53 は癌抑制 遺伝子で,ゲノムの変異を防ぐという重要な役割 を果たしているが,これが変異すると癌細胞が排 除されなくなるという重大な事態が生じる.この ように,MDS の変異遺伝子として NRAS と TP53 が同定され,その後,この変異はかなり白血病に 近い病態で起こる変異であることも分かったが, 2000 年初めになるまで,これ以上の知見がなかな か得られなかった. そして,残念ながら平井教授は,2003 年に 52 歳 で急逝された.平井教授はMDS の研究を自分の ライフワークにしたいと常々私たちに伝えていた. 当時の私は,MDS は未知の疾患なのであまり気が 進まないと言っていた.しかし,約13 年間もの共 同研究者でもあった師が亡くなって以来,その志 を引継ぎ懸命に研究し続けている. 新規遺伝子変異の同定 ( ~ 2010 年 ) 大きな転機は,30 億塩基対の全ゲノムのコピー 数を一度に解析できる処理能力の高い方法が開発 されたことである.ゲノムのコピー数は,最初は 父親と母親から1 コピーずつもらうので 2 である.
それが3 に増えたり,あるいは 1 に減ったり 0 に なったりする.全ゲノム解析は,このようなゲノ ムの増減が生じている全ての場所を知ることがで きる技術である.この技術を使って,2009 年に 我々のグループが,MDS では CBL 遺伝子が変異 することを最初に報告した.3 また,他のグループ が,TET2 遺伝子と EZH2 遺伝子変異を新たに同定 した.4,5 このように全ゲノムを解析する方法によっ て,どの遺伝子が変異するのかが明らかになって きた. 最初のNRAS から 20 数年間が経過したが,2010 年までに10 数個の変異遺伝子が同定された.以 前から分かっていた染色体異常に加えて数種の新 たな遺伝子変異が明らかになったわけだが,ここ には一つの問題があった.これらの遺伝子の変異 は,AML や真性多血症などの,MDS 以外の骨髄 腫瘍でも同様に認められることである.AML では, 特異的な染色体転座によって融合遺伝子を形成す ることが重要であり,融合遺伝子の検出は,現在 ではAML の日常診療にも使われている.一方, MDS を特徴づける遺伝子の変異は,まだ知られて なかった.また,MDS 症例 222 例の染色体や遺伝 子の変異を調べても,10% ~ 15% の症例には我々 が知っている遺伝子の変異は認められず,3 どうして MDS になっているのかを想像するすべもなかった. 技術の進歩 ヒトゲノム全塩基配列の決定 大きな変革は,塩基配列を決定する能力が格段 に上昇して30 億塩基対あるヒトゲノムの全配列 の決定が可能になったことである.私が研究を始 めた頃はMaxam-Gilbert 法という方法で,200 から 300 塩基対の配列を決定するのに 2 ~ 3 週間もか かったが,今日ではその能力は数100 万倍に増強 された.これは,初期のコンピューターに比べる と,現在のコンピューターではクロック周波数が 格段に上昇し,極めて高速に処理できるのと同様 で,DNA の塩基配列決定能力は 30 年前にくらべ ると劇的に進歩したのである.2000 年にはヒトゲ ノムの概要配列 (draft sequence) が解読され ,6,7 続 いて2003 年には精密配列が決定された .8 大量並列シークエンス この遺伝子配列を決定する革命的な技術は,大 量並列シークエンス (massive parallel sequencing) で ある.まず,小さなスライドガラスの上に,塩基 配列を決定したいDNA 断片を大量に撒く.すると, 数億個の分子クラスターができるので,それらの 上で1 つずつ塩基を決定していく.例えば,A と いう塩基を入れたりB という塩基入れたりして, それらを1 回ずつ写真に撮る.これを 100 回繰り 返すと,各フラグメントの100 塩基対が決定する. その100 塩基対が一度に数億個も決まるので,全 て合わせると膨大なDNA の配列量,情報量にな る.現在,平均的な最先端のDNA シークエンサー では,6 日間で 1 テラバイトが解読でき,30 億塩 基対あるヒトゲノムの約200 ~ 300 人分を決定す ることができる.癌はゲノムに変異が起こって発 症するので,どこに異常が起こるのかを調べれば よい.昔は想像もできない非現実的な問題であっ たが,このように革命的に技術が進歩して,現在 は癌の全ゲノムを調べることが可能になった. 癌のゲノム解析 ヒトゲノムのリファレンスシークエンスに,調 べた癌のゲノムの断片を1 個ずつあてはめていく と,1 塩基の置換や欠失,ある領域のホモ欠失・ヘ ミ欠失や増幅,染色体転座などを見出すことがで きる.また,hepatitis virus や human papillomavirus などのヒト以外の塩基配列が入っていれば,ウイ ルスの存在も明らかになる.このように,癌のゲ ノムのほぼすべての情報を網羅的に決定すること ができるようになった.9 現在,世界各国の機関や 組織が参画して,1 種類のがんについて少なくと も500 症例以上を解析してデータ化するという大 規模な癌ゲノム研究プロジェクトが進行している. この国際共同プロジェクトは順調に進んでおり, どのような遺伝子異常が原因となっているのか, 数年後にはヒトの主要な癌について癌ゲノムの様 相を知ることができるだろう.
