8. 臨床試験の試験成績
別添資料ヘ-1-1~3総 括
MDT-1112 経カテーテルペーシングシステムは、心臓に周期的に人工的な電気刺激を 与えることによって、正常に近い心臓の収縮リズムを回復させ、患者を日常生活に復帰さ せることを目的として、カテーテルを用いて経皮的に挿入し、心臓内に留置される植込み 型 心 臓 ペ ー ス メ ー カ で あ る 。 本 品 の 臨 床 試 験 と し て 、 日 本 を 含 む 国 際 共 同 治 験 『MDT-1112 経カテーテルペーシングシステムの臨床試験/Micra Transcatheter Pacing Study (MDT-1112 国際共同治験)』を実施した。MDT-1112 国際共同治験の概要及び治 験から得られた知見について記述する。 MDT-1112 国際共同治験の概要を下表に示す。 試験の種類 前向き、単群、多施設共同、国際共同試験 対象 心室シングルチャンバペースメーカに対する ClassⅠ又は ClassⅡの植込み適応を有 する患者 症例数 全地域:登録(同意取得) 685 例、植込み試行 666 例、植込み成功 660 例 日本:登録 38 例、植込み試行 36 例、植込み成功 36 例 使用方法 植込み手術 検査・観察項目 抗凝固療法を含むシステム及び手技情報、胸部 X 線検査、X 線透視動画、電気的テ スト、短時間運動負荷テスト、QOL、患者満足度、機器操作性、有害事象、機器不具 合、システム変更、ホルター心電図、長時間トレッドミルテスト、その他 治験期間 2013 年 12 月 4 日(最初の登録)~2015 年 5 月 19 日(1 回目の解析のための来院打 切り日となる 300 例目の 6 か月来院日) 代表施設名及び 施設数Allgemeines Krankenhaus der Stadt Linz(オーストリア)を含む合計 55 施設 資料番号 添付資料ヘ-1-1~3 概略 本治験は、MDT-1112 経カテーテルペーシングシステムの安全性及び有効性を評価 するとともに、長期間の機器性能を評価する前向き、多施設共同、単群の国際共同治 験である。本治験は、心室シングルチャンバペースメーカの適応を有する患者を対象 とし、植込みが成功した患者について、退院前、1 か月、3 か月、6 か月及び以降 6 か 月ごとに治験終了までフォローアップを行う。主要評価項目は、植込み後 6 か月にお ける MDT-1112 システム又は手技に関連する主要合併症非発生率、及び適切な PCT を有する被験者の割合であり、両評価項目が満たされた場合に成功と判断され る。また、本治験ではグループ逐次デザインを採用し、全体の第一種の過誤を 0.05 に 維持しつつ、主要評価項目の解析を最大 3 回実施することとしていた。 考察・結論 本治験は、MDT-1112 システムの安全性と有効性を合理的に保証するために、試験 デザイン及び解析方法について、事前に十分な検討及び標準化を行った。治験の実 施に際しては、全地域で統一された治験実施計画書及び症例報告書を使用し、同一 水準のモニタリングを実施することで結果の均一性を担保した。また、日本以外の地 域で実施された試験成績については、外国で実施された臨床試験データの受入れ 要件を満たしていることを確認した。 MDT-1112 経カテーテルペーシングシステムについて、666 例の被験者に対して植込 みを試行し、660 例(99.1%)において植込みが成功した。植込み成功被験者 275 例が 植込み後 6 か月来院を完了した時点で、事前に規定された安全性主要評価項目及 び有効性主要評価項目を満たしたことから、MDT-1112 の安全性及び有効性が証明 された。植込み試行被験者 666 例に対する、合計 2,920.5 か月間のフォローアップに おいて、MDT-1112 システム又は手技に関連する主要合併症が、25 例において 28 件発生した。
考察・結論(続き) 植込み後 6 か月(183 日)における MDT-1112 システム又は手技に関連する主要合併 症非発生率の Kaplan-Meier 推定値は 95.6%(98.66%CI:92.7~97.4%)であり、過去の ペースメーカ臨床試験 6 件(2,667 例)から成る参照データセットのうち、心房細動を有 する被験者集団 977 例に基づいて事前に規定された性能基準である 83%を有意に上 回った(P<0.0001)。また、事後解析によって、参照データセットの全被験者集団(2,667 例)を用いて、MDT-1112 と従来型のリード付きペーシング技術との比較を行った。こ れは、本治験のすべての被験者が心房細動を有していたわけではないことから、最も 保守的な比較と考えられる。その結果、MDT-1112 は従来技術と比較して、6 か月に おける主要合併症発生率を有意に低減させることが証明された(従来技術の 7.4%に対 して MDT-1112 では 4.4%、P=0.018)。MDT-1112 は、参照データセットと比較して、ア クセス部位、ペーシングの問題及びデバイス固定に関連した主要合併症発生率を低 下させると考えられた。参照データセットと比較して、MDT-1112 でリスクが有意に上昇 した主要合併症の領域は認められなかった。加えて、フォローアップ期間において明 らかなデバイスのディスロッジメントの報告はなく、3 か月来院及び 12 か月来院での X 線分析に基づくデバイスの位置変化も観察されていないことから、デバイスの固定が 十分であったことが示唆された。 要約すると、安全性に関する以下の知見が得られた。 参照データセット(2,667 例)と比較して、6 か月における主要合併症発生率が低い (参照データセットにおける発生率 7.4%に対して、MDT-1112 では 4.4%)。 システム変更を含むシステム修正(摘出、位置変更、交換等)の発生割合が低い (666 例のうち 3 例、0.5%)。3 例のうち 1 例は MDT-1112 デバイスの摘出及び新しい MDT-1112 デバイスの植込みが行われ、2 例は体内に MDT-1112 デバイスを留置 したまま従来のペーシングデバイスが植え込まれた。 未知の事象なし(0%)。 デバイステレメトリの問題なし(0%)。 明らかなディスロッジメントなし(0%)。 全身性感染なし(0%)。 植込み後 6 か月時点において、パルス幅 0.24ms で測定した PCT が低値で(2V 以下) 安定している(植込み時からの上昇が 1.5V 以下)被験者の割合は、事前に規定された 有効性主要評価項目の性能目標である 80%を上回る 98.2%(98.66% CI:95.0~99.6%)で あった(P<0.0001)。また、観察された閾値から、6 か月のフォローアップを通じた実際の 機器使用条件に基づいて推定された電池寿命中央値の平均値は 12.5 年であった。 被験者の 99.6%(98.66% CI:97.2~100%)において、植込み後 6 か月時点での VCM PCT が手動測定(自動)PCT から 0.5V 以内であった。これは、事前に規定された性能 目標である 85%を上回っており、MDT-1112 システムの VCM 機能の精度が確認され た。また、30 件のトレッドミルテストによって、レートレスポンス動作が適切であることも 確認された。 植込み手技及び機器操作性は、複数地域の植込み医師及び被験者に好意的に受 け入れられた。55 実施医療機関における 92 名の医師が植込み手技に成功し、その 大部分の手技について、「容易」又は「極めて容易」であったと報告された。 また、日本の実施医療機関で MDT-1112 が植え込まれた被験者 36 例について、日 本以外の地域で MDT-1112 が植え込まれた被験者と試験成績を比較した。日本の被 験者については、他地域の被験者と比較して一部の被験者背景が異なる、手技所要 時間が概して長いといった傾向が示唆されたが、主要評価項目及び副次評価項目に 有意な地域間較差は認められなかった。したがって、MDT-1112 の植込みが試行され た 666 例の被験者集団全体から導かれた安全性及び有効性に関する推論を、日本 に適用することが可能であると判断した。 本治験では、治験機器及び植込み手技について、予定されていた評価項目のすべ てを満たすか、それを上回る結果が得られた。したがって、本治験は、MDT-1112 シス テムの安全性及び有効性を合理的に保証できると考えられた。
8.1 臨床試験成績 別添資料ヘ-1-1~3 8.1.1 外国で実施された臨床試験成績を用いた根拠 MDT-1112 国際共同治験において、日本以外の地域で実施されたものは、外国で実施された 臨床試験データの受入れ要件を満たしている。外国で実施された臨床試験成績を含めて評 価資料とした根拠として、平成 9 年 3 月 31 日付薬発第 479 号『外国で実施された医療用具の 臨床試験データの取扱いについて』、及び平成 18 年 3 月 31 日付薬食機発第 0331006 号『医 療機器に関する臨床試験の試験成績のうち外国で実施したものの取扱いについて』に示され る要件への適合状況を表 8-1 に示す。 表 8-1 外国で実施された臨床試験データ受入れ要件への適合状況 通知 要件 MDT-1112 国際共同治験の適合状況 薬発第 479 号 1 臨床試験の方法、臨床評価の方法 等が、我が国の基準又はガイドライン を満たすものであるか、我が国の医 療実態に適応し得るものであること。 治験計画段階で、ペースメーカ療法、MDT-1112 の使用方法及び本治験の対象患者を含む試験 デザインについて検討を行い、試験方法、評価 方法等が、本邦の医療実態に適応し得るもので あることを確認している。また、以下に述べるよう に受入れ要件を満たしており、試験方法及び評 価方法は本邦の基準及びガイドラインを満たすも のと判断する。 2 試験を適切に実施し得る経験と能力 を有する研究者により、公的機関又 は大学附属医療機関等信頼性ある 医療機関において実施されたもので あること。 治験実施計画書において、本治験の治験責任 医師等は、不整脈を有する被験者の診断及び IPG を用いた不整脈治療の経験を有し、治験実 施計画書に関する教育訓練を受けることが規定 されている。また、すべての実施医療機関は、治 験関連手順を実施する前に倫理委員会及び該 当する関連規制当局の承認(例:所轄官庁の承 認)、並びに当該実施医療機関の準備が整って いることを示すメドトロニック社からの文書を得な ければならない。したがって、本治験は、適切な 研究者によって、信頼性のある医療機関で実施 されていると判断する。
