目次
1 背景及び概観 ... 4 1.1 製剤開発の概要 ... 4 1.2 1.CG.03.SUM.0443 及び RDT.07.SRE.27013 試験の血中メトロニダゾール濃度 の分析方法の概要 ... 6 1.2.1 1.CG.03.SUM.0443 試験の血中メトロニダゾール濃度の分析方法 ... 6 1.2.2 RDT.07.SRE.27013 試験の血中メトロニダゾール濃度の分析方法 ... 7 1.2.3 血漿中及び血清中薬物濃度の差異の検討 ... 7 2 個々の試験結果の要約 ... 9 3 全試験を通しての結果の比較と解析 ... 9 4 付録 ... 9表目次
表 1 臨床試験で使用した製剤の一覧 ... 5 表 2 メトロニダゾールゲル 0.75%と Metrogel 0.75% [Bioglan]の組成の違い ... 5 表 3 1.CG.03.SUM.0443 試験の血中メトロニダゾール濃度の分析方法 - バリデーション 成績のまとめ ... 6 表 4 RDT.07.SRE.27013 試験の血中メトロニダゾール濃度の分析方法 - バリデーショ ン成績のまとめ ... 7略号一覧
略称・略号 省略していない表現又は定義1
背景及び概観
1.1
製剤開発の概要
ガルデルマ社は、現在、有効成分メトロニダゾールを 0.75% 含有するゲル製剤として、2 つの製 品(Rozex gel 及びMetrogel [Bioglan] 1)を販売している。
臨床試験で使用した製剤の一覧を表 1 に示した。メトロニダゾールゲル 0.75% (Rozex gel、市 販予定製剤、以下、「本剤」)と Metrogel [Bioglan] の組成の違いを表 2 に示した。
Metrogel [Bioglan]は、英国において「がん性悪臭の軽減」で承認をされている製剤であり、 この製剤は、Bioglan Laboratory Ltd 社(Bioglan 社)により開発が行われた。酒さの治療薬と して 1989 年にアイルランドで 1991 年に英国で承認を取得した。その後、1994 年にがん性悪 臭の軽減の追加適応症の承認を英国で取得した。 年に Metrogel [Bioglan]の販売権がガル デルマ社に移管された以後は、ガルデルマ社が英国のみで 2 つの適応症で Metrogel [Bioglan] の販売を行っている。表 2 に示した通り、Metrogel [Bioglan] には Bronopol 及び Nipastat が含 まれており、これらは本邦では新添加物に該当し品質および非臨床に関する追加のデータが 必要とされる。
本剤は、欧州地域において 1991 年に最初に酒さの治療薬として承認を取得し、欧州諸国にお ける直近の承認は 2003 年である。ガルデルマ社は、世界的に本剤の販売を精力的に継続して いる。また、本剤に含まれる添加物は、本邦における公定書規格に合致するものと考えられ る(表 2 参照)。 年 月 日に行われた独立行政法人医薬品医療機器総合機構 (PMDA)との治験相談において、ガルデルマ社は、承認申請に必要なデータの質および将 来の安定供給の観点より、本邦におけるがん性悪臭の軽減を適応症とした申請予定製剤とし て、本剤が適切であることを提案し、PMDA より、今後、国内において本剤による患者を対 象とした臨床試験を実施するのであれば、本剤を本邦における申請予定製剤とすることは受 け 入 れ 可 能 と の 見 解 が 示 さ れ た 。 し た が っ て 、 本 邦 に お い て 本 剤 に よ る 臨 床 試 験 (RDT.07.SRE.27013)を実施し、市販予定製剤として申請をする。 1“Metrogel”は、Bioglan 社で開発をされた英国での製品名ですが、ガルデルマ社でも Rozex Gel との同等の製品を“METROGEL”というブランド名で一部の国々で販売しているため、混 乱を避けるために本申請資料では、Bioglan 社で開発をされた製剤については “Metrogel
表 1 臨床試験で使用した製剤の一覧 商品名 適応症 臨床試験 Metrogel 0.75% [Bioglan] 酒さの急性炎症性増悪の治療 がん性悪臭の軽減 Metrogel 037 試験 メトロニダゾールゲル 0.75% 酒さの治療 RDT.07.SRE.27010 試験 1.CG.03.SUM.0461 試験 a 1.CG.03.SUM.0447 試験 a 1.CG.03.SUM.0443 試験 a RDT.07.SRE.27013 試験 a 処方変更前製剤[カルボキシビニルポリマー(ベンゼン含有)による製剤(表 2 を参照)]を使用した試験 表 2 メトロニダゾールゲル 0.75%とMetrogel 0.75% [Bioglan]の組成の違い メトロニダゾールゲル0.75% Metrogel 0.75% [Bioglan] 機能 日本の 公定書 の収載 成分 濃度 (% w/w) 成分 濃度 (% w/w) メトロニダゾール メトロニダゾール 主薬 あり エデト酸ナトリウム水和物 キレート剤 あり カルボキシビニルポリマーa ゲル化剤 あり ヒドロキシエチルセル ロース ゲル化剤 あり プロピレングリコール プロピレングリコール 湿潤剤 /溶剤 あり パラオキシ安息香酸メチル 保存剤 あり パラオキシ安息香酸プロピル 保存剤 あり Bronopol b 保存剤 なし Nipastat c 保存剤 なし 水酸化ナトリウム pH 調整剤 あり リン酸 pH 調整剤 なし 精製水 精製水 基剤 あり 総量 100.00 総量 100.00 a カルボキシビニルポリマー (ベンゼンフリー)。海外の臨床試験(1.CG.03.SUM.0461, 1.CG.03.SUM.0447 及び 1.CG.03.SUM.0443)では、処方変更前製剤[カルボキシビニルポリマー(ベンゼン含有)による製剤]を使用し た(詳細は [2.3.P.2.2 ] 参照)。 b Bronopol: 2-ブロモ-2-ニトロプロパン-1,3-ジオール c Nipastat: パラオキシ安息香酸メチル、パラオキシ安息香酸エチル、パラオキシ安息香酸プロピル及びパラオキ シ安息香酸ブチルのプレミックス品
1.2
1.CG.03.SUM.0443 及びRDT.07.SRE.27013 試験の血中メトロニダゾール濃
度の分析方法の概要
1.2.1 1.CG.03.SUM.0443 試験の血中メトロニダゾール濃度の分析方法
バリデーション成績のまとめを表 3 に示す。 表 3 1.CG.03.SUM.0443 試験の血中メトロニダゾール濃度の分析方法 - バリデーション成績のまとめ 測定物質 血清中メトロニダゾール 測定方法 紫外吸収検出を用いた逆相高速液体クロマトグラフィー 測定施設 (米国) 経皮投与 経口投与 特異性 測定物質を含まないヒト血清による干渉 は観察されない 測定物質を含まないヒト血清による干渉 は観察されない 直線性 検量線の直線性の範囲:12-150 ng/mL 検量線の直線性の範囲:200-15 000 ng/mL 真度及び精度 併行精度 精度範囲:1.5 - 5.0 % 真度範囲:99.9 - 101.5 % 併行精度 精度範囲:1.3 - 2.6% 真度範囲:98.6 - 101.2 % 室内再現精度 精度範囲:1.0 - 6.4 % 真度範囲:100.4 - 102.2 % 室内再現精度 精度範囲:0.6 -3.8 % 真度範囲:98.5 - 101.0 % 定量下限値 9.6 ng/mL 200 ng/mL 検量線の範囲 12 - 150 ng/mL 200 - 15 000 ng/mL 出典: 分析法検討報告書 003-A (1.CG.03.SUM.0443 試験 治験総括報告書 [5.3.3.1.4], p 463 – 502) 分析法検討報告書 003-B (1.CG.03.SUM.0443 試験 治験総括報告書 [5.3.3.1.4], p.312 – 351)1.2.2 RDT.07.SRE.27013 試験の血中メトロニダゾール濃度の分析方法
バリデーション成績のまとめを表 4 に示す。 表 4 RDT.07.SRE.27013 試験の血中メトロニダゾール濃度の分析方法 - バリデーション成績のまとめ 測定物質 血漿中メトロニダゾール 測定方法 タンデム質量分析付き高速液体クロマトグラフィー測定施設 Galderma R and D, Sophia-Antipolis (フランス)
測定範囲 2 - 100 ng/mL 特異性 有意な干渉はない 持ち越し 持ち越しは観察されない 真度及び精度 併行精度 定量下限値付近:平均バイアスは11.7%、精度 11.8% 定量下限値以上:平均バイアス範囲は8.7 - 11.7%、精度 3.9 – 7.1 % 室内再現精度 定量下限値付近:平均バイアスは -1.6%、精度 16.1% 定量下限値以上:平均バイアス範囲は2.3 – 10.0%、精度 5.0 – 10.0 % 希釈率 1/10 平均回収率 メトロニダゾール: 83.1% メトロニダゾール-d4 (内部標準物質): 86.4% マトリックス効果 マトリックス効果 低濃度:94.4% (CV = 4.1%) 高濃度:100.8% (CV = 4.9%) 溶血によるマトリックス効果 低濃度:100.6% (CV = 2.2%) 高濃度:103.0% (CV = 1.9%) 高脂質血によるマトリックス効果 低濃度:100.4% (CV = 1.7%) 高濃度: 98.3% (CV = 1.9%) 出典:分析法検討報告書RDS.03.VRE.34308 [5.3.1.4.1]