Nanopore Sequencing さらに,Nanopore sequencing という技術が開発 されつつある.膜たんぱく質中に形成したナノ ポア (1 メートルの 10 億分の 1 のサイズの超微 細 な 穴 ) に,DNA の 1 本鎖を通していく.する と,通過する塩基の種類 (A, T, G,C) によって 電気抵抗が少しずつ変わる.その微細な電流の変 化をシーモス (CMOS) 半導体チップ上でモニタ ーしていくと,通過した塩基の種類がわかる.つ まり,DNA 分子を 1 塩基ずつ直接見ていく技術 で,一気に10 万塩基対の配列が決まり,数分間で ヒトゲノムを読むことができる.Oxford Nanopore Technologies 社が,近々上梓する予定である.USB ぐらいの小さなサイズでコストも割と安く,将来 的にはベッドサイドで塩基配列を調べられるよう になる可能性が高い.数年後には,癌のゲノムを 読むことがより身近になる日が確実に訪れ,診療 の現場でゲノム情報を得ることができるようにな る.癌だけではなく,遺伝性疾患はもちろんのこと, 糖尿病や高血圧などの疾患も遺伝子レベルで診断 できるようになるだろう. MDS のゲノム解読 大 量 並 列 シ ー ク エ ン ス 技 術 を 使 っ て, ま ず, MDS の様々な病型から幅広く 56 例を選択して全 エクソンを解析した.全ゲノムは30 億塩基対ある が,実際に遺伝子をコードしている領域は約3,000 万塩基対で,ゲノム全体の約1% ~ 2% に相当する. その領域だけを集中的に解読した結果,遺伝情報 が決定的に変わってしまうような遺伝子変異が, MDS では 1 症例あたり平均 9.2 個検出された ( 図 2).肺癌や悪性黒色腫では数百から千個の遺伝子 が変わってしまうので,それに比べると非常に少 ない.さらに,その9.2 個の遺伝子の中で MDS 発 症に寄与する重要な役割を担う変異,これをドラ イバー遺伝子変異というが,そのような変異は数 個であることも明らかになった.MDS の複数の症 例で繰り返し変異が認められた遺伝子の中で,最 も高頻度であったTET2 遺伝子は明らかに MDS の 原因遺伝子であると考えられる.TET2 遺伝子の他 にも,既にMDS で変異することが知られている 遺伝子も認められたが,驚いたことに,高頻度ト ップ5 のうち 4 つの遺伝子 (SRSF2, U2AF1, ZRSR2, 図 2. MDS 症例の全エクソン解析によって検出された遺伝子変異の個数 ■Missense ■Frameshift ■Nonsense ■Splice site ■Nonflameshift 9.2 / sample 1 症例当たりの遺伝子変異数
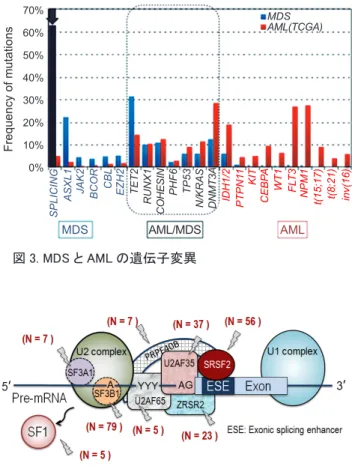
SF3B1) は,予測しなかった新たな遺伝子であった. これらは全て,細胞の遺伝情報の発現にとって基 本的な機能の一つである「RNA スプライシング」 を担う遺伝子であった. MDS における RNA スプライシング遺伝子変異 細胞の遺伝情報は,ゲノムDNA から中間的な メッセンジャーRNA(mRNA) に受け渡される.次 い でmRNA に基づいてたんぱく質が合成され, 様々な細胞の機能が発揮される.その中間段階の mRNA の合成に関与する RNA スプライシング遺 伝子の異常が,MDS の病態に深く関わっているこ とが分かった.1 例だけで変異している遺伝子も 含めると,RNA スプライシング遺伝子の変異の頻 度は約57% であった. さらに,MDS の様々な病型と他の血液腫瘍を 含む合計582 例の遺伝子変異を解析したところ, MDS では RNA スプライシング遺伝子変異が特に 高い頻度で生じていた.不応性貧血 (MDS without RS) で約 44%,慢性骨髄単球性白血病 (CMML) で約55%,鉄芽球性貧血 (RARS/RCMD-RS) では 約85% という極めて高い頻度であった .10 SF3B1 の変異が,RARS; 64~83%,RCMD-RS; 57~76%, RARS-T; 68~73% という報告もある .11 一方,AML や骨髄増殖性疾患 (MPN) など他の血液腫瘍では 10 % 以下と低頻度であった .10 つまり,RNA スプ ライシング遺伝子の変異は,MDS に特異的である. 図3 は MDS と AML において種々の遺伝子が 変異している割合を示したものである.それぞ れに特徴的な変異が認められる中で,MDS では RNA スプライシング遺伝子の変異の頻度が約 6 割を超え,圧倒的に突出している.MDS 発症に はRNA スプライシング遺伝子の変異が関与して いることは明らかである.さらに,その遺伝子変 異は,RNA スプライシングの最初の過程である prespliceosome(complex A) の形成12に関与する因 子に集積していた.図4 に示すように,pre-mRNA の過程で3' スプライシング部位の認識に関わる主 要な因子が,遺伝子変異によって異常を来たして いたのである.さらに興味深いことに,これらの 遺伝子変異は重複することなく,排他的に起こっ ている.10 つまり,RNA スプライシング因子が 1 個壊れると,装置全体が根本的に壊れてしまうの である.では, RNA スプライシング装置が壊れる と,どうしてMDS の病態となってしまうのか.こ れは世界中で懸命に研究中だが,残念ながらまだ 解明されていない. 新規遺伝子変異の同定 (2011 ~ 2014 年 ) 全エクソン解析をすることで,MDS に関わって いる主要な遺伝子変異が同定された.おそらく新 しい遺伝子変異,少なくとも高頻度で現れるよう な遺伝子変異はもう検出されないだろう.現時点 で我々は,MDS は遺伝子変異が原因であり,ど の遺伝子が変異しているかに関して,ほぼ把握で きるようになった ( 図5).さらに最近,友人の牧
島 秀 樹 氏 (Cleveland Clinic, USA) と共に 200 例の MDS の全エクソンを解析した.データをみると数
図 3. MDS と AML の遺伝子変異
図 4. RNA スプライシング装置と MDS における遺伝子変異 〔Yoshida K, et al. Nature 2011;478:64-69. より改変〕 MDS AML/MDS AML
SPLICING
ASXL1 JAK2 BCOR CBL EZH2 TET2 RUNX1
COHESIN
PHF6 TP53
N/KRAS DNMT3A IDH1/2 PTPN1
1 KIT CEBP A WT1 FLT3 NPM1 t(15;17) t(8;21) inv(16) MDS AML(TCGA) 70% 60% 50% 40% 30% 20% 10% 0% Frequency of mutations