通知 要件 MDT-1112 国際共同治験の適合状況 3 適切な手順と方法(世界医師会が定 めたヘルシンキ宣言の遵守、我が国 の医療用具の臨床試験の実施に関 する基準又はこれと同等以上の外国 の基準への適合)で実施されたもの であること。 本治験は、臨床試験の実施に関する基準(GCP) として知られている国際的な倫理的及び科学的 な質に関する基準に準拠して実施されている。ま た、本治験は、ISO 14155:2011 及びその他の国 際臨床試験要件に示されている GCP の原則を 反映するようデザインされている。 全地域のすべての治験実施医療機関が下記 に従い、これらを遵守している。 ヘルシンキ宣言(プライバシー及びデータ保護 法を含む)又は各参加国の法規のいずれかのう ち、本治験被験者にとってより手厚い保護が得 られる方 21 CFR Part 11(電子記録、電子署名) 21 CFR Part 54(治験責任医師等の資産開示) 治験実施計画書に記載されている手順 各地域の倫理委員会の要件 上記の規制要件に加え、本治験は関連する地域 法に準拠して実施している。 本治験は、2007 年 FDA 改正法(FDAAA)及びヘ ルシンキ宣言に従い、初回登録に先立って Clinical Trials.gov に公開登録している。加えて、 地域法によって要求される場合、本治験は地域 の規制当局のデータベースにも登録されている。 薬発第 479 号 (続き) 4 臨床試験データの基礎となった個別 の症例記録・統計解析記録等の生 データ等について、必要に応じ調査 し得るものであること。 本治験では、臨床試験用の電子データ管理シス テムを使用してデータを収集している。治験実施 計画書に定める試験手順には、原資料が必要で あることを定めている。一部の手順については、 被験者の診療記録に来院の証拠があれば、CRF の記載項目を原資料とみなすことができるが、そ の場合にも、別の方法で原資料を作成することを 強く推奨している。 治験実施計画書において、治験依頼者又は FDA を含む規制当局が、治験の実施について評 価し、GCP、治験実施計画書及び適用される規 制要件の遵守状況を確認するために、実施医療 機関の監査を行うことができることを定めている。 また、治験責任医師等/実施医療機関は、原 データ/原資料を直接閲覧に供し、治験関連の モニタリング、監査、倫理委員会による審査、及 び規制当局の調査に協力しなければならないこ とを規定している。
通知 要件 MDT-1112 国際共同治験の適合状況 薬食機発第 0331006 号: 受け入れ可能 な国又は地域 1 臨床試験を実施した国又は地域に おける薬事に関する法令において、 医療機器の臨床試験の実施基準が 定められており、その基準が我が国 の医療機器 GCP と同等以上のもの であって、当該基準に従って実施さ れた臨床試験及びそれと同等と考え られる臨床試験については、その試 験成績に関する資料を承認の申請 書に添付して差し支えないこと。 本治験は、臨床試験の実施に関する基準(GCP) として知られている国際的な倫理的及び科学的 な質に関する基準に準拠して実施する。また、本 治験は、ISO 14155:2011 及びその他の国際臨 床試験要件に概要が示されている GCP を反映 するようデザインされている。 全地域のすべての治験実施医療機関が下記 に従い、これらを遵守している。 ヘルシンキ宣言(プライバシー及びデータ保護 法を含む)又は各参加国の法規のいずれかのう ち、本治験被験者にとってより手厚い保護が得 られる方 21 CFR Part 11(電子記録、電子署名) 21 CFR Part 54(治験責任医師等の資産開示) 本治験実施計画書に記載されている手順 各地域の倫理委員会の要件 上記の規制要件に加え、本治験は関連する地域 法に準拠して実施している。 本治験は、2007 年 FDA 改正法(FDAAA)及びヘ ルシンキ宣言に従い、初回登録に先立って Clinical Trials.gov に公開登録している。加えて、 地域法により要求される場合、本治験は地域の 規制当局のデータベースにも登録されている。 薬食機発第 0331006 号: 臨床試験の実 施及び管理に ついて 1 法第 14 条第 5 項の規定に基づく書 面又は実地の調査に際して確認で きるよう、医療機器の臨床試験の実 施の基準に係る必須文書と同等もし くはそれ以上の文書が作成されてい ること。 治験実施計画書において、医療機器の臨床試 験の実施の基準に係る必須文書と同等の文書を 作成及び保管することを定めている。 2 法第 14 条第 5 項の規定に基づく書 面又は実地の調査に際し、臨床試 験の依頼者、臨床試験実施医療機 関その他の臨床試験に関与した者 に対して、必要な協力が得られるよう にしておくこと。 治験実施計画書において、治験依頼者又は FDA を含む規制当局が、治験の実施について評 価し、GCP、治験実施計画書及び適用される規 制要件の遵守状況を確認するために、実施医療 機関の監査を行うことができることを定めている。 また、治験責任医師等又は実施医療機関は、原 データ又は原資料を直接閲覧に供し、治験関連 のモニタリング、監査、倫理委員会による審査、 及び規制当局の調査に協力しなければならない ことを規定している。 薬食機発第 0331006 号: 臨床試験の実 施及び管理に ついて(続き) 3 監査あるいはその他の方法により、 申請者により臨床試験全体の信頼 性が保証されていること。 メドトロニック社による監査を行っていることを確認 している。 4 試験実施国・地域の基準が、医療機 器 GCP より高い水準の被験者保護 を要求する場合には、それに従って いるものであること。 治験実施計画書において、全地域で遵守する規 制要件に加え、関連する地域法に準拠して実施 することを定めている。 薬食機発第 0331006 号: 承認申請資料 について 1 臨床試験の試験成績原著が日本語 以外の言語で作成されている場合、 添付する資料中の「総括報告書」に ついては、原著の写しにその日本語 による要約を添付することで差し支 えないこと。なお、この場合、独立行 政法人医薬品医療機器総合機構よ り、必要に応じ「総括報告書」の一部 又は全部についての邦文訳を求め られることがあること。 本治験の試験成績原著に該当する「Micra Transcatheter Pacing Study PMA Clinical Report, Version 3, 06 October 2015」及び「Micra
Transcatheter Pacing Study PMA Report Addendum: Japanese Specific Data Analyses, Version 1, 29 September 2015」の邦文訳に、日 本で報告が必要な追加情報を加えたものを総括 報告書として添付する。
通知 要件 MDT-1112 国際共同治験の適合状況 2 「総括報告書」には、臨床試験の依 頼責任者の署名が記されていること。 記載あり。 3 我が国以外の国又は地域による査 察が終了しているときには、原則とし て、その結果を記載した通知文書等 の写しを添付すること。 該当文書なし。 4 我が国の医療機器 GCP と臨床試験 を実施した我が国以外の国又は地 域の基準とで相違点がある場合、そ の相違点をまとめ、資料に添付する こと。さらに、その相違点の当該試験 の信頼性や治験に用いた治験機器 の品質への影響などの有無につい て意見を付すこと。 本治験のデザインに反映されている ISO 14155: 2011 及び米国の GCP と、日本の医療機器 GCP との間の相違点は、本質的なものではないことが 示されている1。したがって、日本以外の地域で実 施された臨床試験データの受入れに際して、問 題となる信頼性又は治験機器の品質に与える影 響はないものと判断する。 5 カルテの保管場所、手術を実施した 場所その他の臨床試験に関わる所 在地が、臨床試験を実施した医療機 関所在地と異なるような場合、その 事実関係に関する資料を添付するこ と。 該当なし。 6 臨床試験に用いた医療機器の品質 に関する資料を添付すること。 本治験に用いた治験機器の品質に関する資料 は、該当する製造所において保管されており、必 要に応じて閲覧又は写しの入手が可能である。 8.1.2 MDT-1112 国際共同治験 本治験についての概略を表 8-2 に示す。なお、本治験における有害事象の定義は以下のとお りである。 全般 有害事象(AE) 治験機器との因果関係の有無に関わらず、被験者、使用者又はその他の者に生じた あらゆる好ましくない医学的事象、意図しない疾患若しくは傷害、又は好ましくない臨 床的徴候(異常な臨床検査所見を含む。) 注 1: 本定義には、被験機器又は対照に関連する事象が含まれる。 注 2: 本定義には、用いられた手技に関連する事象が含まれる。 注 3: 本定義は、使用者又はその他の者については、被験機器に関連する事象に限 定される(ISO 14155:2011、3.2 項)。 報告対象となる有害 事象 本治験では、全地域で共通して、すべての重篤な有害事象及びシステム、アクセサ リー、手技又は心血管に関連する可能性のある有害事象を収集する。 機器による有害作用 (ADE) 治験機器の使用に関連する有害事象 注 1: 本定義は、治験機器の使用、交付、植込み、設置若しくは操作に関する説明書 の不備若しくは不適、又は機器不良に起因する有害事象を含む。 注 2: 本定義は、治験機器の誤使用又は意図的な目的外使用に起因する有害事象 を含む(ISO 14155:2011、3.1 項)。 機器不具合(DD) 識別、品質、耐久性、信頼性、安全性、又は性能に関する医療機器の不備 注: 機器不具合には、機器不良、誤使用、及び不適切なラベリングを含む (ISO 14155:2011、3.15 項)。 関連性
