2007 年度 修士論文
生体分子のフォールディング 解析システムの構築と高速化
提出日 : 2008 年 2 月 4 日 指導 : 村岡 洋一 教授
早稲田大学大学院理工学研究科 情報ネットワーク専攻
学籍番号 : 3606U067-5
徳永 慎一
概 要
近年、計算機の発展によりスーパーコンピューターやPCクラスタを使うこと によって、1フェムト秒ごとの細かいタイムステップでも100ナノ〜1マイクロ 秒単位の生体分子のシミュレーションが短時間で出来るようになった。それに伴 い、過去の微視的な解析手法とは異なる大規模データの解析手法が求められてい る。一般的なシミュレーションデータ解析では、タンパク質の状態遷移を可視化 することが多く、それらはタンパク質の定常状態の振る舞いや、特定構造から別 の構造に遷移する現象を明らかにすることを目指している。本研究では分子動力 学シミュレーションで得られたトラジェクトリデータをいくつかの構造に分類し、
HMM(隠れマルコフモデル)による解析を行うことで、タイムステップ毎の時系
列遷移からは解析が難しい、タンパク質のフォールディング・リフォールディン グ過程を含めた熱揺らぎのパターン解析を提案し、その解析を行うシステムの構 築を行った。
また本研究では、同システムの高速化も目的としている。マイクロ秒単位のシ ミュレーションデータの解析を行うにあたり、メモリ容量がボトルネックとなる。
たとえば、倍精度浮動小数点(64bit=8byte)で表現された50,000×50,000の配列 は約20GBとなり、主成分分析における固有値計算がシステム全体のボトルネッ クとなってしまう。本研究ではシミュレーションで得た膨大なトラジェクトリデー タを扱うにあたり、Quad Core Opteron プロセッサと大容量メモリを積んだ共有 メモリマシン上での高速化を行う。その結果、主成分分析において同マシン上で 動かした数値計算ライブラリOctaveの11.9倍の演算性能を得ることができた。
さらに本研究では、本システムを用いたシミュレーションデータの解析も目的 としており、10残基の世界最小タンパク質であるシニョリン(Chignolin)に関し て1マイクロ秒のシミュレーション、20残基のタンパク質であるTrp-cageに関 して400ナノ秒のシミュレーションを行い、それぞれについて解析を行った。本 システムを2つのタンパク質へ適用することによって、安定構造を発見する(構 造決定を行う)ための指針として有効であることを示した。
i
目 次
第1章 序論 1
1.1 研究の目的と背景 . . . . 1
1.2 本論文の構成 . . . . 3
第2章 熱揺らぎ解析システム 4 2.1 タンパク質の熱揺らぎ . . . . 4
2.2 分子動力学法 . . . . 5
2.3 システム構成 . . . . 6
2.4 DataSet . . . . 7
2.4.1 RMSD . . . . 7
2.4.2 Rg . . . . 7
2.4.3 Cα distance . . . . 8
2.5 Analysis . . . . 8
2.5.1 PCA . . . . 8
2.5.2 Clustering . . . . 9
2.5.3 HMM . . . . 9
2.6 Visualizer . . . . 10
第3章 解析システムの高速化 11 3.1 主成分分析 . . . . 11
3.1.1 Householder変換 . . . . 11
3.1.2 QR法 . . . . 12
3.2 高速化手法 . . . . 13
3.2.1 HouseHolder変換における高速化 . . . . 13
ATLAS . . . . 14
3.2.2 QR法における高速化 . . . . 15
第4章 高速化の評価 17 4.1 実験環境 . . . . 17
4.2 ATLAS . . . . 18
4.3 HouseHolder変換の評価 . . . . 19
4.4 QR法の評価. . . . 20
4.5 主成分分析全体の評価 . . . . 21
第5章 解析結果 23 5.1 シミュレーションに用いた系:Chignolin . . . . 23
5.1.1 主成分分析 . . . . 24
5.1.2 クラスタリング . . . . 25
5.1.3 HMM . . . . 26
5.2 シミュレーションに用いた系:不安定コントロール . . . . 27
5.2.1 主成分分析 . . . . 28
5.2.2 HMM . . . . 29
5.3 シミュレーションに用いた系:Trp-Cage . . . . 30
5.3.1 主成分分析 . . . . 30
5.3.2 HMM . . . . 32
第6章 まとめと今後の課題 33 6.1 まとめ . . . . 33
6.2 今後の課題 . . . . 33
謝辞 35
参考文献 36
付 録A 著者外部発表 38
ii
iii
図 目 次
2.1 folding . . . . 4
2.2 分子動力学法の基本手順 . . . . 5
2.3 システム構成 . . . . 6
2.4 タンパク質同士の重ね合わせ . . . . 7
2.5 タンパク質の構造(参考文献[10]) . . . . 8
3.1 三重対角化 . . . . 11
3.2 1ステップごとの処理 . . . . 15
3.3 並列化 . . . . 16
4.1 NB(ブロックサイズ)と性能 . . . . 18
4.2 行列サイズと性能 . . . . 19
4.3 CPU数と性能 . . . . 19
4.4 行列サイズと性能 . . . . 20
4.5 CPU数と性能 . . . . 21
4.6 Octaveとの比較 . . . . 21
5.1 Cα distanceをPCAにかけた結果 . . . . 24
5.2 kmeansによるクラスタリング . . . . 25
5.3 HMM . . . . 26
5.4 PCA - 不安定コントロール . . . . 28
5.5 HMM - 不安定コントロールとの比較 . . . . 29
5.6 Cα distanceをPCAにかけた結果 . . . . 31
5.7 HMM - Trp-Cage . . . . 32
iv
表 目 次
2.1 出力PDBファイル . . . . 5
2.2 k-means法のアルゴリズム . . . . 9
2.3 HMMの定義 . . . . 9
3.1 高速化部分 . . . . 13
3.2 BLAS . . . . 14
3.3 QR法のアルゴリズム. . . . 15
4.1 実験環境 . . . . 17
4.2 HPLベンチマーク(参考文献[11]) . . . . 18
5.1 データセット . . . . 23
5.2 データセット . . . . 30
1
第 1 章 序論
1.1 研究の目的と背景
