目次
2.4 非臨床試験の概括評価 ...2 2.4.1 非臨床試験計画概略 ...4 2.4.2 薬理試験...5 2.4.3 薬物動態試験 ...11 2.4.4 毒性試験...18 2.4.5 総括及び結論 ...31 2.4.6 参考文献一覧 ...322.4
非臨床試験の概括評価
本項で使用した略号及び用語の定義一覧を表 2.4-1 に示す。 表 2.4-1 略号及び用語の定義一覧 略号及び用語 定義 ALP アルカリホスファターゼ ALT アラニンアミノトランスフェラーゼ APD30 30%再分極時の活動電位持続時間 APD30-90 90%再分極時の活動電位持続時間と 30%再分極時の活動電位持続時 間の差 APD90 90%再分極時の活動電位持続時間 AST アスパラギン酸アミノトランスフェラーゼ AUC 血漿中濃度-時間曲線下面積 AUC24h 時間 0 から投与後 24 時間まで外挿した血漿中濃度-時間曲線下面積 AUCinf 時間 0 から無限時間まで外挿した血漿中濃度-時間曲線下面積 14C 質量数 14 の炭素放射性同位元素 cAMP 環状アデノシン一リン酸 CHO 細胞 チャイニーズハムスター卵巣由来細胞 Cmax 最高血漿中濃度 Cmax, u 最高血漿中非結合型濃度 +dp/dt(max) 左心室内圧の最大立ち上がり速度 EC50 50%有効濃度 GLP 医薬品の安全性に関する非臨床試験の実施の基準hERG ヒト ether-a-go-go-related gene
HPLC-UV 紫外光検出高速液体クロマトグラフィー IC50 50%抑制濃度 ICa カルシウム電流 ICH 日米 EU 医薬品規制調和国際会議 IKr 急速活性型遅延整流カリウム電流 IKs 緩徐活性型遅延整流カリウム電流 INa ナトリウム電流 Ito 一過性外向きカリウム電流 LC-MS/MS 液体クロマトグラフィー-タンデムマススペクトロメトリー M1 代謝物:フェニルグリオキシル酸,M-1 M3 代謝物:馬尿酸,M-3 M5 代謝物:ミラベグロン脱アシル体(M16)-Nw-アセチル体,YM-538852, M-5 M6 代謝物:ミラベグロン-N-脱アルキル体(フェネチルアミン体), YM-538855,M-6 M8 代謝物:ミラベグロン-N-a位酸化体(フェニル酢酸誘導体), YM-538853,M-8 M9 代謝物:4-アセチルアミノフェニル酢酸,YM-340790,M-9 M11 代謝物:ミラベグロン-O-グルクロニド,YM-382984,RB-3 M12 代謝物:ミラベグロンケトン酸化体(M18)-N-COO-グルクロニド, YM-538858,RB-6
表 2.4-1 略号及び用語の定義一覧(続き) 略号及び用語 定義 M13 代謝物:ミラベグロン-N-COO-グルクロニド,YM-538859,RB-9 M14 代謝物:ミラベグロン-Nω-グルクロニド,YM-554028,RB-11 M15 代謝物:ミラベグロン-N-O-グルクロニド,YM-9636324,H1 M16 代謝物:ミラベグロン脱アシル体,YM-208876 M17 代謝物:ミラベグロン脱アシル体-o-O-グルクロニド,H2 M18 代謝物:ミラベグロンケトン酸化体,YM-538856 M19 代謝物:ミラベグロン脱アシル体-Nw-アセチル体-O-グルクロニド, RB-4 M20 代謝物:ミラベグロン-N-a位酸化体(フェニル酢酸誘導体)-N-アセ チル体,YM-345827,RB-5 M21 代謝物:ミラベグロン脱アシル体-Nw-アセチル体-N-COO-グルクロニ ド-ケトン酸化体,RB-7 M22 代謝物:ミラベグロン脱アシル体-Nw-アセチル体-N-COO-グルクロニ ド,RB-10 MAPD90 90%単相性活動電位持続時間 MCH 平均赤血球血色素量 MCV 平均赤血球容積 MDR1 P-糖蛋白 PK Pharmacokinetics(薬物動態) t1/2 消失半減期 tmax 最高血漿中濃度到達時間 Tp-e T 波のピークから終末までの時間
2.4.1
非臨床試験計画概略
膀胱から外尿道口に至る経路は下部尿路と呼ばれ,その機能である蓄尿及び排尿は,主に,交 感神経(下腹神経),副交感神経(骨盤神経)及び体性神経(陰部神経)の三つの神経系により調 節されている。生成された尿を膀胱内に溜める蓄尿期では,膀胱における神経支配は交感神経が 優位となり,下腹神経終末より放出されるノルアドレナリンが膀胱平滑筋に存在する β アドレナ リン受容体を刺激することで膀胱を弛緩させる。膀胱内に貯留された尿を排出する排尿期におい ては,膀胱における神経支配は副交感神経が優位となり,骨盤神経終末より放出されるアセチル コリンが膀胱平滑筋に存在するムスカリン受容体を刺激することで膀胱を収縮させる。 過活動膀胱とは,尿意切迫感を必須とした症状症候群であり,頻尿や切迫性尿失禁を伴う[1]。 現在,過活動膀胱の標準治療薬としてムスカリン受容体拮抗薬が使用されているが,ムスカリン 受容体拮抗薬は,その作用機序から排尿機能を悪化させる懸念を有し,加えて,口内乾燥,便秘, 霧視等の副作用を発現することが知られている[2]。 