1.2.3
血漿中及び血清中薬物濃度の差異の検討
1.CG.03.SUM.0443 試験では血清を用いて、RDT.07.SRE.27013 試験では血漿を用いてメトロニダ ゾールの全身吸収を検討した。メトロニダゾールの血清蛋白質結合率はそれほど高くないとの文 献報告(Ralph et al 1974 [5.4.29])、及び平衡透析法により求められた血清蛋白質結合率が血中濃 度 1 μg/mL の場合に 8.1%、血中濃度 10 μg/mL の場合に 11.2%とのフラジール® の添付文書の記 載(Schwartz, 1976 [5.4.31])から、ガルデルマ社は 2 種の検体によるメトロニダゾールの血中濃 度に差異はないと考えている。メトロニダゾールの血清蛋白質結合率がそれほど高くないことは、Kaye らによっても確認され、 健康成人にフラジール®
400mg を単回経口投与した後に、異なる生体試料(血漿、唾液、血清、 全血)を用いて同一の方法により測定をしたメトロニダゾール濃度の時間・推移は、血清および 血漿で差異が認められなかったことが報告している(Kaye et al 1980 [5.4.25])。
2
個々の試験結果の要約
バイオアベイラビリティ及び生物学的同等性試験は実施していない。3
全試験を通しての結果の比較と解析
該当しない。4
付録
該当しない。目次
1 背景及び概観 ... 5 2 個々の試験結果の要約 ... 6 2.1 臨床薬理試験 ... 6 2.1.1 メトロニダゾールゲル 0.75%の健康被験者を対象とした単回貼布及び 光貼布試験による皮膚安全性の検討:国内第 1 相試験 (RDT.07.SRE.27010 試験) ... 6 2.1.1.1 目的 ... 6 2.1.1.2 試験内容 ... 6 2.1.1.3 結果 ... 7 2.1.1.4 結論 ... 72.1.2 Modified Draize Skin Sensitisation Study with Metronidazole 0.75% Gel in Human Subjects:1.CG.03.SUM.0461 試験 ... 8
2.1.2.1 目的 ... 8
2.1.2.2 試験内容 ... 8
2.1.2.3 結果 ... 8
2.1.2.4 結論 ... 9
2.1.3 Evaluation of Contact Sensitisation Potential of MetroCream 0.75%, MetroCream Vehicle, MetroGel® 0.75% and MetroGel® Vehicle following Repeated Applications to the Skin of Humans:1.CG.03.SUM.0447 試験 ... 10
2.1.3.1 目的 ... 10
2.1.3.2 試験内容 ... 10
2.1.3.3 結果 ... 10
2.1.3.4 結論 ... 12
2.2 薬物動態試験 ... 13
2.2.1 Pharmacokinetic/Bioavailability evaluation of topically administered metronidazole cream, 0.75% and metronidazole lotion 0.75% in healthy adult volunteers:1.CG.03.SUM.0443 試験 ... 13 2.2.1.1 目的 ... 13 2.2.1.2 試験内容 ... 13 2.2.1.3 結果 ... 13 2.2.1.4 結論 ... 15 2.2.2 メトロニダゾールゲル 0.75%のがん性皮膚潰瘍の悪臭に対する治療に おける安全性及び有効性に関する 14 日間のオープン試験:国内第 3 相試験(RDT.07.SRE.27013 試験)... 16 2.2.2.1 目的 ... 16 2.2.2.2 試験内容 ... 16 2.2.2.3 結果 ... 17 2.2.2.4 結論 ... 17
3 全試験を通しての結果の比較と解析 ... 18
3.1 臨床薬理試験 ... 18
3.2 薬物動態 ... 18
4 特別な試験 ... 20
表目次
表 1 ヒト生体試料を用いた非臨床試験の一覧 ... 5 表 2 臨床薬理試験及び薬物動態試験の一覧 ... 5 表 3 パッチテスト判定基準 ... 6 表 4 光パッチテスト判定基準 ... 7 表 5 皮膚刺激指数(国内第 1 相試験) ... 7 表 6 皮膚感作性の判定基準 ... 8 表 7 メトロニダゾールゲル 0.75%に対する反応の要約(1.CG.03.SUM.0461 試験) ... 9 表 8 ゲル基剤に対する反応の要約(1.CG.03.SUM.0461 試験) ... 9表 9 North America Contact Dermatitis Group Grading Scale ... 10
表 10 導入期における被験者の反応(1.CG.03.SUM.0447 試験) ... 11 表 11 誘発期における被験者の反応(1.CG.03.SUM.0447 試験) ... 11 表 12 健康被験者にメトロニダゾールを単回局所投与及び単回経口投与したときの メト ロニダゾールの薬物動態パラメータ(1.CG.03.SUM.0443 試験) ... 14 表 13 メトロニダゾールの薬物動態パラメータの要約(国内第 3 相試験) ... 17
図目次
図 1 メトロニダゾールを経口投与(実測値)及び局所投与したときの 血清中メトロニ ダゾール濃度(平均値)の推移(1.CG.03.SUM.0443 試験) ... 14略号一覧
略称・略号 省略していない表現又は定義AUC Area under the serum (or plasma) concentration-time curve
血中濃度-時間曲線下面積 AUC0-24 Area under the serum (or plasma)
concentration-time curve from zero to 24 hours
0~24 時間までの血中濃度-時間曲線下面 積
AUC0-∞ Area under the serum (or plasma)
concentration-time curve from zero to infinity
0~無限大時間までの血中濃度-時間曲線 下面積
Cmax Maximum serum (or plasma) concentration 最高血中濃度
Ctrough Residual concentration
(trough serum [or plasma] concentration)
トラフ値 CV Coefficient of variation 変動係数
HPLC High performance liquid chromatography 高速液体クロマトグラフィー Tlag Latency period 最初に検出されるまでの時間
1
背景及び概観
ヒト生体試料を用いた非臨床試験の一覧を表 1に示す。メトロニダゾール外用剤の非臨床試験と して、拡散セルを用いたメトロニダゾールゲル 0.75%(Rozex® Gel、市販予定製剤)からのメト ロニダゾールの皮膚浸透性を in vitro で検討した([2.6.4.3.1.1 項]を参照)。 表 1 ヒト生体試料を用いた非臨床試験の一覧 試験の種類 試験番号 内容 他のヒト生体試料を用いた試験 RDS.03.SRE.4720 試験 in vitro 試験 メトロニダゾールゲル 0.75%からのメトロニダゾールの皮膚浸透性の 検討 臨床薬理試験及び薬物動態試験の一覧を表 2に示す。メトロニダゾール外用剤の局所安全性を評 価する臨床薬理試験として健康被験者を対象とした 3 試験(RDT.07.SRE.27010 試験、 1.CG.03.SUM.0461 試験及び 1.CG.03.SUM.0447 試験)を実施した。また、健康被験者を対象とし た薬物動態試験 1 試験(1.CG.03.SUM.0443 試験)を実施し、更に、がん性皮膚潰瘍に伴う悪臭 を有する被験者を対象として実施した第 3 相試験(RDT.07.SRE.27013 試験)の一部としてメト ロニダゾールの薬物動態を検討した。 各試験の詳細は2.7.2.2 項に記載した。 表 2 臨床薬理試験及び薬物動態試験の一覧 試験の種類 試験番号 内容 健康被験者における初期安全性、忍容性試験 RDT.07.SRE.27010 試験 日本人対象(国内) 単回パッチテスト及び光パッチテストによる皮膚刺激性及び光毒性の 検討 1.CG.03.SUM.0461 試験 外国人対象(海外) 修正 Draize 皮膚感作試験による刺激性及び感作性の検討 1.CG.03.SUM.0447 試験 外国人対象(海外) 繰り返し投与によるアレルギー性接触皮膚炎(接触感作性)の検討 健康被験者における薬物動態試験 1.CG.03.SUM.0443 試験 外国人対象(海外) メトロニダゾールゲル 0.75%を単回投与したときの血清中メトロニダ ゾール濃度の測定及び薬物動態パラメータの算出(他の剤形[クリー ム、ローション、錠剤]との比較) 有効性及び安全性試験 RDT.07.SRE.27013 試験 日本人対象(国内) 患者における安全性及び有効性の検討 メトロニダゾールゲル 0.75%を 14 日間投与したときの血漿中メトロニ ダゾール濃度の測定及び薬物動態パラメータの算出 注: 国内試験は市販予定製剤、海外試験は処方変更前製剤[カルボキシビニルポリマー(ベンゼン含有)によ2
