Nonlinear Disposition and Metabolic Interactions
of Cannabidiol Through CYP3A Inhibition In Vivo in Rats
Michiru Nagao,1Yukako Nakano,1Masataka Tajima,1Erika Sugiyama,1Vilasinee Hirunpanich Sato,2 Makoto Inada,3and Hitoshi Sato1,*
Abstract
Introduction:Cannabidiol (CBD) is known to affect the pharmacokinetics of other drugs through metabolic inhi- bition of CYP2C19 and CYP3A4. However, there is a lack ofin vivoevidence for such drug interactions. Therefore, we investigated the saturability of CBD metabolism and CBD-drug interactions through inhibition of CYP3Ain vivo.
Materials and Methods:A nanoemulsion formulation of CBD (CBD-NE) was orally administered to rats at doses of 5, 10, 25, and 50 mg/kg, and plasma concentrations of CBD were measured by using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to examine the dose-dependency of CBD exposure (area under the curve [AUC]). To examine the effect of a CYP3A inhibitor on CBD pharmacokinetics, rats were orally pretreated with 50 mg/kg keto- conazole (KCZ), a strong CYP3A inhibitor, before oral administration of CBD-NE at doses of 10 and 50 mg/kg, and plasma concentrations of CBD were measured using LC-MS/MS. Moreover,13C-erythromycin was orally administered following administration of either NE (without CBD), as a control, or CBD-NE at 1, 10, and 50 mg/kg, and13C-breath response was measured by using an infrared analyzer.
Results:After administration of various doses of the nanoemulsified CBD formulation to rats, the exposure of CBD (i.e., the AUC calculated from the plasma concentration–time profile) increased in a greater than dose- proportional manner, especially at doses above 10 mg/kg. The AUC and maximum plasma concentration (Cmax) of CBD after oral administration of CBD-NE (10 mg/kg) increased approximately three times by the coad- ministration of KCZ. Moreover, according to the CBD-induced changes of13C-breath response, the metabolism of
13C-erythromycin was shown to be inhibited by CBD at doses of 10 and 50 mg/kg, but not at 1 mg/kg.
Conclusions:Nonlinear disposition and CYP-mediated drug interactions of CBD at doses exceeding 10 mg/kg were demonstrated for the first timein vivoin rats. Given the present results, it is proposed that caution for dose- dependent drug interactions should be considered for CBD.
Keywords:cannabidiol; drug interactions; CYP3A; nonlinear pharmacokinetics; metabolism; rats
Introduction
Cannabidiol (CBD) is one of the main cannabinoids contained in cannabis plants. The chemical structure of CBD is similar to that of a psychoactive cannabinoid, D9-tetrahydrocannabinol, whereas CBD has no psycho- tropic effects.1 CBD modulates the activities of various cellular effectors, including the cannabinoid receptors CB1 and CB2,25HT1Areceptors,3l- andd-opioid recep- tors,4TRPV1 cation channels,5fatty acid amide hydro- lase,5GPR3/6/12/55,6,7peroxisome proliferator-activated
receptor c,8 and voltage-dependent anion channel 1.9 CBD has been reported to have clinical effects against ep- ilepsy, anxiety, insomnia, and nausea.1,8,10
Many types of CBD formulations have been available in the market, but oil preparations11are the most popu- lar owing to the water-insoluble nature of CBD. After CBD formulations are orally administered, the intestinal absorption of CBD molecules is limited by their poor solubility. After a proportion of CBD molecules entered into the portal vein, CBD is extensively metabolized in
1Division of Pharmacokinetics and Pharmacodynamics, Department of Pharmacology, Toxicology and Therapeutics, School of Pharmacy, Showa University, Tokyo, Japan.
2Department of Pharmacology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand.
3Development Department Diagnostic Division, Otsuka Pharmaceutical Co., Ltd., Tokushima, Japan.
*Address correspondence to: Hitoshi Sato, PhD, Division of Pharmacokinetics and Pharmacodynamics, Department of Pharmacology, Toxicology and Therapeutics, School of Pharmacy, Showa University, 1-5-8 Hatanodai, Shinagawa-ku, Tokyo 142-8555, Japan, E-mail: [email protected]
1
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
the liver by cytochrome P450 enzymes, mainly CYP3A4 and CYP2C19.1,12
As CBD is not only an inhibitor of CYP3A and CYP2C but also a substrate of these enzymes,1,12,13sat- uration of these enzymatic activities may lead to the nonlinear disposition of CBD as well as CBD-related drug interactions. Therefore, we investigated the correla- tion between the CBD dose and exposure (i.e., the area under the curve [AUC] of plasma CBD concentrations) and the CBD-drug interactions through alterations in CYP3A activity. For these purposes, we employed a highly absorbable nanoemulsion formulation of CBD (CBD-NE) developed previously in our laboratory14to eliminate the absorption factors and to focus on the met- abolic aspects of CBD.
Materials and Methods Materials
CBD powder (purity 99%) was supplied by Endoca (Hoofddorp, The Netherlands).