β アドレナリン受容体の 3 つのサブタイプのうち,ヒト膀胱平滑筋に発現しているサブタイプ は,そのほとんどが β3アドレナリン受容体であることが遺伝子レベルにおいて確認されており[3], 機能的な弛緩反応においても β3アドレナリン受容体が重要な役割を担っていることが確認されて いる[4]。また,β3アドレナリン受容体作動薬が過活動膀胱モデルにおいて膀胱容量増加作用を 示し[5],ムスカリン受容体拮抗薬と異なり,排尿時の膀胱収縮力を低下させない[6]ことが報 告されている。したがって,β3アドレナリン受容体作動薬は既存薬とは異なる新たな作用機序を 有し,さらにムスカリン受容体拮抗薬による副作用を軽減できる新規過活動膀胱治療薬になり得 るとして期待されている。 ミラベグロンは,アステラス製薬株式会社において創製された選択的 β3アドレナリン受容体作 動薬であり,過活動膀胱を適応症として本邦を始め,米国,欧州諸国及びアジア諸国において臨 床開発中である。 今回の申請にあたり,本薬の薬理学的,薬物動態学的及び毒性学的特性を明らかにする目的で 以下の各種試験を実施した。 効力を裏付ける試験として,ミラベグロンの β アドレナリン受容体に対する刺激作用及び特異 性,膀胱弛緩作用,膀胱内圧に対する作用並びに膀胱機能に対する作用を,既存のムスカリン受 容体拮抗薬であるトルテロジン及びオキシブチニンの作用との比較を含め検討した。また,ヒト 血漿中で同定された 8 種の代謝物の β アドレナリン受容体に対する刺激作用及び特異性について も検討を加えた。さらに,安全性薬理試験として,コアバッテリー試験,フォローアップ試験及 び補足的安全性薬理試験を実施した。コアバッテリー試験は,医薬品の安全性に関する非臨床試 験の実施の基準(GLP)適合試験として安全性薬理試験ガイドライン[日米 EU 医薬品規制調和 国際会議(ICH)S7A]に準拠して実施した。 ミラベグロンの非臨床薬物動態に関しては,一連の in vitro 及び in vivo 試験によりその吸収, 分布,代謝及び排泄を評価した。薬理及び毒性試験で使用された動物種のマウス,ラット,ウサギ,イヌ及びカニクイザルにおいてミラベグロンの血漿中濃度を測定し,薬物動態の投与量依存 性及び反復投与による薬物動態への影響を検討した。ラット及びイヌにおいて,ミラベグロンを 経口投与したときのバイオアベイラビリティを検討した。イヌにおいて,吸収に及ぼす食餌の影 響を検討した。また,14C-ミラベグロンをラット及びカニクイザルに経口投与したときの放射能の 組織分布並びに放射能の尿及び糞中への排泄を調べることにより,ミラベグロン由来成分の体内 動態,残留性あるいは蓄積性を検討した。さらに,動物及びヒトの試料を用いた種々の in vitro 代 謝試験並びに in vivo 代謝物検索及び構造推定を実施するとともに,ヒト血漿において同定された すべての代謝物の血漿中濃度を毒性試験で使用された動物種において測定した。 ミラベグロンの非臨床における安全性について,ラット及びイヌを用いた単回投与毒性試験, ラット,イヌ及びカニクイザルを用いた反復投与毒性試験,細菌,ヒトリンパ球及びラットを用 いた遺伝毒性試験,マウス及びラットを用いたがん原性試験,ラット及びウサギを用いた生殖発 生毒性試験,ウサギを用いた局所刺激性試験並びにその他の毒性試験(皮膚感作性試験,不純物 に関する試験及び溶血性試験)により評価した。すべての重要な毒性試験は GLP 適合試験として 医薬品毒性試験法ガイドラインあるいは ICH ガイドラインに準拠して実施した。ミラベグロンの 投与経路は臨床投与経路に準じて経口投与とした。
2.4.2
薬理試験
2.4.2.1
効力を裏付ける試験
β アドレナリン受容体に対する刺激作用及び特異性 ヒト及び各種動物の β アドレナリン受容体の各サブタイプを発現させた CHO 細胞を用い,細胞 内 cAMP 濃度を指標として各 β アドレナリン受容体サブタイプに対する刺激作用を検討した。そ の結果,ミラベグロンはヒト β3アドレナリン受容体に対して刺激作用を示し,その EC50値は 1.5 nmol/L であった(2.6.2.2.1.1 β アドレナリン受容体に対する刺激作用)。ミラベグロンの固有活 性(完全活性薬であるイソプロテレノールの最大反応を 1 としたときの相対値)は,ヒト β1,β2 及び β3アドレナリン受容体においてそれぞれ 0.1,0.2 及び 0.8 であった。ミラベグロンはラット, イヌ及びカニクイザルの β3アドレナリン受容体に対しても刺激作用を示し,固有活性はそれぞれ 1.0,0.8 及び 0.8 であった。ミラベグロンのラット,イヌ及びカニクイザルの β1アドレナリン受 容体に対する固有活性はそれぞれ 0.6,0.3 及び 0.2 であった。また,これら動物種の β2アドレナ リン受容体に対する固有活性はいずれも 0.1 であり,ミラベグロンは β2アドレナリン受容体に対 してほとんど刺激作用を示さなかった。 ヒト β アドレナリン受容体の各サブタイプを発現させた細胞の膜画分を用いた受容体結合実験 において,ミラベグロンはヒト β3アドレナリン受容体に高い親和性を示した(2.6.2.2.1.2 ヒト β アドレナリン受容体に対する親和性)。