2.5.4 有効性の概括評価 トロンボモデュリン アルファの DIC に対する有効性の評価は、前期第 2 相臨床試験(添付資 料番号:5.3.5.2-1)、後期第 2 相臨床試験(添付資料番号:5.3.5.1-1)、第 3 相臨床試験(添付資 料番号:5.3.5.1-2)の 3 試験の成績に基づいて行った。 3 試験の概略は表 2.5.4-1 に示した。 表 2.5.4-1 有効性評価試験の概略 試験 区分 目的 対象 (DIC の基 礎疾患) 施 設 数 実 施 例 数 治験 期間 主要 評価 項目 有効性成績 添付 資料 番号 前期 第 2 相 DIC に対する有効性及 び安全性を検討する DIC 又は DIC の疑い (基礎疾患 を限定せず) 32 施 設 44 例 19 年 月~ 19 年 月 全般改 善度 1,900~19,000 U/人(0.3~ 3.0 mg/人)の用量範囲*で 用量の増加と共に中等度 改善以上率が上昇した。 5.3.5. 2-1 後期 第 2 相 DIC に対する有効性及 び安全性を検討し、用 量-反応関係を検証 する DIC 又は DIC の疑い (基礎疾患 を限定せず) 92 施 設 120 例 19 年 月~ 19 年 月 全般改 善度 中等度改善以上率は、用量 依 存 的 に 上 昇 し 、 380 U/kg(0.06 mg/kg)が十分に 高い効果を有することが 示された。 5.3.5. 1-1 第 3 相 DIC に対する有効性及 び安全性をヘパリン ナトリウムと比較す る DIC (造血器悪 性腫瘍ある いは感染症) 113 施 設 232 例 20 年 月~ 20 年 月 DIC 離脱率 DIC 離脱率において、ヘパ リンナトリウムに対し本 剤 380 U/kg(0.06 mg/kg)の 非劣性が検証され、同時に 優越性が示された。 5.3.5. 1-2 実施例数:治験薬が投与された例数 *体重当たりの投与量で示すと、体重 50 kg として、1,900~19,000 U/人(0.3~3.0 mg/人) は、38~380 U/kg(0.006~0.06 mg/kg)に相当する。 2.5.4.1 試験対象集団の特性 対象患者は、前期第 2 相及び後期第 2 相臨床試験では、厚生省 DIC 診断基準11)に基づき「DIC」 又は「DIC の疑い」と診断された患者とした。第 3 相臨床試験では、DIC としてより確定的な集 団を対象とするため、同診断基準で「DIC」と診断された患者のみを対象とした。なお、厚生省 DIC 診断基準では、白血病及び類縁疾患、再生不良性貧血、抗腫瘍剤投与後などの骨髄巨核球減 少が顕著で、高度の血小板数の低下をみる場合は、血小板数及び出血症状のスコアを DIC 診断に 用いないこととしている。したがって、本剤の第 3 相臨床試験においても、治験開始時に血小板 数低下の主な原因が DIC によるものか否かを、治験実施計画書に予め規定した判断基準(2.7.3.6 項)に基づいて治験担当医師が判断し、白血病群(血小板数の低下の主な原因が DIC 以外の原因 である群)あるいは非白血病群に分類した上で、「DIC」の診断を行った。
DIC の基礎疾患については、前期第 2 相及び後期第 2 相臨床試験では限定しなかった。一方、 第 3 相臨床試験では、医薬品機構との 2 回の治験相談(第 2 相終了後相談、個別相談)における 助言を受けて、基礎疾患は、以下の点で類似している「造血器悪性腫瘍あるいは感染症」に限定 した。 • 固形癌に比べ基礎疾患の治療が可能な場合が多く、抗凝固薬による DIC 治療意義が大きい • 発症する DIC が急性の経過をたどることが多い • 本剤及び対照薬であるヘパリンの至適用量が同様である 年齢については、対象患者における高齢者の割合が高いと考えられたため、3 試験共に上限を 設定しなかった。一方、3 試験共に 15 歳未満の患者を除外したため、小児に対する使用経験はな い。また、第 3 相臨床試験では、対照薬であるヘパリンナトリウムの添付文書で原則禁忌とされ ている「死に至るもしくは生命を脅かすような出血のある患者」などの出血リスクの高い患者や、 「重篤な腎疾患あるいは肝疾患」の合併患者は新たに除外した。そのため、第 3 相臨床試験では、 後期第 2 相臨床試験までの患者集団と一部異なる可能性が考えられた。 実際の患者集団は、「2.7.3.3.1.2 項」の表 2.7.3-23 に示したとおり、前述した DIC 診断、DIC の 基礎疾患を除き、性別、年齢等人口統計学的特性、開始時 DIC スコア、及び基礎疾患の重症度な どの主要な背景因子において、後期第 2 相臨床試験と第 3 相臨床試験で大きな違いは認められな かった。 2.5.4.1.1 市販後に本剤が投与されると予想される患者集団と試験対象集団との間の相違 第 3 相臨床試験の対象集団は、「造血器悪性腫瘍あるいは感染症を直接誘因基礎疾患とするDIC」 に限定したため、「造血器悪性腫瘍あるいは感染症」以外を基礎疾患とする DIC 患者に対する本 剤の使用経験は少ない。しかしながら、「2.5.4.3.5 項」及び「2.7.3.3.3.2 項」に後述のとおり、本 剤の作用機序、薬理試験や臨床薬理試験の結果、及び後期第 2 相臨床試験・第 3 相臨床試験の結 果から、「造血器悪性腫瘍あるいは感染症」以外を基礎疾患とする DIC 患者に対しても、本剤は 有効な薬剤であると考えられた。したがって、[効能・効果]は「汎発性血管内血液凝固症(DIC)」 と設定した。 2.5.4.2 試験方法 2.5.4.2.1 試験デザイン 各試験における主な試験デザイン、用法・用量、投与期間について、表 2.5.4-2 に示した。 前期第 2 相及び後期第 2 相臨床試験は、非盲検下で実施した。2 試験共に試験開始時点では出 血等のリスクに関する情報が十分でなく、なんらかの重篤な基礎疾患を合併する対象患者の安全 性を確保するには、盲検下で本剤を投与することは時期尚早と考えたためである。一方、第 3 相 臨床試験は二重盲検比較対照試験とした。本剤と対照薬のヘパリンナトリウムでは 1 日の投与時 間が異なるため、ダブルダミー法を用いて盲検性を確保した。 前期第 2 相及び後期第 2 相臨床試験では、従来の DIC の臨床試験と同様に基礎疾患を限定しな かった。しかしながら、DIC 治療の成否が基礎疾患の経過に影響を受けることはよく知られてい
上記の 2 試験を含め、従来の DIC の臨床試験においては、DIC の基礎疾患がエントリーしやす い造血器悪性腫瘍に偏り、感染症の症例が少ないという問題点があった22)。前述の医薬品機構と の 2 回の治験相談においては、 といった助言を受けた。 これらの助言に基づいて、第 3 相臨床試験では、基礎疾患を造血器悪性腫瘍、感染症に限定し、 基礎疾患毎に、動的割り付けを実施した。また、DIC の基礎疾患別に試験を行うことが最善では あったが、各基礎疾患単独では、ヘパリンナトリウムとの非劣性検証に必要な症例数を確保する ことが困難と考えられたため、1 つの試験で両方の基礎疾患の DIC に対する効果を検証すること とした。したがって、非劣性検証は、両方の基礎疾患を併合して行うこととした。ただし、併合 のためには薬剤と基礎疾患の間に質的な交互作用がないことが前提となるため、交互作用の有無 の検出が可能と考えられた 100 例以上を各基礎疾患共に集積することとした。 また、担当医師が各症例において、基礎疾患の経過を評価し、基礎疾患の経過と DIC 離脱の関 係を検討できるデザインとした。 本剤の用法は、1 日 1 回 30 分静脈内持続投与とし、3 試験間で変更はなかった。投与期間は 3 試験共に 6 日間とした。試験毎の用量、投与期間の設定根拠については、「2.7.3.1 項」及び「2.7.3.4 項」に記載した。 併用治療については、抗凝固薬、抗血小板薬、線溶系薬剤は有効性評価に影響をおよぼす可能 性があることから、3 試験共に併用禁止とした。 表 2.5.4-2 各試験の主な実施方法 前期第 2 相臨床試験 後期第 2 相臨床試験 第 3 相臨床試験 試験形式 非盲検,用量漸増試験 非盲検,並行群間用量比較試 験 二重盲検実薬比較試験(ダブ ルダミー法) 割り付け 方法 - 動的割り付け(層化因子: 基礎疾患,投与前 FDP 値, 年齢) 基 礎 疾 患 毎 に 動 的 割 り 付 け(層化因子:開始時 DIC スコア,開始時出血症状) 目標症例 数 50 例(各ステップ 10~15 例)( 基礎 疾 患毎 の目 標 設 定はない) 120 例(基礎疾患毎の目標 設定はない) 220 例(基礎疾患毎に最低100 例) 用法 1 日 1 回 30 分静脈内持続投与 1 日 1 回 30 分静脈内持続投与 1 日 1 回 30 分静脈内持続投 与(ヘパリンナトリウム:24 時間静脈内持続投与) 用量 1,900 U/人(0.3 mg/人) ~19,000 U/人(3.0 mg /人)* 低用量:38 U/kg (0.006 mg/kg) 中用量:130 U/kg (0.02 mg/kg) 高用量:380 U/kg(0.06 mg/kg) 380 U/kg(0.06 mg/kg)(ヘパリ ンナトリウム:8 U/kg/hr) 投与期間 6 日間 6 日間 6 日間 *体重当たりの投与量で示すと、体重 50 kg として、1,900~19,000 U/人(0.3~3.0 mg/人) は、38~380 U/kg(0.006~0.06 mg/kg)に相当する。 2.5.4.2.1.1 基礎疾患治療の影響を少なくするための方策 DIC の臨床評価において、基礎疾患治療の影響を完全に排除することは不可能であるが、第 3 相臨床試験では、以下のような方策により、基礎疾患の治療の影響をできるだけ少なくした。