1 Fearnot NE. GCP Convergence Improves Transportability of Medical Device Clinical Data. Regulatory Focus. 2013;1-7.
手技関連 MDT-1112 システムの植込み又は変更に直接的に関連して生じる有害事象 システム関連 MDT-1112 植込みデバイス関連:MDT-1112 デバイスの存在又は性能(意図されるも のかどうかは問わない)に起因する有害事象 デリバリーカテーテル関連:デリバリーカテーテルの存在又は性能(意図されるものか どうかは問わない)に起因する有害事象 ソフトウェア関連:MDT-1112 ソフトウェアの性能(意図されるものかどうかは問わない) に起因する有害事象 注: システム構成品が原因となって事象が発生したが、どの構成品が関連している のかが不明確である場合、基本的には当該事象が認められる前に最後に使用 した構成品とする。 アクセサリー/ツール 関連 プログラマ関連:プログラマの存在又は性能(意図されるものかどうかは問わない)に起 因する有害事象 イントロデューサ関連:イントロデューサの存在又は性能に起因する有害事象 植込み用具関連:植込み用具(デリバリーカテーテル及びイントロデューサを除く)の存 在又は性能に起因する有害事象 抜去用具関連:抜去用具の存在又は性能に起因する有害事象 ホルター心電計関連:ホルター心電計(ケーブルを含む)の存在又は性能に起因する 有害事象 心血管関連 心臓及び血管又は血液循環に関連する有害事象 入院 24 時間以上の治療入院(観察病棟、救急治療室、外来通院は除く。) 重篤性 予測できない機器に よる有害作用 (UADE) 機器に起因若しくは関連して生じた健康又は安全に対するあらゆる重篤な有害作用、 生命を脅かす問題又は死亡のうち、その性質、重症度又は発生頻度について治験実 施計画若しくは申請書類(補足的な計画書又は申請書類を含む。)において事前に特 定されていないもの。あるいは、機器に関連し、被験者の権利、安全又は福祉にかか わるその他のあらゆる予測できない重大な問題[21 CFR 812.3(s)]。 重篤な有害事象 (SAE) 以下の有害事象 a) 死亡に至ったもの。 b) 被験者の健康状態に重大な悪化をもたらし、以下の項目のいずれかの結果に 至ったもの。 1) 生命を脅かす疾患又は傷害 2) 身体構造又は身体機能の永久的障害 3) 入院又は入院期間延長 4) 生命を脅かす疾患若しくは傷害、又は身体構造若しくは身体機能の永久的障 害を避けるための内科的又は外科的介入 c) 胎児の障害、胎児死亡又は先天的異常若しくは先天的欠陥に至ったもの。 注: 以前からの病状のため予定された入院、又は治験実施計画書で規定された処 置は、重篤な健康悪化がない限り、重篤な有害事象とはみなさない(ISO 14155: 2011、3.37 項)。 機器による重篤な有 害作用(SADE) 重篤な有害事象の原因となった機器による有害作用(ISO 14155:2011、3.36 項) 予測できない機器に よる重篤な有害作用 (USADE) 現行のリスク分析報告書において、その性質、発生頻度、重篤性又は転帰が特定され ていない機器による重篤な有害作用 注: 予測される機器による重篤な有害作用(ASADE)は、リスク分析報告書でその性 質、発生頻度、重篤性又は転帰が既に特定されている作用である(ISO 14155: 2011、3.42 項)。
合併症 以下のいずれかの項目に該当する有害事象は合併症であるとみなされる。 a) 死亡に至る。 b) 重要な機器機能の停止を伴う。 c) 侵襲的介入を要する。 非侵襲的[FDA、CFR 21、812.3(k)]:診断的機器又は処置に適用する際、設計又は 意図していないものを意味する:身体の皮膚、粘膜、眼窩若しくは尿道に侵入又は貫 通する、また、耳では外耳道を越えて、鼻では鼻孔を越えて、口腔では咽頭を越え て、肛門管では直腸を越えて、膣では子宮口を越えて侵入する。 侵入=何かの中に入る若しくは何かを通り抜ける、伸長する、又は拡散する。抵抗に 逆らって入る。侵入口を得る。 貫通=力をかけた場所から入りこむ又は通り抜ける。 注: システム又は手技関連の有害事象のみを合併症又は観察事象として分類する。 主要合併症 下記を引き起こす合併症 死亡 機器の機械的又は電気的機能不全(例:ペーシング機能無効化、機器の電気的な 停止)による機器機能の永久的喪失 入院 48 時間以上の入院期間延長 システム変更(摘出、再留置、交換) 注: システム又は手技関連の有害事象のみを主要合併症、軽度の合併症又は観察 事象として分類する。 軽度合併症 主要合併症ではない合併症と分類されるあらゆる有害事象(例:薬剤の静脈内投与の みに基づいて合併症と分類された事象) 注: システム又は手技関連の有害事象のみを主要合併症、軽度合併症又は観察事 象として分類する。 観察事象 合併症ではない有害事象 注: システム又は手技関連の有害事象のみを主要合併症、軽度合併症又は観察事 象として分類する。 時期 植込み前の有害事象 同意説明文書への署名後、植込み中の皮膚切開までの間に発現する有害事象 植込み中の有害事象 植込み中の皮膚切開から皮膚縫合終了までの間に発現する有害事象 植込み後の有害事象 植込み時の皮膚縫合終了後に発現する有害事象 その他 不可避の有害事象 手術手技に固有の有害事象であり、すべての被験者に一定期間発生することが予測 される事象 事象の内容 手術時点を起点とする時間枠(時間) 麻酔に関連する悪心/嘔吐 24 微熱(100ºF すなわち 37.8℃未満) 48 アクセス部位の疼痛 72 軽度~中等度の挫傷/斑状出血 168 睡眠障害(不眠症) 72 手術台に横たわったことに関連する背部痛 72 システム変更 MDT-1112 植込みデバイスの最初の植込み成功後に、MDT-1112 システムの再留 置、摘出又は交換のためにイントロデューサが体内に挿入された場合、再インターベ ンションであると見なされ、システム変更に該当する。 システム修正 システム変更に加えて、MDT-1112 植込みデバイスをオフに設定し、従来型ペーシン グシステムが追加で植え込まれた場合。
表 8-2 MDT-1112 国際共同治験の概略 治験機器 本治験で使用した MDT-1112 の構成を下表に示す。 治験機器 アクセサリー システム/ モデル番号 構成品 MDT-1112 MDT-1112 システム MC1VR01 MDT-1112 植込みデバイス (MDT-1112 デバイス) MDT-1112 経カテーテル ペーシング システム MDT-1112 経大腿静脈カ テーテルデリバリーシステム (MDT-1112 デリバリーカテー テル) SW022 MDT-1112 ソフトウェア MDT-1112 プログラマ MDT-1112 アクセサリー 2090 シリーズ ケアリンク 2090 MI2355A MDT-1112 イントロデューサ ER220 モデル ERX10 拡張レンジ TEL-B アンテナ ER220 拡張 レンジホル ターモニタリ ングシステム DR220 ホルター 試験目的 MDT-1112 経カテーテルペーシングシステムの安全性及び有効性を評価する。 主要評価項目 【安全性】 1. 植込み後 6 か月における MDT-1112 システム及び/又は手技に関連する主要合併 症非発生率が 83%を上回ることを検証する。 【有効性】 2. 植込み後 6 か月における適切な PCT を有する被験者の割合が 80%を超えることを検 証する。 副次評価項目 1. MDT-1112 の VCM PCT の精度を手動で測定した PCT(手動測定 PCT)と比較して 示す。 2. MDT-1112 システムのレートレスポンス動作を示す。 補助的評価項目 1. 6 か月時点の MDT-1112 デバイスの予測電池寿命の要約。 2. 有害事象の要約。 3. MDT-1112 システムのペーシングインピーダンス及びセンシング振幅の特性を明ら かにする。 4. 植込み手技の特性を明らかにする。 5. 携帯型ホルターモニタリング所見による MDT-1112 システム性能の要約。 6. QOL の要約。 7. デバイスの位置変化の要約。 試験の種類 前向き、単群、多施設共同国際試験 対象 選択基準に適合し、除外基準のいずれにも抵触しない患者 選択基準 ACC/AHA/HRS ガイドライン(2008 年度版)及び各国のガイドラインにおいて、心室シン グルチャンバペースメーカに対する ClassⅠ又はⅡの植込み適応を有する患者。 治験で定められた事項を守ることができ、かつ、守る意志がある。また、フォローアップ 期間を通して居住地が変わらないと予想される患者。 18 歳以上の患者(地域法でより高い年齢が要求される場合にはそれ以上)。