個々の試験結果の要約
2.1 臨床薬理試験
2.1.1 メトロニダゾールゲル 0.75%の健康被験者を対象とした単回貼布及び光貼布
試験による皮膚安全性の検討:国内第 1 相試験(RDT.07.SRE.27010 試験)
[5.3.3.1-1]2.1.1.1 目的
日本人の健康成人男性被験者を対象として、メトロニダゾールゲル 0.75%(市販予定製剤)を単 回局所投与したときの皮膚刺激性及び光毒性を、ゲル基剤、日本薬局方精製水及びフィンチャン バー(パッチのみ)と比較し、検討した。2.1.1.2 試験内容
本試験は、単一施設、無作為化、評価者盲検、個体内比較の対照比較試験であった。 単回パッチテストでは、各治験薬約 50 µL をフィンチャンバーに塗布し(フィンチャンバーのみ の貼布を除く)、48 時間密閉貼布した。単回パッチテスト部位は、貼布 48 時間後に全治験薬を 除去し、清拭した後、表 3のパッチテスト判定基準(須貝, 1977 [5.4.13])に従って治験薬除去の 60 分後(貼布開始 49 時間後)及び 24 時間後(貼布開始 72 時間後)に同一評価者により判定し た。 光パッチテストでは、各治験薬約 50 µL をフィンチャンバーに塗布し(フィンチャンバーのみの 貼布を除く)、24 時間密閉貼布し、紫外線 A 波(20 J/cm2)を治験薬除去の 60 分後に照射した。 光パッチテスト部位は、貼布開始 24 時間後に全治験薬を除去し、治験薬除去の 60 分後(貼布開 始 25 時間後[紫外線 A 波照射前])及び照射終了の 60 分後に、パッチテスト判定基準(表 3) に従って判定した。光蕁麻疹反応の有無を確認し、紫外線照射部位をフィンチャンバーで遮光し た。治験薬除去の 24 時間後に遮光用に貼布したフィンチャンバーを除去し、その 60 分後(貼布 開始 49 時間後)及び 24 時間後(貼布開始 72 時間後)に同一評価者により判定した。更に、こ の評価の後、光パッチテスト判定基準(表 4)に従って貼布部位の反応と比較し、光蕁麻疹反応 の有無を確認した。 表 3 パッチテスト判定基準 反応 判定 スコア 反応なし (-) 0 軽度な紅斑 (±) 0.5 紅斑 (+) 1 紅斑+浮腫 (++) 2 紅斑+浮腫+丘疹~小水疱 (+++) 3 大水疱 (++++) 4表 4 光パッチテスト判定基準 反応 判定 反応なし又は貼布部位と同等の反応 (-) 貼布部位と比較してわずかに強い反応 (±) 貼布部位と比較して明らかに強い反応 (+) 貼布部位と比較して 2 段階強い反応 (++) 貼布部位と比較して 3 段階強い反応 (+++) 貼布部位と比較して 4 段階強い反応 (++++)
2.1.1.3 結果
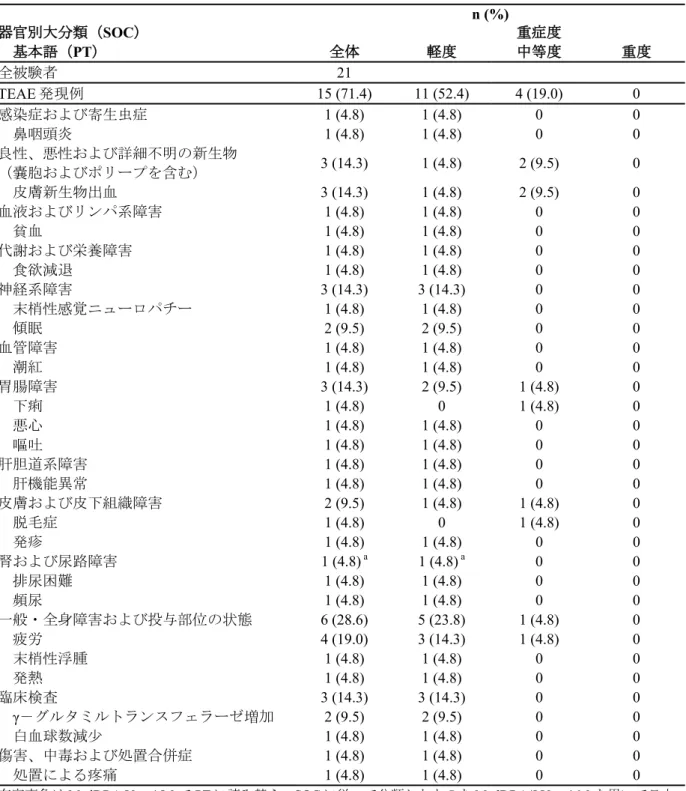
本試験には 20 例が組み入れられ、全 20 例が試験を完了した。被験者は全て日本人男性であり、 被験者の年齢範囲は 20~39 歳であった。 各試験製剤の皮膚刺激指数を表 5に示す。 表 5 皮膚刺激指数(国内第 1 相試験) 試験製剤 皮膚刺激指数 メトロニダゾールゲル 0.75% 10.0 ゲル基剤 10.0 日本薬局方精製水 5.0 フィンチャンバー(パッチのみ) 7.5 皮膚刺激指数 = 除去 60 分後又は 24 時間後の反応の強い方のスコア総和 × 100 被験者数 皮膚刺激スコアの範囲は 0~400 出典: RDT.07.SRE.27010 試験 治験総括報告書 [Table4-5][Section 14](5.3.3.1-1) 光パッチテストで紫外線 A 波照射の 60 分後に光蕁麻疹反応を評価した結果、いずれの治験薬で も光蕁麻疹反応は認められなかった。2.1.1.4 結論
本試験では、メトロニダゾールゲル 0.75%及びゲル基剤には、皮膚刺激性及び光蕁麻疹反応のい ずれも認められなかった。2.1.2 Modified Draize Skin Sensitisation Study with Metronidazole 0.75% Gel in
Human Subjects:1.CG.03.SUM.0461 試験
[5.3.3.1-2]2.1.2.1 目的
健康成人被験者を対象として、修正 Draize 皮膚感作試験によりメトロニダゾールゲル 0.75%(処 方変更前製剤[カルボキシビニルポリマー(ベンゼン含有)による製剤])の刺激性及び感作性 を評価した。2.1.2.2 試験内容
本試験は、修正 Draize 皮膚感作性試験による個体内比較の対照試験であった。 被験者の上腕又は背部に、メトロニダゾールゲル 0.75%又はゲル基剤 0.2 g を含有した検査用 パッチを密閉貼布した。3 週間の導入期には、各被験者は 1 週間で 3 回、同じ部位にパッチを貼 り換えた(パッチは各部位に 48~72 時間貼布し続けた)。導入期の後、2 週間パッチを外した状 態にした。この 2 週間の休薬期間の後、誘発期として各被験者に被験物質を含むチャレンジパッ チを 72 時間貼布した。貼布開始 96 時間後に、各チャレンジ部位のスコアを表 6に従って評価し た。 表 6 皮膚感作性の判定基準 スコア 説明 G 「橙皮状皮膚(peau d’orange)」のような最小限の光沢を帯びた状態 0 陰性 ± 曖昧な反応 1 紅斑 2 紅斑及び硬結 3 紅斑、硬結及び小水疱 4 紅斑、硬結及び大水泡2.1.2.3 結果
本試験には合計 214 例が組み入れられ、そのうちの 24 例は試験を中止した。被験者は男性が 76 例、女性が 138 例であり、被験者の年齢範囲は 18~83 歳であった。 メトロニダゾールゲル 0.75%及びゲル基剤の皮膚感作性の結果を表 7及び表 8に示す。 導入期又は誘発期において、1 以上のスコアを示した被験者はいなかった。表 7 メトロニダゾールゲル 0.75%に対する反応の要約(1.CG.03.SUM.0461 試験) 導入期(n) 誘発期(n) スコア 1 回目 2 回目 3 回目 4 回目 5 回目 6 回目 7 回目 8 回目 9 回目 72 時間後 96 時間後 0 204 200 196 192 189 188 188 188 186 187 190 ± 0 0 0 2 3 3 3 3 5 3 0 1 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 0 0 0 合計 204 200 196 194 192 191 191 191 191 190 190
出典: 1.CG.03.SUM.0461 試験 治験総括報告書 [SUMARY OF RESPONSE TO SITE#1](5.3.3.1-2)
表 8 ゲル基剤に対する反応の要約(1.CG.03.SUM.0461 試験) 導入期(n) 誘発期(n) スコア 1 回目 2 回目 3 回目 4 回目 5 回目 6 回目 7 回目 8 回目 9 回目 72 時間後 96 時間後 0 203 200 196 193 191 190 189 188 189 190 190 ± 0 0 0 1 1 1 2 3 2 0 0 1 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0 0 0 0 0 0 0 合計 203 200 196 194 192 191 191 191 191 190 190
出典: 1.CG.03.SUM.0461 試験 治験総括報告書 [SUMARY OF RESPONSE TO SITE#2](5.3.3.1-2)
2.1.2.4 結論
本試験の条件下では、メトロニダゾールゲル 0.75%又はゲル基剤のいずれでもアレルギー性接触 皮膚炎を示すエビデンスは認められなかった。
2.1.3 Evaluation of Contact Sensitisation Potential of MetroCream 0.75%,
MetroCream Vehicle, MetroGel
®0.75% and MetroGel
®Vehicle following Repeated
Applications to the Skin of Humans:1.CG.03.SUM.0447 試験
[5.3.3.1-3]
2.1.3.1 目的
健康被験者を対象として、メトロニダゾールゲル 0.75%(処方変更前製剤[カルボキシビニルポ リマー(ベンゼン含有)による製剤])、ゲル基剤、メトロニダゾールクリーム 0.75%、及びク リーム基剤がアレルギー性接触皮膚炎を誘発する可能性があるか否かを評価した。 本試験で使用した MetroGel®はメトロニダゾールゲル 0.75%と同じ製剤である。2.1.3.2 試験内容
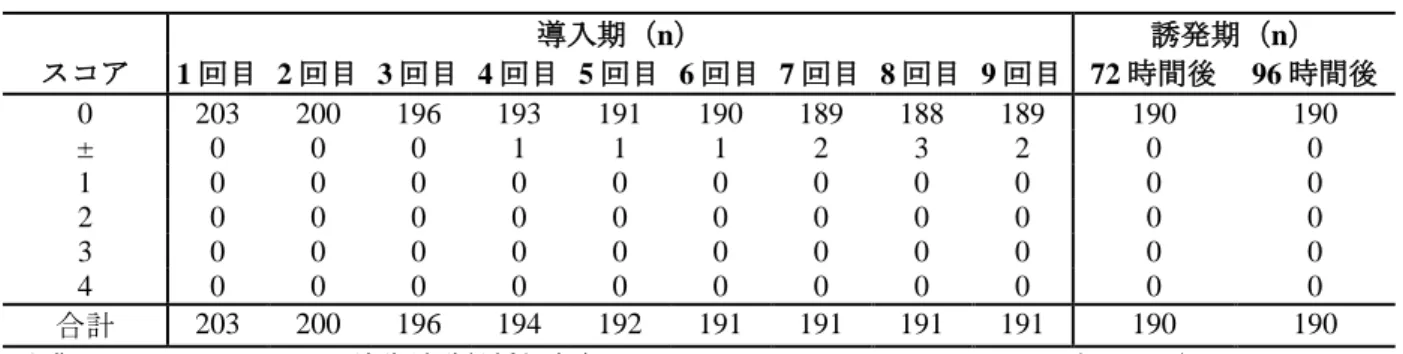
本試験は、個体内比較の二重盲検試験であった。 メトロニダゾールゲル 0.75%、ゲル基剤、メトロニダゾールクリーム 0.75%、及びクリーム基剤 の 4 つの製剤の皮膚感作性を評価するため、各治験薬 200 μL を含有する検査用パッチを繰り返 し密閉貼布した。各被験者は 1 週間に 3 回、3 週間パッチを貼布した。導入期の間は、パッチを 48 時間貼布し、パッチを除去した後に貼布部位を評価した。2 週間の休薬期間の後、誘発期とし て各被験者にチャレンジパッチを 48 時間貼布し、貼布開始 48 及び 96 時間後に各チャレンジ部 位を表 9に従って評価した。表 9 North America Contact Dermatitis Group Grading Scale スコア 説明 0 反応なし +0.5 斑状紅斑 +1 硬結性紅斑 +2 紅斑、浸潤及び小水疱 +3 水疱性の反応又は潰瘍