• 試験デザイン 「2.5.4.2.1 項」に記載のとおり、症例登録の際には、各基礎疾患単独でも評価できるよ うに薬剤の割り付けを基礎疾患別(造血器悪性腫瘍、感染症)に動的に割り付けた。この ように試験をデザインすることにより、基礎疾患治療によるバイアスの介入を回避するよ うにした。 • DIC スコア
DIC 離脱・非離脱の判定には厚生省 DIC 診断基準による DIC スコアを用いた。DIC 離脱・ 非離脱の判定は、医師の総合的な評価による判断ではないため、基礎疾患に対する治療内 容やその成否の印象が判定に加味されることがない。また、本診断基準は、白血病や抗腫 瘍剤等の影響により血小板数が減少した DIC では血小板数と出血症状の点数をスコアに含 めないため、このような基礎疾患や基礎疾患治療の影響を排除するよう考慮されている。 • 基礎疾患の経過 基礎疾患治療の結果については、主要評価項目である DIC 離脱・非離脱の評価時期であ る投与開始 7 日目(又は中止時)に、基礎疾患の経過として「改善」「不変」「悪化」の 3 段階で判定することとした。なお、基礎疾患の経過判定については、開鍵前に全症例に ついて医学専門家がその妥当性確認を行った。また、この基礎疾患の経過毎に DIC 離脱・ 非離脱を集計し、基礎疾患の経過と DIC 離脱・非離脱との関係を検討することを予め計画 した。 2.5.4.2.2 有効性評価項目 2.5.4.2.2.1 主要評価項目 第 3 相臨床試験では、前述の医薬品機構との 2 回の治験相談における助言に基づき、投与 開始 7 日目(又は中止時)の DIC 離脱率を主要評価項目とした。DIC 離脱については、厚生省 DIC 診断基準に基づいて「DIC の可能性が少ない」に患者が該当する場合、一律、DIC 離脱と判定し た。 DIC 離脱率を主要評価項目とした理由は、以下の 3 点である。 • DIC は複雑な病態を呈するため、複数の項目を組み合わせた複合評価指標を用いることで、 DIC 治療薬の DIC におよぼす影響を正確に把握することができると考えられたこと • DIC 離脱・非離脱の判定が凝血学的検査値及び臨床症状から算出可能な DIC スコアに基づ いたものであり、医師の総合的評価ではないため、基礎疾患の治療内容やその成否の印象 が判定に加味されないこと • 医療現場では DIC スコアをもとに抗凝固薬投与の必要性の有無を判断しており、「DIC 離 脱」が DIC の治療目標のひとつであることから、DIC 離脱率が高いことがその薬剤の臨床 的意義に直接結びつくこと
しかしながら、DIC スコア及び DIC スコアに基づく「DIC」「DIC の可能性が少ない」の判定 は、本来 DIC 診断に用いられることを前提に決定されたものであることから、DIC スコアに基づ く DIC 離脱率を評価指標に用いたことの妥当性について、第 3 相臨床試験成績に基づき、事後的
的検査値改善度及び臨床症状改善度によるマトリックスを設定し、評価の客観性を高めた。全般 改善度は第 3 相臨床試験でも、後期第 2 相臨床試験と同一の評価基準により評価が行われた。ま た、後期第 2 相臨床試験では、前述の治験相談の結果に基づき、DIC 離脱率についても追加で集 計した。 2.5.4.2.2.2 副次的評価項目 各試験における副次的評価項目は、出血症状の改善度、臓器症状の改善度、凝血学的検査値改 善度、転帰などとした。 第 3 相臨床試験では、薬剤の特徴を評価するための副次的評価項目として、「出血症状の経過」 を設定した。DIC における出血症状は、さまざまな部位に起こることが知られており8)、これら の出血傾向が持続した場合には、患者の QOL が著しく損なわれる9)。また、全身状態の悪化を招 き、基礎疾患に対する積極的な治療機会が失われることもある。さらに、頭蓋内出血、肺・気管 出血、消化管出血といった重要臓器での出血は、致死的な状況につながる可能性も高い7)。一方、 「2.5.1.1.4.5 項」に記載したとおり、DIC の治療においては、「全身的に高度な凝固系の活性化状 態にある DIC に対し抗凝固療法を施す必要がある一方で、DIC は出血が起こりやすい状態にある ため、十分な抗凝固療法ができない」という問題点があり、出血を悪化させずに十分な抗凝固効果 を発揮できる薬剤が医療現場で求められている。本剤は、作用機序及び後期第 2 相臨床試験まで の結果から、出血傾向の助長作用は少ないと考えられた。さらに、凝固系の活性化を強力に抑制 することで、血小板、凝固因子の消費性低下や二次線溶の活性化を抑制し、DIC に伴う出血症状 を改善することが期待された。以上のことから、「出血症状の経過」は本剤の特徴を評価する項 目として適切と考えられた。 また、凝血学的検査値改善度は、厚生省 DIC 診断基準で用いられている検査項目をベースに、 7 項目の検査データに基づいた評価指標である。そのため、凝血学的検査値改善度は評価者の主 観が全く入らない客観的な評価指標であると同時に、DIC における凝固線溶系の状態を総合的に 評価できる有用な評価指標であると考えられる。前述の全般改善度判定マトリックスに占める本 改善度の比重は大きく、全般改善度との相関性も高い。凝血学的検査値改善度は、全般改善度と 同様に、後期第 2 相臨床試験、第 3 相臨床試験共に同一の評価基準により評価が行われた。評価 方法の詳細は「2.7.3.1 項」に記載した。 2.5.4.2.3 対照群の選択及び非劣性検証試験実施の妥当性 第 3 相臨床試験において、対照薬はヘパリンナトリウムとし、その用量は 8 U/kg/hr とした。 ヘパリンは、血液凝固に関わる種々の適応症で承認されており、高い抗凝固作用を有する。ま た、これまでの多くの使用経験から、標準薬として位置付けられる。DIC に対する用量は、概ね 8 U/kg/hr に相当する 1 日 10,000 U/人程度が推奨されている4,50)。これまで本邦において公表され ている DIC を対象とした比較臨床試験は、いずれもヘパリンを対照薬として実施されており、ヘ パリンの用量は 7~10 U/kg/hr に設定されている46,51,52)。以上のことから、ヘパリンナトリウムを 非劣性検証試験の対照薬として選択したこと、用量を 8 U/kg/hr としたことは適切と考えられた。 また、非劣性の限界値は 5%に設定した。致死的疾患である DIC において、ヘパリンとプラセ ボとの比較試験は実施されておらず、ヘパリンとプラセボとの効力差を示した成績はこれまでに ない。Retrospective な使用成績調査によれば、ヘパリンの DIC に対する臨床効果(ヘパリン未使 用例との差)は 24~30%であり53,54)、ヘパリンとプラセボとの効力差は、DIC 離脱率において 15
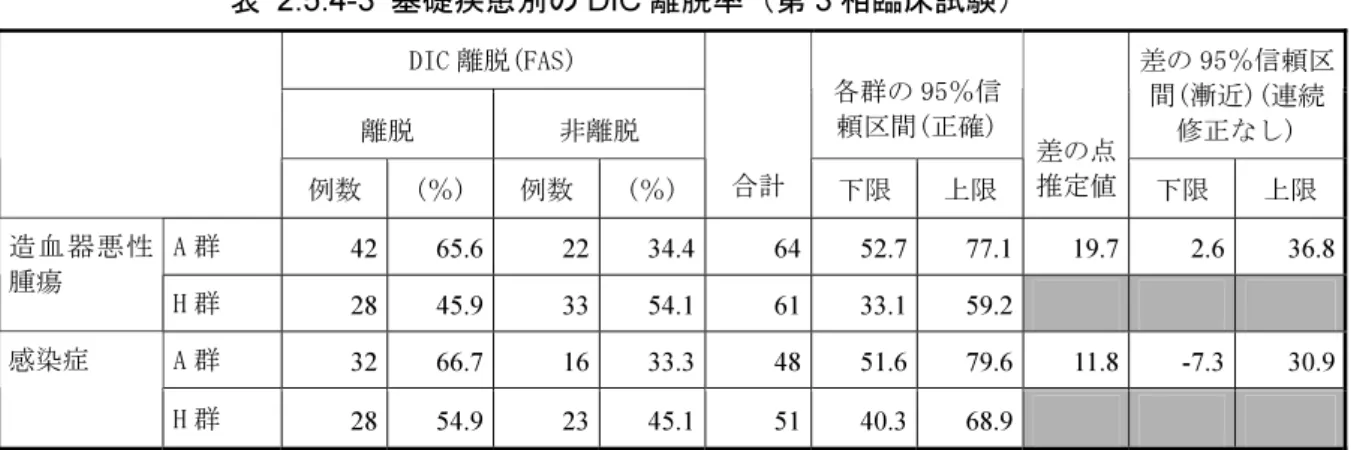