多くの遺伝子が変異していて,1 例 1 例の変異遺 伝子は全く異なるパターンをとっていた.つまり, 同じMDS でも遺伝子変異という観点からすると, それぞれ異なっていて同一ではなく,随分と多様 であることがわかる.将来的には1,000 例を対象 に解析したいと考えている. 技術的な大革新により,我々は,変異頻度の高 い遺伝子を次々と同定することができた.全ての 遺伝子について説明することはできないので割愛 するが,その中で2 つの遺伝子について概説す る.まずSETBP1 遺伝子は,重い奇形を伴う小児 の先天性疾患Schinzel-Giedion 症候群の原因遺伝子 として知られている.このSETBP1 の変異頻度を 他の血液腫瘍を含む727 例で調べた結果,二次性 AML で 16.8%,CMML で 14.5% と高い頻度で変異 が認められた.一方, de novo AML では 0.7% と低 頻度であった.つまり,SETBP1 遺伝子の変異は, MDS から白血病に進行する段階に関わっている .13 次 に,cohesin は 4 つ の 分 子(STAG2,RAD21, SMC1A,SMC3)から構成される複合体で,これ らの分子をコードする遺伝子は,同じく先天性疾 患であるCornelia de Lange 症候群と Roberts 症候群 の原因遺伝子であることがわかっている.さて, DNA は細胞分裂期(S 期)に複製され,複製後に 二つの細胞に分配される(M 期)が,複製 DNA(姉 妹染色体)は,細胞が分裂するまで解離しないよ うにつなぎとめておかなければならない.この役 割を果たす分子がcohesin である.Cohesin の 4 つ の分子はリング状に繋がって姉妹染色体をとりま き,アセチルトランスフェラーゼESCO が鍵をか けて姉妹染色体の接着が完成する.MDS では,こ のように染色体を束ねているcohesin に変異が認め られるのである.14 様々な病型の血液腫瘍610 例 を解析した結果,Cohesin 遺伝子の変異は AML の 12.1%(19/157),MDS の 8.0%(18/224),CMML の10.2%(9/88),CML の 6.3%(4/64),MPN の 1.3% (1/77)に認められた .14 この他にも,BCOR/BCORL1 15などの多くの遺伝 子変異が報告され,2011 年~ 2014 年の数年間に, 急速にほぼ全ての重要な遺伝子変異が同定された. MDS の発症に多くの遺伝子が関わっていることを 理解することは簡単ではないが,これは癌におい てはむしろ一般的なことである.逆に,CML の場 合はシンプルで分かり易い.CML の原因は分子標 的治療薬イマチニブの標的であるフィラデルフィ ア染色体異常,すなわちBCR-ABL 蛋白であるが, 図 5. MDS に関わっている主要な遺伝子変異
このような1 つの遺伝子変異で発症する癌は非常 に稀である. 遺伝子変異と予後の関連性 MDS における遺伝子変異が分かったので,この 結果を治療に役立てたい.そのためには,ある遺 伝子が変異していると予後が良いのか悪いのか, 病態に何か特徴があるのかを知りたい.そのため には関連性を見なければならないので,多数の症 例を調べることが必要である.これはどの臨床試 験もみな同様である. そこで我々は,ドイツの共同研究者と,支持療 法だけで経過観察したMDS 症例の 944 例を対象 に遺伝子変異を徹底的に調べた.16 そして,遺伝子 変異に基づいた予後予測モデルを作った.このよ うな研究では,作ったモデルが,その特定のサン プルセットにだけ成り立つという事態,すなわち オーバーフィッティングがしばしば起こる.そこ で,まずtraining cohort (n = 730) でモデルを作成し, validation cohort (n = 214) でそのモデルが成り立つ かどうかを調べる方法で,二重に検証した.16 次に,変異頻度の高い遺伝子含めた104 個の遺 伝子について,遺伝子をコードしている領域だけ を上手く濃縮して,次世代シークエンサーで塩基 配列を調べた.得られた情報をスーパーコンピュー タで処理すると,合計2,764 個の変異が認められ た.16 昔のシークエンス技術では,何年もかけて 延々と続けなければならない作業であるが,今日 では3 か月程度で完遂できる.さらに,シークエ ンスしたリード数を勘定して正常と比較すると, リード数が少ない箇所(欠失)や多い箇所(増幅) も同時に検出可能である.このように約1,000 例 の症例について104 個の遺伝子が徹底的に調べら れた癌は,おそらく今までにないであろう.MDS は,昔は全くわからない疾患であったが,遺伝子 変異の観点からみると,他の癌と比べても今日で は最もよく解析された癌であると言える. 図6 に 944 例の MDS 症例におけるドライバー 遺伝子変異/欠失の頻度を示す.16 少し残念なこと に,変異が見つかったのは944 例中 864 例 (91.5%) で,8.5% の症例では変異は検出されなかった.遺 伝子104 個を徹底的に調べても原因が判明するの は約9 割にとどまり,残りは分からない.これは MDS がいかに多様な原因によって起こっているか を示唆している.これらを全て調べるには,もっ と多くの症例を対象にする必要があるが,1% の MDS 症例に生じている異常を 95% の信頼区間の 確率で捉まえようとすると1 万症例が必要になる. あまり現実的ではなく,ここにまだ大きな限界が ある.その限界はさておき,逆のポジティブな見 図 6. MDS 症例におけるドライバー遺伝子変異 / 欠失の頻度 864/944 cases (91.5%)
〔Haferlach T, et al. Leukemia 2014;28:241-247. より改変〕
TE T2 SF 3B 1 ASXL 1 SR SF 2 DNM T3 A RUNX 1 U2 AF 1 ZRS R2 STA G2 TP 53 EZ
H2 CBL JAK2 BCOR IDH2 NRAS MPL NF1 ATM IDH1 KRAS PHF6 BRCC3 ET V6 LAM B4 NCO R2 SM C3 RAD2 1 CT CF PT PN 11 FL T3 GNAS FBXW 7 SM C1A CE BP A NP M 1 DCL RE 1C KIT FANCL GAT A2 GP RC5 A U2 AF 2 IR F1 LUC7 L2 GNB1 SF 1 PIGA 35% 30% 25% 20% 15% 10% 5% 0% Frequency 5q-RA RCMD RARS RCMD-RS RARS-T RAEB-1 RAEB-2
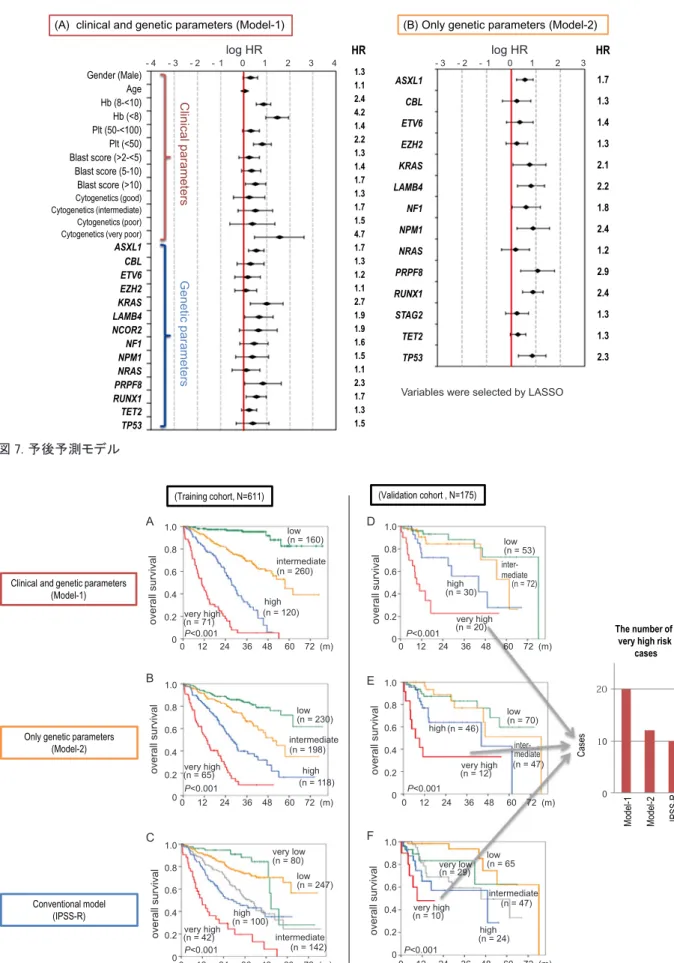