タンパク質は生体分子の中で最も重要な物質の一つであり、その機能の発現に は固有の立体構造の形成が必要である。化学的には鎖状の高分子であるタンパク 質が、どのような状態を経由して折りたたみ、最終的にどのような立体構造をと るのかという疑問はフォールディング問題と呼ばれる。天然状態においてはタン パク質は非常に安定した状態に折りたたまれて(フォールディング)いることが多 く、なんらかの機能をもっていることが多い。つまり、そのタンパク質の安定な 状態や存在確率の高い状態を突き止めることは、タンパク質の構造・機能予測に つながる。これを読み解くために分子動力学シミュレーションが用いられている。
分子動力学法は、生命科学の分野において生体分子のダイナミクス、機能や構 造予測を解析するために用いられている。分子動力学法は主に溶液中の生体分子 をシミュレートするのに用いられるが、一般に生体分子のシミュレーションは、
大規模で複雑な系を扱い、しかも生体内に近い環境での精密な計算が必要である ことから、多大な計算時間を必要とする。こうした分子動力学法のような膨大な 計算時間を必要とするシミュレーションに対して、並列計算は必要不可欠な技術 となっている。
近年、計算機の発展によりスパコンや並列クラスタを使うことによって、ナノ 秒単位のシミュレーションが短時間で出来るようになった。しかし、一般的には 100ナノ秒〜マイクロ秒単位のシミュレーションを行うにはまだまだ時間がかか り、マイクロ秒単位のシミュレーションによる大量データの解析例は少ない。
分子動力学シミュレーションで得られたデータの解析には、各トラジェクトリ
データのCα(またはバックボーン)間の二面角を用いることが多いが、本研究で
はCα間の距離を用いて解析を行う。また、一般的なシミュレーションデータ解析 では、タンパク質の状態遷移を可視化することが多く、それらはタンパク質があ る構造から別の構造に遷移する現象を明らかにすることを目指している。タンパ ク質の熱揺らぎのパターン解析には自由エネルギー地形や統計処理などを用いた 解析手法が存在するが、情報工学の観点から解析を行った例は少ない。したがっ て、分子動力学シミュレーションで得られたトラジェクトリデータをいくつかの 構造に分類し、確率情報処理の観点からトラジェクトリデータの状態遷移を可視 化することは、タンパク質の安定な状態や存在確率の高い状態を明らかにする一
1.1. 研究の目的と背景 2 つの指針として、有用な手段であると考えられる。そこで、本研究ではトラジェ クトリデータをHMM(Hidden Marcov Model)にかけることで、シミュレーショ ンにおけるタンパク質の状態遷移を確率的なモデルとして可視化することにより、
タイムステップ毎の時系列遷移からは解析が難しい、タンパク質の熱揺らぎのパ ターン解析を提案し、その解析を行うシステムの構築を行う。
また本研究では、構築したシステムの高速化も目的としている。マイクロ秒単 位のシミュレーションデータの解析を行うにあたり、メモリ容量がボトルネック となる。たとえば、倍精度浮動小数点で表現された50,000×50,000の配列は約 20GBのメモリ容量を使用するため、解析における大きなボトルネックとなる。
本システムではまず解析しやすい形に変換したシミュレーションデータを主成分 分析にかけるが、大量のシミュレーションデータにより作成される共分散行列は 同様のメモリ容量を必要とする。さらにこの巨大な密行列の固有値計算を行わな ければならないため、主成分分析がシステムのボトルネックとなる。そこで本研 究では、同システムの主成分分析における処理について、Quad Core Opteronプ ロセッサと大容量メモリを積んだ共有メモリマシン上での高速化を行う。
さらに本研究では、同システムを用いたシミュレーションデータの解析も目的 としており、シニョリン(Chignolin)、Trp-cageの二つにタンパク質に関しての解 析を行う。シニョリンはアミノ酸10残基からなる立体構造が決定された世界最 小のタンパク質であり、Trp-cageはアミノ酸20残基からなるタンパク質である。
また、シニョリンに関しては、シニョリンとアミノ酸配列が似ているが、立体構 造が決定されないタンパク質(不安定コントロール)との比較も行う。
したがって本研究では、1.)分子動力学シミュレーションのデータ解析を行う一 貫したシステムの構築、2.)同システムの高速化、3.)同システムを用いたタンパ ク質の解析、の3つを目的とする。
本稿の構成は以下の通りである。2章では解析を行うシステムについて説明す る。次に,3章ではシステムの高速化についての提案を行い、4章では高速化し たシステムの実行速度についての考察を行う。5章では本研究で行った分子動力 学シミュレーションについてと解析結果を述べ、6章で全体をまとめる.
1.2. 本論文の構成 3
1.2 本論文の構成
本論文の構成を以下に示す。
• 第2章
実装した解析システムについて説明する
• 第3章
システム内のモジュールの高速化についての提案を行う
• 第4章
高速化したシステムの実行速度についての考察を行う
• 第5章
本研究で行った分子動力学シミュレーションと解析結果について述べる
• 第6章
まとめと今後の課題について述べる
4
第 2 章 熱揺らぎ解析システム
本章ではタンパク質とその熱揺らぎに関する基礎知識について述べ、実装した 解析システムの構成について説明する。
2.1 タンパク質の熱揺らぎ
タンパク質は生体分子の中で最も重要な物質の一つであり、その機能の発現に は固有の立体構造の形成が必要である。化学的には鎖状の高分子であるタンパク 質が、どのような状態を経由して折りたたみ、最終的にどのような立体構造をと るのかという疑問はフォールディング問題と呼ばれる。2.1に示すようにタンパ ク質構造にはフォールディング(折りたたまれている状態)とアンフォールディン
グ(伸びている状態)がある。天然状態においてはタンパク質はフォールディン
グ(非常に安定した)状態にいることが多く、何らかの機能をもっていることが多
い。つまり、そのタンパク質の安定な状態や存在確率の高い状態を突き止めるこ とは、タンパク質の構造・機能予測につながる。これを読み解くために分子動力 学シミュレーションが用いられており、シミュレーション解析は必要不可欠なも のとなっている。
図 2.1: folding
2.2. 分子動力学法 5
2.2 分子動力学法
分子動力学法(Molcular Dynamics=MD)とは、生体分子のような多分子系に おいて、ニュートンの運動方程式を積分することによって、個々の原子の運動を シミュレーションする手法である。このような積分は多数の原子が互いに相互作 用しながら運動しているので多体問題となり、解析的に解くことが事実上不可能 であるため、有限差分法を用いて数値的に解くことになる。分子動力学法のアル ゴリズムを図2.2に示す。