~20%程度は存在するであろうと推定した。しかしながら、十分な精度に基づく推定ではないた め、「臨床的に許容できる差」は十分に小さくする必要があると考え、非劣性の限界値は 5%に設 定した。 なお、対照薬及び非劣性の限界値は、医薬品機構との 2 回の治験相談における助言を受け て最終的に決定した。 一方、試験の分析感度の存在は、「臨床試験における対照群の選択とそれに関連する諸問題」 (2001 年 2 月 27 日付け、医薬審発第 136 号)55)に記載されているとおり、「薬剤効果に対する感 度の既存の証拠」及び「試験の適切な実施」の 2 つが満たされているかどうかから推測される。 「薬剤効果に対する感度の既存の証拠」については、ヘパリンとプラセボとの比較試験がない ことから、厳密な評価は難しいが、本邦において公表されている DIC を対象とした比較臨床試験 においては、ヘパリンはほぼ一貫して DIC に対する改善効果を示していた46,51,52)。 「試験の適切な実施」については、第 3 相臨床試験の結果、GCP 違反例はなく、選択・除外基 準違反など、重要な逸脱により PPS 不採用となった症例も 232 例中 22 例と、他剤の DIC を対象 とした臨床試験52)と比較して少なかった。また、中止申し出のため、試験から脱落した症例は 2 例、データ欠測により主要評価項目である DIC 離脱が判定不能となった症例は 3 例と共に少な かった(2.7.3.2.3 項)。なお、「試験開始前にヘパリンが使用された患者」は、ヘパリンとの比 較を行う上で試験成績にバイアスを与え、分析感度を低下させる可能性があるため、予め除外し た。以上より、第 3 相臨床試験は、GCP に則って適切に実施されたと考えられた。 以上、第 3 相臨床試験は「薬剤効果に対する感度の既存の証拠」及び「試験の適切な実施」を 満たしていたと考えられ、適切な分析感度を有していたものと推測された。 2.5.4.3 有効性評価の主要な結果 2.5.4.3.1 DIC 離脱率 DIC 離脱率は、第 3 相臨床試験及び後期第 2 相臨床試験で検討された。 第 3 相臨床試験では、治験薬が投与された 232 例のうち、227 例が FAS 採用例、224 例が FAS における DIC 離脱率評価対象例であった。 DIC 離脱率を基礎疾患別にみると、造血器悪性腫瘍では、トロンボモデュリン アルファ投与群 (以下、A 群)65.6%(42/64 例)、ヘパリンナトリウム投与群(以下、H 群)45.9%(28/61 例) であった。また、感染症では A 群 66.7%(32/48 例)、H 群 54.9%(28/51 例)であった。いずれ の基礎疾患においても、DIC 離脱率は A 群が H 群よりも高かった。特に、造血器悪性腫瘍では、 群間差の 95%信頼区間の下限が 2.6%で、0%を上回っていた。また、薬剤と基礎疾患の間に質的 交互作用は認められなかった(表 2.5.4-3)。 造血器悪性腫瘍、感染症の両者を併合した解析の結果、Woolson-Bean 法による層調整後の DIC 離脱率は、FAS において、A 群 66.1%、H 群 49.9%であった。群間差の下側信頼限界値は 3.3%と、 治験実施計画書に予め設定した-5%を上回っており、本剤のヘパリンナトリウムに対する非劣性が 検証された。さらに、下側信頼限界値が 0%を上回っていたことから、本剤の DIC 離脱効果はヘ パリンナトリウムより優れていることが示された(表 2.5.4-4)。
表 2.5.4-3 基礎疾患別の DIC 離脱率(第 3 相臨床試験) DIC 離脱(FAS) 離脱 非離脱 各群の 95%信 頼区間(正確) 差の 95%信頼区 間(漸近)(連続 修正なし) 例数 (%) 例数 (%) 合計 下限 上限 差の点 推定値 下限 上限 A 群 42 65.6 22 34.4 64 52.7 77.1 19.7 2.6 36.8 造血器悪性 腫瘍 H 群 28 45.9 33 54.1 61 33.1 59.2 A 群 32 66.7 16 33.3 48 51.6 79.6 11.8 -7.3 30.9 感染症 H 群 28 54.9 23 45.1 51 40.3 68.9 表 2.5.4-4 Woolson-Bean 法による層調整後の DIC 離脱率(第 3 相臨床試験) DIC 離脱の有無(FAS) -層調整後の離脱率(%)- 各群の 95%信頼 区間 差の 95%信頼区間 各群の 離脱率 下限 上限 差の点推定値 下限 上限 A 群 66.1 57.0 75.2 16.2 3.3 29.1 H 群 49.9 40.7 59.0 PPS を対象に解析した場合、及び共変量による調整を行った場合も同様にヘパリンナトリウム に対する本剤の優越性が示された(2.7.3.3.2.1 項)。また、年齢等、いくつかの患者背景因子で薬 剤群間に偏りが認められたが、偏りを調整した結果及び部分集団別解析の結果から、これらの偏 りは試験の主要な結論に大きな影響をおよぼしていないと考えられた(2.7.3.3.2.1 項)。薬剤群間 に偏りがみられなかった背景因子についても部分集団別に検討した結果、A 群ではいずれの部分 集団でも一貫して DIC 離脱率が高い傾向にあり、ほとんどの部分集団で A 群の DIC 離脱率が H 群を上回っていた(2.7.3.3.2.1 項)。さらに、治験薬投与期間中の基礎疾患に対する治療の影響に ついても検討したところ、治療の実施状況は両薬剤群間に大きな違いはなく、部分集団別の DIC 離脱率は、ほとんどの層において、A 群が H 群よりも高かった(2.7.3.3.2.1 項)。以上のことか ら、上記の検証結果は頑健であると考えられた。 後期第 2 相臨床試験では、治験実施計画書に準拠した集団(以下、対象 II)を主たる解析対象 集団とした。治験薬が投与された 120 例のうち、100 例が対象 II 採用例、94 例が DIC 離脱率評価 対象例であった。 DIC 離脱率は、投与量の増加に伴い高くなる傾向を示し、第 3 相臨床試験と同用量の高用量群 では、55.9%(19/34 例)であった(2.7.3.2.2 項)。基礎疾患別では造血器悪性腫瘍(白血病)が 58.8%(10/17 例)、感染症が 83.3%(5/6 例)と、いずれも高い率であった(2.7.3.3.2.1 項)。 以上、本剤の DIC 離脱率は、後期第 2 相臨床試験及び第 3 相臨床試験の 2 試験で一貫して高く、 本剤の DIC 離脱効果の頑健性が示唆された。 また、第 3 相臨床試験において、基礎疾患の経過と DIC 離脱率との関係を検討したところ、基 礎疾患の経過が「改善」の集団は「悪化」の集団に比べ、両薬剤群共に DIC 離脱率が明らかに高 かった。「2.5.4.2.1 項」に記載したとおり、基礎疾患の経過が DIC 治療の成否におよぼす影響は
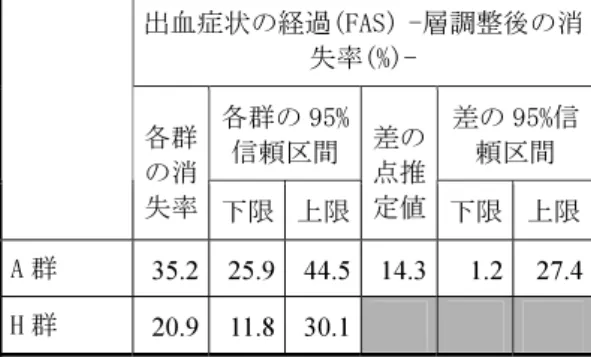
大きいと考えられた。一方、基礎疾患の経過が「改善」「不変」「悪化」の層それぞれのカテゴ リーで、両薬剤群の DIC 離脱率を比較すると、A 群の DIC 離脱率が H 群に比べ高い、もしくは概 ね同程度であった(2.7.3.3.2.1 項)。したがって、「本剤の DIC 離脱効果がヘパリンナトリウム より優れる」という結果は、基礎疾患の経過に依存したものではないことが示唆された。 2.5.4.3.2 出血症状改善度 出血症状の改善、悪化等に関する有効性評価は、第 3 相臨床試験及び後期第 2 相臨床試験の 2 試験で検討された。 第 3 相臨床試験では、FAS 採用例 227 例が出血症状の経過の評価対象例であった。 造血器悪性腫瘍、感染症のいずれの基礎疾患においても、出血症状の「消失」例は A 群に多く、 逆に「悪化」例は H 群に多く認められた。 造血器悪性腫瘍、感染症の両者を併合し、層調整した解析の結果、出血症状の経過は A 群が優 る結果であった(両側:p=0.0271、拡張 Mantel 検定)(表 2.5.4-5)。また、出血症状の「消失」 率においても、A 群 35.2%、H 群 20.9%であり、群間差の点推定値(95%信頼区間)は 14.3%(1.2% ~27.4%)であり、下側信頼限界値は 0%を上回っていた(表 2.5.4-6)。特に、造血器悪性腫瘍で は単独でも、群間差の 95%信頼区間の下限が 2.1%で、0%を上回っていた(2.7.3.3.2.2 項)。 また、開鍵後に、出血症状の経過に対する血液製剤の影響を検討した。その結果、いずれの基 礎疾患でも、本剤の出血症状の経過に対する、血液製剤の併用投与の影響は小さいと考えられた。 特に、造血器悪性腫瘍を基礎疾患とする DIC 患者において、濃厚血小板が使用されなかった症例 は、A 群 22 例、H 群 10 例と A 群で多かった(両側:p=0.0250)。 これらのことから、出血症状の経過で A 群が H 群より優れていたことは、濃厚血小板投与など 血液製剤の併用投与に起因するものではないことが示唆された(2.7.3.3.2.2 項)。 表 2.5.4-5 基礎疾患別の出血症状の経過(第 3 相臨床試験) 出血症状の経過(FAS) 消失 改善 不変 悪化 拡張 Mantel 検定(両 側) 例数 % 例数 % 例数 % 例数 % 合計 症状 なし χ2 値 DF P 値 A 群 14 32.6 9 20.9 10 23.3 10 23.3 43 21 4.8848 1 0.0271 造血器悪 性腫瘍 H 群 6 13.3 12 26.7 10 22.2 17 37.8 45 16 A 群 17 37.8 10 22.2 10 22.2 8 17.8 45 5 感染症 H 群 13 28.3 10 21.7 10 21.7 13 28.3 46 6