方をすれば,約9 割の症例には何らかの変異が見 つかる. た だ し,10% 以 上 の 患 者 に 認 め ら れ た ド ラ イ バ ー 変 異 は,TET2,SF3B1,ASXL1,SRSF2, DNMT3A,RUNX1 の 6 つの遺伝子変異であった. その中で頻度の多いTET2 や SF3B1 遺伝子変異で も頻度は30% ~ 35% 未満であり,その他の遺伝子 変異の頻度は高いものではない.16 やはりMDS は, 遺伝子的にはCML のような単純な癌ではなく,多 様な複雑な癌であるといえるだろう. 我々のグループ16とイギリスのSanger Institute の研究結果17から,変異頻度が高い上位12 の遺伝 子を表1 に記した.1 遺伝子を除いて同じ遺伝子が 列挙されているので,一致率・再現性は非常に高く, この結果は真実を反映していると思われる.MDS は,少なくともトップ12 の遺伝子変異に限れば, 均一な疾患であるといえる.RNA スプライシング に関わる遺伝子が最も多く,次にDNA のメチル化 やクロマチンの修飾などのエピジェネティックス に関わる遺伝子,あるいは転写因子やシグナル伝 達分子など,さまざまな細胞の機能に関わる遺伝 子が変異している.16 一方,1 症例あたりのドライバー遺伝子変異数の 中央値は3 個であった.変異数の平均値と MDS の 各病型との関連をみると,RA や 5q− 症候群などの 予後が良い病型では1.5 個であるのに対して,リ スクが高い病型になるに従って値は増加し,RAEB では4 個であった.つまり,遺伝子変異数と MDS のリスクが相関している.この結果は,MDS がよ りリスクの高い病型に進展するにつれて遺伝子変 異が蓄積していることを反映している.16 次に,それぞれの遺伝子変異が予後に及ぼす影 響を調べた.単変量解析でさまざまなパラメータ 間の相互作用を全く無視して1 個ずつ予後との関 連性をみたところ,唯一の例外であるSF3B1 遺伝 子を除いて,大半の遺伝子変異は予後不良に関連 しており,なかでもTP53 や EZH2 はとりわけ予後 が悪い.16 つまり,遺伝子の変異が予後に及ぼす効 果には強弱があり,明らかに患者の予後,運命を 決定している. 予後予測モデルの構築 MDS の臨床現場では,患者のリスクを評価する ところから診療が始まる.低リスクの患者は,輸 血や補助療法で治療する.一方,高リスクの患者は, まず造血幹細胞移植の適応がある場合は移植をし, 移植の適応がない場合はアザシチジンなどの治療 を選択する.低リスクの患者に移植をすると,移 植関連死のために生命予後が短くなる可能性もあ る.患者のリスクを評価して適切な治療を選択す ることが重要である.従って,リスクを評価する 予後予測モデルは,治療法を決める上で重要な手 段の一つである. そこで我々は,様々なパラメータを用いて2 つ のモデルを構築した.Model-1 は,現在 MDS の予 後予測に用いられているIPSS-R(revised Internation-al Prognostic Scoring System) の臨床情報と染色体異 常 (clinical parameter) に遺伝子変異の情報 (genetic parameter) を追加したモデル ( 図 7A),Model-2 は,遺伝子変異の情報のみで構築したモデルであ る ( 図7B).Model-1 と Model-2 で選ばれた遺伝子
変異は1 個を除いて同じである.これらの 2 つの
モデルを使って予後予測をしたところ,4 つのグ
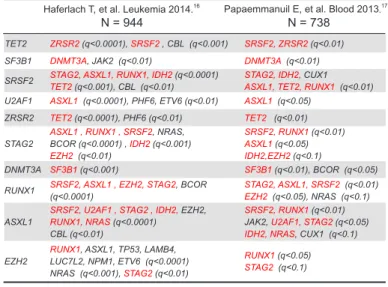
ループ (low, intermediate, high, very high) に層別化 することが最も効率的であることが分かった(図 8).Model-1 によるそれぞれのリスクグループの 表 1. MDS における変異頻度トップ 12 遺伝子 〔Papaemmanuil E, et al. Blood 2013;122:3616-3627〕 〔Haferlach T, et al. Leukemia 2014;28:241-247.〕 TET2 SF3B1 ASXL1 SRSF2 DNMT3A RUNX1 U2AF1 ZRSR2 STAG2 TP53 EZH2 CBL* N = 738 111 genes tested SF3B1 TET2 SRSF2 ASXL1 DNMT3A RUNX1 U2AF1 TP53 EZH2 IDH2* STAG2 ZRSR2 N = 944 104 genes tested (* 不一致)
図 7. 予後予測モデル
図 8. 各予後予測モデルにおけるリスク群別の生存曲線 〔Haferlach T, et al. Leukemia 2014;28:241-247. より改変〕
ASXL1 CBL ETV6 EZH2 KRAS LAMB4 NF1 NPM1 NRAS PRPF8 RUNX1 STAG2 TET2 TP53 C lin ica l p ara m ete rs G en etic p ar am ete rs Gender (Male) Age Hb (8-<10) Hb (<8) Plt (50-<100) Plt (<50) Blast score (>2-<5) Blast score (5-10) Blast score (>10) Cytogenetics (good) Cytogenetics (intermediate) Cytogenetics (poor) Cytogenetics (very poor) ASXL1 CBL ETV6 EZH2 KRAS LAMB4 NCOR2 NF1 NPM1 NRAS PRPF8 RUNX1 TET2 TP53 1.3 1.1 2.4 4.2 1.4 2.2 1.3 1.4 1.7 1.3 1.7 1.5 4.7 1.7 1.3 1.2 1.1 2.7 1.9 1.9 1.6 1.5 1.1 2.3 1.7 1.3 1.5 HR 1.7 1.3 1.4 1.3 2.1 2.2 1.8 2.4 1.2 2.9 2.4 1.3 1.3 2.3 HR
Variables were selected by LASSO
Clinical and genetic parameters (Model-1)
(Training cohort, N=611) (Validation cohort , N=175)
Only genetic parameters (Model-2)
Conventional model (IPSS-R)
The number of very high risk
cases M odel -1 M odel -2 IPSS -R Cas es
(A) clinical and genetic parameters (Model-1) (B) Only genetic parameters (Model-2)
