九州大学学術情報リポジトリ
Kyushu University Institutional Repository
DIF-1ハダイチョウガンサイボウカブニオイテTCF7L2 ノハツゲンヲヨクセイスルコトニヨリ、Wnt/β-
cateninシグナルデンタツケイロヲソガイスル
神宮司, 健太郎
九州大学大学院医学系学府
https://doi.org/10.15017/21727
出版情報:Kyushu University, 2011, 博士(医学), 課程博士 バージョン:
権利関係:(C) 2011 Elsevier Inc.
1
平成 23 年度 医学専攻博士論文
DIF-1 は大腸がん細胞株において TCF7L2 の発現を抑制する
ことにより、Wnt/β-catenin シグナル伝達経路を阻害する
九州大学大学院医学研究院 生体情報科学講座臨床薬理学分野
論文提出者 神宮司 健太郎 指導教員 笹栗 俊之
高橋 富美
2 要約
我 々 は こ れ ま で に 細 胞 性 粘 菌 Dictyostelium discoideum が 分 泌 す る 分 化 誘 導 因 子 Differentiation-inducingfactor-1(DIF-1)がβ-catenin タンパク質の分解を誘導することで
Wnt/β-cateninシグナル伝達経路を阻害し、ヒトがん細胞株の増殖を抑制することを報告し
てきた。β-cateninタンパク質の分解がDIF-1 の効果に不可欠であるか否かを明らかにする ために、Wnt/β-cateninシグナル伝達経路が恒常的に活性化されているヒト由来大腸がん細 胞株(HCT-116、SW-620、DLD-1)に対する DIF-1 の効果を検討した。DIF-1 はβ-catenin タンパク質の分解非依存性にcyclin D1の発現をmRNA、タンパク質レベル共に減少させ、
G0/G1期における細胞周期拘束を起こすことにより、強力に細胞増殖を抑制した。DIF-1に よるtranscription factor 7-like 2(TCF7L2)発現量の抑制によって、TCF依存性転写活性及
びcyclin D1プロモーター活性が抑制されることが明らかとなった。ルシフェラーゼレポー
ター活性測定及びTCF7L2プロモーター断片を用いたEMSAにより、翻訳開始点を起点と して-609から-601 bp上流に位置する転写因子early growth response-1(Egr-1)結合部位が、
DIF-1の効果に関与していることが示された。さらに、RNAi法による内在性TCF7L2の欠
失により cyclin D1 プロモーター活性及びタンパク質量が減少し、TCF7L2 の強制発現は
DIF-1によるTCF依存性転写活性及びcyclin D1プロモーター活性の低下効果を減弱させた。
したがって、DIF-1は大腸がん細胞株において、Egr-1依存性のTCF7L2転写活性を減少さ せることによりTCF7L2発現を抑制し、Wnt/β-cateninシグナル伝達経路を阻害しているこ とが示唆された。我々の結果は、DIF-1による Wnt/β-cateninシグナル伝達経路の阻害機構 に新たな知見をもたらすものである。
3 序論
Wnt/β-catenin シグナル伝達経路は初期発生から成体組織における恒常性維持において重
要な役割を担っている。恒常的な Wnt/β-catenin シグナル伝達経路の活性化はがんにむすび つくことはよく知られている(1-4)。特に、大腸がんにおいてWnt/β-cateninシグナル伝達経 路の関与が数多く報告されている。多くの大腸がんが Wnt/β-catenin シグナル伝達経路の構 成分子であるadenomatous polyposis coli(APC)もしくはβ-cateninに変異を有しており、そ れにより核内にβ-catenin が蓄積し標的遺伝子の転写が活性化されている(5-8)。数多くの
Wnt/β-cateninシグナル伝達経路の標的遺伝子が、がんに直結するがん原遺伝子である(9-11)。
それらにおいてTCF7L2は腫瘍形成に関わることが報告されており、β-catenin/TCF7L2複合 体は小腸上皮細胞の増殖と分化におけるマスタースイッチとして報告されている(12-14)。 したがって、β-catenin/TCF7L2複合体による転写活性を抑制する抗がん薬は大腸がん治療に おいて有望である。
Differentiation-inducing factors(DIFs)は細胞性粘菌Dictyostelium discoideumが分泌し、粘 菌の分化を誘導する物質として同定された(15、16)。DIFファミリーにおいて、DIF-1(1-
(3,5-dichloro-2, 6-dihydroxy-4-methoxyphenyl)-1-hexanone)は初めて同定されたものである。
DIF-1の活性はDictyosteliumに限られたものではなく、ヒト由来細胞に対して強力に増殖を
阻害することが報告されている(17-19)。これまでに我々は様々なヒト由来細胞において DIFがGlycogen synthase kinase-3β (GSK-3β)の活性化を介してβ-cateninの分解を誘導する
ことで Wnt/β-catenin シグナル伝達経路を阻害し、それによる cyclin D1 発現の抑制により
G0/G1 期での細胞周期拘束を起こすことを報告してきた(20-27)。しかしながら、β-catenin 分解機構の異常のため Wnt/β-catenin シグナル伝達経路が恒常的に活性化されている大腸が ん細胞株に対するDIFの効果はまだ明らかとなっていない。
本研究で我々は、β-catenin(HCT-116)もしくはAPC(DLD-1、SW-620)に変異を有する ヒト由来大腸がん細胞株に対するDIF-1の効果を検証した。
4 材料と方法 1. 化合物と抗体
DIF-1は以前に述べた方法で合成された(15、16)。MG132はPeptide Instituteから購入し た。SB216763はBIOMOL internationalから購入した。TOPflash(TCFレポータープラスミド)