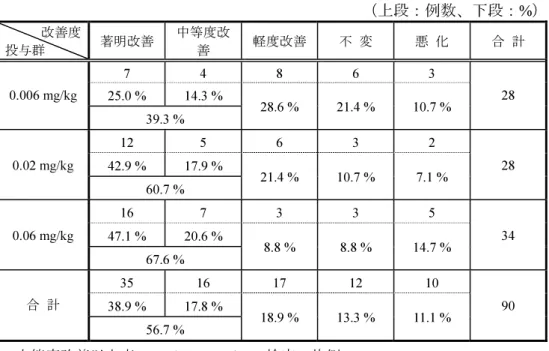
表 2.5.4-6 Woolson-Bean 法による層調整後の出血症状の消失率(第 3 相臨床試験) 出血症状の経過(FAS) -層調整後の消 失率(%)- 各群の 95% 信頼区間 差の 95%信頼区間 各群 の消 失率 下限 上限 差の 点推 定値 下限 上限 A 群 35.2 25.9 44.5 14.3 1.2 27.4 H 群 20.9 11.8 30.1 DIC 離脱・非離脱と出血症状の経過、凝血学的検査値改善度と出血症状の経過との関係を開鍵 後に調べたところ、DIC 離脱例や凝血学的検査値改善度が「著明改善」「中等度改善」の症例で は、出血症状の経過が「消失」「改善」の症例が多く、DIC 離脱・非離脱と出血症状の経過、凝 血学的検査値改善度と出血症状の経過の相関性はいずれも高いと考えられた(2.7.3.3.2.2 項)。 これらの結果から、本剤は DIC における凝固異常を改善させ、凝固因子の消費性低下や二次線 溶活性化を抑制した結果、DIC に伴う出血症状も改善させたと考えられた。したがって、第 3 相 臨床試験の出血症状の経過において、A 群と H 群に差がみられたことは、本剤の凝固異常改善作 用がヘパリンナトリウムよりも強いことを反映した結果と考えられた。 後期第 2 相臨床試験では、対象 II 採用例 100 例が出血症状改善度の評価対象例であった。 後期第 2 相臨床試験の結果、投与量の増加に伴い出血症状の「消失」率は高くなる傾向を示し、 第 3 相臨床試験と同用量の高用量群では、48.1%(13/27 例)であった(2.7.6.4.4.3.1.2 項)。基礎 疾患別では造血器悪性腫瘍(白血病)が 45.5%(5/11 例)、感染症が 80.0%(4/5 例)と、いずれ も出血症状の「消失」率は高かった(2.7.3.3.2.2 項)。 以上、出血症状の「消失」率は、後期第 2 相臨床試験及び第 3 相臨床試験の 2 試験で一貫して 高く、本剤の出血症状改善効果の頑健性が示唆された。 2.5.4.3.3 全般改善度及び凝血学的検査値改善度 全般改善度及び凝血学的検査値改善度は、後期第 2 相臨床試験、第 3 相臨床試験の 2 試験で、 同一の評価基準で検討された。 後期第 2 相臨床試験では、対象 II 採用例のうち、90 例が全般改善度及び凝血学的検査値改善度 の評価対象例であった。 後期第 2 相臨床試験の結果、全般改善度の「中等度改善」以上率は、低用量群 39.3%(11/28 例)、 中用量群 60.7%(17/28 例)、高用量群 67.6%(23/34 例)であり、Cochran-Armitage 法を用いた傾 向検定を行った結果、有意であった(片側:p=0.0188、表 2.5.4-7)。本剤が投与された全例を対 象とした集団でも同様に傾向検定を行った結果、有意であり(2.7.6.4.4.3.1.1 項)、検証結果は頑 健であると考えられた。また、凝血学的検査値改善度の結果も全般改善度と同様であり、「中等 度改善」以上率は、低用量群 35.7%(10/28 例)、中用量群 60.7%(17/28 例)、高用量群 67.6%(23/34 例)と、用量増加に伴って高まった(2.7.6.4.4.3.1.2 項)。 以上のとおり、後期第 2 相臨床試験では、3 用量群間比較における用量依存性の検証により、 本剤の DIC 患者に対する有効性が示された。
表 2.5.4-7 全般改善度(後期第 2 相臨床試験) (上段:例数、下段:%) 改善度 投与群 著明改善 中等度改善 軽度改善 不 変 悪 化 合 計 7 4 8 6 3 25.0 % 14.3 % 0.006 mg/kg 39.3 % 28.6 % 21.4 % 10.7 % 28 12 5 6 3 2 42.9 % 17.9 % 0.02 mg/kg 60.7 % 21.4 % 10.7 % 7.1 % 28 16 7 3 3 5 47.1 % 20.6 % 0.06 mg/kg 67.6 % 8.8 % 8.8 % 14.7 % 34 35 16 17 12 10 38.9 % 17.8 % 合 計 56.7 % 18.9 % 13.3 % 11.1 % 90 中等度改善以上率;Cochran-Armitage 検定 片側:p=0.0188 0.006、0.02、0.06 mg/kg は、それぞれ 38、130、380 U/kg に相当する。 第 3 相臨床試験では、FAS 採用例のうち 208 例が全般改善度及び凝血学的検査値改善度の評価 対象例であった。結果は後期第 2 相臨床試験の結果を再現するものであった。 すなわち、DIC の基礎疾患別に 2 試験の結果を比較すると、全般改善度の「中等度改善」以上 率は、造血器悪性腫瘍(白血病)では、後期第 2 相臨床試験の高用量群で、70.6%(12/17 例)、 第 3 相臨床試験の A 群で、73.3%(44/60 例)、感染症では、後期第 2 相臨床試験の高用量群で、 83.3%(5/6 例)、第 3 相臨床試験の A 群で、71.1%(32/45 例)と一貫して高い改善率を示した。 また、同様に、凝血学的検査値改善度の「中等度改善」以上率は、造血器悪性腫瘍(白血病) では、後期第 2 相臨床試験の高用量群で、70.6%(12/17 例)、第 3 相臨床試験の A 群で、73.3% (44/60 例)、感染症では、後期第 2 相臨床試験の高用量群で、83.3%(5/6 例)、第 3 相臨床試験 の A 群で、71.1%(32/45 例)と、一貫して高い改善率を示した(2.7.3.3.2.3 項)。 第 3 相臨床試験においては、全般改善度、凝血学的検査値改善度のいずれにおいても、造血器 悪性腫瘍、感染症の両者を併合し、層調整した解析の結果、A 群が H 群に比べ優る結果であり (2.7.6.5.3.4.3 項)、DIC 離脱率における両薬剤群の差を裏付けるものであった。 2.5.4.3.4 凝血学的検査値に対する効果 DIC 患者における凝固線溶系の異常の程度、及び本剤の抗凝固作用を検討するため、3 試験共 に凝血学的検査を実施した。 その結果、第 3 相臨床試験の A 群においては、本剤投与後に、ほとんどの凝血学的検査値はそ の異常が是正される方向へ変動した。中でも、TAT、D-ダイマー、プロテイン C、PAI-1、ATIII 、
これらの結果は、本剤が、凝固系の活性化を抑制することにより、DIC 離脱効果及び出血症状 の改善効果を発揮することを客観的に裏付ける結果と考えられた。また、「2.4.2.1 項」に記載し たとおり、本剤の凝血学的検査値異常の改善作用は、in vitro 試験、DIC モデル試験などで確認さ れており、非臨床試験から得られた結果が臨床試験でも再現されたものと思われた。 DIC における凝固・線溶動態と臨床検査項目との関係について、図 2.5.4-1 に示した。また、第 3 相臨床試験において、両薬剤群の変化量(又は変化率)に差があることが示唆された項目のう ち、特に本剤の効果及び特徴の裏付けにおいて重要と考えられる TAT、D-ダイマー、プロテイン C、ATIII について、結果とその考察を記述した。 フィブリノゲン エンドトキシン サイトカイン 血管内皮細胞活性化 単球活性化 組織腫瘍細胞の破壊 など 外因系 内因系 プラスミノゲン 組織腫瘍細胞の崩壊 サイトカイン など プロトロンビン ATIII PC TM 血小板 Xa/Va APC TAT PIC FDP D-ダイマー tPA-PAI-1 FDP フィブリンモノマー soluble fibrin fibrin solid fibrin 線 溶 系 凝 固 系 α2PI tPA PAI-1 プラスミン トロンビン 図 2.5.4-1 DIC における凝固・線溶動態と臨床検査項目56)(一部改変) PC:プロテイン C、Xa:活性化第 X 因子、Va:活性化第 V 因子、 :制御因子 :DIC 発症時に高値となる因子、 ::DIC 発症時に低値となる因子