Clobetasol and acetic acid were purchased from Sigma-Aldrich (St. Louis, MO).
Acetonitrile was purchased from Honeywell Interna- tional (Seelze, Germany). Tween 20 was purchased from MP Biomedicals (Solon, OH). Ketoconazole (KCZ), car- boxymethyl cellulose (CMC), olive oil, vitamin E, and ammonium formate were purchased from FUJIFILM Wako (Osaka, Japan).13C-erythromycin (13C-EM) was obtained from Cambridge Isotope Laboratories (MA).
Preparation of CBD-NE
Pure CBD powder was formulated as a nanoemulsion, as previously described,14which is a ternary phase com- prising vitamin E acetate as an oil, ethanol as a cosurfac- tant, Tween-20 as a surfactant, and distilled water at 1.7, 3.8, 70.0, and 24.5 w/w%, respectively. The prepared CBD-NE formulation was kept at 4C until use.
Animals and procedures
Male Wistar rats (Sankyo Labo Service Corporation, Tokyo, Japan), weighing 210–250 g, were allowed food and waterad libitum. All animal studies were approved and carried out in accordance with the guidelines for animal experimentation in Showa University (Tokyo, Japan). The rats were fasted overnight for 12 h before all experiments. For the pharmacokinetic study, the left femoral artery of each rat was cannulated with silicon tub- ing (SP-31; Natsume Seisakusho Co., Ltd., Tokyo, Japan) to collect blood samples under isoflurane inhalation anes- thesia. Each rat was placed in a Bollman cage, and various
treatments with CBD and other agents were performed after the animals had recovered from anesthesia.
Dose-dependency of the pharmacokinetic profiles of CBD-NE
Four doses of CBD-NE (5, 10, 25, and 50 mg/kg) were administered orally to the rats, and 200lL of blood was sequentially collected at 0.5, 1, 2, 4, 8, and 10 h from the cannulated femoral artery to examine the dose-dependency of CBD pharmacokinetics. Pharma- cokinetic parameters were calculated as described later, and the pharmacokinetic nonlinearity was examined by a power model analysis,15fitting the dose-AUC rela- tionship of CBD to the following equation:
log AUCð Þ = log(a) þ b · log doseð Þ
whereaandbrepresent the intercept and the slope of the regression line, respectively. Both AUC0–8 and AUC0–Nwere applied to this analysis to be correlated with the CBD dose. The proportionality of the relation- ship was evaluated from the 95% confidence interval (CI) ofb, that is, the relationship was indicated to be linear when the CI ofb value contained 1; otherwise, the relationship was considered nonlinear.
Effect of a CYP3A inhibitor on CBD pharmacokinetics KCZ was suspended in 1% CMC with purified water and orally administered at a dose of 50 mg/kg 1 h be- fore oral CBD-NE administration at CBD doses of 10 and 50 mg/kg. Blood samples were collected after 0.5, 1, 2, 4, 8, and 10 h.
Preparation of plasma samples for measurement Plasma samples were obtained from the blood by centri- fugation (at 4C) at 10,000gfor 10 min and processed for quantification of CBD, as previously described.14Briefly, plasma samples were spiked with an internal standard (IS), clobetasol propionate, then extracted through Bond Elut Plexa cartridges (60 mg, 3 mL; Agilent Technologies, Santa Clara, CA), and reconstituted in a mixture of aceto- nitrile: 1 mM ammonium formate (80:20, v/v %). The su- pernatant of the reconstituted solution was then filtered using a syringe filter (Ekicrodisc3CR, diameter 3 mm, 0.45lm, Japan) and stored at23C until analysis.
Quantification of CBD with liquid chromatography- tandem mass spectrometry
Quantification of CBD in plasma was conducted using liquid chromatography-tandem mass spectro- metry, as previously described.14 The MS system (QTRAP5500; ABSCIEX, Framingham, MA) was
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
used in the electrospray ionization mode. The retention times of the IS and CBD were 5.71 and 11.77 min, re- spectively. The calibration curves of plasma CBD con- centrations were prepared separately for the low (0.01–
0.50lg/mL) and high (0.2–2.0lg/mL) ranges. The calibration curves of plasma CBD concentrations were prepared separately for the low (0.01–0.50lg/mL) and high (0.2–2.0lg/mL) ranges using two different concen- trations of IS solution so that the peak ratios (CBD/IS) of the multiple reaction monitoring MS were set to the appropriate range. According to the previous study,14 clobetasol was used as an IS because of its similar high lipophilicity to CBD, which could be adequate for pro- viding the similar extraction property from plasma.