また,ミラベグロンは α1Aアドレナリン受容体,ムスカリ ン M2受容体,ナトリウムチャネルサイト 2,ドパミントランスポータ及びノルエピネフリントランスポータに対して低い親和性を示した(2.6.2.2.1.3 各種受容体,イオンチャネル及びトランス ポータに対する親和性並びに酵素活性に対する作用)。 膀胱弛緩作用及び膀胱内圧に対する作用 ラット摘出膀胱において,ミラベグロンは組織内 cAMP 濃度を上昇させた(2.6.2.2.2.1 ラット 摘出膀胱組織内の cAMP 濃度に対する作用)。カルバコールにより持続性収縮を惹起させたラット (2.6.2.2.2.2 ラット摘出膀胱における弛緩作用)及びヒト(2.6.2.2.2.4 ヒト摘出膀胱における弛緩 作用)摘出膀胱において,ミラベグロンは弛緩作用を示した。ラット摘出膀胱におけるミラベグ ロンの弛緩作用は,選択的 β1アドレナリン受容体遮断薬 CGP-20712A 及び選択的 β2アドレナリン 受容体遮断薬 ICI-118,551 の前処置により影響を受けなかったことから(2.6.2.2.2.3 ラット摘出膀 胱におけるミラベグロンの弛緩作用に対する β アドレナリン受容体遮断薬の影響),ミラベグロン による膀胱弛緩作用は β3アドレナリン受容体を介するものと考えられた。ペントバルビタール麻 酔ラットにおいて,ミラベグロンは静止時膀胱内圧を低下させたが(2.6.2.2.3.1 ラット静止時膀 胱内圧に対する作用),ムスカリン受容体拮抗薬のトルテロジン及びオキシブチニンは明らかな静 止時膀胱内圧低下作用を示さなかった。ペントバルビタール麻酔イヌにおいて,ミラベグロンは カルバコール誘発膀胱内圧上昇を抑制した(2.6.2.2.3.2 イヌ膀胱内圧に対する作用)。 膀胱機能に対する作用 ウレタン麻酔ラットにおいて,ミラベグロンは律動性膀胱収縮の収縮力に影響を及ぼすことな く,収縮頻度を減少させた(2.6.2.2.4.1 律動性膀胱収縮に対する作用)。一方,オキシブチニンは 収縮力の低下と収縮頻度の増加を引き起こした。同試験において,ミラベグロンの 14 日間反復投 与後の作用を確認したところ,作用の減弱は認められなかった(2.6.2.2.4.1.3 律動性膀胱収縮に対 する反復投与後の作用)。無麻酔カニクイザルにおいて,ミラベグロンは平均一回排尿量を増加さ せ,排尿回数を減少させた(2.6.2.2.4.2 無麻酔カニクイザルにおける平均一回排尿量及び排尿回 数に対する作用)。脳梗塞により一回排尿量の減少したラットにおいて,ミラベグロンは平均一回 排尿量を増加させた(2.6.2.2.4.3 過活動膀胱モデルラットにおける平均一回排尿量に対する作用)。 尿道部分閉塞ラットにおいて,ミラベグロンは一回排尿量,排尿圧及び残尿量に影響を及ぼすこ となく,排尿前収縮回数を減少させた(2.6.2.2.4.4 尿道部分閉塞ラットにおける排尿機能に対す る作用)。同試験において,トルテロジン及びオキシブチニンは排尿前収縮回数を減少させず,そ れぞれ一回排尿量減少及び残尿量増加作用を示した。 ヒト血漿中代謝物の薬理作用 ヒト血漿中で同定されたミラベグロンの 8 種の代謝物(M5,M8,M11,M12,M13,M14,M15 及び M16)について,ヒト β アドレナリン受容体各サブタイプに対する刺激作用並びに各種受容 体,イオンチャネル,トランスポータに対する親和性及び酵素活性に対する作用を検討した。M13 のヒトb3アドレナリン受容体に対する固有活性はミラベグロンと同程度であったが,その EC50値 (1.1 mmol/L)はミラベグロンの EC50値(1.5 nmol/L)に比べ高かった(2.6.2.2.5.1 ヒト β アドレ ナリン受容体に対する刺激作用)。その他のヒト血漿中代謝物のヒトb3アドレナリン受容体に対す る固有活性は,いずれも 0.5 以下であった。ヒト血漿中代謝物のヒトb1及びb2アドレナリン受容
体に対する固有活性は,それぞれ 0.1 及び 0.2 以下であった。また,M5 は 10 μmol/L でドパミン トランスポータの特異的リガンド結合を 83%阻害し,M16 は同濃度でドパミントランスポータ及 びノルエピネフリントランスポータの特異的リガンド結合をそれぞれ 73%及び 68%阻害した (2.6.2.2.5.2 各種受容体,イオンチャネル及びトランスポータに対する親和性並びに酵素活性に 対する作用)。それ以外のヒト血漿中代謝物の受容体,イオンチャネル及びトランスポータの特異 的リガンド結合に対する阻害率及び酵素活性に対する阻害率は,10 μmol/L でいずれも 50%未満で あった。ヒト血漿中代謝物の薬理活性及びヒト血漿中における存在量(2.7.2.2.2.3.1 性差及び高齢 者試験[CL-072])から判断して,これらヒト血漿中代謝物がミラベグロン投与時の薬効に寄与す る可能性及び副作用を発現させる可能性は低いと考えられた。 以上の結果より,ミラベグロンはヒト β3アドレナリン受容体に対して選択的な刺激作用を示す とともに,膀胱組織内の cAMP 濃度を上昇させ,膀胱平滑筋を弛緩させること,また,膀胱弛緩 作用に基づき排尿前収縮及び排尿回数を減少させ,平均一回排尿量を増加させることが明らかと なった。さらに,ミラベグロンはムスカリン受容体拮抗薬であるトルテロジンやオキシブチニン と異なり,排尿時膀胱収縮に影響を及ぼしにくいことが示された。