除外基準 ペースメーカ依存患者(補充調律≦30bpm)。 注: ペースメーカ依存患者に対する制限は、ステアリングコミッティ及び DMC が初期の 25 例における 1 か月ホルター検査から得られたホルター心電図及びデバイス診断 データについて安全性を検討したのちに解除される。メドトロニック社は、ステアリン グコミッティの承認を受けてこの除外基準が解除可能となったのちに、米国外の治験 実施医療機関にその旨を通知する。同じ安全性報告書を FDA にも提出する。メドト ロニック社は、ペースメーカ依存患者への拡大に対する FDA の承認を受けてこの除 外基準が解除可能となったのちに、米国の治験実施医療機関に通知を行う。 ペースメーカ、ICD 又は CRT 機器が現在植え込まれている、又は植込みの既往がある 患者。 不安定狭心症を有する、又は適格性確認前 30 日以内の AMI の既往がある患者。 神経刺激装置その他永続的に植え込まれる医療機器が現在植え込まれている患者 (体外式ペースメーカは除く。)。 三尖弁を機械弁に置換している、下大静脈フィルタを留置している、又は LVAD が植 え込まれている患者。 病的肥満患者でプログラミングヘッドを使用して 5 インチ(12.5cm)以内のテレメトリ通信 ができないと医師が判断する患者。 大腿静脈の構造的に、23Fr イントロデューサが挿入できない又は右心に植え込むこと ができない(閉塞、重度の蛇行等)と医師が判断する患者。 緊急時の胸骨切開術に耐容できないと考えられる患者。 ニッケルチタン合金(ニチノール)に対するアレルギーの既往がある患者。 酢酸デキサメタゾン 1.0mg の単回投与が禁忌の可能性がある患者。 余命が 12 か月以内と考えられる患者。 交絡する可能性がある薬剤若しくは医療機器の臨床試験に現在登録されている、又 は本治験期間中に参加が予定されている患者。並行して実施している臨床試験への 同時登録は、事前に治験依頼者から文書による承認を得た場合のみ認められる。 妊娠中又は授乳中の女性。妊娠の可能性がある女性で信頼性のある避妊法又は自 制を行っていない患者。 地域法で要求される除外基準(年齢等)に抵触する患者。 治験責任医師等によって治験参加を妨げる病状を有すると判断される患者。 症例数 全地域: 登録(同意取得) 685 例、植込み試行 666 例、植込み成功 660 例 日本: 登録 38 例、植込み試行 36 例、植込み成功 36 例 使用方法 植込み手術 検査・観察項目 及び時期 被験者の登録及びベースライン来院後に MDT-1112 デバイスの植込みを行う。植込み 後、MDT-1112 デバイスが体内に留置されたすべての被験者について、退院前、1、3、 6 か月及び以降 6 か月ごとにフォローアップを行う。MDT-1112 デバイスが体内に留置さ れなかった場合、当該被験者については 1 か月間のフォローアップを行う。フォローアッ プのフローチャートを図 8-1、検査・観察項目及びその時期を表 8-3 に示す。 評価方法・評価 基準 評価項目別に設定された統計的方法による。 代表施設名及び 施設数
Allgemeines Krankenhaus der Stadt Linz を含む合計 55 施設[米国:30、東ヨーロッパ/中 央ヨーロッパ:3、西ヨーロッパ:9、アジア太平洋(日本除く):7、カナダ:2、日本:4]におい て実施。
試験期間 2013 年 12 月 4 日(最初の登録)~2015 年 5 月 19 日(1 回目の解析のための来院打切り 日となる 300 例目の 6 か月来院日)
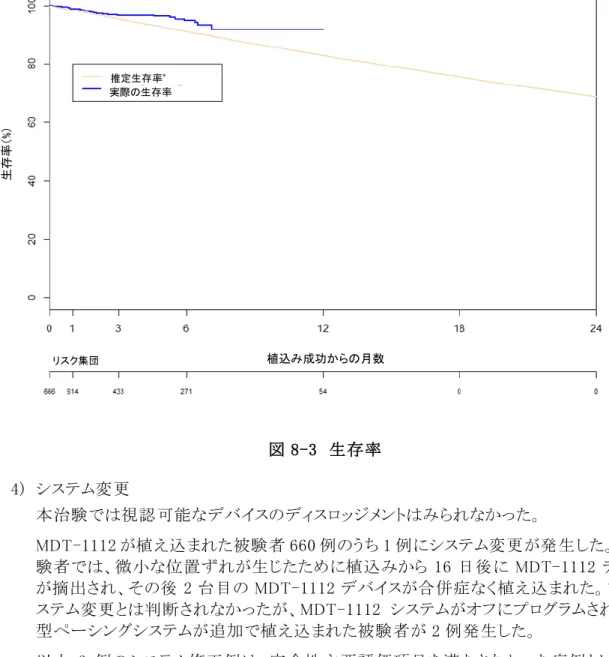
試験成績 2013 年 12 月 4 日に全地域における最初の被験者が MDT-1112 国際共同治験に登録 され、2013 年 12 月 5 日に MDT-1112 システムの植込みが行われた。2015 年 5 月 19 日 に 300 例目の植込み被験者の 6 か月フォローアップ来院が完了し、主要評価項目及び 副次評価項目に関する 1 回目の解析のための来院打切り日となった。本解析に使用した データは 2015 年 5 月 19 日以前に収集されたもので、2015 年 6 月 3 日以前にメドトロニッ ク社が受領したものである。この治験データベースは、解析のため 2015 年 6 月 24 日に 固定された。また本文書作成に使用した解析は、2016 年 10 月 6 日から 2016 年 10 月 13 日の間にメドトロニック社内の規定された調査過程により特定された日本の GCP 省令 遵守に問題のある 59 例を除外した。そのため、2015 年 5 月 19 日時点で 275 例の植込 み被検者の 6 か月フォローアップ来院が完了しており、治験データベースは 685 例の登 録被験者を対象とするコホートに基づく。本治験の DMC による会議が 2015 年 7 月 18 日に開催され、本治験の主要評価項目が満たされた旨がメドトロニック社に対して通知さ れた。来院打切り日である 2015 年 5 月 19 日の時点で、本治験に登録されている被験者 は 685 例であった。MDT-1112 システムの植込みが試行された被験者は 666 例であり、 植込みが成功した被験者は 660 例(99.1%)であった。これらの植込みは、18 か国の 55 施 設において 92 名の医師によって行われた。登録被験者 685 例中、合計で 26 例の被験 者が治験終了又は中止となり、うち 25 例は MDT-1112 の植込み成功前に終了又は中止 となった。 安全性主要評価 項目 評価項目: 植込み後 6 か月における MDT-1112 システム及び/又は手技に関連する主 要合併症非発生率が 83%を上回ることを検証する。 結果: 評価項目を満たした。 MDT-1112 の植込みが試行された被験者 666 例に対する合計 2,920.5 か月 間のフォローアップにおいて、25 例で発生した 28 件の主要合併症が CEC に よって MDT-1112 システム又は手技関連と判定された。植込み後 6 か月(183 日)における MDT-1112 システム又は手技に関連する主要合併症非発生率 の Kaplan-Meier 推定値は 95.6%(98.66% CI:92.7~97.4%)であった。これは事 前に規定された性能基準である 83%を有意に上回った(P<0.0001)。さらに、こ の安全性主要評価項目に関する統計的推論は、欠測データに対しても頑健 であることが示された。 また、参照データセット(心房細動を有する被験者 977 例)との比較から、 MDT-1112 の安全性プロファイルは既承認のペーシングシステムと同等であ り、臨床試験計画段階で予想された成績を上回ることが示唆された。保守的 な参照データセット(全 2,667 例)との比較からも、本治験における主要合併症 発生率が低いことが示された(P=0.018)。なお、地域を日本又は日本以外に 分類したサブグループ解析の結果、日本と他地域との間に異質性は認めら れず、安全性主要評価項目に関する統計的推論を日本に対して外挿するこ とが可能であると判断した。 有効性主要評価 項目 評価項目: 植込み後 6 か月における適切な PCT を有する被験者の割合が 80%を超える ことを検証する。 結果: 評価項目を満たした。 評価が可能であった被験者 272 例のうち 267 例(98.2%)が 6 か月時点で適切 な PCT を有していた(98.66% CI:95.0~99.6%)。適切な PCT とは、6 か月時点 での PCT が 2.0V 以下、植込みから 6 か月時点までの PCT の上昇が 1.5V 以下であり、ペーシング閾値上昇によるシステム修正が不要であったことを指 す。この結果は事前に規定された性能基準である 80%を有意に上回った (P<0.0001)。さらに、この有効性主要評価項目に関する統計的推論は、欠測 データに対しても頑健であることが確認された。 また、地域を日本又は日本以外に分類したサブグループ解析の結果、日本 と他地域との間に異質性は認められず、この有効性主要評価項目に関する 統計的推論を日本に対して外挿することは妥当であると判断した。