The calibration curveR2 values were 0.986–0.998, and the limit of quantification was 10 ng/mL.14
13C-EM breath test
The inhibitory effect of CBD on CYP3A was evaluated using the13C-EM breath test, which is a noninvasive, easy, and rapid method forin vivoevaluation of CYP3A activity.16 CBD-NE (1, 10, and 50 mg/kg) or NE vehicle without CBD (as a control) was orally administered, fol- lowed by oral administration of 13C-EM (200 mg/kg) after 90 min. The13C-breath response [D13CO2(&) val- ues] was measured in the exhaled breath of rats after 0, 5, 10, 20, 30, 45, 60, 90, 120, 150, and 180 min using an infrared analyzer (POC-ONE; Otsuka Electronics Com- pany, Ltd., Tokushima, Japan).16
Pharmacokinetics and statistical analyses
The area under the concentration versus time curve from zero to a particular time (AUC0–t), AUC from zero to in- finity (AUC0–N), terminal half-life (T1/2), total clearance (CLtot/F), volume of distribution (Vdss/F), the maximum plasma concentration (Cmax), time to reach Cmax(Tmax), and mean residence time (MRT) were calculated by moment analysis using Microsoft Excel software.17The obtained PK parameters are expressed as the mean– standard error of the mean (SEM). Statistical analyses for comparison of PK parameters were conducted using Student’st-test between two groups and a one-way anal- ysis of variance (ANOVA) withpost hocDunnett’s test for comparison of three or more groups. Ap-value of <0.05 was considered to indicate statistical significance.
Results
Pharmacokinetic profiles of CBD-NE in rats
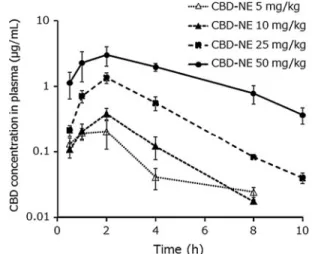
The CBD concentrations in plasma after administration of various doses of CBD-NE are shown in Figure 1, and the pharmacokinetic parameters are presented
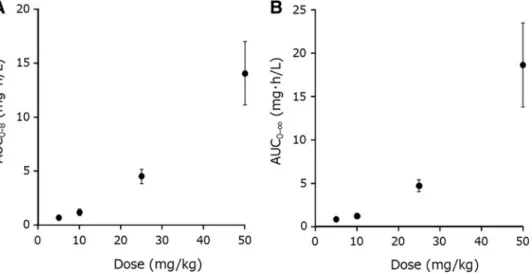
in Table 1. The correlation between the CBD dose and AUC (i.e., AUC0–8and AUC0–N) followed an as- cending curve, as shown in Figure 2. The results from the power model analysis are summarized in Table 2.
The nonlinear pharmacokinetic properties of CBD after oral administration of CBD-NE were demon- strated as each 95% CI did not contain the value of 1.
Effect of KCZ on the pharmacokinetic profiles of CBD-NE
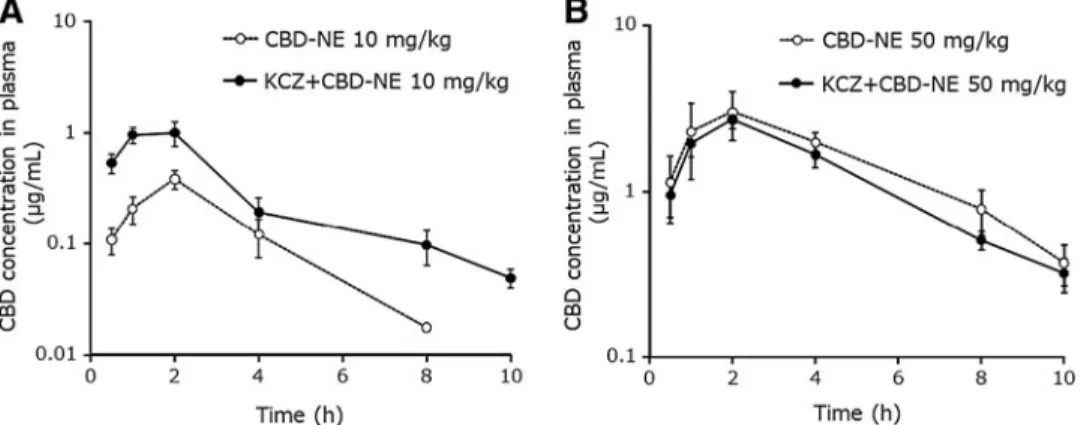
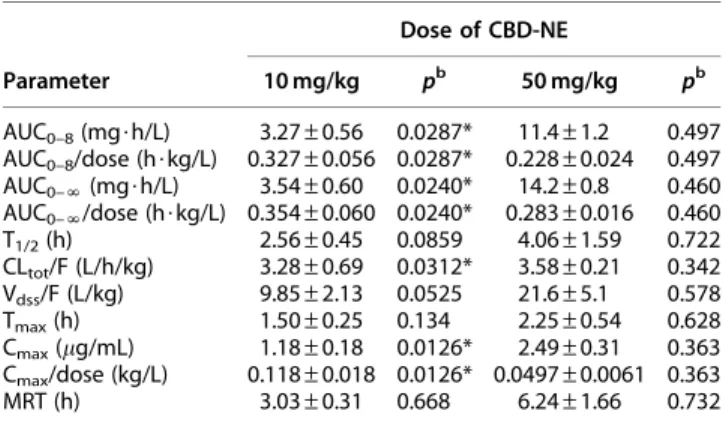
As illustrated in Figure 3A, oral administration of KCZ significantly increased the CBD plasma concentrations at a lower CBD dose (10 mg/kg). The pharmacokinetic parameters of AUC0–8, AUC0–N, and Cmax were in- creased approximately three times and that of CLtot/F was decreased by approximately one-third (Tables 1 and 3). In contrast, in the case of CBD-NE at the high- est CBD dose (50 mg/kg), the pharmacokinetic profiles and parameters of CBD were not altered by KCZ (Fig. 3B, Tables 1 and 3).