2.1.3.3 結果
本試験には 124 例が組み入れられ、104 例が試験を完了した。被験者は、男性が 29 例、女性が 95 例であり、年齢範囲は 18~85 歳(平均年齢:43 歳)であった。 導入期における各試験製剤による反応の程度を表 10に示す。表 10 導入期における被験者の反応(1.CG.03.SUM.0447 試験) 試験製剤 紅斑グレードの範囲 メトロニダゾールゲル 0.75% 0~+1 ゲル基剤 0~+1 メトロニダゾールクリーム 0.75% 0~+1 クリーム基剤 0~+0.5 出典: 1.CG.03.SUM.0447 試験 治験総括報告書 [Table 4](5.3.3.1-3) 誘発期(チャレンジパッチを貼布した後)における各試験製剤による反応の程度を表 11 に示す。 48 時間後又は 96 時間後の評価では、紅斑グレードが+1 以上を示した被験者はいなかった。 表 11 誘発期における被験者の反応(1.CG.03.SUM.0447 試験) メトロニダゾールゲル 0.75% 紅斑グレード 48 時間後の反応(n) (N=106) 96 時間後の反応(n) (N=104) 0 71 83 +0.5 35 21 +1 0 0 +2 0 0 +3 0 0 ゲル基剤 紅斑グレード 48 時間後の反応(n) (N=106) 96 時間後の反応(n) (N=104) 0 69 81 +0.5 37 23 +1 0 0 +2 0 0 +3 0 0 メトロニダゾールクリーム 0.75% 紅斑グレード 48 時間後の反応(n) (N=106) 96 時間後の反応(n) (N=104) 0 77 84 +0.5 29 20 +1 0 0 +2 0 0 +3 0 0 クリーム基剤 紅斑グレード 48 時間後の反応(n) (N=106) 96 時間後の反応(n) (N=104) 0 76 83 +0.5 30 21 +1 0 0 +2 0 0 +3 0 0 出典: 1.CG.03.SUM.0447 試験 治験総括報告書 [Table 6](5.3.3.1-3)を一部改変
2.1.3.4 結論
本試験の条件下では、メトロニダゾールゲル 0.75%、ゲル基剤、メトロニダゾールクリーム 0.75%、又はクリーム基剤のいずれでもアレルギー性接触皮膚炎を示すエビデンスは認められな かった。
2.2 薬物動態試験
2.2.1 Pharmacokinetic/Bioavailability evaluation of topically administered
metronidazole cream, 0.75% and metronidazole lotion 0.75% in healthy adult
volunteers:1.CG.03.SUM.0443 試験
[5.3.3.1-4]2.2.1.1 目的
健康成人被験者を対象として、メトロニダゾールクリーム 0.75%及びメトロニダゾールローショ ン 0.75%を局所投与したときの薬物動態を経口剤であるメトロニダゾール錠 250 mg 及び海外で 販売されているメトロニダゾールゲル 0.75%(処方変更前製剤[カルボキシビニルポリマー(ベ ンゼン含有)による製剤])を投与したときと比較した。2.2.1.2 試験内容
本試験は、単回投与による非盲検、無作為化、4 群クロスオーバー法による試験デザインにより 実施した。 メトロニダゾールクリーム 0.75%、メトロニダゾールローション 0.75%、又はメトロニダゾール ゲル 0.75%の約 1 g(メトロニダゾールとして約 7.5 mg)を正確に量り取った後,各被験者は治 験責任医師の前で顔全体にメトロニダゾールの外用剤を塗布した。 各被験者は、治験責任医師の前でメトロニダゾール錠 250 mg(経口剤)を 200 mL の水とともに 服用した。 血液検体(各採血ポイントで 8 mL)は、12 時間後までの採血ポイントではカテーテルにより採 取し、24 及び 48 時間後の採血ポイントでは直接静脈血を採取した。血液検体の採取は、各メト ロニダゾール製剤を投与する直前、並びに、投与 0.25、0.5、1、1.5、2、3、4、6、8、12、24 及 び 48 時間後に行った。 薬物動態パラメータは、ノンコンパートメント法により算出した。薬物動態パラメータとして、 最高血清中濃度到達時間(Tmax)、最初に検出されるまでの時間(Tlag)、最高血清中濃度 (Cmax)、0 から無限大時間又は 24 時間までの血清中濃度-時間曲線下面積(それぞれ AUC0-∞ 及び AUC0-24)を各投与製剤について算出した。2.2.1.3 結果
本試験には 12 例が組み入れられ、メトロニダゾールの 4 つの製剤による全ての治療群に無作為 化された。全 12 例が試験を完了した。被験者の年齢範囲は 24~34 歳(平均年齢:27.8 歳)であ り、男性が 5 例(42%)、女性が 7 例(58%)であった。 血清中メトロニダゾール濃度の平均値の推移(実測値)を図 1に示す。血清中メトロニダゾール濃度は、経口剤では約 1~1.5 時間後に最高値を示し、3 つの外用剤では 約 8~12 時間後に最高値を示した。ベースライン(0 時間)では、いずれも検体でもメトロニダ ゾールは検出されなかった。また、48 時間後における血清中メトロニダゾール濃度は、48 検体 のうちの 40 検体で検出限界未満(< 9.6 ng/mL)であった。 図 1 メトロニダゾールを経口投与(実測値)及び局所投与したときの 血清中メトロニダゾール濃度(平均値)の推移(1.CG.03.SUM.0443 試験) 出典: 1.CG.03.SUM.0443 試験 治験総括報告書 [Figure 4](5.3.3.1-4) メトロニダゾールの外用剤及び経口剤を投与したときの薬物動態パラメータを表 12に示す。 表 12 健康被験者にメトロニダゾールを単回局所投与及び単回経口投与したときの メトロニダゾールの薬物動態パラメータ(1.CG.03.SUM.0443 試験) パラメータ (平均値 ± 標準偏差) メトロニダゾール 0.75%外用剤 メトロニダゾール 経口剤 ゲル クリーム ローション Tlag (h) 0.89 ± 0.64 0.81 ± 0.44 0.99 ± 0.91 0.09 ± 0.17 Tmax (h) 8.51 ± 2.84 10.62 ± 6.82 9.36 ± 2.47 1.51 ± 1.39 Cmax (ng/mL) 29.1 ± 6.7 32.9 ± 10.6 34.4 ± 11.4 7248 ± 3019 AUC0-24 (ng•h/mL) 555.6 ± 124.2 600.0 ± 185.1 634.1 ± 213.1 58504 ± 11724 AUC0-∞ (ng•h/mL) 814.8 ± 251.4 912.7 ± 379.7 971.1 ± 433.6 67207 ± 15380 略号: AUC0-24 = 0~24 時間までの血清中濃度-時間曲線下面積、AUC0-∞ = 0~無限大時間までの血清中濃度-時
間曲線下面積、Cmax = 最高血清中濃度、Tlag = 最初に検出されるまでの時間、Tmax = 最高血清中濃度到達
時間
出典: 1.CG.03.SUM.0443 試験 治験総括報告書 [Table 6]、[Table 7]、[Table 8]、[Table 9]、[Table 10]及び [Table 11](5.3.3.1-4)
2.2.1.4 結論
メトロニダゾールクリーム 0.75%、メトロニダゾールローション 0.75%、及びメトロニダゾール ゲル 0.75% 1 g(それぞれメトロニダゾールとして 7.5 mg)を単回局所投与したときのメトロニ ダゾールの吸収は、メトロニダゾールを経口投与したときよりも低く、Tmaxはより長くなった。 メトロニダゾールクリーム及びメトロニダゾールローションを局所投与したときのメトロニダ ゾールの吸収は、海外で販売されているメトロニダゾールゲルを投与したときと比較して、大き な違いは認められず、メトロニダゾールを経口投与したときよりも明らかに低かった。2.2.2 メトロニダゾールゲル 0.75%のがん性皮膚潰瘍の悪臭に対する治療における
安全性及び有効性に関する 14 日間のオープン試験:国内第 3 相試験
(RDT.07.SRE.27013 試験)
[5.3.5.2-1]2.2.2.1 目的
がん性皮膚潰瘍の悪臭に対する治療におけるメトロニダゾールゲル 0.75%(市販予定製剤)の安 全性及び有効性を検討した。更に、メトロニダゾールゲル 0.75%を 14 日間投与したときの総曝 露量及び定常状態について検討するために血漿中メトロニダゾール濃度を測定し、薬物動態パラ メータを算出した。2.2.2.2 試験内容