A B C D E F 1.0 0.8 0.6 0.4 0.2 0 1.0 0.8 0.6 0.4 0.2 0 1.0 0.8 0.6 0.4 0.2 0 1.0 0.8 0.6 0.4 0.2 0 1.0 0.8 0.6 0.4 0.2 0 1.0 0.8 0.6 0.4 0.2 0 overall survival overall survival overall survival overall survival overall survival overall survival intermediate high 0 12 24 36 48 60 72 (m) P<0.001 very high low (n = 160) (n = 260) (n = 120) (n = 71) P<0.001 P<0.001 P<0.001 P<0.001 P<0.001 low (n = 53) low (n = 70) low (n = 65 low (n = 230) low (n = 247) intermediate (n = 198) high (n = 118) very high (n = 65) very low (n = 80) very low (n = 29) intermediate (n = 142) high (n = 100) very high (n = 42) very high (n = 10) very high (n = 12) high (n = 24) intermediate (n = 47) inter-mediate (n = 47) high (n = 46) very high (n = 20) high (n = 30) inter-mediate (n = 72) 0 12 24 36 48 60 72 (m) 0 12 24 36 48 60 72 (m) 0 12 24 36 48 60 72 (m) 0 12 24 36 48 60 72 (m) 0 12 24 36 48 60 72 (m) 20 10 0 log HR log HR - 4 - 3 - 2 - 1 0 1 2 3 4 - 3 - 2 - 1 0 1 2 3
〔Haferlach T, et al. Leukemia 2014;28:241-247. より引用〕 3 年生存率は 95.2%, 69.3%, 32.8%, 5.3%,Model-2
で83.3%, 66.4%, 39.7%, 9.5% であり,これらのリ スクグループはvalidation cohort でもおおよそ再現 することができた.予後不良群についてみると, IPSS-R の very high は 611 例中 42 例であるが,遺
伝子変異の情報を追加したModel-1 では 71 例とな
り,予後不良の症例はもっと多いことがわかる(図 8).Validation cohort でみると,低リスク群の再現 性はよいとはいえないが,very high risk の再現性 は良好で,Model-1, Model-2 とも IPSS-R よりも多 くのhigh-risk 症例を予測することができた .16 この ように統計的にも,遺伝子変異の情報を追加する ことによって,予後予測能力が有意に向上するこ とがわかった.つまり,従来のIPSS-R モデルより も優れた予後予測モデルを作ることができるとい うことである.そこで現在,我々の研究に基づい て,全世界で数千例を対象にして,遺伝子変異の 情報を加えた新しいIPSS 分類を構築する試みが始 まっている.数年後にはベッドサイドで,MDS 患 者の遺伝子変異を調べてリスク分類をすることが 日常的になったり,あるいは必須の検査になった りするかもしれない.米国では,TP53 や RUNX1, あるいはEZH2 や ETV6 などの 6 つの遺伝子検査に ついては既に保険適応が認められていて,日本よ り随分と進んでいる.実際,TP53 変異がある症例 の予後は大変に悪いので,造血幹細胞移植をして も救命することができないかもしれない.遺伝子 変異の情報を加えた予後予測モデルは,患者にとっ ても我々にとっても大変重要になる. 遺伝子変異間の相関 多くの遺伝子変異は一見ランダムに生じている ようであるが,実は特定の組み合わせがあること がわかった.図9 は,各遺伝子変異間の相関を示 したものである.16 ある特定の二つの遺伝子の組 み合わせが高い確率で生じている一方,確率の低 い組み合わせもある.たとえば,前述のとおり鉄 芽球性貧血と関連が深いSF3B1 遺伝子変異は,他 の遺伝子変異とは排他的に生じ, RNA スプライシ ングに関わる他の遺伝子変異が生じる確率が低い. SF3B1 変異があると,概して遺伝子変異の数が少 なく予後も良い.変異は無秩序に起こっているの ではなく,特定の二つの遺伝子間には,ある種の 機能的な協調作用,相互作用がある.つまり,あ 図 9. 遺伝子変異間の相関 SF3B1 SRSF2 U2AF1 ZRSR2 LUC7L2 PRPF8 TET2 IDH1 IDH2 DNMT3A ASXL1 EZH2 ETV6 IRF1 RUNX1 NPM1 PHF6 BCOR TP53 BRCC3 STAG2 MPL JAX2 PTRRT KRAS NRAS CBL NF1 LAMB4
SF3B1SRSF2U2AF1 LUC7L2ZRSR2PRPF8TET2IDH1IDH2DNMT3AASXL1EZH2 RUNX1 PHF6BCORTP53BRCC3STAG2 MPLJAX2PTRR T KRASNRASCBLNF1LAMB4 ETV6IRF1 NPM1 Pathways Splicing DNA methylation Chromatin modication Transcription DNA repair Cohesin Receptors/Kinases RAS pathway Others Correlation coefficient q<0.0001 q<0.001 q<0.01 1.0 0.0 -1.0