Effect of CBD on the13C-EM breath test
As shown in Figure 4 and Table 4, the AUCs of
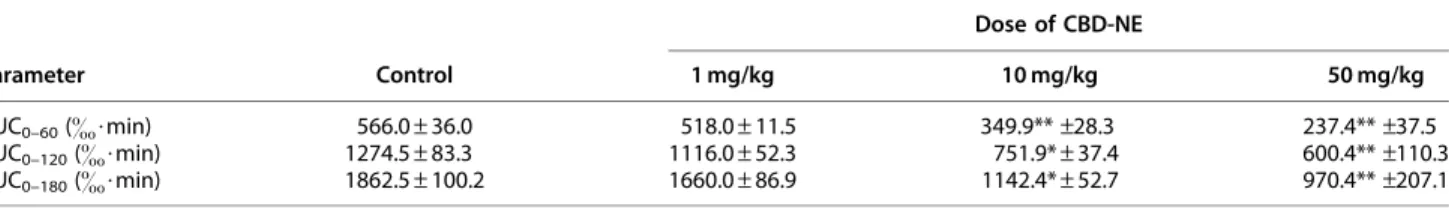
13C-breath responses were not significantly different between the control and lowest CBD dose (1 mg/kg) groups, although there was a significant difference between the control and higher CBD dose (10 and 50 mg/kg) groups.
FIG. 1. Plasma CBD concentration–time curves after oral administration of CBD-NE (5, 10, 25, and 50 mg/kg) to rats.
Each symbol with vertical bars represents the mean–SEM (n=4). CBD was not detected at 10 h after administration of CBD-NE at doses of 5 and 10 mg/kg. CBD, cannabidiol;
CBD-NE, nanoemulsion formulation of CBD; SEM, standard error of the mean.
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
Discussion
The oral absorption of CBD from oil formulations is highly variable1; as such, the dose–exposure (AUC) re- lationship of CBD has not yet been clearly defined. In the present study, we first found that the plasma con- centration of orally administered CBD in rats followed a nonlinear pharmacokinetic profile as the AUC of CBD increased in a greater than dose-proportional manner (p<0.0001), as demonstrated by the power model anal- ysis. As the slope of the exponential was more than 1 (Table 2), the nonlinearity was attributable to the satura- tion of metabolic clearance of CBD, rather than the sat- uration of its intestinal absorption, at higher doses of CBD. Generally, the dose-AUC relationship of a drug
is considered to be less sensitive to variability when the absorption rate is limited by poor solubility, and the pharmacokinetic variability may be largely governed by the absorption process. Therefore, we employed a rap- idly absorbable new formulation of nanoemulsified CBD, which was previously developed in our laboratory.14The absorption rate of CBD from the nanoemulsion formula- tion was rapid, with a Tmaxvalue of 1.50–2.75 h (Table 1), compared with an olive oil formulation in which the Tmax was 8.0 h.14 In the case of solubility-limited absorption, the AUC should increase in a less than dose-proportional manner.18,19In addition, it has been reported that CBD is not a substrate of P-glycoprotein, a drug efflux trans- porter.20Therefore, the observed nonlinear dose-AUC
FIG. 2. Correlations between(A)dose and AUC0–8of CBD and(B)dose and AUC0–Nof CBD. Each symbol with vertical bars represents the mean–SEM (n=4). AUC, area under the curve.
Table 1. Pharmacokinetic Parameters of Cannabidiol After Oral Administration of Nanoemulsion Formulation of Cannabidiol at Various Doses to Ratsa
Parameter
Dose of CBD-NE
5 mg/kg 10 mg/kg 25 mg/kg 50 mg/kg
AUC0–8(mg$h/L) 0.689–0.194 1.18–0.30 4.51–0.67 14.0–2.9
AUC0–8/dose (h$kg/L) 0.138–0.039 0.118–0.030 0.180–0.027 0.280–0.059
AUC0–N(mg$h/L) 0.825–0.190 1.21–0.30 4.72–0.67 18.6–4.8
AUC0–N/dose (h$kg/L) 0.165–0.038 0.121–0.030 0.189–0.027 0.373–0.097
T1/2(h) 3.95–1.14 1.45–0.12 1.64–0.08 3.33–0.61
CLtot/F (L/h/kg) 7.55–1.67 10.1–2.0 5.89–1.06 2.82–0.60
Vdss/F (L/kg) 36.5–10.5 29.4–6.7 19.2–4.3 17.6–2.9
Tmax(h) 1.50–0.25 2.00–0.00 2.00–0.00 2.75–0.65
Cmax(lg/mL) 0.280–0.075 0.386–0.074 1.35–0.24 3.58–0.91
Cmax/dose (kg/L) 0.0560–0.0151 0.0386–0.0074 0.0544–0.0096 0.0716–0.0183
MRT (h) 4.88–1.11 2.85–0.11 3.19–0.12 5.52–0.46
aEach value is presented as the mean–SEM (n=4).