本試験は、多施設共同、非盲検、非対照試験であった。 がん性皮膚潰瘍に伴う悪臭を有する被験者を対象として、メトロニダゾールゲル 0.75%を 1 日 1 回~2 回、14 日間投与した。血中メトロニダゾール濃度は、総曝露量を評価するため、予想され る Tmax近辺で測定した。血液検体は、被験者での曝露量のピークを確実に予測するために Tmax 近辺で数回採取するスケジュールとした。血液検体の採取は、メトロニダゾールゲル 0.75%を 7 日間投与した後(Day 7)に 3 回行った。 ● 投与前の採取(Ctroughの測定用)。 ● 投与後 2~8 時間の間に更に 2 回の採取。 血液検体の採取時刻は治験責任(分担)医師の判断により決定した。 更に、定常状態の評価のため Day 14 に採取した(Ctroughの測定用)。 血漿中メトロニダゾール濃度は、バリデーションされた HPLC 法により測定した(定量限界: 2 ng/mL)。 次の薬物動態パラメータを算出した。 Day 7 ● Ctrough(トラフ値[投与前濃度]) ● Cmax(最高血漿中濃度) ● Tmax(最高血漿中濃度到達時間) ●部分的 AUC(T0から最終の採血時間までの台形法により算出した濃度-時間曲線下面積) Day 14 ● Ctrough(トラフ値)2.2.2.3 結果
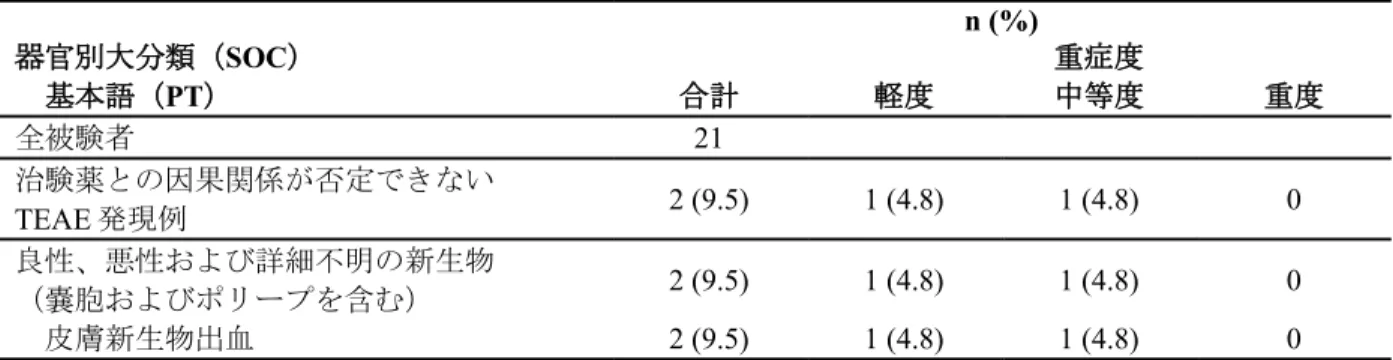
本試験には合計 21 例が組み入れられた。全 21 例に治験薬が投与され、このうちの 20 例が試験 を完了した。被験者の年齢範囲は 39~82 歳(平均年齢:64.4 歳)であり、全例が女性で、乳が ん患者であった。 メトロニダゾールの薬物動態パラメータの要約を表 13に示す。 メトロニダゾールゲル 0.75%を 7 日間投与した後の Cmaxの平均値は 852 ng/mL(範囲:136~ 2872 ng/mL)であり、部分的 AUC の平均値は 2955 ng•h/mL(最終の採血ポイント[2.0~7.2 時 間後]での範囲:382~8373 ng•h/mL)であった。Ctroughの平均値± 標準偏差は Day 7 及び Day 14でそれぞれ 380 ± 281 及び 510 ± 565 ng/mL であった。
Day 7 における Cmaxの変動係数(CV%)は 82%、Ctroughの CV%は 74%、Day 14 における Ctroughの
CV%は 111%と、薬物動態パラメータの CV%が大きく、血漿中メトロニダゾール濃度の被験者 ごとのばらつきが大きいことが示された。これは、被験者ごとの治験薬の投与量が治験責任(分 担)医師の裁量に任されていたこと、採血時間も異なっていたことによると考えられる。 表 13 メトロニダゾールの薬物動態パラメータの要約(国内第 3 相試験) パラメータ 評価日 N 平均値 ± 標準偏差 中央値 最小値, 最大値 Cmax (ng/mL) Day 7 20 852 ± 697 725 136, 2872 Tmax (h) Day 7 20 3.7 ± 1.5 3.3 2.0, 7.2 部分的 AUC (ng•h/mL) Day 7 20 2955 ± 2641 2313 382, 8373 Ctrough (ng/mL) Day 7 20 380 ± 281 375 5, 975 Ctrough (ng/mL) Day 14 20 510 ± 565 303 58, 2410
略号: AUC = 血漿中濃度-時間曲線下面積、Cmax = 最高血漿中濃度、Ctrough = トラフ値、Tmax = 最高血漿中濃度
到達時間 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 12](5.3.5.2-1)
2.2.2.4 結論
メトロニダゾールゲル 0.75%を 14 日間局所投与しがん性皮膚潰瘍の悪臭に対する治療に用いた ときの全身曝露量は低く、個体差が大きかった。メトロニダゾールゲル 0.75%を 7 日間投与した 後の Cmaxの平均値は 852 ng/mL(範囲:136~2872 ng/mL)であった。3
全試験を通しての結果の比較と解析
3.1 臨床薬理試験
国内第 1 相試験(RDT.07.SRE.27010 試験)では、日本人の健康被験者 20 例を対象として、単回 パッチテスト及び光パッチテストを実施した。その結果、メトロニダゾールゲル 0.75%及びゲル 基剤には、皮膚刺激性及び光蕁麻疹反応のいずれも認められなかった(2.7.2.2.1.1 項を参照)。 1.CG.03.SUM.0461 試験では、外国人の健康成人被験者 214 例を対象として、修正 Draize 皮膚感 作試験によりメトロニダゾールゲル 0.75%の刺激性及び感作性を評価した。その結果、メトロニ ダゾールゲル 0.75%又はゲル基剤のいずれにおいてもアレルギー性接触皮膚炎を示すエビデンス は認められなかった(2.7.2.2.1.2 項を参照)。 1.CG.03.SUM.0447 試験では、外国人の健康被験者 124 例を対象として、メトロニダゾールゲル 0.75%及びゲル基剤によりアレルギー性接触皮膚炎を誘発する可能性を評価した。その結果、こ れらの試験製剤のいずれにおいてもアレルギー性接触皮膚炎を示すエビデンスは認められなかっ た(2.7.2.2.1.3 項を参照)。 これら 3 つの試験の結果、メトロニダゾールゲル 0.75%には、皮膚刺激性及び光蕁麻疹反応のい ずれも認められず、アレルギー性接触皮膚炎を示すエビデンスも認められなかった。3.2 薬物動態
メトロニダゾールゲル 0.75%を皮膚潰瘍部位に局所投与したときの全身吸収については、国内第 3 相試験(RDT.07.SRE.27013 試験)で評価した。がん性皮膚潰瘍に伴う悪臭を有する日本人被験 者にメトロニダゾールゲル 0.75%を 7 日間の投与したときの Cmaxの平均値 ± 標準偏差は 852 ± 697 ng/mL(範囲:136~2872 ng/mL)であった(2.7.2.2.2.2 項を参照)。なお、国内第 3 相 試験におけるメトロニダゾールゲル 0.75%の 1 日投与量は、メトロニダゾール錠(250 mg)を経 口投与したときの全身曝露量よりも低いと考えられる 30 g(メトロニダゾールとして 225 mg) までとした。 健康被験者を対象として海外で実施した 1.CG.03.SUM.0443 試験(2.7.2.2.2.1 項を参照)では、メ トロニダゾール 250 mg 錠を単回経口投与した後の Cmaxの平均値は 7248 ng/mL(範囲:4270~ 13970 ng/mL)であり、本試験で見られた全身曝露量は、市販されているメトロニダゾールの経 口製剤で報告されているデータと同様であった。メトロニダゾール 250 mg 及び 500 mg を成人に 単回経口投与したときの Cmaxの平均値は、それぞれ 5.1~6.2 µg/mL(Amon et al, 1978 [5.4.18]、Levison, 1974 [5.4.27]、Ralph et al, 1974 [5.4.29])及び 9~13 µg/mL(Houghton et al, 1979 [5.4.23]、 Mattila et al, 1993 [5.4.28])であった。また、日本人の健康女性被験者 5 例を対象として、メトロ ニダゾール錠 250 mg(フラジール®内服錠)を単回経口投与したときの C
maxの平均値は
1.CG.03.SUM.0443 試験でメトロニダゾール 250 mg を単回経口投与したときの Cmaxの平均値は 7248 ng/mL であり、国内第 3 相試験でメトロニダゾールゲル 0.75%を投与したときに得られた Cmaxの平均値(852 ng/mL)と比較して 8.5 倍高く、国内第 3 相試験で最も曝露量が高かった被験 者の Cmax(2872 ng/mL)よりも 2.5 倍高かった。 以上から、国内第 3 相試験において 1 日 30 g までのメトロニダゾールゲル 0.75%を、4~140 cm2 の大きさの悪臭を伴うがん性皮膚潰瘍に投与したときの全身曝露量は、メトロニダゾール 250 mg を単回経口投与したときを超えることはないことが示された。140 cm2よりも大きい皮膚 潰瘍を有する患者の場合には、1 日に 30 g を超えるメトロニダゾールゲル 0.75%を使用する可能 性が考えられるが、この場合でも、予想される局所投与による全身曝露量は、市販されているメ トロニダゾールの経口製剤での 1 日最大投与量である 2250 mg(750 mg × 3)を投与したときの 全身曝露量を超えることはないと考えられる。
4
特別な試験
特別な試験は実施していない。
5
付録
該当しない。目次
1 背景及び概観 ... 4 1.1 有効性評価の計画 ... 4 1.1.1 有効性評価の対象となった臨床試験の概略 ... 4 1.2 有効性の評価方法 ... 6 1.2.1 評価項目及び評価方法 ... 6 1.2.1.1 国内第 3 相試験(RDT.07.SRE.27013 試験) ... 6 1.2.1.2 海外第 3 相試験(Metrogel 037 試験) ... 8 2 個々の試験結果の要約 ... 9 2.1 国内第 3 相試験(RDT.07.SRE.27013 試験) ... 9 2.1.1 被験者の内訳 ... 10 2.1.2 人口統計学的特性 ... 10 2.1.3 有効性評価 ... 11 2.1.3.1 主要評価項目 ... 11 2.1.3.2 副次評価項目 ... 12 2.1.3.3 その他の評価項目 ... 15 2.2 海外第 3 相試験(Metrogel 037 試験) ... 17 2.2.1 被験者の内訳 ... 17 2.2.2 人口統計学的特性 ... 17 2.2.3 有効性評価 ... 18 2.2.3.1 主要評価項目 ... 18 2.2.3.2 副次評価項目 ... 19 3 全試験を通しての結果の比較と解析 ... 21 3.1 試験対象集団 ... 21 3.1.1 対象及び選択基準・除外基準の比較 ... 21 3.1.2 人口統計学的特性及び他の基準値の特性の比較 ... 21 3.2 全有効性試験の結果の比較検討 ... 22 3.3 部分集団における結果の比較 ... 22 4 推奨用法・用量に関する臨床情報の解析 ... 23 5 効果の持続・耐薬性 ... 23 5.1 効果の持続 ... 23 5.2 耐薬性 ... 23 6 付録 ... 24 6.1 有効性及び安全性試験 ... 24表目次