2.4.2.2
安全性薬理試験
安全性薬理コアバッテリー試験 ミラベグロン(30,100 及び 300 mg/kg)を単回経口投与したラットにおいて(2.6.2.4.1.1 中枢 神経系に及ぼす作用),30 mg/kg 以上で自発運動の低下,100 mg/kg 以上で握力の低下,横臥,眼 瞼閉鎖及び呼吸深大,300 mg/kg で筋緊張の低下(全身及び腹部),腹臥及び正向反射の消失が認 められた。ミラベグロンは 1 回目の hERG 電流測定試験において,30 mmol/L まで hERG 電流を抑制しなかっ た(2.6.2.4.1.2.1 hERG チャネルに対する作用)。2 回目の hERG 電流測定試験において,30 mmol/L で 14.7%の hERG 電流抑制作用を示した[2.6.2.4.1.2.2 hERG チャネルに対する作用(追加試験)]。 ミラベグロンは 30 mmol/L までモルモット摘出乳頭筋の活動電位に影響を及ぼさなかった (2.6.2.4.1.2.4 心筋活動電位に対する作用)。 ミラベグロンの 5 種のヒト血漿中代謝物(M5,M11,M12,M14 及び M16)について,hERG 電流(2.6.2.4.1.2.3 ミラベグロン代謝物の hERG チャネルに対する作用)及びモルモット摘出乳頭 筋の活動電位に対する作用(2.6.2.4.1.2.5 ミラベグロン代謝物の心筋活動電位に対する作用)を検 討した。その結果,M5 及び M16 は hERG 電流を抑制し,IC50値はそれぞれ 21 及び 31 mmol/L で
あった。また,M14 は 30 mmol/L で 17.3%の hERG 電流抑制作用を示した。モルモット摘出乳頭 筋において,M5 は 3 mmol/L で 30%再分極時の活動電位持続時間(APD30)を 6.1%延長させ,90%
再分極時の活動電位持続時間と 30%再分極時の活動電位持続時間の差(APD30-90)を 7.9%短縮さ
延長させた。また,M16 は 30 mmol/L で APD90を 5.0%延長させた。M11 及び M12 は 30 mmol/L ま で hERG 電流及びモルモット摘出乳頭筋の活動電位に影響を及ぼさなかった。 ミラベグロン(3,10,30 及び 100 mg/kg)を単回経口投与した無麻酔カニクイザルにおいて (2.6.2.4.1.2.6 無麻酔カニクイザルの心血管系及び呼吸系に及ぼす作用),10 mg/kg 以上で心拍数 の増加,100 mg/kg で嘔吐及び横臥,PR 及び QRS 間隔の延長が認められた。体温,血圧,血液ガ ス及び血中電解質濃度に変化は認められなかった。 安全性薬理フォローアップ試験 ミラベグロン及びヒト血漿中代謝物(M5,M11,M12,M14 及び M16)について,ナトリウム チャネル[ナトリウム電流(INa)],カルシウムチャネル[カルシウム電流(ICa)],2 種のカリウ ムチャネル[緩徐活性型遅延整流カリウム電流(IKs)及び一過性外向きカリウム電流(Ito)]に対 する作用を検討した(2.6.2.4.2.1.1 心筋イオンチャネルに対する作用)。その結果,ミラベグロン は 10 mmol/L でナトリウム電流を 48.5%抑制し,カルシウム電流を 15.3%抑制した。M16 は 10 mmol/L でナトリウム電流を 10.5%抑制し,カルシウム電流を 8.8%抑制した。M5,M11,M12 及び M14 は 10 mmol/L においていずれのイオン電流に対しても影響を及ぼさなかった。 ミラベグロン及びヒト血漿中代謝物(M5,M11,M12,M14 及び M16)について,イヌ動脈灌 流左室切片に対する作用を検討した(2.6.2.4.2.1.2 イヌ動脈灌流左室切片に対する作用)。その結 果,ミラベグロンは 300 ng/mL で貫壁性双極心電図の QT 間隔及び心内膜下における APD90を軽 度短縮させたが,貫壁性再分極時間のばらつきの指標とされる T 波のピークから終末までの時間 (Tp-e)に影響を及ぼさず,催不整脈作用を示さなかった。M5 は 100 ng/mL で QT 間隔,Tp-e 間 隔及び APD90を軽度短縮させ,催不整脈作用を示さなかった。M11,M12,M14 及び M16 は 100 ng/mL までイヌ動脈灌流左室切片に対して影響を及ぼさなかった。 補足的安全性薬理試験(一般薬理試験を含む) 中枢神経系に対する作用 ミラベグロン(1,3,10,30 及び 100 mg/kg)を単回経口投与したマウスにおいて(2.6.2.4.3.1 中 枢神経系に対する作用),10 mg/kg 以上で腹臥及び直腸温の上昇が認められた。30 mg/kg で自発運 動量の増加,100 mg/kg で自発運動量の減少,警戒性の低下,肢筋緊張度の低下,腹筋緊張度の低 下,懸垂力の低下,体温の軽度低下,皮膚蒼白及び立毛が認められた。 