AUC, area under the curve; CBD, cannabidiol; CBD-NE, nanoemulsion formulation of CBD; CLtot/F, total clearance; Cmax, maximum plasma concen- tration; MRT, mean residence time; SEM, standard error of the mean; T1/2, terminal half-life; Tmax, time to reach Cmax; Vdss/F, volume of distribution.
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
relationship of CBD should have resulted from the sat- uration of metabolic clearance, but not from absorption- related factors.
CBD is known to be metabolized in the human liver by CYP1A1, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5,12with CYP3A4 and CYP2C19 being the predominant responsible isoforms.12 On the other hand, CBD inhibits CYP3A4/5 and CYP2C19.1,13 Therefore, the nonlinear dose-AUC relationship may be the result of self-inhibition of CBD metabolism. The non- linearity of CBD pharmacokinetics after oral administra- tion of CBD-NE was obvious at doses above 10 mg/kg (Fig. 2). Thein vivosaturability of drug-metabolizing en- zymes for CBD prompted us to conduct further CBD- drug interaction experiments.
In general, there are two cases of pharmacokinetic CBD-drug interactions. In case 1, the pharmacokinetics of CBD are influenced by other drugs that inhibit CYP3A or CYP2C. In case 2, CBD affects the pharma- cokinetics of other drugs that are metabolized by CYP3A or CYP2C.In vitro drug interactions between CBD and other drugs have been extensively stud- ied,1,12,13butin vivoevidence for CBD-drug interactions has not been confirmed on a pharmacokinetic basis.
Therefore, in the present study, we investigated the pos- sibility of the above in vivocases of CBD-drug interac- tions in rats based on the pharmacokinetic alterations of CBD and coadministered drugs.
Again, to precisely assess CBD-drug interactions without complications of poor solubility of CBD after oral administration, we employed a rapidly ab- sorbable nanoemulsified formulation of CBD.14
For case 1 (Fig. 3), the CBD concentration after oral administration of 10 mg/kg was significantly increased by KCZ, which indicated that the metabolism of CBD was effectively inhibited by the CYP3A inhibitor. In contrast, at a higher CBD dosage (50 mg/kg), CBD pharmacokinetics were not affected by KCZ, indicat- ing that the metabolism of CBD was not inhibited by the CYP3A inhibitor, most likely because the meta- bolic enzyme CYP3A was already inhibited by the high dose of CBD itself. This result was consistent with the saturable disposition (i.e., nonlinear pharmacokinetics) of CBD observed at higher doses. Interestingly, the in- crease in the Cmaxvalue of CBD after oral administration of 10 mg/kg CBD-NE in the presence of KCZ (50 mg/kg) coadministration and the increase in Cmaxfrom the low to high doses of CBD (10 vs. 50 mg/kg) turned out to be similar (an increase of approximately three times), which suggested that the inhibitory effect of CBD on CYP3A was sufficiently strong compared with that of KCZ.
For case 2 (Fig. 4), we examined whether CBD inhibited the metabolism of erythromycin, which is a selective CYP3A substrate, using a 13C-EM breath test,16 where N-demethylation of 13C-EM was medi- ated by CYP3A and the desorbed methyl radical was fi- nally exhaled as13CO2in breath, which is presented as
Table 2. Power Analysis of the Dose–Area Under the Curve Proportionality of Cannabidiola
Parameter b(slope) SE 95% CI p
AUC0–8(mg$h/L) 1.36–0.14 1.06–1.67 <0.0001 AUC0–N(mg$h/L) 1.37–0.15 1.05–1.70 <0.0001
aDose-AUC relationships of CBD were fitted to log(AUC)=log(a)+b· log(dose).
CI, confidence interval; SE, standard error.
FIG. 3. Plasma CBD concentration–time curves after oral administration of CBD-NE at CBD doses of 10 mg/kg(A)and 50 mg/kg(B)to rats, 1 h after oral administration of ketoconazole. Each symbol with vertical bars represents the mean–SEM (n=4).