とFOPflash(TCFレポータープラスミドのネガティブコントロール)はUpstate Biotechnology から購入した。cyclin D1 レポータープラスミド(野生型と変異型)はカリフォルニア大学 のDr. O.TestuとDr. F. McCormickに供与していただいた。抗cyclin D1ポリクローナル抗体 は Santa Cruz Biotechnology から購入した。抗 Egr-1 モノクローナル抗体は Cell Signaling Technologyから購入した。抗β-cateninモノクローナル抗体はBD Bioscienceから購入した。
抗α-tubulinモノクローナル抗体は Calbiochemから購入した。抗 GAPDHモノクローナル抗
体はAbcamから購入した。抗TCF7L2モノクローナル抗体はMILLIPOREから購入した。
2. 細胞培養
ヒト由来大腸がん細胞株HCT-116細胞(β-cateninに変異)、DLD-1及びSW-620細胞(APC に変異)はDulbecco’s modified Eagle’s medium(DMEM)に10% Fetal bovine serum(FBS)、
100 U/ml penicillin G、0.1µg/ml streptomycinを含むmediumで培養した。
3. 細胞増殖測定
HCT-116細胞(3×104細胞/well)、DLD-1細胞(5×104細胞/well)そしてSW-620細胞(7.5
×104 細胞/well)は 24 穴プレートに播き、様々な濃度の DIF-1 で処理した。細胞は
trypsin/EDTA処理によって回収し、Coulter Counter(Beckman Coulter)を用いて細胞数の測 定を行った。
4. フローサイトメトリー解析
細胞は50 µg/ml propidium iodide(PI)、0.1% sodium citrate、0.1% Triton X-100を含む低張 性溶液を用いて懸濁した。PIによって染色したサンプル(1×105細胞)はBecton-Dickinson FACSCalibur(Becton-Dickinson)を用いて蛍光を測定した。
5. ウェスタンブロット法
ウェスタンブロット法は以前に述べた方法で行った(21)。サンプルは12% SDS-PAGEに よって分離しその後polyvinylidene difluorideメンブレンにセミドライ方式(1時間、12V)
で転写した。免疫反応性タンパク質は検出試薬(LumiGLO; Cell Signaling Technology)によ って視覚化した。濃度測定にはNIH Image Jソフトウエアを用いた。
6. 核内タンパク質抽出
核内タンパク質は60-mmディッシュに播いた細胞からNE-PERTM nuclear and cytoplasmic
extraction reagents(Pierce)を用いて核内タンパク質を精製した。精製した核内タンパク質は
その後ウェスタンブロット法とゲルシフトアッセイに用いた。
5 7. RT-PCR法
TRIzol® Reagent(Invitrogen)を用いてトータルRNAを抽出した。1 µg のRNAを用いて
cyclin D1、TCF7L2、Egr-1、GAPDH mRNA量をRT-PCRによって解析した。以下のプライ
マーを用いた。TCF7L2 (forward: 5’-ACG AGG GCG AAC AGG AGG AG-3’, reverse: 5’-TGG GCG AGA GCG ATC CGT TG-3’)、Egr-1 (forward: 5’-GGT CAG TGG CCT AGT GAG C-3’, reverse: 5’-TGC TGT CGT TGG ATG GCA C-3’)。他のプライマーセットは以前に述べた方法 で行った(21)。
8. RNA干渉
TCF7L2 Select Stealth™ RNAiはInvitrogenから購入した。製造元のプロトコールに従って、
Lipofectamine™ RNAiMAX (Invitrogen)を用いてdsRNA(100 nM)をトランスフェクショ ンした。Stealth™ RNAi negative control(Invitrogen)はネガティブコントロールとして用い た。
9. TCF7L2レポータープラスミドの作成
ヒトゲノムDNAからPCR法によって、ヒトTCF7L2遺伝子の5’上流領域(開始コドンを 基点とした-1306/-1 bp を含む領域)を増幅し、ホタルルシフェラーゼレポーターベクター
(pGL3-Basic) に組込んだ。続いてこれを鋳型として、異なった長さの TCF7L2 レポータ ープラスミド(-869/-1 bp、-629/-1 bp、-604/-1 bp、-578/-1 bp、-434/-1 bp、-223/-1 bp)を作 成した。
10. TCF7L2強制発現用プラスミドの作成
HCT-116細胞からトータルRNAを抽出しRT-PCRによってTCF7L2 cDNAを得た。得ら
れたcDNA(GenBank accession number FJ010167)はDNAシークエンスによって確認し、
pcDNA3(Invitrogen)に組み込んだ。
11. ルシフェラーゼレポーター活性測定
レポーターベクターとpRL-SV40をLipofect Amine Plus reagent(Invitrogen)を用いてトラ ンスフェクションした。24 時間培養後、示した時間 DIF-1 によって刺激した。ルシフェラ ーゼ活性をルミノメーター(Lumat LB 9507)にて測定した。また、同時にウミシイタケル シフェラーゼ活性をコントロールとして測定した。
12. ゲルシフトアッセイ
TCF7L2プロモーター領域の-634/-605 bpと-618/-589 bpに相当する相補的なオリゴヌクレ
オチドを合成し(5’-GGC GCC CGA AAG GAT CAT TGT TAG CCG CCC-3’と5’-CAT TGT TAG CCG CCC CCG CCC CGC CCA CCC-3’)、Biotin 3’ End DNA Labeling Kit(Pierce)を用