心血管系及び呼吸系に対する作用 ミラベグロン(3,10,30,60 及び 100 mg/kg)を単回経口投与した無麻酔カニクイザルにおい て(2.6.2.4.3.2.1 心血管系及び呼吸系に対するミラベグロンの作用),30 mg/kg 以上で心拍数増加, 100 mg/kg で QRS 間隔の延長及び 3 例中 2 例で嘔吐が認められた。 ミラベグロン(0.01,0.03,0.3,3 及び 10 mg/kg)を単回経口投与した無麻酔イヌにおいて (2.6.2.4.3.2.1 心血管系及び呼吸系に対するミラベグロンの作用),0.03 mg/kg 以上で心拍数の増 加,PR 間隔の短縮,0.3 mg/kg 以上で呼吸数の増加,収縮期血圧及び平均血圧の低下,10 mg/kg で血液中二酸化炭素分圧の低下が認められた。ミラベグロンは,10 mg/kg まで QTc 間隔に影響を 及ぼさなかった。
ミラベグロン(10 mg/kg)を単回静脈内投与した無麻酔イヌにおいて(2.6.2.4.3.2.1 心血管系及 び呼吸系に対するミラベグロンの作用),平均血圧の低下,心拍数の増加,P 波の消失,QRS 間隔 の延長及び心室頻拍が認められ,4 例中 2 例は心室細動により死亡した。 ハロセン麻酔イヌにおいて,心室筋の単相性活動電位持続時間に対するミラベグロン(0.03, 0.3,3,10 及び 30 mg/kg)の単回静脈内投与による作用を検討したところ(2.6.2.4.3.2.1 心血管系 及び呼吸系に対するミラベグロンの作用),正常リズム(洞調律)条件下において 3 mg/kg で心拍 数の増加,90%単相性活動電位持続時間(MAPD90)の短縮,QT 間隔の短縮及び T 波の増高が認 められた。ペーシング刺激(刺激間隔:300 ms)条件下において,0.3 mg/kg 以上で MAPD90の短 縮が認められた。 ミラベグロン(0.1,0.3 及び 1 mg/kg)を麻酔下に単回静脈内投与したウサギにおいて(2.6.2.4.3.2.1 心血管系及び呼吸系に対するミラベグロンの作用),1 mg/kg で心拍数及び心筋酸素消費量の指標 である二重積(心拍数´収縮期血圧)の増加が認められた。 ミラベグロンの心血管系に対する作用の機序検討 無麻酔イヌにおいて(2.6.2.4.3.2.2 ミラベグロンの心血管系に対する作用の機序検討),ミラベ グロン 100 mg/kg の経口投与直後に非選択的 β アドレナリン受容体遮断薬であるプロプラノロー ル(1 mg/kg)を静脈内投与した場合,ミラベグロンの呼吸数及び心拍数増加作用並びに血圧低下 作用は 100 mg/kg 単独経口投与時の作用に比べ軽度であった。また,ミラベグロン 10 mg/kg の単 回静脈内投与により,3 例中 3 例で心室頻拍が認められた。その後プロプラノロールを静脈内投 与したが,3 例中 1 例では心室細動に移行し,死亡した。残りの 2 例では期外収縮,房室ブロッ ク等の不整脈が認められたが,その後回復した。一方,プロプラノロールの静脈内投与後 5 分に ミラベグロン 10 mg/kg を単回静脈内投与した場合,期外収縮及び房室ブロック等の不整脈が認め られたが,心室頻拍が認められたのは 3 例中 1 例のみで,その後全例回復した。 ペントバルビタール麻酔イヌにおいて(2.6.2.4.3.2.2 ミラベグロンの心血管系に対する作用の機 序検討),ミラベグロン 0.001 mg/kg 以上の静脈内投与により,拡張期血圧,平均血圧及び左心室 内圧の低下並びに心拍数及び左心室内圧の最大圧立ち上がり速度[+dp/dt(max)]の増加が認めら れた。一方,神経節遮断薬であるヘキサメトニウム及び迷走神経活性を抑制するアトロピンの静 脈内投与後においては,検討したミラベグロンの最低用量である 0.01 mg/kg の静脈内投与により 血圧の低下が認められたが,心拍数及び+dp/dt(max)の増加は 0.1 mg/kg 以上の静脈内投与後でのみ 認められた。ヘキサメトニウム,アトロピン及び選択的 β1アドレナリン受容体遮断薬であるメト プロロールの静脈内投与後においては,ミラベグロンの心拍数及び+dp/dt(max)の増加作用は,ほ ぼ完全に抑制された。これらのことから,低用量(0.001~0.03 mg/kg)静脈内投与後に認められ たミラベグロンのイヌにおける心拍数増加作用は,血管の β3アドレナリン受容体刺激による血圧 低下に伴う反射性頻脈によるものであると考えられた。また,ミラベグロンはイヌにおいて, 0.1 mg/kg 以上の静脈内投与により心臓の β1アドレナリン受容体に対して直接的な刺激作用を示 すことが考えられた。