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
the13C-breath response. The use of13C-labeled probes for assessment ofin vivometabolic activity has been ex- tensively studied by our research group.16,21–23In par- ticular, we have demonstrated the utility of the13C-EM breath test for the quick and noninvasive evaluation of CYP3A-mediated drug interactions in vivo.16 As summarized in Table 4, the AUCs of the 13C breath response decreased by approximately half in the CBD- NE coadministration groups (10 and 50 mg/kg) com- pared with the control, indicating suppression of
13C-EM metabolism due to CBD-mediated inhibition of CYP3A. In contrast, there was no difference in the
13C-breath response between the control group and the CBD-NE coadministration group at 1 mg/kg CBD (Fig. 4), indicating little or no inhibition of CYP3A ac- tivity caused by the low dose of CBD. The Cmaxvalues of CBD in plasma after oral CBD-NE administration at 10 and 50 mg/kg were determined to be 0.386 and 3.58lg/mL (Table 1), which correspond to molar con- centrations of 1.23 and 11.4lM, respectively. In addi- tion, the Cmax of CBD in plasma at the 1 mg/kg dose was estimated to be *0.12lM, as extrapolated from the 10 mg/kg dose. Therefore, the threshold plasma concentration of CBD required to inhibit the metabo- lism of EM was estimated to be in the range from 0.12 to 1.23lM. This finding is of great interest in regard to in vivo CYP3A inhibition by CBD because this threshold range for inhibition of EM clearance in vivowas similar to the reported Ki value of CBD in- hibition against CYP3Ain vitro(1lM).13
Atsmon et al.24developed a new formulation for CBD by embedding purified CBD in a seamless gelatin matrix and used this formulation in humans. In their study, in- testinal absorption of CBD from the gelatin matrix pellets was found to be faster than that from the conventional oromucosal spray, and the resultant AUCs of CBD at the single oral doses of 10 and 100 mg were determined to be 10.31–4.14 and 153.0–34.7 ng$h/mL (mean–SD;
n=14), respectively,24indicating that the increase in dose resulted in an increase of the AUC in a greater than dose- proportional manner. Therefore, it is suggested that the pharmacokinetic nonlinearity of CBD may also be ob- served in humans when the absorption process does not limit its bioavailability.
There is a clinical report about the interaction be- tween CBD and warfarin,25 in which the prothrombin time-international normalized ratio increased from 2.96 to 6.86 when the CBD dose was titrated from 5 up to 10 mg/kg/day. In this case, CBD may have inhibited CYP3A4 and CYP2C9, which metabolize S- and R-warfarin, respectively, to such an extent that the effects of warfarin were enhanced. In addition, results of the13C-breath test demonstrated that the metabolism of 13C-EM was inhibited by CBD doses of 10 and 50 mg/kg, but not at 1 mg/kg. Collectively, these results suggested that the proposed caution related to CYP3A- mediated drug interaction should be considered at the relatively higher doses of CBD with a CYP3A substrate.
There is another clinical case report of the drug in- teraction between tacrolimus and CBD.26 Tacrolimus is known to be metabolized by CYP3A4 and CYP3A5 in humans and its pharmacokinetics could be affected
Table 3. Effect of Ketoconazole on Pharmacokinetic Parameters of Nanoemulsion Formulation
of Cannabidiol in Ratsa
Parameter
Dose of CBD-NE
10 mg/kg pb 50 mg/kg pb AUC0–8(mg$h/L) 3.27–0.56 0.0287* 11.4–1.2 0.497 AUC0–8/dose (h$kg/L) 0.327–0.056 0.0287* 0.228–0.024 0.497 AUC0–N(mg$h/L) 3.54–0.60 0.0240* 14.2–0.8 0.460 AUC0–N/dose (h$kg/L) 0.354–0.060 0.0240* 0.283–0.016 0.460 T1/2(h) 2.56–0.45 0.0859 4.06–1.59 0.722 CLtot/F (L/h/kg) 3.28–0.69 0.0312* 3.58–0.21 0.342 Vdss/F (L/kg) 9.85–2.13 0.0525 21.6–5.1 0.578 Tmax(h) 1.50–0.25 0.134 2.25–0.54 0.628 Cmax(lg/mL) 1.18–0.18 0.0126* 2.49–0.31 0.363 Cmax/dose (kg/L) 0.118–0.018 0.0126* 0.0497–0.0061 0.363
MRT (h) 3.03–0.31 0.668 6.24–1.66 0.732
aEach value is presented as the mean–SEM (n=4).
bCompared with CBD-NE at corresponding doses of 10 and 50 mg/kg without administration of ketoconazole (Table 1).
*p<0.05.
FIG. 4. Effect of CBD on the time profiles of breath response [D13CO2(&) values] after oral administration of
13C-EM. CBD-NE (1, 10, and 50 mg/kg) or NE vehicle without CBD (as a control) was orally administered, followed by oral administration of13C-EM (200 mg/kg) after 90 min. Each value with vertical bars is expressed as the mean–SEM (n=3).13C-EM,13C-erythromycin.