図 2.2: 分子動力学法の基本手順
基本手順は粒子の位置や速度を短い時間刻みΔtで離散的に計算していくとい うものである。すなわち、ある時刻において原子間に働く力を計算し、それに基 づいてΔt後の原子の位置を求め、その求めた位置で新たに原子間に働く力を計 算する、という操作の繰返しである。温度や圧力を一定にする場合、その調整の ための操作も繰返しに含まれる。こうして得られた原子の座標の軌跡をもとに、
種々の物理量を計算する。
また、実際にMDシミュレーションにより実際に出力されるファイルの内容は 以下のようなものである。
表 2.1: 出力PDBファイル
ATOM 1 N GLY 1 -5.667 -2.804 -9.928 0.00 0.00 ATOM 2 H1 GLY 1 -5.723 -2.016 -10.558 0.00 0.00 ATOM 3 H2 GLY 1 -5.960 -3.597 -10.480 0.00 0.00
・
・
・
ATOM 138 OXT GLY 10 11.061 -0.489 4.474 0.00 0.00
分子動力学法における1タイムステップごとに、このファイルが生成される。
2.3. システム構成 6 左から順に、原子の順番、原子名、残基の種類、残基番号、x座標、y座標、z座 標、占有率、温度因子を表している。本研究において、占有率、温度因子に関し ては基本的に0.00なので考慮しない。このようなMDシミュレーションを用いて 得られた時系列ごとに変化する一連の構造データを、トラジェクトリデータ(生 体分子がシミュレーション中にどのように変化していくかの軌跡)と呼ぶことに する。
2.3 システム構成
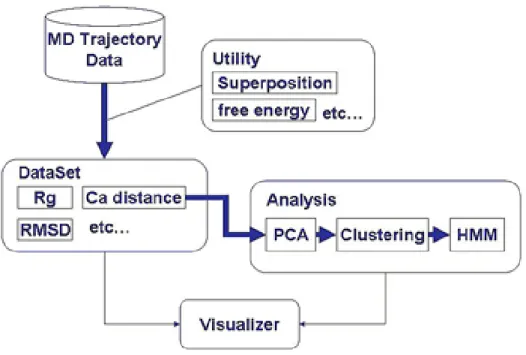
MDにより得られたトラジェクトリデータを、自由エネルギーの地形や、生体 分子の周りの水分子の動き、PDB(Protein Data Bank)構造とのRMSDなど様々 なデータとして可視化することができるシステムを考案する。図2.3に示すよう に全体のシステムは、DataSet、Analysis、Visualizerの大きく3つのモジュール に分けることができる。
図 2.3: システム構成
2.4. DataSet 7
2.4 DataSet
主に入力データと出力データを保持するモジュールである。MDで得たトラ ジェクトリデータは、座標データの他にも以下のような様々なデータとして保持 される。
2.4.1 RMSD
2つのタンパク質構造がどの程度違うのかを表す値である。図2.4に示すよう に、タンパク質同士の重ね合わせ処理[3]を行った後、以下の式で求めることが できる。ここでnは原子数、dnは重ね合わせたタンパク質同士における各原子の 距離である。
RM SD= s1
n X
n
d2n (2.1)
図 2.4: タンパク質同士の重ね合わせ
2.4.2 Rg
タンパク質のコンパクトさを表す値であり、以下の式で表される。ここで、N は残基の数、−→r は各残基のCα原子の座標である。
Rg = vu ut 1
2N2 XN
i,j
(−→ri − −→rj) (2.2)
2.5. Analysis 8
2.4.3 Cα distance
図2.5に示すように、Cαとはアミノ酸の中心部分を表す炭素(C)であり、Cα
distanceとはタンパク質内のCα間距離のことである。本研究杖はタンパク質内
のCαの全対全の組み合わせを入力データとして用いる。
図 2.5: タンパク質の構造(参考文献[10])
2.5 Analysis
解析を実際に行うモジュールである。Cα間距離を主成分分析することによっ て、シミュレーション中に存在確率の高かった構造を抽出することができる。ま た、第1〜第3主成分を座標軸としたPCA空間をクラスタリングによってグルー プ分けすることにより各クラスターを一つの状態とみなし、シミュレーション内 におけるタンパク質をいくつかの構造に分類する。さらに、各クラスター(構造) を入力としてHMMにかける。
2.5.1 PCA
2.4.3で述べたCα distanceを入力として、トラジェクトリデータの各構造に ついて主成分分析を行い、第1〜3主成分までを抽出する。主成分分析(Principal
Component Analysis)とは、多くの変量の値をできるだけ情報の損失なしに,1
個または少数個の主成分を抽出するテクニックである。変量(次元)の観測値を主 成分に縮約するという意味で、次元を減少させる目的で使用されることが多い。
主成分分析ではさまざまな手法が用いられるが、本研究で用いた主成分分析の手 法については3.1で詳しく述べる。
2.5. Analysis 9
2.5.2 Clustering
k-means法による非階層化クラスタリングを行う。クラスタリング手法は大き
く、階層化クラスタリングと非階層化(分割最適化)クラスタリングに分けられる が、本研究ではより高速な解析を行うため非階層化クラスタリングを用いている。
入力をデータ集合Xi、クラスタ数k、反復回数の上限をmとすると、k-means 法のアルゴリズムは表2.2のようになる。
表 2.2: k-means法のアルゴリズム
(1) Xをランダムにk個のクラスタ分割し、初期クラスタを得る。
(2) 各クラスタについて中心(セントロイド) xi = |X1
i|
Px∈Xiを計算する。
(3) 全てのデータを各セントロイドとの距離が最小になるような クラスタに割り当てる。
(4) 1ステップ前のクラスタから変化がない場合、または反復回数がmの場合 アルゴリズムを終了する。
(5) (2)に戻る。
2.5.3 HMM
クラスタリングによって分類されたクラスター一つ一つを状態とみなして、
HMM(Hidden Markov Model)を用いた解析を行う。HMMの定義を以下に示す。
表 2.3: HMMの定義
・ 潜在変数である状態{Si}Ni
・ 遷移確率分布をA (時刻tで状態Sjへ遷移する確率) P[qi =Sj|qi−1 =Si,· · ·qi−n=Sk]
・ 観測シンボル確率分布をB (状態Siでシンボルvkが出力される確率) P[bj(k)=P[vk|qk =Sj]
・ 初期状態分布をπ (時刻t= 1で状態Siにある確率) πi =P[q1 =Si]
ここで、A、B、π の要素をまとめたモデルλを 定義する。つまり、λ= (A, B, π)である。