(1) TAT TAT は、血中のトロンビン生成量を反映する検査値であり、凝固系の活性化の程度を反 映する。本剤は「2.5.1.2 項」に記載のとおり、トロンビンの生成を抑制することで、DIC に対して有効性を示すと考えられている。 図 2.5.4-2 に示したとおり、第 3 相臨床試験において、造血器悪性腫瘍、感染症のいずれ の基礎疾患でも、本剤、対照薬共に投与後の TAT は投与前に比べ減少した。TAT 減少率の 薬剤群間差の両側 95%信頼区間は 0 を跨いでおらず、A 群は H 群に比べ TAT 減少率が大き いことが示された。 これらの結果は、基礎疾患によらず、本剤が DIC 患者におけるトロンビンの生成を抑制 すること、及びその効果はヘパリンナトリウムよりも強いことを示唆しており、「本剤の DIC 離脱効果がヘパリンナトリウムよりも優れること」を裏付ける結果と考えられた。 前期第 2 相、後期第 2 相臨床試験でも、本剤投与により、TAT は投与前に比べ減少し、 その効果は 3 試験で共通して認められた(2.7.3.3.2.4 項)。 (n=60) (n=58) (n=45)(n=45) (%) A群 H群 A群 H群 (n=105) (n=103) (%) TAT変化率 -150.0 -100.0 -50.0 0.0 50.0 100.0 150.0 造血器悪性腫瘍 感染症 TAT変化率 -150.0 -100.0 -50.0 0.0 50.0 100.0 150.0 A群 H群 投与群間差 差の 95%信頼区間 (A 群-H 群) 下限 上限 TAT 変化率(%) -15.9 -27.1 -5.5
(2) D-ダイマー D-ダイマーは、血液凝固の最終産物である安定化フィブリンが分解された際に生じる物 質である。D-ダイマーは DIC で特異的な二次線溶の程度のみを反映するという特徴があり、 DIC に対する特異度が高いと考えられる。 図 2.5.4-3 に示したとおり、第 3 相臨床試験において、造血器悪性腫瘍、感染症のいずれ の基礎疾患でも、本剤、対照薬共に投与後の D-ダイマーは投与前に比べ減少した。また、 D-ダイマー減少率の薬剤群間差の両側 95%信頼区間は 0 を跨いでおらず、A 群は H 群に比 べ D-ダイマー減少率が大きいことが示された。 これらの結果は、基礎疾患によらず、DIC 患者において、本剤の方がヘパリンナトリウ ムより凝固系の活性化を抑制する作用が強く、その結果、二次線溶も強く抑制することを 示唆しており、「本剤の DIC 離脱効果や出血症状改善効果がヘパリンナトリウムよりも優 れること」を裏付ける結果と考えられた。 前期第 2 相、後期第 2 相臨床試験でも、本剤投与により、D-ダイマーは投与前に比べ減 少し、その効果は 3 試験で共通して認められた(2.7.3.3.2.4 項)。 (n=60) (n=58) (n=45)(n=45) (%) A群 H群 A群 H群 (n=105) (n=103) (%) D-ダイマー変化率 -300.0 -200.0 -100.0 0.0 100.0 200.0 300.0 造血器悪性腫瘍 感染症 D-ダイマー変化率 -300.0 -200.0 -100.0 0.0 100.0 200.0 300.0 A群 H群 投与群間差 差の 95%信頼区間 (A 群-H 群) 下限 上限 D-ダイマー変化率(%) -14.6 -28.4 -4.2 図 2.5.4-3 投与開始 7 日目(又は中止時)の投与前からの変化率(D-ダイマー)a) a) {投与前後の D-ダイマーの変化量(µg/mL)/投与前の D-ダイマー(µg/mL)}から算出
(3) プロテイン C 本剤は薬理学的特性上、トロンビンと共同してプロテイン C を活性化させることによっ て薬効を発現するため、薬効発現にプロテイン C を必要とする。また、DIC においてプロ テイン C は、血液中でのトロンビンの生成と共に消費性に減少することが知られている57) 。 図 2.5.4-4 に示したとおり、第 3 相臨床試験において、造血器悪性腫瘍、感染症のいずれ の基礎疾患でも、本剤、対照薬共に投与後のプロテイン C は投与前に比べ上昇した。また、 プロテイン C 変化量の薬剤群間差の両側 95%信頼区間は 0 を跨いでおらず、A 群の変化量 と H 群の変化量には差があることが示された。前期第 2 相、後期第 2 相臨床試験でも、本 剤投与により、プロテイン C は投与前に比べ上昇し、その効果は 3 試験で共通して認めら れた(2.7.3.3.2.4 項)。 以上の結果から、本剤投与によるプロテイン C の消費性低下の危惧はなく、本剤がトロ ンビンの生成を強力に抑制することによって、正常方向へプロテイン C を回復させたもの と考えられた。ただし、「2.5.4.4 項」に記載のとおり、投与前からプロテイン C が高度に 低下している場合は本剤の薬効低下に注意する必要があると考えられた。 (n=60) (n=58) (n=45)(n=45) (%) A群 H群 A群 H群 (n=105) (n=103) (%) プロテインC変化量 -150 -100 -50 0 50 100 150 造血器悪性腫瘍 感染症 プロテインC変化量 -150 -100 -50 0 50 100 150 A群 H群 投与群間差 差の 95%信頼区間 (A 群-H 群) 下限 上限 プロテイン C(%)変化量 11 4 17
(4) ATIII ATIII は生体内でトロンビンなどの凝固因子を阻害し、血栓形成の制御に重要な役割を果 たしている。プロテイン C と同様に、特に感染症を基礎疾患とする DIC で減少することが 知られている58)。また、ヘパリン類が ATIII の抗凝固作用を促進することにより薬効を発 現するのに対し、本剤は、薬効発現に ATIII を必要としない。 図 2.5.4-5 に示したとおり、第 3 相臨床試験において、造血器悪性腫瘍、感染症のいずれ の基礎疾患でも、A 群では ATIII は投与前に比べ上昇したのに対し、H 群では ATIII は減少 した。ATIII 変化量の薬剤群間差の両側 95%信頼区間は 0 を跨いでおらず、A 群の変化量と H 群の変化量には差があることが示された。前期第 2 相、後期第 2 相臨床試験でも、本剤 投与により ATIII は投与前に比べ上昇し、その効果は 3 試験で共通して認められた (2.7.3.3.2.4 項)。 A 群では、本剤がトロンビンの生成を抑制した結果、ATIII の消費も抑制されたため、 ATIII が上昇したものと考えられる。一方、H 群では、本剤と比較してヘパリンナトリウム の凝固線溶系の異常を是正する作用が弱かったことに加え、薬効発現に伴い ATIII の消費 が進んだことから、ATIII の生成と消費のバランスを生成量優位に傾けるまでに至らなかっ たものと考えられた。 (n=60) (n=58) (n=45)(n=45) (%) A群 H群 A群 H群 (n=105) (n=103) (%) ATIII(集中測定)変化量 -80.0 -60.0 -40.0 -20.0 0.0 20.0 40.0 60.0 80.0 造血器悪性腫瘍 感染症 ATIII(集中測定)変化量 -80.0 -60.0 -40.0 -20.0 0.0 20.0 40.0 60.0 80.0 A群 H群 投与群間差 差の 95%信頼区間 (A 群-H 群) 下限 上限 ATIII(集中測定)(%)変化量 16 11 21 図 2.5.4-5 投与開始 7 日目(又は中止時)の投与前からの変化量(ATIII 集中測定)
2.5.4.3.5 「造血器悪性腫瘍あるいは感染症」以外の疾患を基礎疾患とする DIC 患者における有効性 「2.5.1.1.2 項」に記載したとおり、DIC の本態は基礎疾患によらず共通であり、「造血器悪性 腫瘍あるいは感染症」以外を基礎疾患とする DIC の本態も、「造血器悪性腫瘍あるいは感染症」 を基礎疾患とする DIC と同様に、血液凝固系の過度な活性化によるトロンビンの過剰生成である。 したがって、トロンビンの生成を抑制する本剤は、「造血器悪性腫瘍あるいは感染症」以外を基 礎疾患とする DIC に対しても効果を示すと考えられる。このことは、以下に示す薬理試験及び臨 床薬理試験の結果からも示唆されている。 • In vitro において血漿に TF を添加し血液凝固系を活性化させる系で、本薬が凝固系活性化 に対する抑制作用を示すことが確認されている(2.6.2.2.4.4 項)。 • In vivo において動物(ラット、サル)に TF を投与し血液凝固系を活性化し DIC を発症さ せるモデルで、本薬が DIC 発症抑制効果を示すことが確認されている(2.6.2.2.6.1.1 項及び 2.6.2.2.6.1.5 項)。さらに、TF とは血液凝固活性化の機序が異なるリポポリサッカライド を投与するモデル(ラット)においても、本薬が DIC 発症抑制効果を示すことが確認され ている(2.6.2.2.6.1.4 項)。 • 後期第 2 相臨床試験において、本剤投与開始前、投与開始 4 日目、7 日目にプロトロンビ ナーゼ活性*を ex vivo にて測定した結果、「造血器悪性腫瘍あるいは感染症」以外を基礎疾 患とする DIC においても「造血器悪性腫瘍あるいは感染症」を基礎疾患とする DIC と同様 に、本剤投与後、プロトロンビナーゼ活性の顕著な低下が認められた{添付資料番号 5.3.5.1-1(後期第 2 相臨床試験総括報告書)図 11.4.1.4o}。 *プロトロンビナーゼ活性:本薬は、トロンビンと結合することによりプロテイン C を活性 化し、プロトロンビナーゼ複合体の形成阻害作用及び活性阻害作用を示す また、本剤の「造血器悪性腫瘍あるいは感染症」以外を基礎疾患とする DIC に対する臨床試験 の成績については、「造血器悪性腫瘍あるいは感染症」以外を基礎疾患とする DIC の中で、最も 患者数が多い固形癌を直接誘因基礎疾患とする DIC(以下、固形癌 DIC)について、後期第 2 相 臨床試験の成績を中心に検討した。なお、検討結果の詳細は「2.7.3.3.3.2 項」に記載した。 その結果、以下の点から、本剤は固形癌 DIC に対しても有効であり、本剤投与による DIC 改善 効果の臨床的意義は造血器悪性腫瘍を基礎疾患とする DIC(以下、造血器悪性腫瘍 DIC)や感染 症を基礎疾患とする DIC(以下、感染症 DIC)と同様に大きいと考えられた。 • 固形癌 DIC に対する本剤の成績と、症例数が多く結果の信頼性が高いと考えられる造血器 悪性腫瘍 DIC に対する本剤の成績を比較した結果、全体として固形癌 DIC における成績は 造血器悪性腫瘍 DIC における成績に比べ低いものの、固形癌 DIC でも用量の増加と共に改 善度が高まる傾向が認められた。また、本剤投与後のDICスコアやTATの推移についても、 造血器悪性腫瘍 DIC における結果と同様であった。さらに、造血器悪性腫瘍 DIC と同様に、 固形癌 DIC においても、DIC 離脱が 28 日目の生命予後の改善に寄与していると考えられ た。 • 固形癌 DIC において、本剤の成績と既存薬で報告されている成績を間接的ながら比較した 結果、本剤の 380 U/kg(0.06 mg/kg)群の固形癌 DIC に対する改善率は、既存薬に対し少なく
以上、本剤の作用機序、非臨床試験の結果、及び臨床試験の結果から、本剤は「造血器悪性腫 瘍あるいは感染症」以外を基礎疾患とする DIC に対しても有効であると考えられた。 2.5.4.4 部分集団における結果の類似性、相違について 第 3 相臨床試験で主要評価項目とした DIC 離脱率について、部分集団における結果を、第 3 相 臨床試験及び後期第 2 相臨床試験の 2 試験で比較した。部分集団別 DIC 離脱率の結果の詳細は、 「2.7.3.3.3 項」に記載した。また、第 3 相臨床試験における DIC の基礎疾患毎の、部分集団別 DIC 離脱率の結果は、「2.7.3.3.2.1 項」に記載した。 その結果、本剤の 380 U/kg(0.06 mg/kg)群では、いずれの部分集団でも一貫して DIC 離脱率が高 い傾向にあった。また、第 3 相臨床試験においては、ほとんどの部分集団で A 群の DIC 離脱率が H 群を上回っており、「本剤の DIC 離脱効果がヘパリンナトリウムより優れる」という主要な結 論の頑健性は高いと考えられた。 部分集団別解析結果のうち、注目すべき部分集団に関して以下に考察する。 (1) 投与開始時プロテイン C 値 前述のとおり、本剤はトロンビンと共同してプロテイン C を活性化させることによって 薬効を発現する。このため血漿中のプロテイン C 値によって本剤の効果が影響を受ける可 能性が考えられた。部分集団別解析の結果、投与開始時プロテイン C 値が 20%以上 50%未 満の層においては、50%以上の層と比較して DIC 離脱率が低くなることはなかった。しか し、第 3 相臨床試験では、20%未満の層において本剤投与群の DIC 離脱率は他の層と比較 して低かった。ただし、H 群においても同様に低く、本剤においてのみ特異的に有効性が 低下する可能性は低いと考えられた。また、in vitro における試験結果から、プロテイン C 値が 10%以上であれば、臨床試験での薬物濃度と同程度の 1 µg/mL の濃度において本剤の 薬効発現に対する影響は少ないことが確認されている(2.4.2.1.4.3 項)。以上より、プロテ イン C が検出限界である 10%より高値であれば、本剤の薬効はほとんど低下しないと考え られた。 一方、開始時プロテイン C 値が検出限界である 10%以下と高度に低下した症例が頻度は 少ないものの、後期第 2 相臨床試験で 4 例(低用量群 1 例、中用量群 1 例、高用量群 2 例)、 第 3 相臨床試験の A 群で 4 例の計 8 例に認められた。これらの症例の DIC 離脱については、 後期第 2 相臨床試験の 4 例中、高用量群である 380 U/kg (0.06 mg/kg)群の 1 例で離脱した以 外、3 例は非離脱もしくは判定不能であった。第 3 相臨床試験の 4 例は全て非離脱であっ た。開始後 28 日目の転帰は、両試験合わせた 8 例中 4 例が生存例であり、生存例は後期第 2 相臨床試験の高用量群の 1 例、第 3 相臨床試験の 3 例と、いずれも 380 U/kg(0.06 mg/kg) が投与された症例であった。ただし、H 群の該当症例は 1 例と少なく、10%以下で本剤の 薬効が特異的に低下するかどうかは確定的ではなかった。 以上の臨床成績と、in vitro における試験結果を考え合わせると、開始時プロテイン C 値 が 10%以下の症例に対して本剤の効力が減弱する可能性は否定できなかった。したがって、 プロテイン C 値が 10%以下と高度に低下する可能性の高い以下の患者においては、可能な 限り本剤投与前にプロテイン C 値を測定すべきであると考えられた。 - 肝臓における蛋白合成能が著しく低下している患者 - プロトロンビン時間が高度に延長しており、ビタミン K 欠乏が疑われる患者
しかしながら、プロテイン C 値の測定に関して、院内検査で実施できない場合、採血か ら検査結果の確認まで2~3日を要する場合もあるため、緊急を要するDICの治療において、 本剤投与開始前にプロテイン C 値を予め確認することは困難な場合があると想定される。 したがって、上記のようなプロテイン C 値が高度に低下する可能性の高い患者に本剤を投 与する場合は、可能な限り早期にプロテイン C 値を測定し、10%以下の低値であり、かつ DIC に改善が見られない場合は速やかに他剤での治療に切り替える必要があると考える。 以上のことより、添付文書(案)の[重要な基本的注意]に「プロテイン C 濃度が高度 に低下している可能性が高い患者に本剤を投与する場合は、可能な限り本剤投与前、又は 投与開始後早期にプロテイン C 濃度を測定し、10%以下の低値であり、かつ DIC の改善が みられない場合は速やかに他剤での治療に切り替えること。」を記載し、注意を喚起する こととした。 (2) 投与開始時 ATIII 値 ATIII 値は、特に感染症 DIC で低下しやすいことが知られている58)。一方、本剤は薬理 学的特性上、薬効発現に ATIII を必要としない。In vitro の試験でも、ATIII 低下は本薬の薬 効に影響しないことが確認されている(2.4.2.1.4.3 項)。投与開始時 ATIII 値別の検討の結 果、第 3 相臨床試験において、本剤投与群は ATIII 値によらず、高い DIC 離脱率を示した。 以上のことから、本剤は DIC 患者に対し、ATIII 値によらず効果を発揮すると考えられた。 さらに、図 2.5.4-5 に示したとおり、ATIII の投与前後の変動を比較した解析では、本剤投 与群では投与後、ATIII は増加した。これらのことから、本剤は、ATIII 低下患者に対して も、乾燥濃縮人アンチトロンビン III を補充せずに治療効果を得られる可能性があることが 示唆された。 2.5.4.5 推奨される用法・用量 添付文書(案)において、用法・用量は以下のとおり設定した。 • 用法・用量:通常、成人には、トロンボモデュリン アルファとして 1 日 1 回 380 U/kg を約 30 分かけて点滴静注する。なお、症状に応じ適宜減量する。 各試験の用法・用量を設定するに至った経緯及び根拠は、「2.7.3.4 項」に記載した。 本項では、これらの試験結果をまとめ、以下に、推奨される用法・用量の設定根拠について記 載した。 2.5.4.5.1 用法についての設定根拠 • DIC の治療には緊急性を要する場合が多いため、本剤の投与法としては、短時間で有効血 漿中濃度に到達させる静脈内投与が望ましいと考えられた。 • 2 時間静脈内持続投与による第 1 相臨床試験の成績から、ヒトにおける本剤の血漿中消失 半減期が約 20 時間であることが明らかとなり、1 日 1 回短時間静脈内投与の用法が選択可