る遺伝子変異が起こると次に選ばれる変異は 決まっている.ある条件のもと,次にどうい う変異があるとMDS に進展しやすいかが決ま り,互いに協調しあってMDS を発症する.我々 の結果16 と別の研究の結果17 と比較すると, 協調的な遺伝子が驚くほど一致した ( 表2). 全く別なセットで検証しても再現されるので, この結果の信頼性は高いといえるだろう. 腫瘍内の多様性 もう一つ,癌を理解する上で重要な話をし よう.癌は,癌細胞が増大して最後に固体を 死に至らしめる病気である.イメージ的には 全て同じ癌細胞が塊を作っているようだが, 癌細胞は全部が同じではなく全く異なること が,最近ますます明らかになってきた.たと えば,癌細胞が人類全体だとすると,同じ人 間でも日本人やアメリカ人,フランス人など 違う集団が存在するように,癌細胞は単一な 集団ではない.つまり,肺癌や乳癌あるいは 白血病などそれぞれの腫瘍内には,違った性 質を持ついくつかの亜集団が存在する.これ を“腫瘍内の多様性”と言う.癌においては, この多様性がほぼ常に認められる.これは, 癌の発症を考える上でもちろん重要だが,癌 の再発や治療抵抗性と深い関係がある.たと えば,ペストが全ヨーロッパを蔓延しても, 何人かの人は耐性を持っているために生き残 る.これは細菌でも同様で,抗生物質で細菌 を殺そうとしても,必ずや耐性菌が現れる. 多様性があるために,ある種の治療をしても その薬に対して抵抗性を持っている細胞は殺 すことができない.治療に対してもともと抵 抗性のあるクローンが発生してくることが, 再発の主要な原因と考えられる.この腫瘍内 多様性をみる上で最も簡単でわかりやすい方 法は遺伝子変異を調べることである.たとえ ば図10 に示すように,腫瘍細胞全部に A の変 異があり,B は全部にはないが二つに共通,C, D,E はそれぞれ固有だとすると,ここから, どういう順番で遺伝子変異が起こったかを推測するこ とができる.合理的には,全部にあるA が最初に起こり, A から B と C が分岐して,さらに B の集団の中に E と D が分かれてきたという系統樹になる.これは進化論 の絵と同様で,人類あるいは生物の種が進化するよう に,癌は時々刻々進化をしている.これが,癌が治り にくい原因の一つである.このように遺伝子変異を比 較すると,腫瘍内の多様性が常に生じ,様々な癌細胞 が生じることが分かる. MDS におけるサブクローン性変異 そこで,MDS 症例 16 例の遺伝子変異を調べたとこ ろ,主要な集団全てにあるクローン性変異と分岐した サブクローン性変異があり,腫瘍内多様性が認められ た.16 MDS は単一な腫瘍細胞が増大した単純な構造で はなく,徐々に進化を繰り返しながらバラエティに富 んだ腫瘍クローンがあることが分かってきた.では, なぜサブクローンの同定が重要なのか.サブクローン 図 10. 腫瘍内多様性 TET2 ZRSR2 (q<0.0001), SRSF2 , CBL (q<0.001) SRSF2, ZRSR2 (q<0.01) SF3B1 DNMT3A, JAK2 (q<0.01) DNMT3A (q<0.01)
SRSF2 STAG2, ASXL1, RUNX1, IDH2TET2 (q<0.001), CBL (q<0.01) (q<0.0001) STAG2, IDH2,ASXL1, TET2, RUNX1 CUX1 (q<0.01) U2AF1 ASXL1 (q<0.0001), PHF6, ETV6 (q<0.01) ASXL1 (q<0.05)
ZRSR2 TET2 (q<0.0001), PHF6 (q<0.01) TET2 (q<0.01) STAG2 ASXL1 , RUNX1 , SRSF2BCOR (q<0.0001) , IDH2 (q<0.001), NRAS,
EZH2 (q<0.01)
SRSF2, RUNX1 (q<0.01) ASXL1 (q<0.05) IDH2,EZH2 (q<0.1)
DNMT3A SF3B1 (q<0.001) SF3B1 (q<0.01), BCOR (q<0.05) RUNX1 SRSF2, ASXL1 , EZH2, STAG2(q<0.0001) , BCOR STAG2, ASXL1, SRSF2 EZH2 (q<0.05), NRAS (q<0.1) (q<0.01) ASXL1 SRSF2, U2AF1 , STAG2 , IDH2, RUNX1, NRAS (q<0.0001) EZH2,
CBL (q<0.01)
SRSF2, RUNX1 (q<0.01) JAK2, U2AF1, STAG2 (q<0.05) IDH2, NRAS, CUX1 (q<0.1) EZH2 RUNX1LUC7L2, NPM1, ETV6 (q<0.0001), ASXL1, TP53, LAMB4,
NRAS (q<0.001), STAG2 (q<0.01)
RUNX1 (q<0.05) STAG2 (q<0.1) 表 2. MDS の遺伝子変異における協調的な遺伝子
Papaemmanuil E, et al. Blood 2013.17
Haferlach T, et al. Leukemia 2014.16
N = 738 N = 944
は,腫瘍全部にあるわけではなく,一部だけにあ る変異である.しかし,たとえ10% しかないサブ クローン変異でも,予後にはクローン変異と同じ 影響を及ぼす.これは大変重要なことで,たとえ ばRUNX1 や AXL1,あるいは TP53 の変異は,わ ずか数% の集団にあるだけでも予後が悪くなる .16 したがって,精密に遺伝子変異を調べることが重 要であり,その目的には高速シークエンサーは最 適な手段である.このような方法は,数年後には 臨床診断に必ず導入されてくるであろう. さて,このような観点で944 例をさらに検証す ると,腫瘍のサブクローンが456 例 (48.3%) に認 められた.16 わずか104 遺伝子だけを調べても明ら かにサブクローン変異があり,MDS が白血病に近 づくにつれて,ますます多様なクローンが増えて いた.また,クローンとサブクローン内のアレル 頻度は,遺伝子ごとに異なっていた.16 つまり,遺 伝子の変異が起こる場合,先述の特定の組み合わ せと共に,変異が生ずる順番もあることが分かっ た. では,MDS はどういう遺伝子変異がどういう順 番で蓄積して起こるのか.最初にRNA スプライシ ング遺伝子やDNA のメチル化に関わるような遺 伝子,次にRUNX1 や EZH2 のような転写因子やエ ピジェネティックスに関わるような遺伝子の変異 が次々と蓄積してMDS を発症する.さらに,RAS pathway の遺伝子,加えて SETBP1 遺伝子が変異 して白血病に進展するとういう順番が存在する.18 完全に正確とまではいえないが,遺伝子変異の順 番がおおよそ分かるようになった. 遺伝子変異の起源 遺伝情報を持つ受精卵からヒトが形成される過 程で,細胞は幾度も分裂する.分裂時に遺伝情報 は全ての細胞に等しく分配されるのが生命の基本 原理である.それを担っているのが遺伝物質DNA 分 子 で,30 億塩基対もある.1 回分裂するたび に30 億塩基対が間違いなく複製することは大変 難しく,少しは間違える.やはり自然は間違え る.もし間違えなければ,進化は起こらない.完 全に正確に遺伝子が複製されれば,種はそのまま であり,猿は猿のまま,大腸菌は大腸菌のままで 進化など起こらない.少し間違えることが大変重 要なわけで,少なくともアウストラロピテクス (Australopithecus afarensis) から人類になるまでの 間においても,相当な数の間違いが起こって,進 化や多様性がおこった.最近,驚くべきことに金 髪はたった一つの遺伝子の変異であることが報告 された.19 このように非常にドラスティックな人類 の形態でさえ,30 億塩基対あるうちから,たった 1 個の塩基の違いで起こる.1 個であっても,変異 が起こる場所によっては大変重要なインパクトを 及ぼすわけである.しかしながら幸か不幸か,た んぱく質をコードしている領域は1 ~ 2% に過ぎ ないので,ほとんどの変異は無害である.1 回に 起こる変異は数個で,これは細胞の種類によって 随分と違う.生殖細胞を作るprimordial germ cell で