2.6. Visualizer 10 本研究で用いるHMMの学習アルゴリズムはBaum-Welchアルゴリズムであ る。Baum-Welch アルゴリズムは、モデルλ が出力した配列から、モデルパラ メータを推定するアルゴリズムである。このアルゴリズムは、前向き(Forward) アルゴリズム、後ろ向き(Backward)アルゴリズム、EMアルゴリズムから構成 される。前向きアルゴリズム、および後ろ向きアルゴリズムは動的計画法の一つ であり、ある時点における各状態にいる確率を求めるアルゴリズムである。この アルゴリズムの考え方は、モデルλが観測系列O=o1, o2,· · ·oT を生成する場合 において、時刻tで状態iから状態jに遷移する確率ξt(i,j)を定義し、その後のシ ンボルの生成過程において、時刻tで状態jにいる確率γt(j)を定義する。この二 つの確率からモデルλ= (A, B, π)の推定を行なうというものである。λを用いて 与えられたサンプルに対する遷移確率を計算し、この遷移確率を確率的な回数と 仮定し、学習用サンプルに対する出現頻度の最尤推定を行って、初期確率、遷移 確率、出現確率を更新する。実際には、すべてのサンプルに対して先に述べた計 算を行ってから、パラメータを更新する工程を値が収束するまで繰り返し、最終 的なパラメータを決定する。
2.6 Visualizer
各解析結果を表示するモジュールである。
• 第1〜第3主成分のPCA空間の投射
• 各保持データを座標軸としたヒストグラム
• 時系列に沿った状態遷移図
• 確率的(HMMによる)状態遷移図
クラスタリングの結果より、トラジェクトリデータの状態が時系列に沿ってど の状態により多く存在しているのか、どの状態からどの状態により多く遷移して いるのかを可視化することができる。また、HMMにかけた結果により確率的な 状態遷移を可視化できる。解析によって可視化した結果については、5で詳しく 述べる。
11
第 3 章 解析システムの高速化
本章では解析システムのボトルネックとなる主成分分析についてと、その高速 化について述べる。
3.1 主成分分析
本研究では固有値計算にQR法を用いた。主成分分析のアルゴリズムは主に以 下の2ステップからなる。
(1) 対象データの分散共分散行列(もしくは相関行列)を求める
(2) (1)で求めた行列の固有値,固有ベクトルを求める
ここでは、(2)において本研究で用いた固有値計算アルゴリズムについて述べる。
3.1.1 Householder 変換
共分散行列を三重対角化するアルゴリズムである。鏡像変換により作成された ベクトルをかけることによって、対象となる密対象行列の三重対角化を行う。図 3.1に第kステップにおける三十対角化を示す。
図 3.1: 三重対角化
図のdで表されたベクトルの第1成分だけを抽出する。そのためには第kステッ プにおける対象行列Akに
H=I−αuut (3.1)
3.1. 主成分分析 12 で表される鏡像変換ベクトルを左からかける。ここで
δ=√
dtd (3.2)
u= (d1−sgn(d1)δ, d2,· · · , dn−k) (3.3) α= 2
|u|2 (3.4)
である。したがって第kステップにおける処理は、Akに鏡像変換ベクトルを左右 からかけるので
Ak+1 =HkAkHk
= (I−αuut)Ak(I−αuut)
=Ak−αuutAk−αAkuut+α2uutAkuut
=Ak−upt−put+ 1
2αuutput+1
2αuptuut (p≡αAku) (3.5)
=Ak−uqt−qut (q≡p− 1
2α(ptu)u) (3.6)
となる。
3.1.2 QR 法
対角化を行うアルゴリズムである。
第kステップにおける行列Akに対し、直交行列Pを次々と左から掛けて上三
角行列R(対角成分より下が全てゼロ)を作ったとする。
Pn−kPn−k−1Pn−k−2· · ·P1Ak =R (3.7) このとき、単位行列に右から同じものを掛けていったものを
Pn−kPn−k−1Pn−k−2· · ·P1 =QT (3.8) とすると、Qは直交行列であり、
Ak =QR (3.9)
行列Akを直交行列と上三角行列に分解することが出来る。このとき、行列Qと Rを入れ換えた
A′ =RQ (3.10)
という新たな行列を考えたとき、
A′ =QTAQ (3.11)
3.2. 高速化手法 13 と変換出来ることから、この新たな行列A′の固有値は元の行列Aの固有値と等 しくなる。これを何ステップも繰り返し対角化すれば、その対角成分より行列A の固有値を求めることができる。一般化すると以下のようになる。
Ak =QkRk (3.12)
Ak+1 =RkQk (3.13)
Ak+1 =QTkAkQk (3.14) これでQR法により対角化することができるが、対角化が進むにつれて対角成分 が固有値に近付いて行くと対角成分と副対角成分の差が大きくなり、副対角成分 の値がゼロに近付きにくくなるという問題がおきる。そこで、全ての対角成分か らある定数分だけ引いておき、後から加えるという作業を行う。
Ak−µI =QkRk (3.15)
Ak+1 =RkQk+µI (3.16)
(3.17) この定数µIの値は、行列Akの右下2×2行列の固有値を使用する。対角化が進 んで副対角成分がゼロに近付いても、同じく対角成分も2×2行列の固有値を引 いているので一緒にゼロに近付くため、高速なまま対角化がすすめられる。
3.2 高速化手法
本研究では共有メモリマシンにおける高速化を行った。
3.2.1 HouseHolder 変換における高速化
3.1.1で示したアルゴリズムにおいて、もっとも演算量が集中する部分は
表 3.1: 高速化部分 (3.5) 行列積p≡αAku (3.6) 行列Akのrank(次元)更新
であり、この部分を高速化対象とする。本研究では、ATLASによりチューニ ングした行列演算ライブラリを使用することにより高速化を行う。
3.2. 高速化手法 14 ATLAS
数値計算ライブラリATLAS(Automatically Tuned Linear Algebra Software)[12]
とは、AEOS(Automated Empirical Optimization of Software)と呼ばれるパラダ イムに基づいて、自動的にチューニングされた線形代数ライブラリを生成するソ フトウェアである。またATLASの生成するソフトウェアはBLAS(Basic Linear Algebra Subprograms)[13]に準拠している。
BLASとは、行列とベクトルの基本演算を行うルーチンを集めたライブラリで、
Level 1 BLAS(ベクトル- ベクトル演算)、Level 2 BLAS(行列- ベクトル演算)、
Level 3 BLAS(行列- 行列演算) で構成されている。
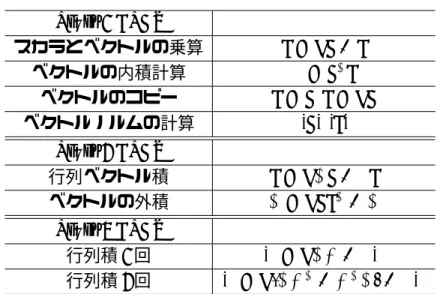
表 3.2: BLAS Level 1 BLAS
スカラとベクトルの乗算 y=αx+y ベクトルの内積計算 β =xTy
ベクトルのコピー y=x, y =αx ベクトルノルムの計算 |x|,|y|
Level 2 BLAS
行列ベクトル積 y=αAx+βy ベクトルの外積 A=αxyT +A
Level 3 BLAS
行列積1回 C =αAB+βC 行列積2回 C=α(ABT +BTA) +βC