いて3’末端をbiotinでラベルした。ラベルしたオリゴヌクレオチドと核内タンパク質を混合
した。100倍量の標識していないオリゴヌクレオチドをコンペティターとして用いた。DNA
6
結合タンパク質を同定するために、核内タンパク質と抗 Egr-1 抗体(500 ng)もしくは抗 α-tubulin抗体(500 ng)をラベルしたオリゴヌクレオチドを加える前に混合した。抗α-tubulin 抗体はネガティブコントロールとして用いた。サンプルは7% native polyacrylamide gelを用 いて電気泳動し、Phototope®-Star Kit(New England Bio Labs)によってラベルしたオリゴヌ クレオチドを検出した。
13. 統計
結果は平均値± S.E.で示した。値間の差は Student’s t-test もしくは one-way ANOVA with Bonferroni post-hoc tests(GraphPad Prism 5.0, GraphPad Software)を用いて統計的に解析した。
P value < 0.05を統計学的に有為であるとした。
7 結果
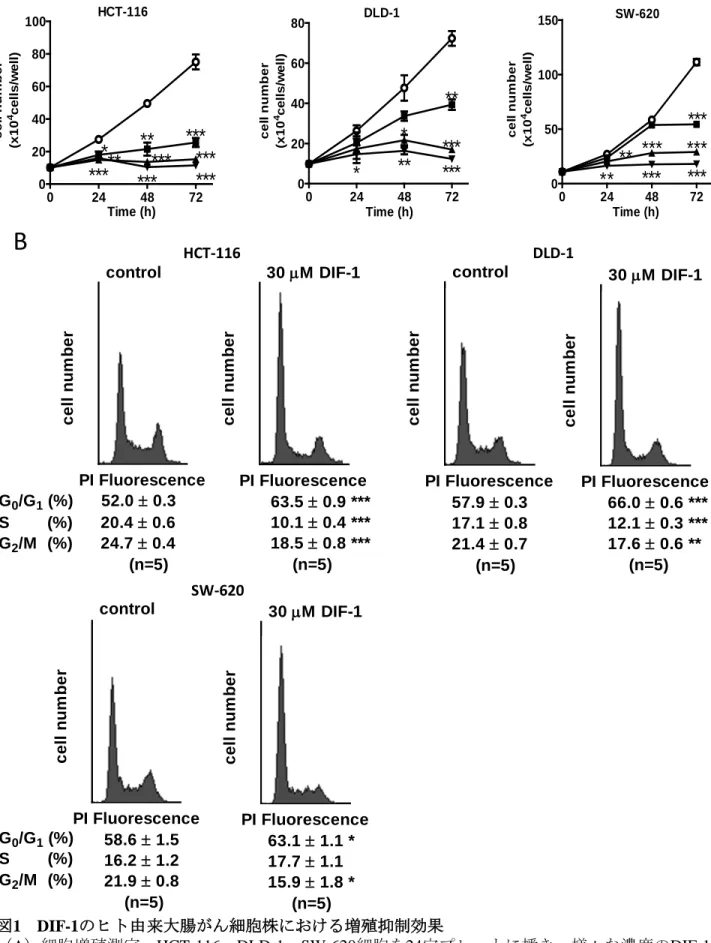
1. ヒト由来大腸がん細胞株に対するDIF-1の増殖抑制効果
DIF-1は様々なヒト由来細胞において強力な抗増殖作用をもち、Wnt/β-cateninシグナル伝
達経路を阻害することが報告されている(20-27)。したがって最初に、Wnt/β-cateninシグナ ル伝達経路が恒常的に活性化されているヒト由来大腸がん細胞株(HCT-116、DLD-1、
SW-620)に対してもDIF-1が増殖抑制作用をもつかどうかを調べた。図 1Aに示すように、
細胞株間で反応性は異なったが、DIF-1は強力にかつ濃度依存性に細胞増殖を抑えた。次に フローサイトメトリー解析を用いて細胞周期に与える影響について調べた。DIF-1によって G0/G1期の細胞が上昇し、G2/M期の細胞が減少していた(図1B)。このことから、DIF-1は ヒト由来大腸がん細胞株に対してG0/G1期で細胞周期を拘束したことが示された。これらの 結果はこれまでの我々の報告に一致しており、DIF-1はヒト由来大腸がん細胞株においても 増殖抑制作用をもつことが示唆された。
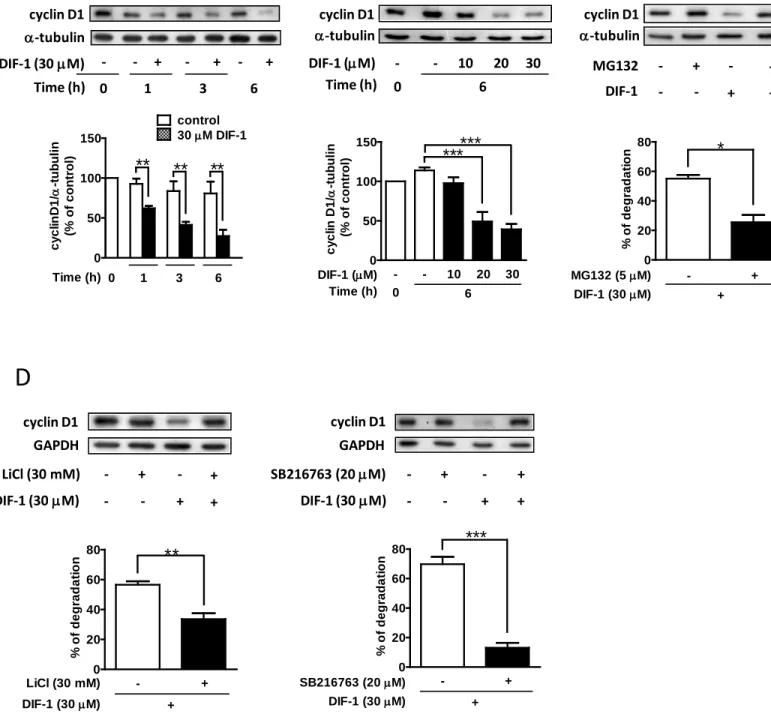
2. DIF-1は大腸がん細胞においてGSK-3βの活性化により cyclin D1のタンパク質分解を誘
導する
DIF-1はGSK-3βの活性化を介してcyclin D1発現を抑制し、G0/G1期で細胞周期を拘束す ることが報告されている(22、23、27)。したがって、HCT-116細胞におけるcyclin D1に対
するDIF-1の効果を調べた。図2Aと2Bに示すように、DIF-1は時間、濃度依存性にcyclin
D1 タンパク質量を減少させた。ユビキチンプロテアソーム阻害剤 MG132 は DIF-1 による cyclin D1タンパク質量の減少を減弱させた(図2C)。GSK-3βがcyclin D1のタンパク質分解 を引き起こすことが報告されている(28、29)。したがって、GSK-3βの阻害剤(LiCl、SB216763)
を用いてDIF-1によるcyclin D1タンパク質分解にGSK-3βが関与しているか否かを調べた。
図2Dに示すように、LiCl(30 mM、3時間)もしくはSB216763(20 µM、3時間)による 前処置によってDIF-1の効果は減弱した。このことから、HCT-116細胞においてもGSK-3β
はDIF-1によるcyclin D1タンパク質分解に関与していることが示された。DIF-1の効果発現
までの時間は細胞株間で異なったが、DLD-1細胞とSW-620細胞においてもDIF-1はGSK-3β
を介してcyclin D1タンパク質分解を誘導していた(データ非掲載)。
解析した3種類の細胞株においてHCT-116細胞が最もDIF-1に対して感受性が高かった為、
以下の実験ではHCT-116細胞を用いた。
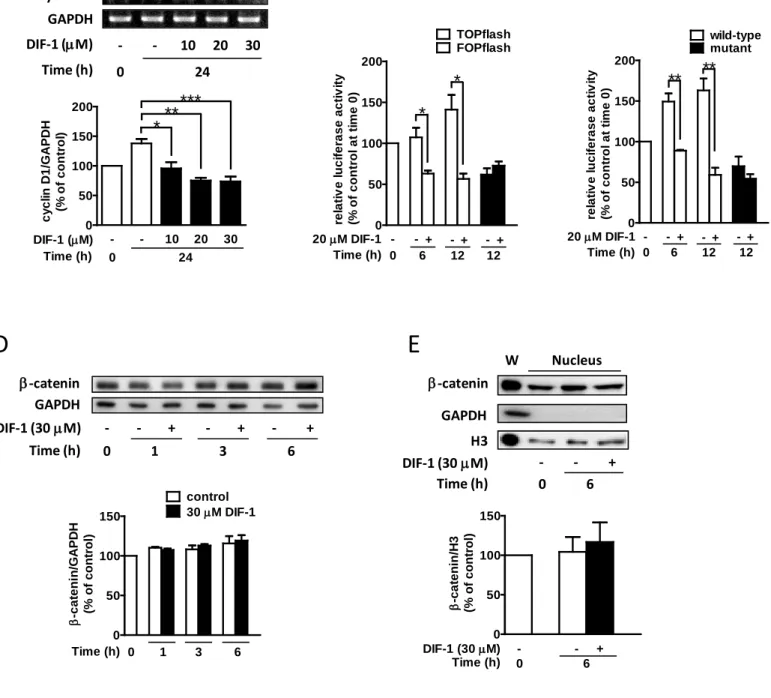
3. DIF-1はHCT-116細胞においてβ-cateninタンパク質量非依存性にcyclin D1プロモーター 活性を減少させる
次にcyclin D1 mRNA量に対するDIF-1の効果を調べた。DIF-1は濃度依存性にcyclin D1 mRNA量を減少させた(図3A)。cyclin D1はWnt/β-cateninシグナル伝達経路の標的遺伝子 であることが報告されている(30)。そこで、TOPflashとFOPflash(TCFレポータープラス ミドとそのネガティブコントロール)を用いて TCF 依存性の転写活性に対する DIF-1 の効 果を調べた。図3Bに示すように、DIF-1はFOPflash活性には影響を与えなかったが、TOPflash 活性はDIF-1刺激後6時間に著しく減少した。また、野生型のcyclin D1プロモーターと-81/-75
8
bpに存在する(23、30)TCF結合部位に変異を生じさせた変異型のプロモーターを用いて、
DIF-1のcyclin D1プロモーター活性に与える影響も調べた。DIF-1は変異型のプロモーター
活性には影響を与えなかったが、野生型のプロモーター活性はDIF-1によって著しく減少し た(図3C)。これまでに我々は、DIF-1がβ-cateninの分解を介してTCF依存性の転写活性を 減少させるという報告をしてきた(23)。そこで、β-cateninタンパク質量に対するDIF-1 の 効果を調べた。図3D と3Eに示すように、全体、核内のβ-cateninタンパク質量はDIF-1 に よる影響を受けなかった。このことから、DIF-1 はβ-catenin タンパク質量非依存性に TCF 依存性の転写活性とcyclin D1プロモーター活性を抑制したことが示唆された。