ペントバルビタール麻酔ラットにおいて(2.6.2.4.3.2.2 ミラベグロンの心血管系に対する作用の 機序検討),ミラベグロン 0.1 mg/kg の静脈内投与の心拍数増加作用は,ヘキサメトニウム,アト ロピン及び交感神経終末におけるカテコールアミン枯渇薬であるレセルピン処置下においても認 められたが,さらにメトプロロールを前処置することによりほぼ完全に抑制された。これらのこ とから,麻酔ラットにおけるミラベグロンの心拍数増加作用は,心臓の β1アドレナリン受容体に 対する直接的な刺激によるものと考えられた。 その他の補足的安全性薬理試験 ミラベグロン(1,3,10,30 及び 100 mg/kg)を単回経口投与した生理食塩水負荷ラットにお いて(2.6.2.4.3.3 尿排泄に対する作用),10 mg/kg 以上で投与後 0~3 時間までの尿量及び電解質 (ナトリウム,カリウム,塩素)排泄量の減少,30 mg/kg 以上で投与後 3~6 時間のカリウム排泄 量の増加及び塩素排泄量の減少が認められた。 モルモット摘出回腸において,ミラベグロンは 10 nmol/L 以上でヒスタミン,塩化バリウム及び セロトニン誘発収縮を,100 nmol/L 以上でアセチルコリン誘発収縮を抑制した(2.6.2.4.3.4 自律神 経系に対する作用)。β3アドレナリン受容体はモルモット回腸の平滑筋弛緩反応に関与しているこ とが報告されていることから[7],この作用は本薬の薬効に基づく作用である可能性が考えられ た。しかしながら,ミラベグロンは 100 mg/kg までマウスの胃腸管内輸送能に影響を及ぼさなかっ た(2.6.2.4.3.5 消化器系に対する作用)。
2.4.3
薬物動態試験
2.4.3.1
分析法
14C-標識したミラベグロンを用いて,マウス,ラット及びカニクイザルにおける吸収,分布,代 謝又は排泄を検討した。14C-ミラベグロン投与時の生体試料中の放射能濃度は,試料に直接又はサ ンプルオキシダイザーにより生じた14CO2を捕集したものに液体シンチレーターを加え,液体シ ンチレーションカウンターで測定した。なお,14C-ミラベグロンの血液及び血漿中並びに組織内の 放射能濃度はすべてミラベグロン当量として表示した。 マウス,ラット,ウサギ,イヌ及びカニクイザルにおける血漿中ミラベグロン濃度は,紫外光 検出高速液体クロマトグラフィー(HPLC-UV)又は液体クロマトグラフィー-タンデムマススペ クトロメトリー(LC-MS/MS)法により測定した。代謝物の血漿中濃度に関しても,マウス,ラッ ト,ウサギ及びカニクイザルにおいて M5,M8,M11,M12,M13,M14,M15 及び M16 をそれ ぞれ,LC-MS/MS 法で測定した。いずれの定量法も良好にバリデートされており,各結果は,薬 物動態試験概要表にまとめて掲載した(概要表 2.6.5.2 分析方法及びバリデーション試験)。2.4.3.2
吸収
ラットに14C-ミラベグロン(10 mg/kg)を単回経口投与したときの吸収率は 66.7%であった(概 要表 2.6.5.20 尿・胆汁中累積排泄)。ラット(3~30 mg/kg)及びイヌ(0.25~1 mg/kg)にミラベ グロンを経口投与したとき,いずれもミラベグロンの血漿中濃度は投与後 0.1~4 時間に Cmaxに達 した。絶対バイオアベイラビリティは,ラットで投与量 3,10 及び 30 mg/kg において 23.0%,48.4% 及び 75.7%,イヌで 0.25,0.5 及び 1 mg/kg において 41.8%,64.6%及び 77.1%であり,投与量の増 加に伴い増大した(概要表 2.6.5.5 単回経口投与後の血液・血漿中濃度)。ヒトにおける絶対バイ オアベイラビリティも投与量の増加に伴い増大した(2.7.1.2.6 絶対バイオアベイラビリティ試験 [CL-033],2.7.1.2.4 IVIVC 試験[CL-076])。ミラベグロンの全身クリアランスはラット及びイヌ のいずれにおいても比較的大きく(それぞれ 47.4 及び 37.2 mL/min/kg),かつ14C-ミラベグロンを ラットに経口投与したときの血漿,尿及び胆汁中放射能の半分以上を代謝物が占めていたことか ら(概要表 2.6.5.15 In vivo での代謝),これらのクリアランスには主として代謝クリアランスが寄 与していると考えられた。さらに,絶対バイオアベイラビリティの投与量の増加に伴う増大は, 小腸又は肝初回通過時の代謝能あるいは排出能の飽和による可能性が考えられたが,代謝物を測 定したラット及びヒトの PK 試験においては,測定したいずれの代謝物の血漿中濃度も投与量の 増加に伴い非線形に増大した(概要表 2.6.5.17 反復経口投与後の血漿中代謝物濃度,2.7.2.2.2.3.1 性差及び高齢者試験[CL-072],2.7.2.2.2.5.1 QT/QTc 評価試験[CL-037])。代謝能の飽和を示唆す るような変化は観察されなかったことから,排出能の飽和が投与量比以上の増大の原因と推察さ れた。ヒトにおいては 300 mg 以上の高投与量では Cmax及び AUCinfは投与量比に伴って上昇する飽和して,その寄与が無視できるようになり,線形な薬物動態が観察された可能性が考えられた (2.7.2.2.2.1.1 第Ⅰ相単回及び反復投与試験[CL-034])。なお,ミラベグロンは小腸に発現してい る排出トランスポータである P-糖蛋白(MDR1)の基質になることが明らかになっている
(2.7.2.2.1.4.1 P-糖蛋白(MDR1)に対するミラベグロンの基質性の検討[ME-031])。