表4.3で示した演算は、ほとんどがLevel 2 BLASの演算となるため、ATLAS で自動的にチューニング(パラメータ指定は手動)したライブラリを使用する。
ATLASによるチューニングを行う際には、以下の点に関して検討を行う。
• 問題サイズ(N)
問題サイズ(N)は解く問題の大きさである。一般的にNの値が大きくなる ほど良い結果が得られるが、Nの増加に伴いメモリの使用量が増える。
• ブロックサイズ(NB)
ブロックサイズ(NB)は、解く問題の粒度である。NBが大きくなると通信 量は減少する一方ロードバランスが悪くなる。NBが小さくなると通信量は 増加するがロードバランスは良くなる。NBがうまくL1キャッシュに乗る ように、チューニングすることが重要である。
3.2. 高速化手法 15 問題サイズに関しては、解析に用いるトラジェクトリデータの大きさに合わせて 動的に変化する。したがってチューニングの際、問題サイズに関しては固定値を 用い、ブロックサイズに関しては、3.2.1で詳しく検討する。
3.2.2 QR 法における高速化
3.1.2で述べたアルゴリズムを表3.3に示す。
表 3.3: QR法のアルゴリズム
(1) Akの1番下の副対角成分が許容誤差範囲ならrankを小さくする (2) 右下の2×2行列部分の固有値muを引いておく
(3) cを直行行列Qと上三角行列Rに分解する (4) RQを計算することによってAk+1を計算する (5) (2)で引いておいたmuを足す
(6) 対角化が終了するまで(1)〜(5)を繰り返す
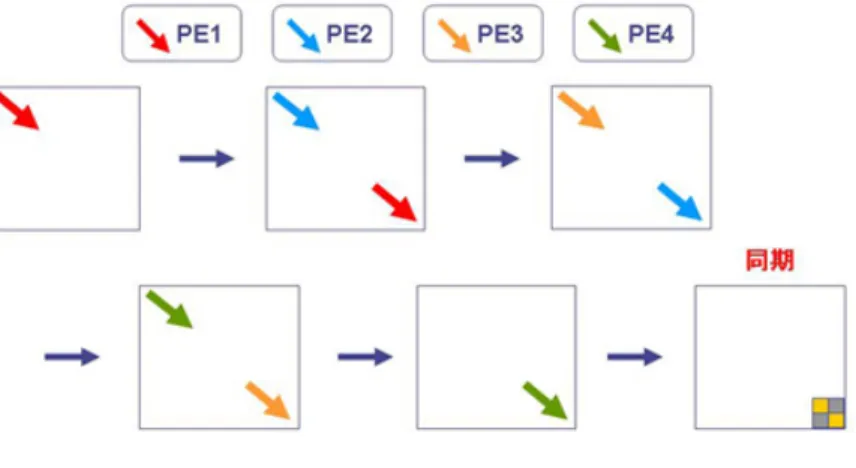
(1)における判定処理において、対象としている行列Akの一番下の副対角成分 を、より速く0に近づけることが高速化につながる。(3)における処理について 図3.2に示す。
図 3.2: 1ステップごとの処理
この部分はAkの副対角成分を左上から右下まで0にしながら上三角行列Rを 作成するという処理である。そして分解した直行行列Qに左からRをかけるこ
3.2. 高速化手法 16 とによって、再び三重対角化する。この部分を複数PEによって並列に行うこと により、収束を早める(図3.3)。
図 3.3: 並列化
また、(5)における処理で複数PE間の同期を行う。
17
第 4 章 高速化の評価
本研究で高速化した主成分分析についての性能評価を行う。
4.1 実験環境
本研究で使用した共有メモリマシンのスペックを表4.1に示す。
表 4.1: 実験環境
CPU Quad-Core Opteron × 2
クロック 2.3GHz
L1キャッシュ 512 KB L2キャッシュ 2 MB
メモリ 16GB
理論性能は
1node 1CPU 2.3(GHz)×4(FPUs/core)
= 9.2GFLOPS
2node 4CPU 2.3(GHz)×4(FPUs/core)×4(CPUs/node)×2(nodes)
= 73.6GFLOPS となる。
4.2. ATLAS 18
4.2 ATLAS
図 4.1: NB(ブロックサイズ)と性能
3.2.1で述べたATLASチューニングの際に使用する問題サイズ(行列サイズ)は
20000を固定値とした。図4.1はブロックサイズを変化させたときの、ATLASに
よってチューニングしたBLASライブラリの性能を示している。
ATLASをチューニングする際に使用したNBのサイズは、ATLASがCPUの
キャッシュサイズを測定する際に導き出した値である28を基にしている。ピーク 性能はNB=224のときの6.16GFLOPSであり、1node1CPUでの理論性能の66.9
%の性能を引き出すことができた。ここで、Visual Technology社によるHPL(High Performance Linpack)の結果について表4.2に示す。
表 4.2: HPLベンチマーク(参考文献[11])
CPU名 1CPU 4CPU 8CPU
AMD Opteron 2356(Barcelona, 2.3GHz) x 2 6.75 - 54 Intel Xeon X5365(Clovertown, 3.00GHz) x 2 6.15 - 49.2 Intel Core 2 Extreme QX6700(2.66GHz) x 1 6.1 24.4 -
全てQuad CoreのCPUベンチマークであり、AMD Opteron 2356については 本研究の実験環境と同じCPUである。問題サイズ(N)、ブロックサイズ(NB)の パラメータは参考文献からは得られなかったが、さらにATLASのチューニング による性能向上ができると考えられる。またXeon、Core 2 Extremeよりも高い 性能を得られた。
4.3. HouseHolder変換の評価 19
4.3 HouseHolder 変換の評価
図 4.2: 行列サイズと性能
図 4.3: CPU数と性能
図4.2は、8CPUで並列化したHouseholder変換(ほぼBLAS 2による演算)を、
問題サイズごとに浮動小数点演算性能を比較した結果である。一般的にLinpack やHPLによるベンチマークは、問題サイズの値が大きくなるほど良い結果が得 られるが、この場合は行列サイズが大きくなるほど、表に示した演算部分の比率 が大きくなり性能が向上したと考えられる。計測している行列サイズが50000で
はなく45000が最大となっているのは、50000ではメモリの使用量の限界を超え
てしまうためである。
4.4. QR法の評価 20 図4.3は、並列化したHouseholder変換を、用いたCPU数ごとに浮動小数点演 算性能を比較した結果である。図4.2において最も良い性能が得られた問題サイ ズである、40000を行列サイズとして用いた。線形に近い性能向上が見られたが、
これは単純なBLAS 2による演算の比率が大きかったため、並列効率の良い結果 が得られたと考えられる。
実測性能は34,98GFLOPSである。これは理論性能の48%であり、表4.2にお ける同CPUでのHPLベンチマーク結果の64.7%の性能である。また、8CPUで の性能は1CPUの5.35倍の性能となった。
4.4 QR 法の評価
図 4.4: 行列サイズと性能
図4.4は、8CPUで並列化したQR法を、問題サイズごとに浮動小数点演算性 能を比較した結果である。Householder変換ほどは問題サイズによるプログラム への影響が少ないことがわかる。
図4.5は、並列化したQR法を、用いたCPU数ごとに浮動小数点演算性能を比 較した結果である。8CPUによる演算性能は1CPUによる場合の2.47倍であり、
並列化効率は良くない。これはCPU数が増加するにつれて、並列化時の同期ま でのCPUの待ち時間も増加しているためである。図3.3で示した並列化手法で は、CPU数の増加と共に、一番最初にスタートしたCPUの待ち時間も増加して いく。収束性の面で考えると速くなっているといえるが、待ち時間がボトルネッ クとなるため、これ以上CPUを増やしても効率は良くないと考えられる。