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
by CYP3A5 genotypes.27CBD has been reported to in- hibit diltiazem N-demethylase activity of both CYP3A4 and CYP3A5, with the in vitro Ki values of CBD for these enzymes being 1 and 0.195lM, respectively.13 In contrast, CBD has been reported to have no effect on the plasma concentrations of midazolam, a sensitive substrate of CYP3A4.28 It has been reported that the pharmacokinetics of midazolam were not affected by CYP3A5 genotypes.29,30 Therefore, considering the lower Ki value of CBD for CYP3A5 compared with CYP3A4, it could be speculated that the risk of interac- tions between CBD and CYP3A substrates increases when the contribution of CYP3A5 is relatively higher than that of CYP3A4.
Nevertheless, the clinical impact of drug interactions should be evaluated not only by pharmacokinetic fac- tors but also by pharmacodynamic factors because the drug concentration in plasma is hazardous only when it reaches the threshold required to elicit adverse effects. Overlapping effects of CBD with other drugs may also contribute to CBD-induced drug interactions, such as for antiepileptic effects.31 Moreover, 6a- and 6b-hydroxy metabolites of CBD were also reported to have the inhibitory effect on CYP3A using the mouse liver microsome.32There is a possibility that similar in- hibition could also be caused by CBD metabolites in humans, but no such evidence is available to our best knowledge.
Conclusions
In this study, the nonlinear pharmacokinetics and drug interactions of CBD were demonstrated for the first time in vivo in rats, which could also be manifested when a rapidly absorbable formulation of CBD was ad- ministered to humans. Moreover, it was proposed that caution for dose-dependent drug interactions should be considered with CBD, although pharmacodynamic considerations are also required to assess the clinical importance of drug interactions.
Acknowledgments
This work was supported by Showa University and Otsuka Pharmaceutical Co., Ltd. The authors wish to thank ENDOCA for kindly supplying the pure CBD crystals for the research.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research received no specific grant from any fund- ing agency in the public, commercial, or not-for-profit sectors.
References
1. World Health Organization Expert Committee on Drug Dependence.
Cannabidiol (CBD) Pre-review Report Agenda Item 5.2 and Peer Review.
World Health Organization: Geneva, Switzerland, 2017.
2. Laprairie RB, Bagher AM, Kelly MEM, et al. Cannabidiol is a negative al- losteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. 2015;
172:4790–4805.
3. Russo EB, Burnett A, Hall B, et al. Agonistic properties of cannabidiol at 5- HT1a receptors. Neurochem Res. 2005;30:1037–1043.
4. Kathmann M, Flau K, Redmer A, et al. Cannabidiol is an allosteric modu- lator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:354–361.
5. Bisogno T, Hanus L, Petrocellis LD, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol.
2001;134:845–852.
6. Laun AS, Shrader SH, Brown KJ, et al. GPR3, GPR6, and GPR12 as novel molecular targets: their biological functions and interaction with canna- bidiol. Acta Pharmacol Sin. 2019;40:300–308.
7. Ryberg E, Larsson N, Sjo¨gren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101.
8. Campos AC, Moreira FA, Gomes FV, et al. Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Phil Trans R Soc B. 2012;367:3364–3378.
9. Rimmerman N, Ben-Hail D, Porat Z, et al. Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: a novel mechanism for cannabinoid-induced cell death. Cell Death Dis. 2013;4:471–481.
10. Bruni N, Pepa CD, Oliaro-Bosso S, et al. Cannabinoid delivery systems for pain and inflammation treatment. Molecules. 2018;23:2478–2503.
11. Pavlovic R, Nenna G, Calvi L, et al. Quality traits of ‘‘cannabidiol oils’’: can- nabinoids content, terpene fingerprint and oxidation stability of European commercially available preparations. Molecules. 2018;23:1230–1251.
12. Jiang R, Yamaori S, Takeda S, et al. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver mi- crosomes. Life Sci. 2011;89:165–170.
Table 4. 13C-Breath Response (Area Under the Curve) After Oral Administration of Nanoemulsion without Cannabidiol (Control) and Nanoemulsion Formulation of Cannabidiola
Parameter Control
Dose of CBD-NE
1 mg/kg 10 mg/kg 50 mg/kg
AUC0–60(&$min) 566.0–36.0 518.0–11.5 349.9**–28.3 237.4**–37.5
AUC0–120(&$min) 1274.5–83.3 1116.0–52.3 751.9*–37.4 600.4**–110.3
AUC0–180(&$min) 1862.5–100.2 1660.0–86.9 1142.4*–52.7 970.4**–207.1
aEach value is presented as the mean–SEM (n=3).
*p<0.05 and **p<0.01 versus control.
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.
13. Yamaori S, Ebisawa J, Okushima Y, et al. Potent inhibition of human cy- tochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011;88:730–736.
14. Nakano Y, Tajima M, Sugiyama E, et al. Development of a novel nanoe- mulsion formulation to improve intestinal absorption of cannabidiol. Med Cannabis Cannabinoids. 2018;2:35–42.
15. Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–1283.
16. Sugiyama E, Kikuchi A, Inada M, et al. The use of13C-erythromycin as an in vivoprobe to evaluate CYP3A-mediated drug interactions in rats.