イヌにミラベグロン(0.5 mg/kg)を非絶食下で経口投与したとき,Cmax及び AUCinfは絶食下投
与時と比較してそれぞれ 78.5%及び 65.8%に低下し,食餌によって血漿中ミラベグロン濃度が低下 することが示された(概要表 2.6.5.7 食餌の影響)。
イヌにミラベグロン(0.5 mg/kg/day)を 1 日 1 回 15 日間反復経口投与したとき,投与初日,8 日目及び 15 日目の血漿中ミラベグロン濃度はいずれもほぼ一定の値を示した(概要表 2.6.5.6 反 復経口投与後の血漿中濃度)。算出したいずれの薬物動態パラメータ(Cmax,tmax,AUC 及び t1/2)
においても反復投与後と投与初日との間で有意な差は認められなかった。また,毒性試験におけ るトキシコキネティクス測定の結果から,マウス,ラット,ウサギ及びカニクイザルにおいても, 血漿中ミラベグロン濃度は,反復投与により大きな影響を受けないものと考えられた(概要表 2.6.7.7 反復投与毒性試験:重要な試験,2.6.7.10 がん原性試験,2.6.7.13 生殖発生毒性試験:胚・ 胎児発生に関する試験)。
2.4.3.3
分布
ミラベグロンの in vitro 血漿蛋白結合率は,いずれの動物種においても 200~5000 ng/mL の濃度 範囲でほぼ一定であり,マウスで 76.7%~77.7%,ラットで 78.5%~79.5%,ウサギで 87.2%~88.2%, イヌで 61.1%~62.0%,カニクイザルで 53.3%~56.4%及びヒトで 72.2%~76.9%(日本人で 76.3% ~76.9%及び白人で 72.2%~73.3%)であった(概要表 2.6.5.11 血漿蛋白との結合)。また,ヒト 血漿中における主結合蛋白はアルブミンで,次いでa-酸性糖蛋白であると推定された (2.7.2.2.1.1.2 血漿中主要結合蛋白の推定[ME-044])。ミラベグロンは血球に移行し,in vitro 血 球移行率は,100~2500 ng/mL の濃度範囲で,ラットで 55.56%~59.70%,イヌで 66.46%~66.99%, カニクイザルで 57.96%~59.36%及びヒトで 60.39%~60.82%であった(概要表 2.6.5.12 血球移行 性)。 ラット及びイヌにミラベグロンを静脈内投与したとき,分布容積はそれぞれ,10.3 及び 14.3 L/kg といずれの動物種においても大きかった(概要表 2.6.5.4 単回静脈内投与後の血漿中濃度)。また, ラットに14C-ミラベグロン(10 mg/kg)を経口投与したとき,ほぼすべての組織において放射能 が検出され,ミラベグロン由来成分は種々の組織へ分布し易いことが示された(概要表 2.6.5.8 単 回経口投与後の組織分布)。 白色及び有色ラットに14C-ミラベグロン(10 mg/kg)を単回経口投与したときの組織内放射能 濃度は,投与部位である消化管以外では,ほとんどの組織で投与後 4 時間に最高値を示した。放 射能の分布は,白色ラットでは肝臓及び腎臓において最も高く,大脳及び小脳で最も低かった。 有色ラットにおける組織内放射能濃度は白色ラットにおけるそれと類似していたが,眼球内放射 能濃度は投与後 24 時間に最高値に達し,その値は白色ラットの最高値の約 18 倍であった(概要表 2.6.5.8 単回経口投与後の組織分布)。白色及び有色ラットにおける血漿中及び脳内の放射能は 速やかに消失したが,その他の組織からの放射能の消失は血漿に比べて緩やかであった。有色ラッ トに投与後 360 時間ではほとんどの組織で放射能濃度は最高値の 10%未満に減少したが,精巣及 び眼球ではそれぞれ最高値の 23%及び 68%を維持していた。有色ラットの眼球内放射能の消失は 緩徐(t1/2:157 日間)であり,ミラベグロン由来成分が眼内メラニンに結合している可能性が示 唆された(2.6.4.4.5 有色ラットにおける眼内分布)。メラニンへの結合は,bアドレナリン受容体 遮断薬やベンゾジアゼピン系薬物のような種々の塩基性薬物に共通の性質として知られている [8]。有色ラットのオートラジオグラムにおいて,眼球内放射能はメラニン高含有組織(毛様体, 脈絡膜及び結膜)と思われる部分に高濃度に分布していた(2.6.4.4.5 有色ラットにおける眼内分 布)。眼球に分布した放射能を 1 mol/L 塩酸─メタノールに抽出して分析したところ,眼球に分布 したミラベグロン由来成分(代謝物)の中で量が最も多かったのはフェネチルアミン体代謝物の M6 で,次いで未変化体であった。白色ラットに14C-ミラベグロン(10 mg/kg)を単回経口投与し たときに血漿中で量が最も多かった代謝物は M6 であり,投与後 1~6 時間において 32.1%~47.1% の存在比率を占めた(概要表 2.6.5.15 In vivo での代謝)。カニクイザルにおいては,14C-ミラベグ ロン(10 mg/kg)を単回経口投与後の血漿中に M6 は認められず,眼球内放射能濃度は投与後 168 時間において肝臓,胆汁に次いで 3 番目に高い値を示したが,その濃度は同じ 10 mg/kg を有色ラッ トに経口投与したときの投与後 168 時間における眼球内放射能濃度の約 6%であった(概要表 2.6.5.8 単回経口投与後の組織分布)。