4.5. 主成分分析全体の評価 21
図 4.5: CPU数と性能
実測性能は16.14GFLOPSである。これは理論性能の22%であり、表4.2にお ける同CPUでのHPLベンチマーク結果の29.9%の性能である。また、8CPUで の性能は1CPUの2.47倍の性能となった。
4.5 主成分分析全体の評価
図 4.6: Octaveとの比較
図4.6は、8CPUで並列化した主成分分析(Householder変換とQR法を合わせ た結果)を、数値計算ライブラリOctaveを用いて作成した単純な主成分分析プ
4.5. 主成分分析全体の評価 22 ログラムと比較したものである。問題サイズごとに浮動小数点演算性能を比較し ている。Octave PCAにおいて、問題サイズが45000のデータが表示されていな いのは、セグメントエラーによりプログラムが実行できなかったためである。本 研究でチューニングした主成分分析プログラムのピーク性能は22.948GFLOPS、
libOctaveによって作成したプログラムは1.562GFLOPSであり、11.9倍の演算性 能を達成した。
23
第 5 章 解析結果
本章では実装したシステムを用いた、MDシミュレーションデータの解析例を 示す。
5.1 シミュレーションに用いた系 :Chignolin
本研究では、分子動力学シミュレーションプログラムSANDER (Simulated An- nealing with NMR-Derived Energy Restraints)[5] を産業技術総合研究所生命情 報工学研究センターのIBM Blue Gene/L上で実行した。解析例としたシニョリ ンタンパク質[6]は、産業技術総合研究所の本田真也博士らによって設計・合成 された世界最小のタンパク質(表5.1)である。ポテンシャル関数にはAMBER のff99[7]を用い、二面角にはSimmerling[8]の補正を行っている。水の作用には、
Generalized Bornモデル[9]、温度は300K、MDの1stepを1フェムト秒とし、
NPTアンサンブルで1マイクロ秒のシミュレーションをBlue Gene/Lの32ノー ド(64CPU)を用いて1433時間行った。
表 5.1: データセット シニョリンタンパク質
(PDBID:1UAO) 10 residues (138 atoms)
5.1. シミュレーションに用いた系:Chignolin 24
5.1.1 主成分分析
図 5.1: Cα distanceをPCAにかけた結果
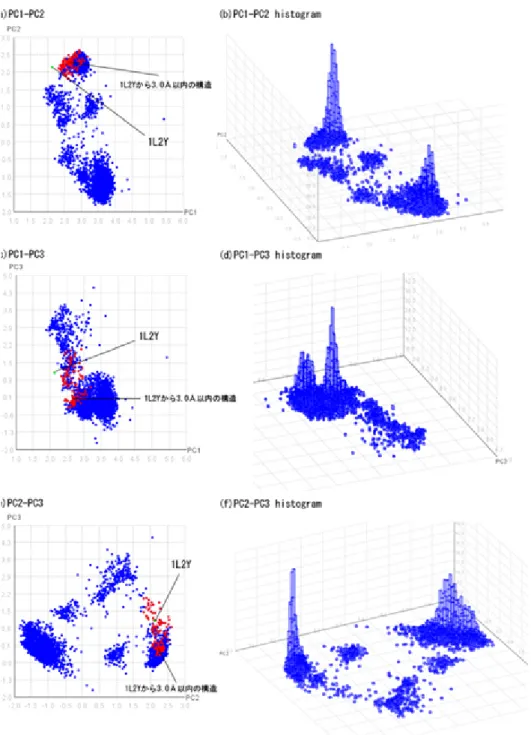
図5.1は、各トラジェクトリデータのCα 間距離を主成分分析し、第1、第2、
第3主成分抽出したものである。図(a)、(c)、(e)については、シニョリンのPDB 構造である1UAOと、1UAOからRMSDが1.0˚A以下の構造についても示してあ る。それぞれのヒストグラムと比較してみると、シミュレーション中もっとも存 在確率の高いタンパク質構造ではなく2番目に存在確率の高い場所に、PDB構 造とそれに近い構造が存在しているという結果が得られた。
5.1. シミュレーションに用いた系:Chignolin 25
5.1.2 クラスタリング
図 5.2: kmeansによるクラスタリング
図5.2は第3主成分まで分析した結果をk-meansによりクラスタリングした結果 である。一つ一つのクラスターは円で表されており、円の大きさが各クラスター に含まれるトラジェクトリデータの個数を表している。円と円の間に結ばれてい る線は、各クラスター内のあるトラジェクトリから、他のクラスター内のトラジェ クトリへの時系列遷移を表している。線の太さが遷移数の多さ、青い線は双方向 リンク、赤い線は片方向リンクである。この図ではクラスタ数を100個に設定し てクラスタリングの可視化を行っている。
5.1. シミュレーションに用いた系:Chignolin 26
5.1.3 HMM
図 5.3: HMM
図5.3にクラスタリングした結果をさらにHMMで学習させた結果を示す。図 (a)はクラスタ数を5にした状態でHMMにかけた確率状態遷移図を表している。
円の大きさが各クラスタに含まれる状態数の大きさを表しており、円から円への 矢印に書かれている数字が遷移確率を表している。各状態の横にはタンパク質の 3次元構造が表示されているが、これはその状態(クラスタ)の代表構造を表して いる。5つの中で2つの大きなクラスタが存在するが、それぞれの構造は類似して おり、安定した構造が存在していることがわかる。また図(b)はクラスタ数を10 にした場合の図であり、同様に2つの安定した構造が存在していることがわかる。
5.2. シミュレーションに用いた系:不安定コントロール 27
5.2 シミュレーションに用いた系 : 不安定コントロール
5.1で用いた系と比較のために用意した系であり、chignolinとほぼ同じ配列を している。しかし実際には以下のような違いがある。
• オリジナルの配列 PDB名 - 1UAO
配列- GYDPETGTWG
• 不安定コントロール PDB名 - 1PGA(の1部分) 配列- GYDDATKEFG
この不安定コントロールを、5.1におけるシミュレーション環境と同じ条件で 同じ時間だけ、シミュレーションを行った。
5.2. シミュレーションに用いた系:不安定コントロール 28
5.2.1 主成分分析
図 5.4: PCA -不安定コントロール
5.1.1同様、図5.4は、各トラジェクトリデータのCα 間距離を主成分分析し、
第1、第2、第3主成分抽出したものである。図(a)、(c)、(e)については、シニョ リンのPDB構造である1UAOと、1UAOからRMSDが1.0˚A以下の構造につい ても示してある。それぞれのヒストグラムと比較してみると、シニョリンのPDB 構造はまったく違う構造が、シミュレーション中に多く発現していることがわか る。ヒストグラムからは存在確率が高い部分が見られるが、安定構造が存在して いるかどうかは次節で述べるHMMの結果と合わせて見るとよくわかる。
5.2. シミュレーションに用いた系:不安定コントロール 29
5.2.2 HMM
図 5.5: HMM - 不安定コントロールとの比較
図5.5にクラスタリングからHMMまで解析を行い、状態遷移図を可視化した 結果を示す。図(a)は図5.3におけるシニョリンをクラスタ数5でHMMにかけ たものと同じものであり、(b)が不安定コントロールをHMMにかけたものであ る。シニョリンと不安定コントロールを比べてみると、それぞれ大きな構造が2 つ存在しているのがわかる。それぞれのクラスタの代表構造を見てみると、(a) に比べて(b)は一定の構造をとっていない。主成分分析の結果と合わせて考える