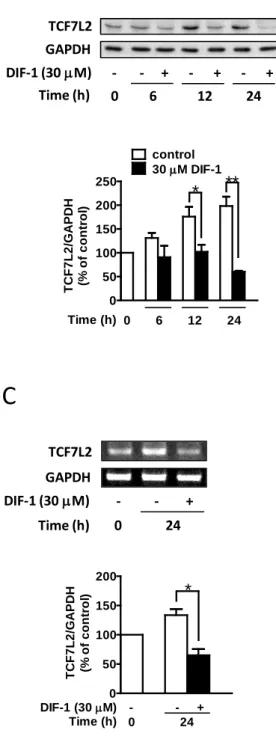
4. DIF-1はHCT-116細胞においてTCF7L2の転写を抑制する
DIF-1がどのようにβ-cateninタンパク質量非依存性にTCF依存性の転写活性を抑制したの
かを明らかにするために、次にTCF7L2の発現に対するDIF-1の効果を調べた。Wnt/β-catenin シグナル伝達経路においてTCF7L2は主要な転写因子として働くことが知られている。コン トロール条件下でのTCF7L2タンパク質量は細胞の培養時間依存性に上昇したが、DIF-1に
よってTCF7L2タンパク質量は時間、濃度依存性に著しく減少した(図4A、B)。またTCF7L2
mRNAに対するDIF-1の影響も調べた。DIF-1によってTCF7L2 mRNAは濃度依存性に減少
した(図4C)。
次にTCF7L2プロモーター活性へのDIF-1の影響を調べた。PCR法によってヒトTCF7L2
遺伝子の5’上流領域を増幅し、ルシフェラーゼレポーターベクター(pGL3-Basic)に組込ん
だ。図4Dに示すように、TCF7L2プロモーター活性は細胞培養時間依存性に上昇したが(24 時間で約 2 倍)、DIF-1 はこの上昇を抑えコントロール値以下まで減少させた。これらの結
果よりHCT-116細胞においてDIF-1は、TCF7L2の転写を抑制することでTCF7L2タンパク
質量を減少させたことが示された。
5. DIF-1はHCT-116細胞においてEgr-1依存性の転写活性を抑制することで、TCF7L2プロ モーター活性を抑制する
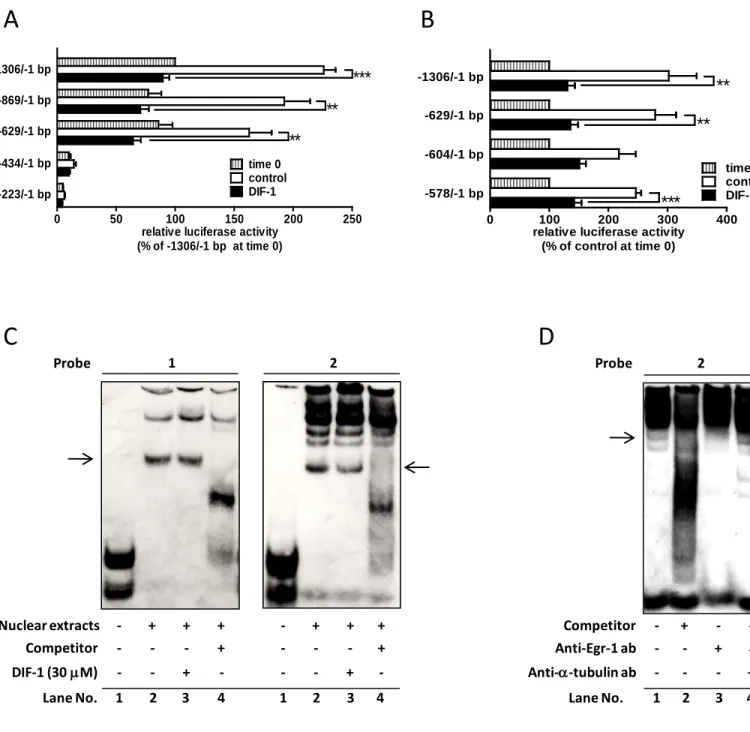
TCF7L2プロモーター上におけるDIF-1の応答配列を決定するために、異なる長さのプロ
モーターを用いて解析をおこなった。図5Aに示すように、629 bpより長いTCF7L2プロモ ーター(−1306/−1 bp, −869/−1 bp, and −629/−1 bp)を含むレポータープラスミドを導入した細 胞では、24時間の培養でプロモーター活性が0時間の1.5~2倍に上昇するのに対し、DIF-1 で刺激した場合はこの活性の上昇がほぼ抑えられていた。さらに、TCF7L2プロモーター上 の必須応答配列を明らかにするために、-629/-1 bp領域をさらに細かく分けたレポータープ ラスミド(−604/−1 bpと−578/−1 bp)を用いてTCF7L2プロモーター活性に対するDIF-1の 効果を調べた。図5Bに示すように、DIF-1の効果は−604/−1 bpを含むレポータープラスミ ドで減弱した。このことから、DIF-1の応答配列は-629~-604 bpに存在することが示唆され た。
次に-629~-604 bpに結合する核内タンパク質に対するDIF-1の影響を調べるために、2種 類のプローブ(プローブ1 : −634/−605 bp、プローブ2 :−618/−589 bp)を用いてゲルシフトア
9
ッセイをおこなった。Egr-1がTCF7L2プロモーター活性を調節するという報告があること から(31)、特に−609/−601 bpに位置するEgr-1結合様配列に注目した。図5Cに示すように、
どちらのプローブも核内タンパク質と結合し(レーン2)、100倍量のコンペティターによっ て著しいバンドの減弱がみられた(レーン4)。DIF-1によって刺激した細胞から抽出した核 タンパク質とEgr-1結合様配列を含むプローブ2の組み合わせにおいて、著しいバンドの減 弱がみられた。このことより、DIF-1はEgr-1のDNA結合能を抑制したことが示唆された。
Egr-1 の関与を検証するために、抗 Egr-1 抗体を用いたスーパーシフトアッセイをおこなっ
た。図5Dに示すように、スーパーシフトしたバンドは検出できなかったが、抗Egr-1抗体 を加えることで明らかなバンドの減弱が認められた。
引き続き、ウエスタンブロットとRT-PCRによってEgr-1タンパク質量とmRNA量に与え
る DIF-1 の影響を調べた。DIF-1 によって細胞全体(図 5E)および核内(図 5F)の Egr-1
タンパク質量は著しく減少し、またEgr-1 mRNA量も減少していた(図5G)。これらのこと
より、DIF-1はEgr-1 mRNA量を抑制することによりEgr-1タンパク質量を減少させ、TCF7L2
プロモーター活性を減少させていることが示された。
6. DIF-1はHCT-116細胞においてTCF7L2の発現を抑制することで、Wnt/β-cateninシグナ ル伝達経路を阻害する
さらに、cyclin D1の発現に対するTCF7L2の関与について検討するために、内在性TCF7L2
をRNAiによって欠失させた。図6Aに示すように、TCF7L2を欠失させることによりcyclin D1タンパク質量は著しく減少した。また、野生型のcyclin D1プロモーターとTCF結合部 位に変異を生じさせた変異型のプロモーターを用いて、TCF7L2 欠失の cyclin D1 プロモー ター活性に与える影響も調べた。変異型のプロモーター活性には変化はみられなかったが、
野生型のcyclin D1プロモーターはTCF7L2欠失によって著しくその活性が抑制された(図
6B)。これらの結果よりHCT-116細胞においてcyclin D1発現はTCF7L2によって促進され ていることが示された。次に、TCF7L2を強制発現させたHCT-116細胞を用いてDIF-1の効 果を調べた。図6Cに示すように、pcDNA3 のみをトランスフェクションさせた細胞におい
ては12時間DIF-1処理によりTCF7L2レベルは減少したが、pcDNA3/TCF7L2をトランスフ
ェクションさせた細胞においてはその効果が減弱した。その後、DIF-1がTCF依存性の転写
活性及びcyclin D1プロモーター活性に与える影響を調べた。pcDNA3のみをトランスフェ
クションさせた細胞と比較して、TCF7L2を強制発現させた細胞においてDIF-1がTOPflash
活性及びcyclin D1プロモーター活性に与える影響は減弱した(図6D)。これらの結果から