は複製は非常に正確であるが,体外培養すると10 倍から100 倍ぐらいの高い確率で間違いが起こる. 癌の起源はこのようなランダムに起こる変異で,1 つの細胞が分裂してできた2 つの細胞は決して同 じではない.細胞が1 回分裂すると,ハプロイド ゲノムあたり0.2 から 4.5 個の変異が生じると見積 もられている.20 遺伝子変異の蓄積 健常人の造血前駆細胞における遺伝子変異の数 を調べた報告がある.21 臍帯血を1 個ずつバラバラ の細胞にして培養した後,それらを全てエクソン 解析する.同様に,20 ~ 70 代の人から骨髄を採 取し,細胞1 個ずつについて由来するクローンを 調べた.その結果,遺伝子変異の数は臍帯血サン プルが最も少なく,骨髄サンプルでは年齢にほぼ 比例して変異数が増加した.20 代でもいくつかの 変異があり,加齢とともに増え,50 代で平均約 7 個, 70 代ではどのクローンを調べても 10 個前後の遺 伝子変異があった.最近,世界最高齢に近い115 歳の女性の血液細胞のゲノムが調べられた.もと もとヒトの造血幹細胞は1 万個~ 2 万個あると言 われているが,それが白血球や赤血球などをつく
り,一生の間造血を担う. ところが,115 歳まで に様々な変異が起きて多くの造血幹細胞が消失し, たった2 個しか残っていなかったと報告された .22 加齢とともに我々の細胞は変化する.その大きな 変化の一つは,ゲノムが正確に複製されずに少し ずつ変わってしまうことである. ここで興味深いのは,MDS の平均発症年齢が 65 歳前後で高齢者に多い疾患であることだ.先に述 べたように,MDS の遺伝子変異の数は平均 9.2 個 であるが,ほとんどはどの細胞にも生じるランダ ムな変異で,MDS 発症に直接関与する遺伝子変異 は2,3 個である.2 万個の造血幹細胞が 1 年間に 4 回分裂すると仮定すると,70 代になるまでに 280 回分裂していることになる.その間に少しずつ遺 伝子変異が蓄積して,場合によってはその中に非 常に都合の悪い変異が起こることがある.ニュー ヨークにいる私の友人Ross Levine のグループの報 告では,健常な80 歳以上の女性の 1 ~ 2% に,白 血病やMDS で変異する頻度の高い TET2 遺伝子の 変異が骨髄に認められた.23 ヒトは加齢とともに, このような都合の悪い変異を起こしているかもし れない. MDS は突然に起こるわけではなく,非常に長い 年月をかけて遺伝子変異が蓄積し,偶発的に発症 するのではないかと類推される.現在は一人のゲ ノムをシークエンスするのに約千ドルもかかり, それでもこれほど詳しく調べられない.しかし, 近い将来,シークエンスが簡便にできるようにな ると,一般検診でTET2 変異あるいは他の遺伝子 変異が既にある人が見つかり,その人は精密にモ ニターするようになるかもしれない.数年後には, 検診の仕方も大きく変わってくるだろう. 再生不良性貧血と MDS 再生不良性貧血は,自己免疫が働いて造血幹細 胞が激減し,血液中の白血球,赤血球,血小板の 全てが減少する疾患である.そのために貧血症状 や感染による発熱,あるいは出血などをきたす. 約7 ~ 8 割の患者は免疫抑制療法で改善するが, ときにMDS や AML に移行する.再生不良性貧血 の患者は,他の人達よりもMDS や AML を発症 する確率が高い.そこで,アメリカ国立衛生研究 所 (NIH) と日本の二つのコホートで,再生不良性 貧血症例を対象に造血器腫瘍関連の106 遺伝子を 調べたところ,両コホートともに約3 割に遺伝子 変異が認められた.その中でも発症頻度が特に高 かった遺伝子は,PIGA, BCOR /BCORL1, DNMT3A,
ASXL1 であった ( 図 11). Var iant al lel e f requenc ies PI GA BCOR /BC OR L1 DN M T3 A ASXL 1 CO HESI N Sp lic ing TE T2 RUNX 1 GN AS PRC2 TP53 LAMB4 PR PF 8 CBL TERF1 /TE RT CS M D1 JAKs IDH2 CUX1 RBBP4 ATRX PH F6 SETBP1 AT M KRAS MPL NF1 POT1 RAP1 A WT 1 PEG3 STAT 3 CD AN 1 DIS3 GAT A1 SH 2B3 others splicing frameshift nonsense missense multiple 図 11. 再生不良性貧血における造血器腫瘍関連遺伝子の変異頻度
Common targets in both cohorts included 4 genes, i.e., PIGA, DNMT3A, BCOR, ASXL1. Clonality cannot be explained only by bottleneck effects!
Darwinian selection plays a role.
14.0% 12.0% 10.0% 8.0% 6.0% 4.0% 2.0% 0.0%
1 例を紹介する.患者は 75 歳男性で,12 年前に
再生不良性貧血と診断された.NIH の Neal Young
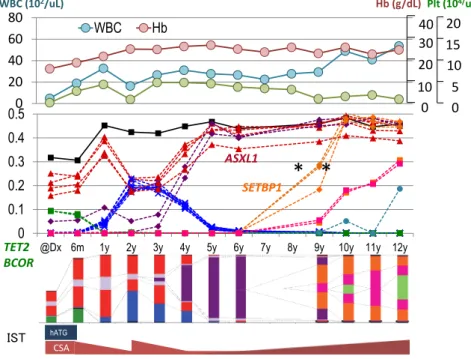
先生が保存されていた初診時から12 年間の検体 を,全てのポイントで全エクソン解析をして詳細 に調べた.その結果,図12 に示すように,初診 時から既に数種の遺伝子変異が認められる.初診 から約1 年後には免疫抑制療法が奏効して TET2, BCOR 変異クローンは消失したが,他のクローン が増えている.その後,あるクローンが大半を占 めたりSETBP1 変異が現れたりと,12 年間にいろ いろな変化が起こっていることが解る. 一方,ヘモグロビン,白血球,血小板の値には 大きな変化はない.血小板値はずっと5 × 104 μL 前後で低いままなので,再生不良性貧血の寛解は むずかしいと思われていた.初診9 年後ぐらいか ら血小板値は1 ~ 2 × 104 μL になり非常に悪い状 況になったので,サイクロスポリンを増量するが 全く効かない.詳細にみると,様々な遺伝子変異 が認められクローン進化が起こっていて, MDS に 関わる遺伝子変異パターンを形成している.この ような症例は,臨床的には,再生不良性貧血なの かMDS なのか鑑別が難しい.