副次評価項目#1 評価項目: 植込み後 6 か月来院時の VCM PCT が正確(手動測定 PCT から 0.5V 以内) であった被験者の割合が 85%を上回ることを示す。 結果: 評価項目を満たした。 6 か月時点で対データ(VCM PCT 及び手動測定 PCT)が利用可能であった 被験者 255 例のうち 254 例(99.6%)の VCM PCT の値が手動測定 PCT(パルス 幅 0.24ms)から±0.5V 以内であった(98.66% CI:97.2~100.0%)。この割合は 事前に規定された評価項目である 85%を有意に上回った(P<0.00001)。さら に、この副次評価項目#1 に関する統計的推論は、欠測データに対しても頑 健であることが確認された。 また、地域を日本又は日本以外に分類したサブグループ解析の結果、日本 と他地域との間に異質性は認められず、この評価項目に関する統計的推論 を日本に対して外挿することは妥当であると判断した。 副次評価項目#2 評価項目: Kay-Wilkoff モデルによる勾配パラメータが、0.65~1.35 の同等性マージンに 収まることを検証することによって、MDT-1112 システムのレートレスポンス動 作を確認する。 結果: 評価項目を満たした。 Kay-Wilkoff モデルによる勾配パラメータが、0.65~1.35 の同等性マージン内 であることを検証し、MDT-1112 システムのレートレスポンス動作を確認した。 3 又は 6 か月来院のいずれかで実施された M-PREP テストで使用可能な 29 件全体について、推定勾配パラメータは 0.862 であった(90% CI:0.760~ 0.964、TOST 法による P 値<0.001)。 補助的評価項目 #1 評価項目: 6 か月来院時における MDT-1112 の予測電池寿命を要約する。 結果: 6 か月来院を完了した被験者 275 例のうち 274 例が 6 か月時点での電池寿 命推定に使用可能なデータを有していた。6 か月来院でのデバイス使用状況 に基づいた予測電池寿命は 12.5 年(範囲:6.0~14.6 年)であり、現行のリード 付きシングルチャンバペーシングシステムと同程度であった。 補助的評価項目 #2 評価項目: 治験期間中に収集したすべての有害事象を要約する。 結果: 登録被験者 685 例において、321 例で 585 件の有害事象が認められた。574 件が植込み試行時又は試行後に発生しており、MDT-1112 システム又は手 技に関連する有害事象は 128 件(109 例)であった。MDT-1112 システム又は 手技に関連する合併症は 54 件(48 例、7.2%)で、大部分は観察事象であっ た。また、未知の事象、明らかな MDT-1112 デバイスのディスロッジメント及び 全身性感染は認められなかった。 MDT-1112 システム又は手技に関連する合併症と MDT-1112 デバイス留置 部位又はタイン再展開回数との間に関連性は認められなかった。ただし、外 傷性心臓損傷リスクは再展開回数に比例して上昇する可能性があることが示 唆された。 日本においては、登録被験者 38 例のうち、27 例で 60 件の有害事象が観察 された。2 件は植込み試行前に、58 件は植込み試行時又は試行後に発生し ており、MDT-1112 システム又は手技に関連する有害事象は 22 件(16 例)で あった。MDT-1112 システム又は手技に関連する合併症は 5 件(5 例)であり、 すべてが軽度合併症と判定された。全地域と比較して、日本における MDT-1112 システム又は手技に関連する有害事象の発生割合が高いことが 示唆されたが、主要合併症と判定される事象は認められなかった。 補助的評価項目 #3 評価項目: MDT-1112 システムのペーシングインピーダンス及びセンシング振幅の特性 を明らかにする。 結果: ペーシングインピーダンスの平均値は、植込み時 714.2Ω、6 か月来院時 623.3Ω であり、R 波振幅の平均値は、植込み時 11.2mV、6 か月来院時 15.1mV であった。以上のことから、植込み後のデバイス固定状態及びセンシ ングが良好であることが示唆された。この結果は日本の被験者においても同 様であった。
補助的評価項目 #4 評価項目: 植込み手技の特性を明らかにする。 結果: 本治験において、666 例が MDT-1112 の植込み試行を受け、そのうち 660 例 で植込みが成功した(植込み成功率 99.1%)。MDT-1112 イントロデューサ挿入 から抜去までの時間(平均±標準偏差)は 34.6±23.1 分であり、X 線透視時間 (平均±標準偏差)は 8.7±9.0 分であった。植込みが成功した 660 例のうち 648 例(98.2%)について、タグテスト中の X 線透視動画によって 2 本以上のタイ ンの固定が確認された。また、植込み試行例 666 例の 59.3%は MDT-1112 デ バイスの再展開を必要とせず、94.9%は再展開が 4 回以下であった。 MDT-1112 デバイスの留置位置は、63.3%が右室心尖部、25.6%が右室中隔 (中隔又は中隔中央部)であった。植込み時の抗凝固療法は様々なアプロー チが用いられていた。 日本における植込み手技特性は他地域と比較して、抗凝固療法、再配置の回 数及び植込み時間について異なる傾向が認められた。また、植込み手技の所 要時間が長い傾向が認められた。これらの差異の多くは、施設又は地域固有 の医療環境及び医療行為に起因するものと考えられる。 補助的評価項目 #5 評価項目: 携帯型ホルターモニタリングで収集した ER220 ホルター心電図所見を要約 する。 結果: 1 か月来院時において 23 件及び 6 か月来院時において、24 件の使用可能 な 24 時間携帯型ホルターモニタリング記録が得られた。ホルター記録の評価 の結果、2 回の心臓周期を超えるペーシング停止は認められず、早期性能評 価項目を満たした。MDT-1112 デバイスは意図したとおりに機能しており、観 察された PVC アンダーセンシング及び体位感度に対しては適切な対応を 行った。 補助的評価項目 #6 評価項目: QOL を要約する。 結果: ベースライン時及び 3 か月来院時における対データを有する植込み成功被験 者について、ベースラインから 3 か月来院までに、SF-36 総合身体的健康スコ ア(平均±標準偏差)で 1.9±7.9 ポイント(P<0.0001)、SF-36 総合精神的健康ス コア(平均±標準偏差)で 3.5±11.5 ポイント(P<0.0001)、EQ-5D VAS スコア(平 均±標準偏差)で 6.2±20.9 ポイント(P<0.0001)の増加が認められた。 補助的評価項目 #7 評価項目: デバイスの位置変化を要約する。 結果: 退院前及び 3 か月来院時に X 線画像が得られている被験者 447 例につい て、デバイスの位置変化が認められた被験者はいなかった。同様に、退院前 及び 12 か月来院時に X 線画像が得られている被験者 54 例についても、デ バイスの位置変化が認められた被験者はいなかった。 追加解析:機器 操作性 評価項目: 機器操作性に関する質問票に対する植込み医師からの回答を要約する。 結果: 植込みが成功した手技 660 件において、大部分(88%)が、植込み手技に関す る全体的な印象を「容易」又は「極めて容易」と回答した。これは日本におい ても同様であった(92%が「容易」又は「極めて容易」と回答)。 追加解析:患者 満足度 評価項目: 3 か月来院での被験者へのアンケート結果を要約する。 結果: 植込み後 3 か月来院を完了したすべての被験者 461 例から満足度質問票へ の回答を収集した。被験者の大多数は、自身の回復について「満足」又は「と ても満足」と回答しており(90%)、デバイス植込みに伴う美容的外見についても 「満足」又は「とても満足」と回答していた(96%)。自身の回復について「とても 不満」と回答した被験者 6 例のうち 3 例については、3 か月来院の前に心不 全関連の有害事象が報告されていた。 日本では、植込み後 3 か月来院を完了した全 26 例から回答を収集した。自 身の回復については被験者の 69%が「満足」又は「とても満足」と回答し、美容 的外観については 92%が「満足」又は「とても満足」と回答した。
図 8-1 フォローアップのフローチャート 登録 (インフォームド コンセント) ベースライン MDT-1112 の 植込みを試みたか? (MDT-1112 イントロデューサ シースを MDT-1112 ペースメーカを植え込む目的 で挿入したか?) 有害事象及び 逸脱の更新 治験終了又は中止 MDT-1112 デバイスの 植込みに成功 (体内に留置) したか? 植込み、退院前、1 か月来 院時に有害事象を評価 手技、システム及びアクセ サリー関連の有害事象が消 失している場合には、30 日 後に治験を終了又は中止 退院前、1、3、6 か月及び 以後 6 か月ごと、治験終了 までフォローアップを行う いいえ いいえ はい はい