HCT-116細胞において、DIF-1によるTCF7L2の減少はWnt/β-cateninシグナル伝達経路及び
cyclin D1プロモーター活性の阻害に結びついていることが示された。
7. DLD-1細胞におけるTCF7L2発現に対するDIF-1の効果
他のヒト由来大腸がん細胞株においても同じ機序がDIF-1によるcyclin D1の転写抑制に 関与しているかどうかを調べるために、DLD-1 細胞を用いて TCF7L2 発現に対する DIF-1 の影響を調べた。HCT-116細胞が野生型APCと変異型β-cateninを有するのに対して、DLD-1
10
は変異型APCと野生型β-cateninを有する。DIF-1により時間、濃度依存性にTCF7L2タンパ ク質量は減少した(図8A、B)。また、DIF-1はTCF7L2 mRNA量も抑制した(図8C)。こ れらの結果から DIF-1は、DLD-1 細胞においても TCF7L2 発現を mRNA、タンパク質レベ ルで抑制することが示された。
11 考察
我々はこれまでにDIF-1がGSK-3βを活性化させることによりβ-catenin分解を誘導し、cyclin
D1発現及びWnt/β-cateninシグナル伝達経路の阻害を引き起こし、ヒト由来がん細胞株の増
殖を抑制することを報告してきた。本研究では、β-catenin分解機構関連タンパク質に変異が 生じることで Wnt/β-catenin シグナル伝達経路が恒常的に活性化されているヒト由来大腸が ん細胞株(SW-620、DLD-1、HCT-116)に対する DIF-1の効果を調べた。DIF-1は強力に細 胞増殖を阻害し GSK-3βの活性化を介して cyclin D1 のタンパク質分解を誘導した。また、
DIF-1はβ-catenin量非依存性にcyclin D1 mRNA量及びプロモーター活性を抑制した。さら
に DIF-1 は核内 Egr-1 タンパク質量を減少させることで、強力に TCF7L2 発現を抑制し
Wnt/β-cateninシグナル伝達経路を阻害した。したがって、これらの結果よりヒト由来大腸が
ん細胞においてDIF-1はGSK-3βの活性化によってcyclin D1のタンパク質分解を誘導し、ま
た、Egr-1を介したTCF7L2発現の抑制によってWnt/β-cateninシグナル伝達経路を阻害する
ことによりcyclin D1 mRNA量を減少させていることが示唆された。
Egr-1遺伝子は初期応答遺伝子グループに属しており、増殖因子やホルモンなど数多くの
環境因子によるシグナルがEgr-1遺伝子の発現を誘導することが報告されている(32)。Egr-1 は増殖因子、サイトカイン受容体、接着分子そしてプロテアーゼなどの遺伝子発現を調節す ることにより生体内での様々な機能を担っている。がん形成におけるEgr-1の機能はまだ明 確にはなっていない。例えば、Egr-1はがん抑制遺伝子を調節しており、Egr-1の発現はヒト 由来がん細胞の増殖を阻害するという報告がある(33-35)。しかしながらEgr-1が、がん細 胞の増殖と生存を促進するという報告もある(36-39)。ヒト由来大腸がん細胞株においては、
curcuminが Egr-1 活性を減少させることにより EGF 受容体発現を抑制し細胞増殖を阻害す
るという報告がある(40)。さらに Fahmy らは、DNAzyme を用いた Egr-1 の阻害がラット 角膜における血管新生を抑制するということから、Egr-1 が血管内皮細胞の増殖とがんの血 管新生において重要な役割を担っていることを見出している(41)。これらの知見は、DIF-1
がin vitroとin vivoにおいて血管内皮細胞の増殖と血管新生を阻害するという我々の報告と
一致している(27)。今回、どのようにDIF-1がEgr-1 mRNAレベルを抑制したのかは明ら かにできなかったが、Egr-1転写活性の阻害がDIF-1 による抗腫瘍効果の機序のひとつなの かもしれない。
DIF-1の応答配列を同定するために、様々な長さのヒトTCF7L2遺伝子の5’上流領域を含
むレポータープラスミドを作成した。そして、-609~-601 bp に存在する Egr-1 結合配列が DIF-1の応答配列であることを見出した。SaegusaらはEgr-1が-786~-778 bpに存在するEgr-1 結合配列を通して TCF7L2 プロモーター活性を調節しているという報告をしている(31)。 我々は、なぜTCF7L2プロモーター上の異なる領域がEgr-1に応答したのか明らかにするこ とができなかった。しかしながら、Saegusa らは子宮体がん細胞株を用いていたことから、
TCF7L2転写調節機構はがん細胞株間において異なる可能性がある。これについては更なる
研究が必要である。
Wnt/β-cateninシグナル伝達経路の恒常的活性化は大腸がんの形成につながりうる。したが
って、Wnt/β-cateninシグナル伝達経路の転写活性を抑制するような抗がん薬は大腸がん治療
12
において有望であろう。このため、複数の Wnt/β-catenin シグナル伝達経路を阻害する化合 物が開発されその効果が調べられているが、その多くが前臨床開発段階にある(42-46)。本 研究で我々は、DIF-1がTCF7L2発現を抑制することによりWnt/β-cateninシグナル伝達経路 の転写活性を著しく抑制することを示した。このことから、β-catenin 調節機構の異常に伴 う大腸がんに対する抗がん薬として DIF-1 は期待できることが示唆された。さらに我々は、
以前の報告で DIF-1 が Wnt/β-cateninシグナル伝達経路非依存性に抗血管新生作用を発揮す ることを報告している。したがって、DIF-1はWnt/β-cateninシグナル伝達経路の阻害作用と 抗血管新生作用によって大腸がんに対する有望な化合物である可能性がある。
13 引用文献
[1] Clevers H. Wnt/β-catenin signaling in development and disease. Cell 2006;127:
469-80.