表 1 臨床的有効性に記載した臨床試験のデザイン上の特徴 ... 5 表 2 においの評価基準 ... 6 表 3 潰瘍部位の臨床所見の評価基準 ... 6 表 4 においの評価基準 ... 8 表 5 潰瘍部位の臨床所見の評価基準 ... 8 表 6 試験計画の要約(国内第 3 相試験) ... 9 表 7 人口統計学的特性及び他の基準値の特性(国内第 3 相試験) ... 10 表 8 医師による Day 14(又は治験中止時)のおいスコア(LOCF 法により欠測値を補完) (国内第 3 相試験) ... 11 表 9 改善率(LOCF 法により欠測値を補完) (国内第 3 相試験) ... 11 表 10 医師、看護師及び被験者によるにおいスコアの要約(国内第 3 相試験) ... 13 表 11 潰瘍部位の臨床所見(国内第 3 相試験)... 14 表 12 VAS による疼痛評価(国内第 3 相試験) ... 15 表 13 細菌学的検査のシフトテーブル(国内第 3 相試験) ... 15 表 14 被験者の QOL の全般改善度(国内第 3 相試験) ... 16 表 15 試験計画の要約(海外第 3 相試験) ... 17 表 16 においスコア(海外第 3 相試験) ... 18 表 17 蜂巣炎スコア(海外第 3 相試験) ... 19 表 18 浸出液スコア(海外第 3 相試験) ... 19 表 19 疼痛(VAS 値のカテゴリ別)(海外第 3 相試験) ... 20 表 20 臨床的有効性及び安全性試験の要約 ... 24図目次
図 1 医師、看護師及び被験者によるにおいスコア(国内第 3 相試験) ... 12略号一覧
略称・略号 省略していない表現又は定義CRC Clinical research coordinator 治験コーディネーター
LOCF Last observation carried forward 欠測値をその前のデータで補完する方法 PMDA Pharmaceuticals and Medical Devices Agency 独立行政法人医薬品医療機器総合機構
QOL Quality of life 生活の質
1
背景及び概観
1.1 有効性評価の計画
1.1.1 有効性評価の対象となった臨床試験の概略
本項では、国内第 3 相試験(RDT.07.SRE.27013 試験)の結果をもとに有効性を評価した。更に、 参考資料として、英国で「がん性悪臭の軽減」に対する適応の承認を取得した際に使用した海外 第 3 相試験(Metrogel 037 試験)の結果についても要約した。 ● 国内第 3 相試験(RDT.07.SRE.27013 試験) RDT.07.SRE.27013 試験は、がん性皮膚潰瘍に伴う悪臭を有する患者を対象として、メトロ ニダゾールゲル 0.75%(Rozex® Gel、市販予定製剤、以下、「本剤」)を 1 日 1 回又は 2 回、 14 日間投与したときの安全性及び有効性を評価した。本試験は国内の 4 つの医療機関で 21 例を対象に実施した。 ● 海外第 3 相試験(Metrogel 037 試験) Metrogel 037 試験は、嫌気性菌の感染又は感染が疑われる悪臭を伴う潰瘍部位を有する患者 を対象として、メトロニダゾールゲル 0.75%(Metrogel® [Bioglan]1)を 1 日 1 回、14 日間投 与したときの有効性を評価した。本試験は英国及びポーランドの 4 つの医療機関で実施した。 各試験のデザイン上の重要な特徴を表 1に示す。 1 “Metrogel®”は、Bioglan 社で開発をされた英国での製品名ですが、ガルデルマ社は本剤(Rozex® Gel)との同等の製品を“METROGEL®”というブランド名で一部の国々で販売しているため、混 乱を避けるために本申請資料では Bioglan 社で開発をされた製剤について “Metrogel® [Bioglan 社]” を使用します。表 1 臨床的有効性に記載した臨床試験のデザイン上の特徴 試験番号 (日本/海外) 目的 試験デザイン 治験薬 投与方法 被験者数 有効性の評価項目 試験報告書 添付場所 第 3 相試験 RDT.07.SRE.27013 (日本) がん性皮膚潰瘍の悪臭に対する 治療におけるメトロニダゾール ゲル 0.75%の安全性及び有効性 を検討する。 多施設共同 非盲検 非対照試験 メトロニダゾールゲル 0.75% (市販予定製剤) 治験薬を投与する前に患部を 十分に清浄し、非粘着性のド レッシング材(ガーゼ、シリ コンガーゼ、創傷被覆・保護 材など)に治験薬を塗り皮膚 潰瘍部位を覆う。必要に応じ て、1 日 1 回から 2 回使用す る(1 日 30 g まで)。 投与期間は 14 日間 21 例 (有効性の評価 対象は 21 例) におい 潰瘍部位の臨床所見 (分泌物の程度及び 性状) 疼痛 細菌学的検査 QOL の全般改善度 [5.3.5.2-1] Metrogel 037 (海外) 嫌気性菌感染による悪臭を伴う 皮膚潰瘍に対するメトロニダ ゾールゲル 0.75%(Metrogel® [Bioglan])の消臭効果を検討す る。 多施設共同 非盲検 非対照試験 メトロニダゾールゲル 0.75% (Metrogel® [Bioglan]) 治験薬を投与する前に皮膚潰 瘍部位を 0.9%生理食塩水で 洗浄し、メトロニダゾールゲ ル 0.75%を薄く塗った滅菌済 みの非接着性シリコンドレッ シング材を皮膚潰瘍部位に直 接適用する(ドレッシング材 は 24 時間そのままにし た)。 14 日間の投与期間中、毎日こ の手順を繰り返す。 48 例 (47 例に治験薬 が投与された。 有効性の評価 対象は 46 例 [Day 14 での有 効性の評価対象 は 43 例]) におい 潰瘍部位の臨床所見 (周囲の蜂巣炎及び 浸出液) 疼痛 細菌学的検査 [5.3.5.2-2]
1.2 有効性の評価方法
1.2.1 評価項目及び評価方法
1.2.1.1 国内第 3 相試験(RDT.07.SRE.27013 試験)
[5.3.5.2-1]1.2.1.1.1 におい
皮膚潰瘍のにおいは、治験責任(分担)医師、看護師(又は CRC)及び被験者が以下の基準 (表 2)に従って評価した。評価時期:スクリーニング、ベースライン(Day 0)、Day 7 及び Day 14(又は治験中止時) 表 2 においの評価基準 スコア 定義 0 においがない 1 においはあるが不快ではない (皮膚潰瘍の近傍[20 cm]でわずかに臭う) 2 軽度に不快なにおい(皮膚潰瘍の近傍で明らかに臭う) 3 中等度に不快なにおい(ベッドサイドで臭う) 4 非常に不快なにおい(部屋に入ると臭う) 本試験では、においの「改善率」を主要評価項目とした。本試験では、においスコアが 2 以上の 被験者を組み入れるため、改善率は治験責任(分担)医師による Day 14(又は治験中止時)のに おいスコアが 0 又は 1 と評価された被験者の割合と定義した。
1.2.1.1.2 潰瘍部位の臨床所見
潰瘍部位の臨床所見は、治験責任(分担)医師が以下の基準(表 3)に従って評価した。 評価時期:スクリーニング、ベースライン(Day 0)、Day 7 及び Day 14表 3 潰瘍部位の臨床所見の評価基準 スコア 定義 分泌物(ドレッシング材の交換頻度) 0 なし 分泌物なし/1 日 1 回ドレッシング材交換 1 軽度 1 日 2 回ドレッシング材交換 2 中等度 1 日 3 回ドレッシング材交換 3 重度 1 日 4 回以上ドレッシング材交換/出血 分泌物の程度は、1 日のドレッシング材の交換頻度により評価し、分泌物の性状(膿性、漿液性 又は血液を含む)を記録した。
1.2.1.1.3 疼痛
皮膚潰瘍に関連した過去 24 時間の痛みについて被験者が評価した。評価は 100 mm の直線 VAS を用いて行った(0 mm:痛みなし~100 mm:最も強い痛み)。
評価時期:スクリーニング、ベースライン(Day 0)、Day 7 及び Day 14
1.2.1.1.4 細菌学的検査
細菌学的検査のために、Day 0 の投与前及び Day 14 のドレッシング材の取換え時に潰瘍部位の最 も炎症と浸出液の強いところ(通常、潰瘍部位の中心)から検体を採取した。検査では、細菌の 同定及び菌数(半定量)を測定した。1.2.1.1.5 被験者のQOLの全般改善度
治験終了時(Day 14)に被験者及び/又はその家族に対して、被験者における満足度質問票の作 成を依頼した。看護師又は医療従事者に対しても、医療従事者における満足度質問票の作成を依 頼した。 満足度質問票の結果に基づき、治験責任(分担)医師は、投与前と比較した被験者の QOL の改 善度を、以下の 5 分類により評価した。 著明改善 改善 やや改善 不変 悪化1.2.1.2 海外第 3 相試験(Metrogel 037 試験)