図 12. 重症再生不良性貧血 (severe aplastic anemia; SAA) の 1 症例
Plt (104/uL) WBC (102/uL) 0 20 40 60 80 WBC Hb 0 10 20 30 40 0 5 10 15 20 0 0.1 0.2 0.3 0.4 0.5
@Dx 6m 1y 2y 3y 4y 5y 6y 7y 8y 9y 10y 11y 12y
CSA hATG IST
* *
Hb (g/dL) ASXL1 TET2 BCOR SETBP1 現在は,再生不良性貧血患者の塩基配列の解析 は行っていないが,MDS を発症する前のこのよう な経緯も考えなければならない.前例で言えば, 初診から約4 ~ 5 年後にシークエンスをすれば, クローン性造血や1 個の造血幹細胞からクローン が再構築されていることがわかったであろう.精 密なモニターが必要であることが早期にわかり, 血小板が激減する前に対応することが充分可能と なる.このように遺伝子を調べることによって, 我々の疾患に対する考え方も随分と変わってくる だろう. おわりに 近年,DNA のシークエンス技術が革命的に進化 し,我々は,ゲノムの病気としての癌を正確に調 べることができるようになった.そして,MDS 発 症に関わっている主要な遺伝子をほぼ全て同定す るに至った.さらに,MDS には腫瘍内多様性があ ることがわかり,遺伝子変異にはヒエラルキーが あることもわかった.また,遺伝子変異の情報を 加えたモデルは,IPSS モデルよりも正確に予後を 予測することができる.たとえば,monosomy 7 とCleveland Clinic Taussig Cancer Institute Hideki Makishima, Kwok Peng Ng, Mikael A. Seaker, Jaroslow P. Maciejewsk Chang Gung Memorial Hospital,
Chang Gung University Lee-Yung Shih
Tokyo Metropolitan Ohtsuka Hospital Ken Ishiyama, Shuichi Miyawaki
Tsukuba University Mamiko Sakata-Yanagimoto,Shigeru Chiba
Showa University Hiraku Mori
University of Tokyo
Cancer Genomics Project Kenichi Yoshida, Yasunobu Nagata, Ayana Kon, Masashi Sanada, Yusuke Sato,
Aiko Matsubara, Yusuke Okuno
Human Genome Center Yuichi Shiraishi, Satoru Miyano
Center for Stem Cell Biology and Regenerative Medicine Ryo Yamamoto, Makoto Otsu, Hiromitsu Nakauchi
Department of Pathology Aiko Nishimoto, Shunpei Ishikawa
Cedars-Sinai Medical Center H Phillip Koeffler
Uniformed Services University of
the Health Sciences Yang Du, Nhu Nguyen
MLL Laboratory Alexander Kolemann, Vera Grossman,Torsten Haferlach, Claudia Haferlach
University Hospital Mannheim Wolf-Karsten Hofmann, Florian Nolte,Daniel Nowak
Deapartment of Pediatrics
Nagoya University Hitoshi Sakaguchi, Hideki Muramatsu, Seiji Kojima
Hirai H, Kobayashi Y, Mano H, et al. A point mutation at codon 13 of the N-ras oncogene in myelodysplastic syn-drome. Nature 1987;327:430-432.
Sugimoto K, Hirano N, Toyoshima H, et al. Mutations of the p53 gene in myelodysplastic syndrome (MDS) and MDS-derived leukemia. Blood 1993;81:3022-3026. Sanada M, Suzuki T, Shih LY, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neo-plasms. Nature 2009;460:904-908.
Delhommeau F, Dupont S, Della Valle V, et al. Muta-tion in TET2 in myeloid cancers. N Engl J Med 2009; 360:2289-2301.
Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010;42:665-667. International Human Genome Sequencing C. Initial sequencing and analysis of the human genome. Nature 2001;409:860-921.
Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001;291:1304-1351. International Human Genome Sequencing C. Finishing the euchromatic sequence of the human genome. Nature 2004;431:931-945.
Meyerson M, Gabriel S, Getz G. Advances in under-standing cancer genomes through second-generation sequencing. Nat Rev Genet 2010;11:685-696.
Yoshida K, Sanada M, Shiraishi Y, et al. Frequent path-way mutations of splicing machinery in myelodysplasia. Nature 2011;478:64-69.
Malcovati L, Papaemmanuil E, Bowen DT, et al. Clini-cal significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neo-plasms. Blood 2011;118:6239-6246.
Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009;136:701-718.
Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet 2013;45:942-946.
Kon A, Shih LY, Minamino M, et al. Recurrent muta-tions in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013;45:1232-1237. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related dis-orders. Blood 2013;122:3169-3177.
Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 参考文献 5q−, 17p−, 12p− があり TP53 変異を持っているハイ リスク症例は,現在の医療では救命することは不 可能であろう.こういう場合に造血幹細胞移植の ような強い治療を実施すると,かえって副作用の ために,患者のQOL を大きく低下させる可能性も ある.遺伝子変異の情報で今より正確な予後予測 ができるので,我々はより早期に治療方針を決定 することができる.遺伝子情報を加えた新しい予 後予測モデルは,近い将来,多大な応用用途があ ると考える. 本日お話した研究は,多くの共同研究者 (表3 ) による成果である.たとえば,研究に必要な検体 を集めるために同意書をとる人,あるいは採血を する人,毎日のデータのチャート作成をする人な ど様々な医療従事者が関わっている.また,DNA のシークエンス解析には膨大なデータを扱う.東 京大学医科学研究所の宮野悟先生や白石友一先生 は数学分野の研究者である.MDS の克服は,共同 研究者をはじめMDS の診療に関わる全ての人々の 大きな希望,目標であり,重要な課題の一つである. 表 3. 共同研究者 (敬称略)
小川誠司教授 (Ogawa Seishii, MD, PhD) 略歴 1988 年 東京大学医学部卒業 1993 年 東京大学医学部附属病院第三内科 非常勤医員 1996 年 日本学術振興会 特別研究員 1997 年 東京大学医学部附属病院第三内科 助手 2002 年 東京大学大学院医学系研究科 造血再生医療寄付講座 客員助教授 2006 年 東京大学 21 世紀 COE プログラム 特任准教授 2008 年 東京大学 「大規模ゲノムミクスによるオーダーメイドがん診療技術の開発」 特任准教授 2013 年 9 月~京都大学大学院 医学研究科腫瘍生物学講座 教授 syndromes. Leukemia 2014;28:241-247.
Papaemmanuil E, Gerstung M, Malcovati L, et al. Clin-ical and biologClin-ical implications of driver mutations in myelodysplastic syndromes. Blood 2013;122:3616-3627; quiz 3699.
Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 2012;366:1090-1098.
Guenther CA, Tasic B, Luo L, et al. A molecular basis for classic blond hair color in Europeans. Nat Genet 2014;46:748-752.
Gundry M, Vijg J. Direct mutation analysis by
high-through-put sequencing: from germline to low-abundant, somatic variants. Mutat Res 2012;729:1-15.
Welch JS, Ley TJ, Link DC, et al. The origin and evo-lution of mutations in acute myeloid leukemia. Cell 2012;150:264-278.
Holstege H, Pfeiffer W, Sie D, et al. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res 2014;24:733-742.
Busque L, Patel JP, Figueroa ME, et al. Recurrent so-matic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012;44:1179-1181. 17. 18. 19. 20. 21. 22. 23.