[2] Nelson WJ, Nusse R. Convergence of Wnt, β-catenin, and cadherin pathways.
Science 2004;303:1483-7.
[3] Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through β-catenin. Science 2002;296:1644-6.
[4] Akiyama T. Wnt/β-catenin signaling. Cytokine Growth Factor Rev. 2000;11:273-82.
[5] Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al.
Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997;275:1787-90.
[6] Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000;24:245-50.
[7] McDonald SA, Preston SL, Lovell MJ, Wright NA, Jankowski JA. Mechanisms of disease: from stem cells to colorectal cancer. Nat. Clin. Pract. Gastroenterol Hepatol. 2006;3:267-74.
[8] Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway.
Oncogene 2006;25:7531-7.
[9] Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982;31:99-109.
[10] Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat. Rev.
Cancer 2008;8:387-98.
[11] Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007;56:417-25.
[12] Shitashige M, Hirohashi S, Yamada T. Wnt signaling inside the nucleus. Cancer Sci.
2008;99:631-7.
[13] Tannishtha R, Hans C. Wnt signalling in stem cells and cancer. Nature 2005;434:843-50.
[14] Waterman ML. Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev. 2004;23:41-52.
[15] Morris HR, Taylor GW, Masento MS, Jermyn KA, Kay RR. Chemical structure of the
morphogen differentiation inducing factor from Dictyostelium discoideum. Nature 1987;328:811-4.
[16] Morris HR, Masento MS, Taylor GW, Jermyn KA, Kay RR. Structure elucidation of two differentiation inducing factors (DIF-2 and DIF-3) from the cellular slime mould Dictyostelium discoideum. Biochem. J. 1988;249:903-6.
[17] Kubohara Y, Saito Y, Tatemoto K. Differentiation-inducing factor of D. discoideum raises intracellular calcium concentration and suppresses cell growth in rat pancreatic AR42J cells. FEBS Lett. 1995;359:119-22.
14
[18] Kubohara Y. DIF-1, putative morphogen of D. discoideum, suppresses cell growth and promotes retinoic acid-induced cell differentiation in HL-60. Biochem. Biophys. Res. Commun.
1997;236:418-22.
[19] Kubohara Y. Effects of differentiation-inducing factors of Dictyostelium discoideum on human leukemia K562 cells: DIF-3 is the most potent anti-leukemic agent. Eur. J. Pharmacol.
1999;381:57-62.
[20] Miwa Y, Sasaguri T, Kosaka C, Taba Y, Ishida A, Abumiya T, et al.
Differentiation-inducing factor-1, a morphogen of dictyostelium, induces G1 arrest and differentiation of vascular smooth muscle cells. Circ. Res. 2000;86:68-75.
[21] Takahashi-Yanaga F, Taba Y, Miwa Y, Kubohara Y, Watanabe Y, Hirata M, et al.
Dictyostelium differentiation-inducing factor-3 activates glycogen synthase
kinase-3β and degrades cyclin D1 in mammalian cells. J. Biol. Chem. 2003;278:9663-70.
[22] Mori J, Takahashi-Yanaga F, Miwa Y, Watanabe Y, Hirata M, Morimoto S, et al.
Differentiation-inducing factor-1 induces cyclin D1 degradation through the phosphorylation of Thr286 in squamous cell carcinoma. Exp. Cell Res. 2005;310:426-33.
[23] Yasmin T, Takahashi-Yanaga F, Mori J, Miwa Y, Hirata M, Watanabe Y, et al.
Differentiation-inducing factor-1 suppresses gene expression of cyclin D1 in tumor cells. Biochem.
Biophys. Res. Commun. 2005;338:903-9.
[24] Matsuzaki E, Takahashi-Yanaga F, Miwa Y, Hirata M, Watanabe Y, Sato N, et al.
Differentiation-inducing factor-1 alters canonical Wnt signaling and suppresses alkaline phosphatase expression in osteoblast-like cell lines. J. Bone Miner. Res. 2006;21:1307-16.
[25] Takahashi-Yanaga F, Mori J, Matsuzaki E, Watanabe Y, Hirata M, Miwa Y, et al.
Involvement of GSK-3β and DYRK1B in differentiation-inducing factor-3-induced phosphorylation of cyclin D1 in HeLa cells. J. Biol. Chem. 2006;281:38489-97.
[26] Matsuda T, Takahashi-Yanaga F, Yoshihara T, Maenaka K, Watanabe Y, Miwa Y, et al.
Dictyostelium differentiation-inducing factor-1 binds to mitochondrial malate dehydrogenase and inhibits its activity. J. Pharmacol. Sci. 2010;112:320-6.
[27] Yoshihara T, Takahashi-Yanaga F, Shiraishi F, Morimoto S, Watanabe Y, Hirata M, et al.
Anti-angiogenic effects of differentiation-inducing factor-1 involving VEGFR-2 expression inhibition independent of the Wnt/β-catenin signaling pathway. Mol. Cancer
2010;10.1186/1476-4598-9-245.
[28] Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957-72.
[29] Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499-511.
[30] Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999;398:422-6.
[31] Saegusa M, Hashimura M, Kuwata T, Hamano M, Watanabe J, Kawaguchi M, et al.
15
Transcription factor Egr1 acts as an upstream regulator of β-catenin signaling through up-regulation of TCF4 and p300 expression during trans-differentiation of endometrial carcinoma cells. J. Pathol.
2008;216:521-32.
[32] Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J.
Cell. Physiol. 2002;193:287-92.
[33] Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFβ1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115-24.