表 8-3 収集したデータの概要 治験手順 ベース ライン 植込み 退院前 1 か月来院 3 か月 来院 6 か月来 院 6 か月ごと (治験終了まで) システムに 起因する 予定外来院 全被験者 インフォームドコンセント 選択除外基準の確認 問診、患者背景、心血管病歴及び心血管薬の服薬状況 抗凝固療法を含むシステム及び手技情報 胸部 X 線検査(2 方向、右前斜位+左前斜位及び 30~50 度推奨) 12 か月のみ X 線透視動画記録(メドトロニック社に送付する) 最終タグテスト時 デリバリーシステム抜去後 電気的テスト プログラマストリップ 短時間運動負荷テスト RPO を有効にで きるまでは必要 RPO を有効にできる までは 3 回のテスト、 RPO を有効にした のちは 1 回のテスト 機器のインテロゲーションファイル(開始時及び終了時) (終了時) 開始時又は終了時 インテロゲーション インテロゲー ション推奨 QOL 質問票(EQ-5D 及び SF-36) 12 か月ごと 患者満足度調査 医師への質問票 有害事象、機器不具合、システム変更、治験実施計画書からの 逸脱、終了若しくは中止又は死亡 発生時 最初の約 50 例のみ―追加のデータ収集 追加の電気的テスト:0.4 及び 1.0ms での自動デクリメント閾 値テスト プログラマ ストリップ プログラマ ストリップ 16~24 時間の携帯型心電図(ER220 ホルター心電図)及び患 者日誌(DR220 が市販又は登録されている米国及び EU 諸国で のみ収集) 長時間トレッドミルテスト―プログラムされたベクトル(1 テスト+イ ンテロゲーションファイル)
表 8-4 各地域別の症例数 地域 施設数 登録例数 植込み試行 例数 6 か月来院 完了例数 米国 30 295 282 99 東ヨーロッパ/中央ヨーロッパ 3 106 105 48 西ヨーロッパ 9 177 176 100 アジア太平洋(日本除く) 7 54 52 13 日本 4 38 36 14 カナダ 2 15 15 1 合計 55 685 666 275 1. 試験デザイン設定の根拠 本治験の試験デザイン及び解析方法については、十分な検討及び標準化を行った。これに よって、MDT-1112 システムの安全性及び有効性を合理的に保証でき、製造販売承認申請に おけるピボタル試験として妥当であると判断した。 1) 対象症例 (1) 選択基準 MDT-1112 システムによる治療は、心臓に周期的に人工的な電気刺激を与えること によって正常に近い心臓の拍動リズムを回復させるものであり、その原理及び目的は 従来の植込み型ペースメーカシステムと同一である。したがって、実際に MDT-1112 シ ス テ ム の 適 応 を 有 す る 一 般 的 な 患 者 集 団 を 用 い て 評 価 を 行 う た め 、 ACC/AHA/HRS ガイドライン2及び各国のガイドラインにおいて、心室シングルチャン バペースメーカに対する ClassⅠ又はⅡの植込み適応を有する患者 を選択基準とし た。なお、心室シングルチャンバペースメーカの適応については、国際的なコンセンサ スが得られており3、ACC/AHA/HRS ガイドラインと日本で準用されている JCS ガイドラ イン4との間にも大きな推奨適応の違いは認められない。したがって、全地域において 標準的な心室シングルチャンバペースメーカの適応を有する患者が登録され、全地 域の患者集団をひとつの集団とみなして評価できると判断した。 (2) 除外基準 交絡因子の可能性、MDT-1112 システムとの電気的又は機械的干渉など、本治験に おける評価を妨げる要因となり得る事項 を回避するため、また被験者の安全性保護 の観点から考えられる基準を設定した。なお、ペースメーカ依存患者については、被 験者の保護を最大化し、患者に対する潜在的 危害のリスクを最小化するため、保守 的な措置として、機器の信頼性が確認されるまでは完全にペースメーカに依存してい る患者を初期コホートから除外した。本治験実施中に、早期性能評価において、初 期の安全性評価項目の両方を満たしたため、ペースメーカ依存患者に対する制限を
2 Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of CardiacRhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;51(21):e1-62.
3 Gillis AM, Russo AM, Ellenbogen KA, et al. HRS/ACCF expert consensus statement on pacemaker device and mode selection. Developed in partnership between the Heart Rhythm Society (HRS) and the American College of Cardiology Foundation (ACCF) and in collaboration with the Society of Thoracic Surgeons. Heart Rhythm. 2012;9(8):1344-1365. 4 不整脈の非薬物治療ガイドライン(2011 年改訂版), http://www.j-circ.or.jp/guideline/pdf/JCS2011_okumura_h.pdf
解除することをすべての実施医療機関に通知し、2014 年 7 月 23 日以降は、完全な ペースメーカ依存患者(補充調律≦30bpm)についても本治験への登録が可能となっ た。早期性能評価報告書については、FDA、本治験のステアリングコミッティ及び本 治験の DMC が確認した。日本においては、2014 年 8 月 1 日に、本事項に関するス テアリングコミッティ、並びに DMC 及び FDA の見解を記載した除外基準の解除に関 する報告及び早期性能評価報告書を PMDA に報告した。 2) 評価項目 (1) 安全性主要評価項目本評価項目の性能要件として、植込み後 6 か月(183 日)におけ る MDT-1112 システム及び/又は手技に関連する主要合併症非発生率について、両 側信頼区間の下限(適用水準はα消費関数によって決定される。) が 83%を上回る場 合、帰無仮説を棄却すると設定した。 MDT-1112 システムは新しい医療機器ではあるが、小型のシングルチャンバペース メーカに類似している。メドトロニック社では、シングルチャンバペースメーカの臨床試 験を近年実施しておらず、シングルチャンバペーシングシステムの安全性に関する最 近のエビデンスも限られていることから、右室ペーシングリード及びデュアルチャンバ ペーシングシステムに関する 6 件の臨床試験5、6、7、8、9、10 (参照データセット)に基づき、 植込み後 6 か月における性能要件を決定した。メドトロニック社の臨床試験を性能要 件の基準として選択したのは、これらの各臨床試験がメドトロニック社の厳密なモニタ リングプロセスに従ったものであったためである。 現在、シングルチャンバペースメーカが植え込まれる患者については、植込み医師の 間で意見が一致しており、大部分が心房細動患者である 3。したがって、参照データ セットにおける植込み後 6 か月の手技及び/又は機器に関連した主要合併症発生率 の推定は、ベースライン時に心房細動が記録されていた被験者におけるペースメーカ、 右室リード若しくは手技、又はそのすべてに関連する主要合併症のみを対象として算 出した。参照データセットの被験者 2,667 例のうち 977 例において、ベースライン時の 心房細動が報告されたていたことから、大規模なデータセットに相当 した。 5 EnRhythm:メドトロニック EnRhythm (承認番号 21900BZX00058000)の製造販売承認申請資料
6 3830:IDE number G020043, SelectSecure リード(承認番号 22400BZX00005000)の製造販売承認申請資料 7 5076:DE number G990022
8 EnRhythm MRI:IDE number G070056、メドトロニック Advisa MRI (承認番号 22400BZX00131000)の製造販売承認申請 資料
9 Advisa MRI:IDE number G110046
10 SAVEPACe: Sweeney MO, Bank AJ, Nsah E, et al. Minimizing ventricular pacing to reduce atrial fibrillation insinus-node disease. N Engl J Med. 2007;357(10):1000-8.