J Pharm Sci. 2011;100:3995–4005.
17. Kenji T. Kyoto University graduate school pharmacy graduate course clinical condition information pharmacy. Available at www.pharm.kyoto-u .ac.jp/byoyaku/Kinetics/download.html (last accessed November 13, 2019).
18. Takano R, Furumoto K, Shiraki K, et al. Rate-limiting steps of oral ab- sorption for pooly water-soluble drugs in dogs; prediction from a minis- cale dissolution test and a physiologically-based computer simulation.
Pharm Res. 2008;25:2334–2344.
19. Willmann S, Schmitt W, Keldenich J, et al. A physiological model for the estimation of the fraction dose absorbed in humans. J Med Chem. 2004;
47:4022–4031.
20. Brzozowska N, Li KM, Wang XS, et al. ABC transporters P-gp and Bcrp do not limit the brain uptake of the novel antipsychotic and anticonvulsant drug cannabidiol in mice. Peer J. 2016;4:2081–2097.
21. Tobita K, Inada M, Sato A, et al. Estimation of gastric pH in cynomolgus monkeys, rats, and dogs using [13C]-calcium carbonate breath test. Dig Liver Dis. 2016;48:1035–1040.
22. Inada M, Kunizaki J, Tobita K, et al. Calcium [13C]carbonate breath test for quantitative measurement of total gastric acid in rats. Scand J Gastro- enterol. 2012;47:148–154.
23. Sugiyama E, Inada M, Kunizaki J, et al. Desirable pharmacokinetic prop- erties of13C-uracil as a breath test probe of gastric emptying in com- parison with13C-acetate and13C-octanoate in rats. Scand J Gastroenterol.
2009;44:1067–1075.
24. Atsmon J, Heffetz D, Deutsch L, et al. Single-dose pharmacokinetics of oral cannabidiol following administration of PTL101: a new formulation based of gelatin matrix pellets technology. Clin Pharmacol Drug Dev.
2018;7:751–758.
25. Grayson L, Vines B, Nichol K, et al. An interaction between warfarin and cannabidiol, a case report. Epilepsy Behav Case Rep. 2018;9:10–11.
26. Leino AD, Emoto C, Fukuda T, et al. Evidence of a clinically significant drug-drug interaction between cannabidiol and tacrolimus.
Am J Transplant. 2019;19:2944–2948.
27. Marfo K, Altshuler J, Lu A. Tacrolimus pharmacokinetic and pharmaco- genomic differences between adults and pediatric solid organ transplant recipients. Pharmaceutics. 2010;2:291–299.
28. Morrison G, Taylor L, Crockett J, et al. A phase 1 investigation into the potential effects of cannabidiol on CYP3A4-mediated drug-drug inter-
actions in healthy volunteers. In: Presented at the The American Epilepsy Society Annual Meeting; November 30 to December 4, 2018; New Orleans, LA.
29. Yu KS1, Cho JY, Jang IJ, et al. Effect of the CYP3A5 genotype on the pharmacokinetics of intravenous midazolam during inhibited and in- duced metabolic states. Clin Pharmacol Ther. 2004;76:104–112.
30. Fromm MF, Schwilden H, Bachmakov I, et al. Impact of the CYP3A5 genotype on midazolam pharmacokinetics and pharmacodynamics during intensive care sedation. Eur J Clin Pharmacol. 2007;63:1129–
1133.
31. Klein P, Tolbert D, Gidal BE. Drug–drug interactions and pharmacody- namics of concomitant clobazam and cannabidiol or stiripentol in re- fractory seizures. Epilepsy Behav. 2019;99:106459.
32. Bornheim LM, Everhart ET, Li J, et al. Characterization of cannabidiol- mediated cytochromeP450 inactivation. Biochem Pharmacol. 1993;45:
1323–1331.
Cite this article as:Nagao M, Nakano Y, Tajima M, Sugiyama E, Sato VH, Inada M, Sato H (2020) Nonlinear disposition and metabolic in- teractions of cannabidiol through CYP3A inhibitionin vivoin rats, Cannabis and Cannabinoid ResearchX:X, 1–8, DOI: 10.1089/
can.2019.0098.
Abbreviations Used ANOVA¼analysis of variance
AUC¼area under the curve CBD¼cannabidiol
CBD-NE¼nanoemulsion formulation of CBD
13C-EM¼13C-erythromycin CI¼confidence interval CLtot/F¼total clearance
Cmax¼maximum plasma concentration CMC¼carboxymethyl cellulose
KCZ¼ketoconazole
LC-MS/MS¼liquid chromatography-tandem mass spectrometry MRT¼mean residence time
SE¼standard error
SEM¼standard error of the mean T1/2¼terminal half-life
Tmax¼time to reach Cmax
Vdss/F¼volume of distribution
Downloaded by SHOWA UNIVERSITY from www.liebertpub.com at 03/04/20. For personal use only.