[34] Yu J, Baron V, Mercola D, Mustelin T, Adamson ED. A network of p73, p53 and Egr1 is required for efficient apoptosis in tumor cells. Cell Death Differ. 2007;14:436-46.
[35] Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor
formation. Int. J. Cancer 1997;72:102-9.
[36] Yang SZ, Abdulkadir SA. Early growth response gene 1 modulates androgen receptor signaling in prostate carcinoma cells. J. Biol. Chem. 2003;278:39906-11.
[37] Virolle T, Krones-Herzig A, Baron V, De Gregorio G, Adamson ED, Mercola D. Egr1 promotes growth and survival of prostate cancer cells. Identification of novel Egr1 target genes. J. Biol. Chem.
2003;278:11802-10.
[38] Abdulkadir SA, Qu Z, Garabedian E, Song SK, Peters TJ, Svaren J, et al. Impaired prostate tumorigenesis in Egr1-deficient mice. Nat. Med. 2001;7:101-7.
[39] Zheng C, Ren Z, Wang H, Zhang W, Kalvakolanu DV, Tian Z, et al. E2F1 Induces
tumor cell survival via nuclear factor-κB-dependent induction of EGR1 transcription in prostate cancer cells. Cancer Res. 2009;69:2324-31.
[40] Chen A, Xu J, Johnson AC. Curcumin inhibits human colon cancer cell growth by
suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene 2006;25:278-87.
[41] Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat. Med. 2003;9:1026-32.
[42] Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010;16:3153-62.
[43] Garber K. Drugging the Wnt pathway: problems and progress. J. Natl. Cancer Inst.
2009;101:548-50.
[44] Liu C, He X. Destruction of a destructor: a new avenue for cancer therapeutics targeting the Wnt pathway. J. Mol. Cell. Biol. 2010;2:70-3.
[45] Dihlmann S, von Knebel Doeberitz M. Wnt/β-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int. J. Cancer 2005;113:515-24.
[46] Verkaar F, Zaman GJ. New avenues to target Wnt/β-catenin signaling. Drug Discov. Today
16 2011;16:35-41.
図1 DIF-1のヒト由来大腸がん細胞株における増殖抑制効果
(A)細胞増殖測定。HCT-116、DLD-1、SW-620細胞を24穴プレートに播き、様々な濃度のDIF-1で培養し た。細胞を示された時間後trypsin/EDTA処理によって回収し、細胞数の測定を行った。(B)フローサイト メトリー。HCT-116、DLD-1、SW-620細胞をDIF-1(30 µM)で24時間刺激後、trypsin/EDTA処理によって回
収した。propidium iodide(PI)で染色後、核内蛍光を測定した。各細胞周期にある細胞をパーセントで表示
している。値は3回の実験結果の平均値±S.E.で表示した。*P<0.05; **P<0.01; ***P<0.001 vs. control.
0 24 48 72
0 20 40 60 80 100
***
***
**
***
***
***
***
***
HCT-116
cell number (x104cells/well)
Time (h)
0 24 48 72
0 20 40 60 80
*
*
* * **
Time (h)
*
***
**
DLD-1
cell number (x104cells/well)
0 24 48 72
0 50 100
150 control
10µM 20µM 30µM
*
*
* *
*
* * *
* * *
* *
SW-620
*
*
* *
* *
Time (h) cell number (x104cells/well)
PI Fluorescence control
G0/G1 (%) 52.0± 0.3 S (%) 20.4 ± 0.6 G2/M (%) 24.7± 0.4 (n=5)
cell number
PI Fluorescence 30µM DIF-1
63.5 ± 0.9 ***
10.1 ± 0.4 ***
18.5 ± 0.8 ***
(n=5)
cell number
HCT-116
B
PI Fluorescence 57.9± 0.3 17.1± 0.8 21.4± 0.7 (n=5) control
cell number
PI Fluorescence 66.0± 0.6 ***
12.1± 0.3 ***
17.6± 0.6 **
(n=5) 30µM DIF-1
cell number
DLD-1
PI Fluorescence 58.6± 1.5 16.2± 1.2 21.9± 0.8
(n=5) control
G0/G1 (%) S (%) G2/M (%)
cell number
PI Fluorescence 63.1± 1.1 * 17.7± 1.1 15.9± 1.8 *
(n=5) 30µM DIF-1
cell number
SW-620
A
図2 ヒト由来大腸がん細胞株においてDIF-1はcyclin D1のタンパク質分解を誘導する
(A)経時的変化。HCT-116細胞を表示された時間DIF-1(30 µM)で刺激した。(B)濃度依存性。HCT- 116細胞を様々な濃度のDIF-1で6時間刺激した。(C)プロテアソーム阻害剤MG132の効果。HCT-116細胞 をMG132で1時間前処置後、DIF-1(30 µM)で6時間刺激した。(D)HCT-116細胞を30 mM LiClもしくは20 µM SB216763で3時間前処置後、DIF-1(30 µM)で6時間刺激した。回収したサンプルは抗cyclin D1抗体と抗 α-tubulin抗体もしくは抗GAPDH抗体を用いてウエスタンブロット法にて解析した。タンパク質バンドを定 量化し、0時間に対するパーセンテージ(AとB)もしくは減少量のパーセンテージ(CとD)で表示した。
値は3回の実験結果の平均値±S.E.で表示し、Student’s t-test(A、C、D)もしくはone-way ANOVA with Bonferroni post-hoc test(B)で統計的に解析した。*P<0.05; **P<0.01; ***P<0.001 vs. control.
cyclin D1 α-tubulin Time (h) DIF-1 (30 µM)
0 1 3 6
- - + - + - +
A B
DIF-1 (µM)
Time (h) 0 6
- - 10 20 30 cyclin D1
α-tubulin
0 50 100 150
control 30µM DIF-1
0 1 3 6 Time (h)
* * *
cyclinD1/α-tubulin (% of control)
* * *
0 50 100 150
* *
Time (h)
DIF-1 (µM) - 20 30
6 - 0
10
**
cyclin D1/α-tubulin (% of control)
* *
cyclin D1 α-tubulin
C
0 20 40 60 80
MG132 (5µM) - +
*
% of degradation
DIF-1 (30µM) +
D
0 20 40 60
80 *
LiCl (30 mM) - +
*
DIF-1 (30µM) +
% of degradation
cyclin D1 GAPDH
DIF-1 (30 µM) LiCl (30 mM) -
-
- + +
-
+ +
0 20 40 60
80 *
SB216763 (20µM) - +
* *
% of degradation
DIF-1 (30µM) +
cyclin D1 GAPDH
DIF-1 (30 µM) SB216763 (20 µM) -
-
- + +
-
+ +
+ - + -
DIF-1 MG132
+ - +
-
図3 HCT-116細胞におけるcyclin D1 mRNA量に対するDIF-1の効果
(A)様々な濃度のDIF-1で24時間刺激した。トータルRNA(1 µg)を用いてRT-PCRによりcyclin D1と GAPDHの発現を解析した。PCRのサイクル数はcyclin D1が26、GAPDHが20である。mRNA発現レベルを定 量化し、0時間に対するパーセンテージで表示した。one-way ANOVA with Bonferroni post-hoc testで統計解析 した。(B)TOPflashもしくはFOPflashとpRL-SV40を導入した。ルシフェラーゼ活性は0時間に対するパー センテージで表示した。(C)ルシフェラーゼレポータープラスミド(野生型cyclin D1もしくは変異型cyclin D1プロモーターを含むpGL3)とpRL-SV40を導入した。24時間の培養後、DIF-1(20 µM)で表示された時 間刺激した。ルシフェラーゼ活性は0時間に対するパーセンテージで表示した。(D)表示された時間DIF-1
(30 µM)で刺激した。(E)DIF-1(30 µM)で6時間刺激し、核内タンパク質を抽出した。回収したサンプ ルは抗β-catenin抗体、抗GAPDH抗体と抗histone H3抗体を用いてウエスタンブロット法にて解析した。タン パク質バンドを定量化し、0時間に対するパーセンテージで表示した。値は3回の実験結果の平均値±S.E.で ある。*P<0.05; **P<0.01; ***P<0.001 vs. control.