と、Cαdistanceで分類すると存在確率の高い部分は確かに存在しているが、その
部分の中でもかなりの構造の差異があると考えられる。したがって不安定コント ロールは、シミュレーション中に様々な構造を取り、安定な構造が存在していな いということが分かる。つまり、不安定コントロールはシニョリンと同じシミュ レーション時間では構造決定することが出来ないと考えられる。
5.3. シミュレーションに用いた系:Trp-Cage 30
5.3 シミュレーションに用いた系 :Trp-Cage
ポテンシャル関数にはAMBERのff99[7]を用い、二面角にはSimmerlingの補 正を行っている。水の作用には、Generalized Bornモデル、温度は315K、MDの
1stepを1フェムト秒とし、NPTアンサンブルで400ナノ秒のシミュレーション
をBlue Gene/Lの32ノード(64CPU)を用いて687時間行った。
表 5.2: データセット Trp-Cage (PDBID:1L2Y) 20 residues (304 atoms)
5.3.1 主成分分析
5.1.1同様、図5.6は、各トラジェクトリデータのCα間距離を主成分分析し、第 1、第2、第3主成分抽出したものである。図(a)、(c)、(e)については、Trp-Cage のPDB構造である1L2Yと、1L2YからRMSDが3.0˚A以下の構造についても示 してある。それぞれのヒストグラムと比較してみると、シミュレーション中もっ とも存在確率の高いタンパク質構造が存在する場所に、PDB構造に近い構造が いくつか存在していることが分かる。しかし、それ以外の場所にもPDB構造に 近い構造がまばらにサンプリングされている。これはRMSDを3.0˚A以下という 少し大きめの値に設定しているからであり、シミュレーションが1マイクロ秒程 度まで進めば2.0˚A程度まで狭められると考えられる。
5.3. シミュレーションに用いた系:Trp-Cage 31
図 5.6: Cα distanceをPCAにかけた結果
5.3. シミュレーションに用いた系:Trp-Cage 32
5.3.2 HMM
図 5.7: HMM - Trp-Cage
クラスタリングによって5つのクラスタ分類し、HMMによる解析を行った。状 態遷移図を可視化した結果を図5.7に示す。2つの大きな円に注目すると、構造は それほど似ていないが、要所要所で表5.2で示したTrp-CageのPDB構造に似て いる部分がある。また上から2番目と4番目の構造に注目してみると、この2つの 構造は似ているが、βヘリックスが2つ存在するPDB構造とは少し違い、βシー トが2つ存在している。つまり、2つの大きな円が示す構造は、シミュレーショ ンにおいてシート構造からヘリックス構造に変化していく途中の構造を表してい ると考えられる。また、このシミュレーションはタンパク質が伸びた(unfolding) 状態からはじめているので、400ナノ秒ではシミュレーション時間が足りないと 考えられる。
33
第 6 章 まとめと今後の課題
本章では本論文の結論と今後の課題について述べる。
6.1 まとめ
本稿では生体分子のフォールディング(熱揺らぎ)解析を行うシステムの開発を 行った。このシステムではMDシミューレヨンにより得られたトラジェクトリデー タを、DataSetモジュールによってRg、自由エネルギー、RMSDなどの解析しや すいデータとして保持しておく。Analysisモジュールによって主成分分析、クラ スタリング、HMMによる解析を行い、VisualizerモジュールによってDataSet、
Analysisで得たデータを可視化することができる。
また、本システムにおける主成分分析の処理について、Quad-Core Opteron(大 容量メモリ)マシンでの最適化を行った。主成分分析では三重対角化にHouseholder 変換、対角化にQR法を用いた。8CPU並列化時において、Householder変換で は1CPUの5.35倍、QR法では2.47倍の浮動小数点演算性能を達成した。また、
同マシンで実行した数値計算ライブラリOctaveによる主成分分析との比較にお いて、本研究において高速化した主成分分析のプログラムは11.9倍の浮動小数点 演算性能を達成した。
そして最後に、本システムを用いて、実際にMDシミュレーションを行ったタ ンパク質であるChignolinとTrp-cageについての解析を行った。Chignolinにつ いては、1マイクロ秒に及ぶシミュレーション結果から、PDB構造とのRMSD が1.0˚A以下となる安定な構造が存在し、もっとも存在確率の高い構造ではなく、
2番目に存在確率の高い構造が安定構造であることが分かる。Trp-cageについて は、400ナノ秒のシミュレーションではPDB構造とのRMSDが3.0˚A程度まで の構造を発現させることが出来ている。しかし主成分分析の結果から、さらにシ ミュレーションが進めばPDB構造により近い安定した結果を得ることが出来る と考えられる。
6.2 今後の課題
1.) 他のタンパク質シミュレーションへの適用
本研究ではChignolinとTrp-cageについての解析を行った。Chignolinに関 しては1マイクロ秒のシミュレーションで十分な解析結果が得られたと言え る。しかしTrp-cageについては400ナノ秒程度ではシミュレーション時間 が十分ではなく、少なくとも1マイクロ秒以上のシミュレーションデータが 必要であると考えられる。また、本システムの精度をあげるためには、10 残基(Chignolin)〜20残基(Trp-cage)程度のタンパク質だけでなく、残基数 がさらに多く、生体分子がより大きく揺らぐようなタンパク質の解析も必 要だと考えられる。
2.) 様々なデータを入力とした解析モジュールの実装
本研究ではではCα distanceを用いた解析システムの実装について述べた
が、Cα間の角度(二面角)や、主鎖間の角度を用いた解析も選択的に行える
ようにしたい。
3.) 他のアーキテクチャへの適用
本研究では大容量メモリのOpteronマシンで最適化を行ったが、Core2Duo やXeonなどへの最適化や、分散メモリマシンへの最適化なども行い、さま ざまなアーキテクチャでへの適用を考えている。また主成分分析だけでな く、クラスタリング、HMMの部分に関しても高速化を行うことが有用であ ると考えられる。
35
謝辞
本修士論文を作成するにあたってたくさんの方にお世話になりました。まず、
最適な研究環境を御用意してくださった村岡洋一教授に深く感謝いたします。産 業技術総合研究所での研究環境を整えてくださった産業技術総合研究所生命情報 工学研究センター 副センター長 野口保氏には大変お世話になりました。産業総 合研究所での並列計算機環境を御用意してくださると同時に分子動力学法とその 解析手法の提案についてご指導してくださった産業技術総合研究所生命情報工学 研究センター分子機能チーム研究員 関嶋政和氏に深く感謝いたします。関嶋氏の 協力なくしては、本論文は完成しえなかったと思います。また並列計算機folon、