• 点滴時間については、DIC 治療における緊急性及び臨床現場での利便性から可能な限り短 いことが望まれる。「2.7.2.2.1.1.2 項」に記載したとおり、2 時間の静脈内持続投与と 30 分 の静脈内持続投与の推定最高血漿中濃度がほとんど同等であり、30 分に点滴時間を短縮し ても安全性に問題ないと考えられ、前期第 2 相臨床試験の用法として、1 日 1 回 30 分の静 脈内持続投与を選択した。 • 臨床現場における用法の選択のひとつとして静脈内急速投与も可能と考えられたため、前 期第 2 相臨床試験の開始後ではあったが、その安全性と薬物動態を調べる目的で、静脈内 急速投与による第 1 相臨床試験を実施した。その結果、「2.7.2.2.1.1.3 項」に記載したとお り、薬物速度論的パラメータには 2 時間静脈内持続投与と静脈内急速投与では差がないこ とが確認された。また安全性にも 2 時間静脈内持続投与時と同様に問題がないことが確認 された。 • 万一投与中に不都合が生じた際に即座に投与を中止できる「静脈内持続投与の安全性」と 「静脈内急速投与の利便性」のバランスを考慮し、安全性を優先させるべきと考え、「30 分の静脈内持続投与」を後期第 2 相及び第 3 相臨床試験における本剤の用法として選択し た。 上記のとおり、用法については、本剤の血漿中濃度半減期が約 20 時間であること、並びに 2 時 間以内の持続投与であればいずれの用法も選択可能であると想定されたことから、「1 日 1 回の 短時間静脈内投与」が適切であると考えられた。一方、DIC 治療における緊急性や安全性の観点 から選択した「30 分の静脈内持続投与」による前期第 2 相臨床試験、後期第 2 相臨床試験、及び 第 3 相臨床試験において、本剤の有効性と安全性が確認されたことから、DIC 患者への使用経験 を有する「1 日 1 回の 30 分の静脈内持続投与」が本剤の用法として妥当であると考えられた。 2.5.4.5.2 用量についての設定根拠 2.5.4.5.2.1 体重当たりの用量設定について • 後期第 2 相臨床試験は、投与量と効果の相関性を検証することが主な目的であり、より正 確に有効性の用量相関性をみることができるように血漿中濃度のばらつきの要因はできる 限り少なくすることが望ましいと考えた。そこで用量は、前期第 2 相臨床試験まで採用し たヒト当たりの設定に代えて、体重当たりの設定を採用することとした。 • 第 1 相臨床試験並びに後期第 2 相臨床試験時の血漿中濃度データを用いた PPK 解析の結果、 本剤の血漿中動態は背景因子により大きく変動しないものと考えられた。ただし、本剤の 分布容積及びクリアランスは体重に比例することから、体重当たりの投与が必要であると 考えられた(2.5.3.1.6 項)。したがって、第 3 相臨床試験においても後期第 2 相臨床試験 と同様に、体重当たりの用量設定を採用した。 2.5.4.5.2.2 推奨用量を 380 U/kg(0.06 mg/kg)と設定した根拠 • 本剤の用量反応関係を検討した後期第 2 相臨床試験の最高用量は、以下の理由より設定し た。すなわち、前期第 2 相臨床試験のステップ 3{13,000 U/人(2.0 mg/人)~19,000 U/人 (3.0 mg/人)}における全般改善度の「中等度改善」以上率は 83.3%(10/12 例)であり、DIC における他の抗凝固薬の成績と比較して同等以上に高く十分な有効性を示しているものと
判断した。また、この用量での安全性に特に問題は認められなかった。一方、サルでの反 復投与毒性試験において出血の毒性発現が認められた 3,800 U/kg(0.6 mg/kg)を、薬物動態の 差を考慮してヒトに外挿すると、1,300 U/kg(0.2 mg/kg)、すなわち 64,000 U/人(10 mg/人) *となる。前期第 2 相臨床試験の最高用量 19,000 U/人(3.0 mg/人)は、その約 1/3 量に相当 するので、個人間の血漿中濃度のばらつきを考慮すると、安全性の観点からこれ以上の高 い用量は設定するべきでなく、19,000 U/人(3.0 mg/人)、すなわち 380 U/kg(0.06 mg/kg)* が後期第 2 相臨床試験の最高用量として適切と考えられた(2.7.3.1.2 項)。 *体重を 50 kg として換算した • 後期第 2 相臨床試験の結果、全般改善度の「中等度改善」以上率は、低用量群 39.3%(11/28 例)、中用量群 60.7%(17/28 例)、高用量群 67.6%(23/34 例)であり、有意な用量相関性 が認められた(p=0.0188)。しかし、中用量群である 130 U/kg(0.02 mg/kg)群と高用量群であ る 380 U/kg(0.06 mg/kg)群の間で改善率に大きな差はなく、用量反応曲線の傾きは比較的緩 やかであった(2.7.3.2.2 項)。本剤の用量反応曲線の傾きが緩やかであることは、ヒト血 漿を用いたトロンビン生成阻害作用(2.4.2.1.4.1 項)並びにサルの DIC モデルを用いた DIC 発症抑制効果の成績(2.4.2.1.5.1 項)でも同様に認められており、これ以上投与量を増やし ても改善率が大きく上昇することはないと推察された。一方、出血の随伴症状発現率には 用量依存的な傾向は認められず(2.5.5.4.6 項)、380 U/kg(0.06 mg/kg)の用量は、安全性に 関して大きな問題はないと考えられた。 • 後期第 2 相臨床試験において、血漿中濃度と有効性の関係を分析した結果、高用量群であ る 380 U/kg(0.06 mg/kg)群では初回投与終了時から最終投与後 24 時間にかけて、本剤の血 漿中濃度は、十分な有効性が期待される濃度範囲(300~900 ng/mL)を超えて維持されており、 380 U/kg(0.06 mg/kg)は、臨床上十分な効果を示す用量であると考えられた(2.5.3.2.1 項)。 一方、血漿中濃度と安全性の関係では、死亡率や出血に関連する随伴症状の発現頻度にお いて、血漿中濃度との相関は認められなかった(2.7.4.2.1.2 項及び 2.7.4.2.1.5.1.2 項)。 • 後期第 2 相臨床試験における主要評価項目及び対象患者は、次相の第 3 相臨床試験のそれ らと異なるため、第 3 相臨床試験への外挿性を確認する目的で、第 3 相臨床試験の主要評 価項目である DIC 離脱率についての検討を行った。その結果、DIC 離脱率においても、全 般改善度と同様に用量依存的に離脱率が高くなる傾向がみられた(2.7.3.2.2 項)。また、 第 3 相臨床試験の対象が造血器悪性腫瘍、感染症に限定されることから、基礎疾患別にみ たところ、いずれの基礎疾患別の結果においても用量の増加と共に DIC 離脱率は高くなる 傾向がみられた。特に高用量群である 380 U/kg(0.06 mg/kg)群では、造血器悪性腫瘍、感染 症のいずれにおいても本剤の高い DIC 離脱効果が認められた(2.7.3.3.2.1 項)。これらの 結果から、本剤 380 U/kg(0.06 mg/kg)は、第 3 相臨床試験における対象患者、主要評価項目 においても、後期第 2 相臨床試験と同様な有効性を示すものと予測された。 • 380 U/kg(0.06 mg/kg)の用量で実施した第 3 相臨床試験の結果、主要評価項目である DIC 離 脱率において、A 群は、H 群より優れていることが示された。部分集団別解析でも、ほと んどの層で A 群の DIC 離脱率が H 群を上回っていた。また、後期第 2 相臨床試験の主要評 価項目であった全般改善度の「中等度改善」以上率は、A 群 72.4%(2.7.6.5.3.4.3.3 項)と、
A 群の出血症状に関連する有害事象発現率は、投与開始 7 日目まで、投与開始後 14 日目 までのいずれの時点でも H 群に比べ低かった。出血症状に関連する有害事象発現率の部分 集団別解析では、各部分集団で A 群の発現率に大きな違いは認められなかった。また、重 篤な有害事象、副作用の発現率も A 群は H 群に比べ、同程度もしくはそれ以下であった (2.5.5.4 項)。 以上の結果から、本剤の 380 U/kg(0.06 mg/kg)の用量は、十分な有効性を示し安全性にも 問題がない用量であることが確認された。 以上の結果及び DIC のように致死的な疾患においては、リスクが許容される範囲で可能な限り 強力な薬効が発揮することが求められることから、「1 日 380 U/kg(0.06 mg/kg)」が臨床推奨用量 として適切であると考えられた。また、PPK 解析及び部分集団別解析の結果から、高齢者等、特 定の集団で用量を調節する必要はないと考えられた。 ただし、「頭蓋内出血、肺出血、消化管出血(継続的な吐血・下血、消化管潰瘍による出血)の ある患者」は、臨床での投与経験が少なく、また、これらの臓器での出血は転帰に重大な影響をお よぼすと考えられるため、本剤による出血のリスクを最小限に抑える目的から、添付文書(案) では「禁忌」とした。