A B C
cyclin D1
Time (h) 0 24 GAPDH
DIF-1 (µM) - - 10 20 30
0 50 100 150 200
Time (h)
DIF-1 (µM) - 20 30
24 - 0
10
****
*
cyclin D1/GAPDH (% of control)
*
0 50 100 150 200
TOPflash FOPflash
- + - + - + 20µM DIF-1
Time (h) 0 6 12 12
-
*
*
relative luciferase activity (% of control at time 0)
0 50 100 150 200
*
wild-type mutant
*
- + - + - +
20µM DIF-1
Time (h) 0 6 12 12
-
relative luciferase activity (% of control at time 0) * *
D E
β-catenin
Nucleus W
GAPDH H3 DIF-1 (30 µM) Time (h)
- - +
0 6
0 50 100 150
control 30µM DIF-1
0 1 3 6 Time (h)
β-catenin/GAPDH (% of control)
0 50 100 150
Time (h) DIF-1 (30µM)
0
- - + 6 β-catenin/H3 (% of control)
- - + - + - + GAPDH
β-catenin
Time (h) 0 1 3 6 DIF-1 (30 µM)
図4 HCT-116細胞においてDIF-1はTCF7L2の発現を抑制する
(A)表示された時間DIF-1(30 µM)で刺激した。回収したサンプルは抗TCF7L2抗体と抗GAPDH抗体を用 いてウエスタンブロット法にて解析した。タンパク質バンドを定量化し、0時間に対するパーセンテージで 表示した。(B)様々な濃度のDIF-1で24時間刺激した。タンパク質バンドは定量化され、0時間に対するパ ーセンテージで表示した。one-way ANOVA with Bonferroni post-hoc testで統計解析した。(C)様々な濃度の DIF-1で24時間刺激した。トータルRNA(1 µg)を用いてRT-PCRによりTCF7L2とGAPDHの発現を解析した
。PCRのサイクル数はTCF7L2が23、GAPDHが20である。mRNA発現レベルは定量化され、0時間に対する パーセンテージで表示した。one-way ANOVA with Bonferroni post-hoc testで統計的に解析した。(D) TCF7L2プロモーターコンストラクト(-1306/-1 bp)とpRL-SV40を導入した。24時間の培養後、DIF-1(20 µM)で表示された時間刺激した。ルシフェラーゼ活性は0時間に対するパーセンテージで表示した。値は3 回の実験結果の平均値±S.E.である。*P<0.05; **P<0.01; ***P<0.001.
Time (h)
DIF-1 (30 µM) - - + - + - + 0 6 12 24 GAPDH
A
C
TCF7L2 GAPDH Time (h)
DIF-1 (µM) - - 10 20 30 0 24
B
TCF7L2
0 100 200 300 400
control 30µM DIF-1
0 6 12 24 Time (h)
*
*
*
TCF7L2/GAPDH (% of control)
0 100 200 300
Time (h)
DIF-1 (µM) - 20 30
24 - 0
10
* *
*
TCF7L2/GAPDH (% of control) ** ** * *
0 50 100 150 200
Time (h)
DIF-1 (µM) - 20 30
24 - 0
10
* **
TCF7L2/GAPDH (% of control) ** ** **
D
0 50 100 150 200 250
Time (h) 0 3 6 9 12 24
* *
* * *
*
relative luciferase activity (% of control)
* *
*
*
control 20µM DIF-1
Time (h)
DIF-1 (µM) - - 10 20 30 0 24
TCF7L2 GAPDH
図5 HCT-116細胞におけるTCF7L2プロモーター上のDIF-1応答配列の同定
(A)表示されたTCF7L2遺伝子の5’上流領域を含むコンストラクト(-1306/-1, -869/-1, -629/-1, -434/-1, -223/- 1 bp)とpRL-SV40を導入した。24時間の培養後、DIF-1(20 µM)で24時間刺激した。ルシフェラーゼ活性 は-1306/-1 bpコンストラクトの0時間に対するパーセンテージで表示した。(B)表示されたTCF7L2遺伝子 の5’上流領域を含むコンストラクト(-1306/-1, -629/-1, -604/-1, -578/-1 bp)とpRL-SV40をHCT-116細胞に導入 した。24時間の培養後、DIF-1(20 µM)で24時間刺激した。ルシフェラーゼ活性は0時間に対するパーセン テージで表示した。(C)DIF-1(30 µM)で24時間刺激したHCT-116細胞から核内タンパク質を抽出し、ゲ ルシフトアッセイをおこなった。4回の実験を代表する結果を示している。特異的バンドは矢印で示した。
(D)スーパーシフトアッセイ。核内タンパク質を抗Egr-1抗体もしくは抗α-tubulin抗体と混合した。3回の 実験を代表する結果を示している。特異的バンドは矢印で示した。値は3回の実験結果の平均値±S.E.であ る。**P<0.01; ***P<0.001.
Nuclear extracts - + + + - + + +
Competitor - - - + - - - +
DIF-1 (30 µM) - - + - - - + -
Lane No. 1 2 3 4 1 2 3 4
B A
0 100 200 300 400
time 0 control DIF-1 relative luciferase activity
(% of control at time 0) -1306/-1 bp *
-629/-1 bp
-604/-1 bp
-578/-1 bp
*
**
*
*
*
0 50 100 150 200 250
relative luciferase activity (% of -1306/-1 bp at time 0) -1306/-1 bp
-869/-1 bp
-434/-1 bp -223/-1 bp -629/-1 bp
*
*
*
DIF-1 control
*
time 0
*
*
*
1 2
Probe 2
Competitor Anti-Egr-1 ab Anti-α-tubulin ab
- +
Lane No. 1 2
- -
-
-
3 + - -
-
4 - + Probe