評価期間である 6 か月は、MDT-1112 システムの安全性プロファイルの特性を明らか にするために選択した。参照データセットの試験を含む多数の臨床試験 において、 ペースメーカシステムに関連した合併症の大部分が植込み直後に発現することが示 されている11、12、13、14、15、16、17。参照データセットにおいても、大部分の合併症は植込み後 早期に発生し、3 か月以降の発生は少ないことが認められている。以上のことから、6 か月(183 日)という評価期間は、MDT-1112 システムの全般的な安全性プロファイル に対して、保守的な推定を示すと判断した。また、メドトロニック社は本治験の開始前 に、植込み後 6 か月の MDT-1112 システム及び/又は手技に関連する主要合併症の 真の非発生率が 90%を下回ることはないと判断した。この仮定は、参照データセットに おける心房細動患者の主要合併症発生率 だけでなく、デュアルチャンバ及びシング ルチャンバペースメーカの大規模レジストリ試験17で報告されている 12.4%の周術期合 併症発生率とも一致するものである。さらに、許容可能な性能の検証可能性を維持し つつ、治験機器植込み被験者数との均衡を保つために、性能要件 (すなわち、信頼 区間の下限)は、予想される MDT-1112 の非発生率を 7%下回る 83%に設定した。 合併症の種類について従来のペースメーカシステムと MDT-1112 システムとを直接比 較 す る こ と は で き な い が 、 MDT-1112 シ ス テ ム 又 は 手 技 関 連 の 主 要 合 併 症 は 、 MDT-1112 システムの安全性プロファイルの特徴を示す。したがって、植込み後 6 か 月における MDT-1112 システム及び/又は手技に関連する主要合併症非発生率が 83%を上回ることを検証することで、類似した患者集団に植込みを施行した場合に、 MDT-1112 システムの安全性プロファイルが現行のシングルチャンバペースメーカ技 術と同等であることを確認できる。 (2) 有効性主要評価項目 本評価項目の性能要件として、適切な 6 か月 PCT を有する被験者の割合について、 両側信頼区間の下限(適用水準はα消費関数によって決定される。) が 80%を上回る 場合、帰無仮説を棄却すると設定した。
11 Ellenbogen KA, Hellkamp AS, Wilkoff BL, et al. Complications arising after implantation of DDD pacemakers: the MOST experience. Am J Cardiol. 2003;92(6):740-749.
12 Pakarinen S, Oikarinen L, Toivonen L. Short-term implantation-related complications of cardiac rhythm management device therapy: a retrospective single centre 1-year survey. Europace. 2010;12:103–108.
13 Aggarwal RK, Connelly DT, Ray SG, Ball J, Charles RG. Early complications of permanent pacemaker implantation: no difference between dual and single chamber systems. Br Heart J. 1995;73:571–575.
14 Parsonnet V, Bernstein AD, Lindsay B. Pacemaker-implantation complication rates: an analysis of some contributing factors. J Am Coll Cardiol. 1989;13:917–921.
15 Moller M, Arnsbo P, Asklund M, et al. Quality assessment of pacemaker implantations in Denmark. Europace. 2002;4:107–112.
16 Wiegand UK, Bode F, Bonnemeier H, Eberhard F, Schlei M, Peters W. Longterm complication rates in ventricular, single lead VDD, and dual chamber pacing. Pacing Clin Electrophysiol. 2003;26:1961–1969.
17 Udo EO, Zuithoff NP, van Hemel NM, et al. Incidence and predictors of short- and long-term complications in pacemaker therapy: the FOLLOWPACE study. Heart Rhythm. 2012;9(5):728-735.
有効性評価項目として選択した PCT は、ペーシング有効性に関する一般的な電気 的測定値であり、近年の臨床試験 8、9、18、19、20とも一貫している。PCT は一般的に植込 み後 60 日以内に安定となり、その閾値が長期間継続することが知られている。ケアリ ンクデータベース21においても、植込み後 30 日以内に PCT が 2.0V 以下であった患 者の割合は、植込み後 630 日においても大きな変化がなく、安定していることが示さ れている。 パルス幅 0.24ms における 2V という PCT 要件は、同一のパルス幅に標準化した場合、 規 制 当 局 による評 価 が行 われた右 室 ペーシング /除 細 動 リードよりも厳 しい要 件 と なっている。しかしながら、MDT-1112 は新しいバッテリー技術が用いられており、ノミ ナルではより狭いパルス幅(0.24ms 対 0.4ms)でペーシングを行う。また、MDT-1112 シ ステムのキャプチャマネジメント機能では 1 時間ごとに PCT の確認が行われ、設定さ れるセーフティマージンのノミナル値は PCT を 0.5V 上回る値である。以上のことから、 大部分の被験者において PCT が 2V を下回ることを検証することで、MDT-1112 シス テムが現行のペーシングシステムと同様のバッテリー寿命を有することが示めされる。 また、6 か月時点の PCT が植込み時に測定された PCT より増加幅が 1.5V を超えな いというもうひとつの要件によって、PCT が植込み後も合理的に安定しており、植込み 時点でデバイスの寿命が予測可能であることを保証することができる。 また、適切な PCT を有する被験者の割合については、ケアリンクデータベースを参照 して決定した。治験計画段階で、ケアリンクデータベースにおいて植込み後 30 日以 内及び植込み後 180 日の PCT データが利用可能な 322 台のデバイスについては、6 か月時点で 93%が本治験の有効性主要評価項目を満たす PCT を有していた。しかし ながら、ケアリンクデータベースには多種類のデバイス及びリードが含まれており、デ バイスのプログラム要件も一定ではないこと、植込み当日の PCT データが利用可能な 患者は 13%(43/322)のみであったこと(一般的に植込み後数日間の PCT は不安定) から、これらのリミテーションを考慮のうえ、有効性主要評価項目を満たす被験者の割 合 に つ い て は 、 93% よ り 4% 低 い 89% と 仮 定 し た 。 ま た 、 95% 信 頼 区 間 の 幅 は 、 MDT-1112 の真の性能及びサンプルサイズに依存する。したがって、信頼区間の下 限は、許容可能な MDT-1112 の性能の検証可能性及び予定される症例数を考慮し て、仮定した真の性能(89%)から 9%低い 80%と設定した。この設定は、MDT-1112 の真 の性能が 89%であった場合、予定される 1 回目の解析(300 例)においても 90%以上の 検出力を確保できるものである。 (3) 副次評価項目#1 本評価項目の性能要件として、植込み後 6 か月来院時の VCM PCT が手動測定 PCT(パルス幅 0.24ms)から 0.5V 以内であった被験者の割合の両側信頼区間(α水 準はα消費関数及び Holm 法によって決定)の下限値が 85%を上回る場合、帰無仮 説を棄却すると設定した。 この性能要件は、右室(又は左室)キャプチャマネジメント機能(Kappa900 臨床試験及 び CAPTURE 臨床試験)及び左室キャプチャマネジメント機能(LVCM 臨床試験)の評 価を行った過去の 3 件の臨床試験に基づいている。3 件の臨床試験の結果から、6 か月来院における VCM PCT が手動測定 PCT から 0.5V 以内である被験者の真の 18 4195:IDE Number G040036、アテインスターフィックスリード(承認番号 22100BZX00070000)製造販売承認申請資料 19 4196:IDE Number G060193、アテインアビリティリード(承認番号 22000BZX01079000)製造販売承認申請資料 20 Attain Performa Quadripolar Lead:IDE Number G120213、ClinicalTrials.gov Identifier NCT01751022