mettonを御用意してくださった上田研究室修士課程の方々には大変お世話にな
りました。ゼミにおいて数々の御助言・御意見を下さった村岡研究室のみなさま に深く感謝いたします。そして最後に、心身面から私を支えてくださった家族に 深く感謝いたします。
2008年2月 徳永 慎一
36
参考文献
[1] Sergei V. Krivov and Martin Karplus:Hidden complexity of free energy surfaces for peptide (protein) folding,PNAS,Vol. 101, No. 41, pp. 14766–
14770 (2004).
[2] David A. Evans and David J. Walesa:Folding of the GB1 hairpin peptide from discrete path sampling,J.Chem.Phys., Vol. 121, No. 2, pp. 1080–
1090 (2004).
[3] Kelly L. Damm and Heather A. Carlson: Gaussian-Weighted RMSD Su- perposition of Proteins: A Structural Comparison for Flexible Proteins and Predicted Protein Structures, Biophys.J., Vol. 90, No. 12, pp. 4558–4573 (2006).
[4] Kauyoshi Ikeda, Kentaro Tomii, Tsuyoshi Yokomizo, Daisuke Mitomo, Kei- ichiro Maruyama, Shinya Suzuki and Junichi Higo:Visualization of confor- mational distribution of short to medium size segments in globular proteins and identification of local structural motifs,Protein Sci, Vol. 14, No. 5, pp.
1253–1265 (2005).
[5] D.A. Case, T.A. Darden, T.E. Cheatham, III, C.L. Simmerling, J. Wang, R.E. Duke, R. Luo, K.M. Merz, D.A. Pearlman, M. Crowley, R.C. Walker, B. Wang, S. Hayik, A. Roitberg, G. Seabra, X. Wu, S. Brozell, V. Tsui, H.
Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, P. Beroza, D.H.
Mathews, C. Schafmeister, W.S. Ross, and P.A. Kollman, 2006, AMBER 9, University of California, San Francisco.
[6] S. Honda, K. Yamasaki, Y. Sawada, and H. Morii, 10-residue folded peptide designed by segment statistics, Structure, 12, 2004, pp. 1507-1518.
[7] Wang, J., Cieplak, P. and Kollman, P. A. (2000) J. Comput. Chem. 21, pp.
1049-1074
[8] Simmerling, C., Strockbine, B. and Roitberg, A. E. (2002) J. Am. Chem.
Soc. 124, pp. 11258-11259
[9] Tsui, V. and Case, D. A. (2001) Biopolymers 56, pp. 275-291
[10] 藤本万里子,廣安知之,三木光範: タンパク質の基礎, ISDL Report, No.20030704001.
[11] Visual Technology: Linpack(High-Performance Linpack) ベンチマーク結 果, http://www.v-t.co.jp/jp/benchmarks/bench test/hpl.php
[12] R. Clinton Whaley: Automatically Tuned Linear Algebra Soft- ware(ATLAS), http://math-atlas.sourceforge.net/
[13] BLAS(Basic Linear Algebra Subprograms), http://www.netlib.org/blas/
[14] 石崎淳也: 線形代数ライブラリ自動チューニングソフトウェアATLASの改 良,早稲田大学大学院理工学研究科情報・ネットワーク専攻修士論文, 2006.
38
付 録 A 著者外部発表
徳永 慎一, 関嶋 政和, 村岡 洋一, 野口 保, ”確率的情報処理による生体分子の 熱揺らぎ解析に関する研究”,情報処理学会 バイオ情報学研究会(Bio 11), 2007.
![表 2.3: HMM の定義 ・ 潜在変数である状態 { Si } N i ・ 遷移確率分布を A (時刻 t で状態 S j へ遷移する確率) P [q i = S j | q i − 1 = S i , · · · q i − n = S k ] ・ 観測シンボル確率分布を B (状態 S i でシンボル v k が出力される確率) P [b j(k) = P [v k | q k = S j ] ・ 初期状態分布を π (時刻 t = 1 で状態 S i にある確率) π i = P [q 1 = S](https://thumb-ap.123doks.com/thumbv2/123deta/9785856.1869111/15.892.137.769.432.635/HMM定義潜在変数ある状態SiN遷移確率分布時刻シンボルシンボル.webp)
![表 4.2: HPL ベンチマーク (参考文献 [11])](https://thumb-ap.123doks.com/thumbv2/123deta/9785856.1869111/24.892.200.688.254.549/表42HPLベンチマーク参考文献11.webp)