[5.3.5.2-2]
1.2.1.2.1 におい
においは、3 名の評価者(治験責任医師、看護師及び被験者)が以下の 0~4 で評価した(表 4)。 評価時期:Day 0、Day 7 及び Day 14
表 4 においの評価基準 スコア 定義 0 においがない 1 においはあるが不快ではない 2 軽度に不快なにおい 3 中等度に不快なにおい 4 非常に不快なにおい 本試験では、においを主要評価項目とし、3 名の評価者(治験責任医師、看護師及び被験者)の 合計を「においスコア」とした。
1.2.1.2.2 潰瘍部位の臨床所見
潰瘍部位の臨床所見として、周囲の蜂巣炎及び浸出液を治験責任(分担)医師が以下の 0~3 で 評価した(表 5)。評価時期:Day 0、Day 7 及び Day 14
表 5 潰瘍部位の臨床所見の評価基準 スコア 定義 0 なし 1 軽度 2 中等度 3 重度
1.2.1.2.3 疼痛
被験者は 100 mm の直線 VAS を用いて痛みを評価した(0 mm:痛みなし~100 mm:最も強い痛 み)。評価時期:Day 0、Day 7 及び Day 14
1.2.1.2.4 細菌学的検査
Day 0 及び Day 15 に、治験責任医師が炎症及び浸出液の強い部分から細菌学的検査用の検体を採 取した。
2
個々の試験結果の要約
2.1 国内第 3 相試験(RDT.07.SRE.27013 試験)
[5.3.5.2-1] 日本で実施した RDT.07.SRE.27013 試験の試験計画の要約を表 6に示す。 表 6 試験計画の要約(国内第 3 相試験) 試験の目的 がん性皮膚潰瘍の悪臭に対する治療におけるメトロニダゾールゲル 0.75%の安 全性及び有効性を検討する。 試験デザイン 多施設共同、非盲検、非対照試験 対象 がん性皮膚潰瘍に伴う悪臭を有する患者 主要な選択基準 1. 20 歳以上の男性又は女性。入院患者又は外来患者 2. がん性皮膚潰瘍を有し、感染を示唆する皮膚潰瘍の悪臭を有する者。治験責 任(分担)医師の判定で、においスコア 0~4 のうち、2[軽度に不快なにお い]以上との者 主要な除外基準 1. 現在、抗生剤の全身投与を受けている、又は過去 2 週間以内に投与された者 2. 現在、メトロニダゾールの局所製剤を投与されている、又は全身投与を受け ている、若しくは過去 1 週間以内に投与された者 目標被験者数 約 25 例 治験薬 被験薬 メトロニダゾールゲル 0.75%(市販予定製剤) 治験薬コード :GK567 剤形 :ゲル 濃度 :0.75% 用量 :1 日 30 g まで 投与経路 :局所 投与期間 :14 日間 用法 :1 日 1 回から 2 回、必要に応じて適宜。14 日間。 塗布部位 :がん性皮膚潰瘍 治験薬を投与する前に患部を十分に清浄し、非粘着性のドレッシング材(ガー ゼ、シリコンガーゼ、創傷被覆・保護材など)に治験薬を塗り皮膚潰瘍部位を 覆う。必要に応じて、1 日 1 回から 2 回使用する。 有効性の評価項目 ○ 主要評価項目 改善率 改善率は、治験責任(分担)医師による Day 14(又は治験中止時)のにおい スコアが 0 又は 1 と評価された被験者の割合と定義した。 (臨床仮説は、本試験での改善率が 70%を下回らないこととした。) ○ 副次評価項目 におい、潰瘍部位の臨床所見、疼痛 ○ その他の評価項目 細菌学的検査、被験者の QOL の全般改善度2.1.1 被験者の内訳
本試験には合計 21 例が組み入れられた。全 21 例に本剤が投与され、このうちの 20 例が試験を 完了した。1 例は、潰瘍部位からの滲出液が増加したため、被験者が本剤の投与中止を希望し、 Day 7 に試験を中止した。2.1.2 人口統計学的特性
被験者の人口統計学的特性及び他の基準値の特性を表 7に示す。全体では、被験者の年齢範囲は 39~82 歳(平均年齢:64.4 歳)であり、全例が女性で、アジア人であった。全 21 例が乳がんの患者であり、Stage III が 5 例(23.8%)及び Stage IV が 16 例(76.2%)であっ た。皮膚潰瘍の大きさの平均値 ± 標準偏差は 68.6 ± 35.4 cm2(範囲:4~140 cm2)であった。ま た、16 例(76.2%)はがん性悪臭に対する治療を受けたことがない被験者であった。 表 7 人口統計学的特性及び他の基準値の特性(国内第 3 相試験) 項目[単位] カテゴリ n (%) 全被験者 - 21 性別 男性 0 女性 21 (100.0) 人種 アジア人 21 (100.0) 年齢[歳] N 21 平均値 ± 標準偏差 64.4 ± 12.2 中央値 65.0 最小値, 最大値 39, 82 悪臭に対する前治療 なし 16 (76.2) あり 5 (23.8) がんのステージ Stage III 5 (23.8) Stage IV 16 (76.2) がんの種類(部位) 乳がん 21 (100.0) その他のがん 0 皮膚潰瘍の大きさ[cm2] N 21 平均値 ± 標準偏差 68.6 ± 35.4 中央値 75.0 最小値, 最大値 4, 140 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 3](5.3.5.2-1)
2.1.3 有効性評価
本試験での有効性は、本試験に組み入れられた全 21 例を対象として評価した。2.1.3.1 主要評価項目
2.1.3.1.1 改善率
有効性の主要評価項目は改善率とした。改善率とは、治験責任(分担)医師による Day 14(又は 治験中止時)のにおいスコアが 0(においがない)又は 1(においはあるが不快ではない)と評 価された被験者の割合と定義した。医師による Day 14(又は治験中止時)のにおいスコア (LOCF 法により欠測値を補完)及び改善率をそれぞれ表 8及び表 9に示す。 主要評価項目である改善率は 95.2%であり、その 90%信頼区間(正確法)は 79.3%~99.8%で あった。この結果から、臨床仮説である「本試験での改善率が 70%を下回らない」ことが確認さ れた。 表 8 医師によるDay 14(又は治験中止時)のおいスコア(LOCF法により欠測値を補完) (国内第 3 相試験) Day 14(又は治験中止時) n (%) 全被験者 21 医師評価によるスコア 0 13 (61.9) 1 7 (33.3) 2 0 3 1 (4.8) 4 0 平均値 ± 標準偏差 0.5 ± 0.7 中央値 0.0 最小値, 最大値 0, 3 においスコア: 0 = においがない、1 = においはあるが不快ではない、2 = 軽度に不快なにおい、3 = 中等度に不 快なにおい、4 = 非常に不快なにおい 略号: LOCF = 欠測値をその前のデータで補完する方法 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 4](5.3.5.2-1) 表 9 改善率(LOCF法により欠測値を補完) (国内第 3 相試験) n (%) 改善率の 90%信頼区間 全被験者 21 改善 20 (95.2) Wald 法 :87.6~100.0 正確法 :79.3~99.8 非改善 1 (4.8) 略号: LOCF = 欠測値をその前のデータで補完する方法 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 5](5.3.5.2-1)2.1.3.2 副次評価項目
2.1.3.2.1 におい
治験責任(分担)医師、看護師及び被験者は、ベースライン(Day 0)、Day 7 及び Day 14 に、 においスコアを 0(においがない)~4(非常に不快なにおい)の 5 段階で評価した。各評価者 によるにおいスコアの推移及びにおいスコアの要約をそれぞれ図 1及び表 10に示す。 医師によるにおいスコアの平均値 ± 標準偏差は、ベースライン(Day 0)の 2.6 ± 0.9 から、Day 7 の 0.9 ± 1.1、及び Day 14 の 0.5 ± 0.8 に低下した。看護師によるにおいスコアの平均値でも医師評 価と同様の結果が認められた。被験者によるにおいスコアは、全ての評価時点で医師評価及び看 護師評価よりもやや低かったが、経時的なプロファイルは類似していた。 図 1 医師、看護師及び被験者によるにおいスコア(国内第 3 相試験) 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Figure 1](5.3.5.2-1)