割合(計画時点では未知)が、少なくとも 93%であると仮定した。また、信頼区間の下限 は、許容可能な MDT-1112 の性能の検証可能性及び予定される症例数を考慮して 85%と設定した。これによって、少なくとも 85%の被験者について、VCM PCT が手動測 定で得られた PCT から 0.5V 以内であることが検証でき、MDT-1112 の VCM 機能に よって PCT が正確に測定されることを確認することができ、1 回目の解析において 90% を上回る検出力を確保できる。 (4) 副次評価項目#2 本評価項目の性能要件として、Kay-Wilkoff モデルによる勾配パラメータが、0.65~ 1.35 の同等性マージンに収まる(TOST 法によって、Holm 調整 P 値が 0.05 未満)こと と設定した。 0.65 及び 1.35 の境界は、主に Corbelli らが収集した実験データ22、23に基づいている。 また、この性能要件は、これまでにその他複数のメドトロニック社のペースメーカ臨床 試 験 に お い て 機 器 の レ ー ト レ ス ポ ン ス 成 績 の 評 価 に 用 い ら れ て い る ( 例 : Sigma 300/303 ペースメーカ、Vitatron DA+検証試験、Kappa700 臨床試験)。Holm 調整 P 値が 0.05 未満であれば帰無仮説を棄却することができ、Kay-Wilkoff の勾配パラメー タが 1 に近いこと、すなわち、MDT-1112 システムが適切なレートレスポンスを行うこと を検証できる。 3) 症例数 (1) 安全性主要評価項目 最大 720 例の植込みを伴う最大 780 例の登録被験者とすることで、安全性主要評価 項目の検定に際して 90%を上回る検出力が得られる。安全性主要評価項目を検定す るための経験的検出力が約 92%となることを証明するため、シミュレーション試験を実 施した。詳細については治験実施計画書の 12.4.6 項を参照のこと。 (2) 有効性主要評価項目 この評価項目が独立して検定された場合、適正な PCT を有する被験者の真の割合 が 89%であれば、植込み時及び植込み後 6 か月の PCT データが利用可能な 173 例 の植込み成功被験者において、第一種の過誤の確率(両側)0.05 でこの評価項目を 検定するために 90%検出力を有する。しかしながら、安全性主要評価項目によって解 析時期及び症例数が決定されることから、この評価項目を検定するための検出力は、 二項分布及び Hwang-Shih-DeCani のα消費関数に基づいて算定した。6 か月来院 を完了した被験者の最大 10%が欠測、又は植込み時及び/又は 6 か月来院時に利 用可能な PCT データが無効であると保守的に仮定する。この仮定に基づいた場合、 適切な PCT を有する被験者の割合を 87%と仮定すると、有効性主要評価項目を満た すための検出力は 99%を上回る。また、適切な PCT を有する被験者の割合が計画値 の 89%と仮定した場合、1 回目の解析で有効性主要評価項目を満たす確率は 90%を 上回る。詳細については治験実施計画書の 12.5.6 項を参照のこと。
22 Corbelli R, Masterson M, Wilkoff BL. Chronotropic response to exercise in patients with atrial fibrillation. Pacing Clin Electrophysiol. 1990;13(2):179-87.
23 Ellenbogen KA, Kay GN, Wilkoff BL, editors.: Clinical Cardiac Pacing and Defibrillation. 2nd ed. WB Saunders Company; 2000.
(3) 国内症例数 本治験では、日本における実施可能性 (参加施設数、治験開始時期及び登録率予 測)から、登録例数 38 例、植込み成功例数 35 例を日本における目標症例数とした。 国際共同治験における各地域の症例数を適切に計画する方法は確立されていない が、新しい使用方法を必要とする MDT-1112 の評価に際しては、少なくとも国内の複 数施設において複数の患者を登録することが必要と考えられることから、このような実 施可能性に基づく目標症例数の設定は妥当であると考えた。また、日本 及び日本以 外の地域における試験成績のプール可能性を検討するために、主要評価項目につ いて地域を日本又は日本以外に分類したサブグループ解析を行うこととした。 4) 多施設共同試験 本治験は、妥当と考えられる期間内に、試験目的を満たすために十分な被験者を登録 する手段として、多地域・多施設において実施することとした。全地域で治験実施計画 書及び症例報告書を統一し、同一水準のモニタリングを実施することで結果の均一性 を担保した。多施設共同試験とすることで、本治験から得られた結果を一般化するため に適切な根拠を与えるものと考える。なお、施設 当たりの被験者数が過度に異なること がないよう、各施設での症例数について上限を設定した。 5) 中間解析及び第一種の過誤の制御(グループ逐次計画) 本治験では中間解析を実施するためにグループ逐次計画を採用した。中間解析の検 討には DMC を利用し、被験者の利益を保護するとともに試験全体の実施を監視するこ ととした。 本治験では最大 3 回の主要評価項目の解析を計画しており、安全性及び有効性主要 評価項目の両方が満たされた最初の解析で治験成功と判断することを定めた。3 回の 解析の予定は、6 か月来院を完了した植込み成功被験者が少なくとも 300 例となった時 点(1 回目)、6 か月来院を完了した植込み成功被験者が少なくとも 450 例となった時点 (2 回目)、6 か月来院を完了した植込み成功被験者が少なくとも 600 例となった時点(3 回目:最終解析)とした。各評価項目について全体の α 水準を 5%(両側;片側 2.5%)に 調整するために、γ パラメータが-2 である Hwang-Shih-DeCani の α 消費関数を利用 した24、25。この関数は、O'Brien-Fleming の棄却限界と Pocock の棄却限界とのほぼ中間 の水準で α を消費する。試験が成功となるには、両主要評価項目について、α 消費関 数に基づく各々の α 水準で帰無仮説が棄却される必要がある。また、本治験の主要評 価項目が満たされた場合、全体の第 一種の過誤を制御し、有意性が統計的に妥当で あることを示すために、Holm 法を用いて副次評価項目の検定を行うこととした。 6) 中央判定 評価者によるバイアス及び評価者間の評価のばらつきを最小化するため、報告された 有害事象のうち、少なくとも死亡及び MDT-1112 システム又は手技に関連する有害事 象は CEC による判定を行うこととした。
24 Tang D and Geller NL. Closed testing procedures for group sequential clinical trials with multiple endpoints. Biometrics. 1999; 55:1188-1192.
25 Glimm. E, Maurer, W, and Bretz, F. Hierarchical testing of multiple endpoints in group-sequential trials. Statistics in Medicine. 2010;29:219-228.





![表 8-7 死亡要約 死亡件数[例、被験者の割合(%)] 登録(N=685) 植込み(N=660) 死亡合計 28(28、4.1%) 28(28、4.2%) 死亡分類 心臓突然死 4(4、0.6%) 4(4、0.6%) 非突然心臓死 9(9、1.3%) 9(9、1.4%) 非心臓死 14(14、2.0%) 14(14、2.1%) 分類不明 1(1、0.1%) 1(1、0.2%) 手技との関連性 関連あり 1(1、0.1%) 1(1、0.2%) 植込み 1(](https://thumb-ap.123doks.com/thumbv2/123deta/7876458.1725788/26.892.109.786.152.842/死亡要約死亡件数例被験者割合登録N植込みN死亡合計死亡植込み.webp)
![表 8-9 ベースライン時の背景データ 被験者特性 登録 (N=685) 植込み (N=660) 性別[例(%)] 男性 407(59.4%) 397(60.2%) 女性 277(40.4%) 263(39.8%) 無回答 1(0.1%) 0(0.0%) 年齢(歳) 平均値±標準偏差 75.3±11.3 75.4±11.2 中央値 77.0 77.0 人種[例(%)] 地域の法規制のため報告なし 124(18.1%) 122(18.5%) 被験者/医師](https://thumb-ap.123doks.com/thumbv2/123deta/7876458.1725788/30.892.173.719.139.568/ベースラインデータ植込み性別例無回答年齢歳平均値中央値人種例.webp)
![表 8-12 心血管系病歴 被験者特性[例(%)] 登録 (N=685) 植込み (N=660) 全般的な心血管疾患の既往なし 32(4.7%) 31(4.7%) 心停止 15(2.2%) 14(2.1%) 心室性不整脈によるもの 2(0.3%) 2(0.3%) 一過性又は可逆性の原因によるもの 8(1.2%) 7(1.1%) 原因不明 6(0.9%) 6(0.9%) 心筋症 62(9.1%) 59(8.9%) 拡張型/うっ血性 8(1.2%) 8(1.](https://thumb-ap.123doks.com/thumbv2/123deta/7876458.1725788/32.892.155.727.158.873/心血管系病歴被験特性登録植込み心停止によるによる心筋症拡張型.webp)