本剤投与中にこれらの症状が認められた場合には、本剤の投与を中止する べきであると考えた。また、本剤投与中に「本剤によると考えられる出血症状の発現・増悪がみ られた場合」や「重篤な腎機能障害を認め、それに伴い出血症状の発現・増悪がみられた場合」 も同様に、本剤の投与を中止するべきであると考え、添付文書(案)の[重要な基本的注意]に、 上記のような場合は投与を中止する旨を記載した。 一方、PPK 解析の結果では腎機能障害患者において投与量調整が必要なほどの影響はなかった ものの、「重篤な腎機能障害のある患者」については、本剤が腎排泄型の薬剤であること、重篤 な腎機能障害患者での投与経験も少ないことなどを考慮すると本剤の投与量が過量になる可能性 を完全に否定できないため、凝血学的検査、臨床検査、及び臨床症状を参考に減量を考慮する必 要があると考えた。特に「血液透析療法中の患者」については腎機能障害の程度が高度であるた め、一律に減量する必要があると考えた。以上の理由から、添付文書(案)の[用法・用量]に 「症状に応じ適宜減量する。」旨を記載し、添付文書(案)の[慎重投与]にも「重篤な腎機能 障害のある患者(患者の症状に応じ適宜 130 U/kg に減量して投与すること。なお、血液透析療法 中の患者には 130 U/kg に減量して投与すること。)」と記載した。 なお、減量の程度に関しては、過度な減量の結果、効力が不十分となることは DIC のような重 篤疾患の場合、死亡等の重大なリスクの増大につながるので、後期第 2 相臨床試験から有効性が 著しく減弱しないことが明らかとなっている 130 U/kg (0.02 mg/kg)とすることが妥当と考えた。 2.5.4.6 観察された効果の臨床的意義及びその限界 2.5.4.6.1 本剤と既存薬の臨床試験実施状況 DIC は、致死的疾患であるため、治療レベルの向上に対するニーズは非常に大きい。しかしな がら、本疾患領域は、以下の理由により、臨床試験の実施が容易ではないことから、エビデンス に基づいた治療レベルの向上には大きな制約がある。 • DIC は重症疾患であるため、選択基準・除外基準に合致し、さらに臨床試験参加への同意 を取得できる患者が少なく、一試験に多数例の症例を集積することが困難である。 • DIC は単一の疾患でなく、様々な種類の基礎疾患に合併するため、多様性に富んでいる。
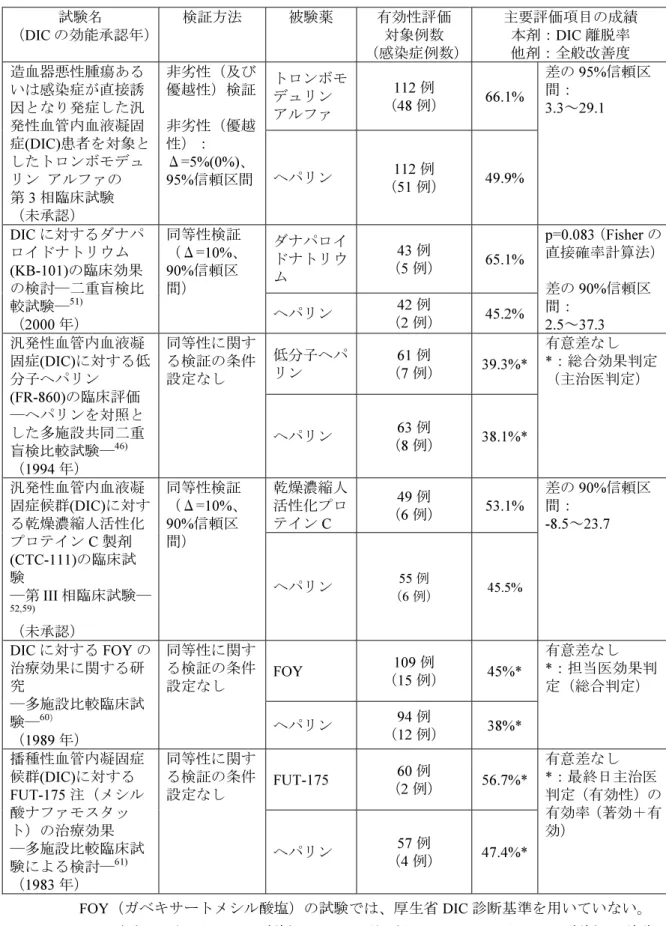
• DIC は致死的な疾患であるために、プラセボ比較試験は倫理的理由により実施困難である。 (1) 既存薬の比較検証試験の概要 このような状況の中、本邦では既存の抗凝固薬においては全てヘパリンを対照薬とした 臨床試験が実施されてきた。それらの検証試験の実施状況を表 2.5.4-8 に示した。なお、乾 燥濃縮人アンチトロンビン III に関しては、造血器悪性腫瘍あるいは感染症を基礎疾患とす る DIC の領域では、本邦において並行群間比較臨床試験が実施されていないので、表には 記載しなかった。
表 2.5.4-8 本剤及び既存薬の第 3 相臨床試験概要 試験名 (DIC の効能承認年) 検証方法 被験薬 有効性評価 対象例数 (感染症例数) 主要評価項目の成績 本剤:DIC 離脱率 他剤:全般改善度 トロンボモ デュリン アルファ 112 例 (48 例) 66.1% 造血器悪性腫瘍ある いは感染症が直接誘 因となり発症した汎 発性血管内血液凝固 症(DIC)患者を対象と したトロンボモデュ リン アルファの 第 3 相臨床試験 (未承認) 非劣性(及び 優越性)検証 非劣性(優越 性): Δ=5%(0%)、 95%信頼区間 ヘパリン (51 例) 112 例 49.9% 差の 95%信頼区 間: 3.3~29.1 ダナパロイ ドナトリウ ム 43 例 (5 例) 65.1% DIC に対するダナパ ロイドナトリウム (KB-101)の臨床効果 の検討―二重盲検比 較試験―51) (2000 年) 同等性検証 (Δ=10%、 90%信頼区 間) ヘパリン (2 例) 42 例 45.2% p=0.083(Fisher の 直接確率計算法) 差の 90%信頼区 間: 2.5~37.3 低分子ヘパ リン (7 例) 61 例 39.3%* 汎発性血管内血液凝 固症(DIC)に対する低 分子ヘパリン (FR-860)の臨床評価 ―ヘパリンを対照と した多施設共同二重 盲検比較試験―46) (1994 年) 同等性に関す る検証の条件 設定なし ヘパリン (8 例) 63 例 38.1%* 有意差なし *:総合効果判定 (主治医判定) 乾燥濃縮人 活性化プロ テイン C 49 例 (6 例) 53.1% 汎発性血管内血液凝 固症候群(DIC)に対す る乾燥濃縮人活性化 プロテイン C 製剤 (CTC-111)の臨床試 験 ―第 III 相臨床試験― 52,59) (未承認) 同等性検証 (Δ=10%、 90%信頼区 間) ヘパリン (6 例) 55 例 45.5% 差の 90%信頼区 間: -8.5~23.7 FOY 109 例 (15 例) 45%* DIC に対する FOY の 治療効果に関する研 究 ―多施設比較臨床試 験―60) (1989 年) 同等性に関す る検証の条件 設定なし ヘパリン (12 例) 94 例 38%* 有意差なし *:担当医効果判 定(総合判定) FUT-175 60 例 (2 例) 56.7%* 播種性血管内凝固症 候群(DIC)に対する FUT-175 注(メシル 酸ナファモスタッ ト)の治療効果 ―多施設比較臨床試 験による検討―61) (1983 年) 同等性に関す る検証の条件 設定なし ヘパリン (4 例) 57 例 47.4%* 有意差なし *:最終日主治医 判定(有効性)の 有効率(著効+有 効) FOY(ガベキサートメシル酸塩)の試験では、厚生省 DIC 診断基準を用いていない。 FOY(ガベキサートメシル酸塩)、FUT-175 注(ナファモスタットメシル酸塩)の試験は非 盲検試験。乾燥濃縮人活性化プロテイン C は効能・効果に DIC を有していない。
(2) 本剤と既存薬における臨床試験実施状況の比較 本剤の検証試験・用量反応試験の計画・結果に関しては、「2.5.1.4 項」及び「2.5.4.3 項」に 記載したとおりであり、それらを既存薬の検証試験との比較の観点からまとめると以下の とおりである。 • 主要評価項目の検証: - 主要評価項目で、対照薬ヘパリンに対して「臨床試験のための統計的原則」(1998 年 11 月 30 日付け、医薬審発第 1047 号)62)、「臨床試験における対照群の選択とそれに関 連する諸問題」(2001 年 2 月 27 日付け、医薬審発第 136 号)55)に基づき、「Δ=5%、 95%信頼区間」の条件で非劣性を示したのは、本剤のみである。同時に優越性を示した のも、本剤のみである。 - 本剤では、DIC 離脱率という臨床的意義が明確で、客観的な評価指標で有効性に関する 結論を得ることができた。 - 本剤の試験では比較可能性を高めるため、対照薬である未分画ヘパリンが前投与されて いる患者を除外した。 • 感染症 DIC での評価: - 既存薬の臨床試験では、造血器悪性腫瘍 DIC における有効性・安全性の知見は多かった が、感染症 DIC に関する知見は非常に乏しかった。それに対し、本剤では、感染症 DIC を 100 例規模で集積した。 - その結果、感染症 DIC においても造血器悪性腫瘍 DIC と同様に、本剤がヘパリンとの 比較の上で有効かつ安全な薬剤であることが確認できた。 • 基礎疾患の影響: - 本剤では、基礎疾患の経過を評価項目とし、DIC 離脱率との関係を検討した。その結果、 両薬剤群間の DIC 離脱率の差が基礎疾患の経過に依存していないことが示された。 • 用量反応関係の検証: - 本剤の有効性が用量反応性の検証によっても示された。 • 有害事象による評価: - 既存薬の臨床試験においては、主に副作用と臨床検査値異常で安全性が評価されている。 一方、本剤の第 3 相臨床試験では、有害事象で安全性を評価することによって、本剤と ヘパリンのリスクを正確に把握できた。 - 転帰(生命予後)に関しては、既存薬では「治験薬投与期間中の転帰」のみ報告されてい るものが多いが、本剤では全ての臨床試験で「治験薬投与開始後 28 日目の転帰」を全例 で調査した。 • 濃厚血小板、新鮮凍結血漿の併用の影響: - DIC の薬効評価には、濃厚血小板、新鮮凍結血漿の併用投与が影響をおよぼすことが知 られている。既存薬の臨床試験においても、これらの併用が抗凝固薬の薬効評価に対し バイアスとなることが指摘されてきた。 - 濃厚血小板、新鮮凍結血漿は、抗凝固療法と並行して実施されるため、本剤の臨床試験 でも、併用可とした。本剤においても濃厚血小板、新鮮凍結血漿の使用量を治験実施計