表 10 医師、看護師及び被験者によるにおいスコアの要約(国内第 3 相試験) ベースライン Day 0 n (%) Day 7 n (%) Day 14 n (%) 全被験者 21 21 20 医師評価によるスコア 0 0 11 (52.4) 13 (65.0) 1 0 5 (23.8) 6 (30.0) 2 13 (61.9) 2 (9.5) 0 3 3 (14.3) 3 (14.3) 1 (5.0) 4 5 (23.8) 0 0 平均値 ± 標準偏差 2.6 ± 0.9 0.9 ± 1.1 0.5 ± 0.8 中央値 2.0 0.0 0.0 最小値, 最大値 2, 4 0, 3 0, 3 ベースラインからの変化量 (平均値 ± 標準偏差) – -1.8 ± 0.8 -2.2 ± 0.7 看護師評価によるスコア 0 1 (4.8) 12 (57.1) 15 (75.0) 1 3 (14.3) 4 (19.0) 4 (20.0) 2 9 (42.9) 2 (9.5) 1 (5.0) 3 3 (14.3) 2 (9.5) 0 4 5 (23.8) 1 (4.8) 0 平均値 ± 標準偏差 2.4 ± 1.2 0.9 ± 1.2 0.3 ± 0.6 中央値 2.0 0.0 0.0 最小値, 最大値 0, 4 0, 4 0, 2 ベースラインからの変化量 (平均値 ± 標準偏差) – -1.5 ± 0.9 -2.1 ± 1.1 被験者評価によるスコア 0 1 (4.8) 9 (42.9) 16 (80.0) 1 8 (38.1) 12 (57.1) 4 (20.0) 2 6 (28.6) 0 0 3 4 (19.0) 0 0 4 2 (9.5) 0 0 平均値 ± 標準偏差 1.9 ± 1.1 0.6 ± 0.5 0.2 ± 0.4 中央値 2.0 1.0 0.0 最小値, 最大値 0, 4 0, 1 0, 1 ベースラインからの変化量 (平均値 ± 標準偏差) – -1.3 ± 1.2 -1.7 ± 1.1 においスコア: 0 = においがない、1 = においはあるが不快ではない、2 = 軽度に不快なにおい、3 = 中等度に不 快なにおい、4 = 非常に不快なにおい 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 6](5.3.5.2-1)
2.1.3.2.2 潰瘍部位の臨床所見
治験責任(分担)医師は、ベースライン(Day 0)、Day 7 及び Day 14 に潰瘍部位の臨床所見を 0 (なし)~3(重度)の 4 段階で評価した。潰瘍部位の臨床所見を表 11に示す。
潰瘍部位の臨床所見スコアの平均値 ± 標準偏差は、ベースライン(Day 0)、Day 7 及び Day 14 でそれぞれ 1.6 ± 0.9、1.1 ± 1.0 及び 0.9 ± 0.9 であり、ベースラインからの変化量の平均値 ± 標準 偏差は、Day 7 及び Day 14 でそれぞれ-0.4 ± 0.7 及び-0.6 ± 0.9 であった。 分泌物の性状は、ベースライン(Day 0)では膿性が 6 例(28.6%)、漿液性 + 血液を含むが 5 例 (23.8%)及び漿液性が 4 例(19.0%)であったのに比較し、Day 7 及び Day 14 では漿液性が多く の被験者で見られた(Day 7 で 12 例[57.1%]及び Day 14 で 10 例[50.0%])。全体では膿性が 減少し、漿液性が増加する傾向が認められた。 表 11 潰瘍部位の臨床所見(国内第 3 相試験) ベースライン Day 0 n (%) Day 7 n (%) Day 14 n (%) 全被験者 21 21 20 潰瘍部位の臨床所見スコア 0 1 (4.8) 5 (23.8) 7 (35.0) 1 12 (57.1) 12 (57.1) 10 (50.0) 2 3 (14.3) 0 1 (5.0) 3 5 (23.8) 4 (19.0) 2 (10.0) 平均値 ± 標準偏差 1.6 ± 0.9 1.1 ± 1.0 0.9 ± 0.9 中央値 1.0 1.0 1.0 最小値, 最大値 0, 3 0, 3 0, 3 ベースラインからの変化量 (平均値 ± 標準偏差) – -0.4 ± 0.7 -0.6 ± 0.9 分泌物の性状 膿性 6 (28.6) 1 (4.8) 1 (5.0) 漿液性 4 (19.0) 12 (57.1) 10 (50.0) 血液を含む 1 (4.8) 1 (4.8) 3 (15.0) 膿性 + 血液を含む 0 1 (4.8) 1 (5.0) 膿性 + 漿液性 2 (9.5) 1 (4.8) 0 漿液性 + 血液を含む 5 (23.8) 3 (14.3) 1 (5.0) 膿性 + 漿液性 + 血液を含む 3 (14.3) 2 (9.5) 4 (20.0) 潰瘍部位の臨床所見スコア: 0 = なし、1 = 軽度、2 =中等度、3 = 重度 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 7](5.3.5.2-1)
2.1.3.2.3 疼痛
被験者は、ベースライン(Day 0)、Day 7 及び Day 14 に皮膚潰瘍に関連した過去 24 時間の痛み について 100 mm の直線 VAS を用いて評価した(0 mm:痛みなし~100 mm:最も強い痛み)。 VAS による痛みの評価を表 12に示す。
VAS の平均値 ± 標準偏差は、ベースライン(Day 0)、Day 7 及び Day 14 でそれぞれ 28.3 ± 29.1、 25.9 ± 27.1 及び 22.2 ± 25.3 mm であり、ベースラインからの変化量の平均値 ± 標準偏差は、Day 7 及び Day 14 でそれぞれ-2.4 ± 21.8 及び-3.7 ± 23.0 mm であった。VAS の平均値にはベースライン から Day 7 又は Day 14 までに変化は認められなかった。 表 12 VASによる疼痛評価(国内第 3 相試験) VAS 値[mm] ベースライン
Day 0 Day 7 Day 14
全被験者 21 21 20 平均値 ± 標準偏差 28.3 ± 29.1 25.9 ± 27.1 22.2 ± 25.3 中央値 13.0 20.0 14.5 最小値, 最大値 0, 84 0, 89 0, 93 ベースラインからの変化量 (平均値 ± 標準偏差) – -2.4 ± 21.8 -3.7 ± 23.0 出典: RDT.07.SRE.27013 試験 治験総括報告書 [Table 8](5.3.5.2-1)
 誘発期(チャレンジパッチを貼布した後)における各試験製剤による反応の程度を 表 11 に示す。 48 時](https://thumb-ap.123doks.com/thumbv2/123deta/6029048.582649/20.893.97.795.140.247/メトロニダゾールゲルメトロニダゾールクリームチャレンジパッチ.webp)
![表 1 臨床的安全性に記載した臨床試験のデザイン上の特徴(続き) 試験番号 (日本/海外) 目的 デザイン 治験薬 投与方法 被験者数 安全性の評価項目 試験報告書添付場所 第 1 相試験 RDT.07.SRE.27010 (日本) 健康成人男性被験者を対象とし て、メトロニダゾールゲル 0.75%を単回局所投与したとき の皮膚刺激性及び光毒性を、ゲ ル基剤、日本薬局方精製水及び フィンチャンバー(パッチの み)と比較し、検討する。 単一施設無作為化 評価者盲検個体内対象比較試験 [被験薬] メト](https://thumb-ap.123doks.com/thumbv2/123deta/6029048.582649/60.1262.89.1166.134.502/デザインメトロニダゾールゲルフィンチャンバーパッチ単一施設無.webp)
![表 7 人口統計学的特性及び他の基準値の特性(国内第 3 相試験) 項目[単位] カテゴリ n (%) 全被験者 - 21 性別 男性 0 女性 21 (100.0) 人種 アジア人 21 (100.0) 年齢[歳] N 21 平均値 ± 標準偏差 64.4 ± 12.2 中央値 65.0 最小値 , 最大値 39, 82 身長[ cm] N 21 平均値 ± 標準偏差 154.51 ± 5.44 中央値 154.10 最小値 , 最大値 145.0, 16](https://thumb-ap.123doks.com/thumbv2/123deta/6029048.582649/67.893.96.796.142.846/人口統計学相試験カテゴリ平均値中央値平均値中央値最小値最大値.webp)
![表 14 重篤な有害事象の一覧(国内第 3 相試験) 被験者 識別番号 人種性別年齢 a 有害事象 器官別大分類( SOC) 基本語(PT) 医師記載用語 発現時の投与量[ g/日] 発現日 [日目] b 発現期間[日間] c 重症度 転帰 治験薬に 対する処置 治験薬との因果関係 アジア人 女性 胃腸障害 下痢(Diarrhoea) Diarrhea 30 12 5 中等度 後遺症なく回復 変更なし 合理的な 関連性なし 一般・全身障害](https://thumb-ap.123doks.com/thumbv2/123deta/6029048.582649/75.1262.59.1162.144.338/相試験転帰治験薬対する処置治験因果関係アジア胃腸中等度障害.webp)
![表 1-3 皮膚刺激指数 試験製剤 皮膚刺激指数 メトロニダゾールゲル 0.75% 10.0 ゲル基剤 10.0 日本薬局方精製水 5.0 フィンチャンバー(パッチのみ) 7.5 皮膚刺激指数 = 除去 60 分後又は 24 時間後の反応の強い方のスコア総和 × 100 被験者数 皮膚刺激スコアの範囲は 0~400 出典: RDT.07.SRE.27010 試験 治験総括報告書 [Table4-5][Section 14](5.3.3.1-1)](https://thumb-ap.123doks.com/thumbv2/123deta/6029048.582649/103.893.88.797.979.1086/メトロニダゾールゲル日本薬局方フィンチャンバーパッチスコア.webp)