上記のように,カニクイザル及び有色ラット間の眼球内放 射能濃度の差は,ミラベグロン代謝物プロファイルの種差に由来するものと推察される。ヒトに おいては,カニクイザル同様,14C-ミラベグロン(160 mg)を空腹時に単回経口投与したときの血 漿中に M6 は認められなかった(2.7.2.2.2.2.3 マスバランス試験[CL-007])。ラット,イヌ及びカ ニクイザル反復経口投与毒性試験(2.4.4.2 反復投与毒性試験)の眼科検査及び眼の病理組織学的 検査において,薬物投与に関連した変化は認められなかったことから,ミラベグロン由来成分の 眼内からの緩徐な消失は眼組織に対して影響を及ぼさないものと考えられた。 白色ラットに14C-ミラベグロン(10 mg/kg)を 1 日 1 回 21 日間反復経口投与したときの放射能 の組織内分布パターンは単回投与時のそれと類似していたが,ほとんどの組織において投与後 24 時間の組織内放射能濃度は投与回数に伴い徐々に上昇した(概要表 2.6.5.9 反復経口投与後の組 織分布)。しかしながら,投与後 24 時間における組織内放射能濃度/血漿中放射能濃度比は投与 14 日目と 21 日目とでほぼ同等であり,組織内放射能濃度は投与 14 日目までにほぼ定常状態に達 したものと推察された。最終投与後の組織内放射能濃度はいずれの組織においても投与後 4 ある いは 24 時間に最高値を示した後,経時的に減少した。最終投与後 360 時間において,腎臓,甲状 腺,肝臓及び副腎で比較的高い放射能が認められ,これらの組織における放射能の消失は緩徐で あった。ラット及びカニクイザルの反復経口投与毒性試験の病理組織学的検査において,甲状腺 及び副腎に薬物投与に関連した変化は認められなかった(2.4.4.2 反復投与毒性試験)。腎臓及び 肝臓では,薬物投与に関連した変化が認められた(2.4.4.8 安全性に関する考察)。
ラットの全身オートラジオグラフィーの結果は組織内放射能濃度測定の結果と良く一致してい たが,後者の試験において測定を実施していない褐色脂肪及びハーダー腺において,ミラベグロ ン由来成分の高濃度の分布が認められた。また,薬効部位である膀胱に放射能が血液と同程度以 上に分布することが確認された。 妊娠ラットに14C-ミラベグロンを経口投与したとき,ミラベグロン由来成分は胎盤を通過し, 胎児へ移行することが示された(概要表 2.6.5.13 妊娠又は授乳動物における試験)。また,哺育 中のラットに14C-ミラベグロンを経口投与したときの乳汁,いくつかの哺乳児組織及び哺乳児の 胃内乳塊において放射能が検出されており(概要表 2.6.5.13 妊娠又は授乳動物における試験), ミラベグロン由来成分は乳汁を介して哺乳児の組織へ分布することが示唆された。
2.4.3.4
代謝
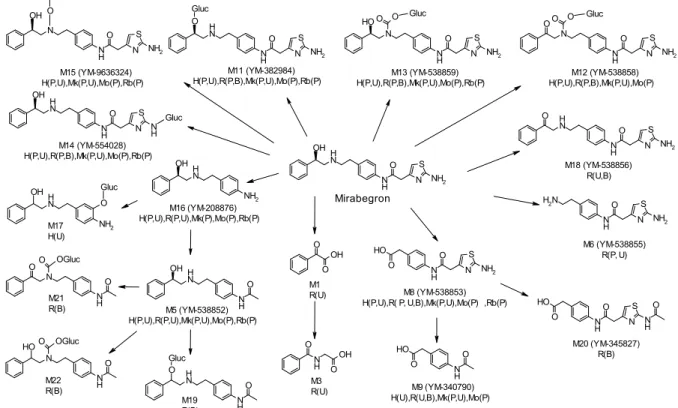
代謝速度及び代謝物プロファイルをヒトと安全性試験に用いた動物種との間で比較する目的で, in vitro 代謝試験を実施した。ミラベグロンをラット,イヌ,カニクイザル及びヒト肝ミクロソー ムとインキュベートしたときの代謝速度を比較したところ,動物種間で大きな差は見られなかっ たが,ヒトにおいて最も遅かった(概要表 2.6.5.14 In vitro での代謝)。また,マウス,ラット, イヌ,カニクイザル及びヒト肝ミクロソームによる代謝物プロファイルに顕著な種差は認められ ず,ヒトで認められた代謝物は検討したほかのいずれかの動物種においても認められた(2.6.4.5.1 In vitro 代謝プロファイル)。 ミラベグロンをマウス,ラット,ウサギ,イヌ,カニクイザル及びヒト血漿中においてエステ ラーゼ阻害剤であるジクロルボスの非存在下及び存在下でインキュベートして in vitro 代謝速度を 比較検討した。ジクロルボス非存在下では,ミラベグロンはヒト血漿中において最も速い代謝速 度で消失し,マウス及びカニクイザル血漿中においても消失が認められた(概要表 2.6.5.14 In vitro での代謝)。一方,ラット,ウサギ及びイヌ血漿中においては未変化体の減少は認められなかった。 ジクロルボス存在下ではいずれの動物種の血漿中においても未変化体の減少は認められなかった ことから,マウス,カニクイザル及びヒト血漿中におけるミラベグロンの代謝は,いずれもエス テラーゼによるものと推察された。 マウス,ラット,カニクイザル及びヒトの in vivo 試料を用い,種々の分析法に基づき,ミラベ グロンの代謝物検索及び構造推定を実施した。その結果,M1,M3,M5,M6,M8,M9,M11, M12,M13,M14,M15,M16,M17,M18,M19,M20,M21 及び M22 の計 18 種の代謝物が同 定あるいは構造推定された。ヒトの尿中においては M5,M8,M9,M11,M12,M13,M14,M15, M16 及び M17 の 10 種の代謝物が認められ,またそのうちの 8 種(M5,M8,M11,M12,M13, M14,M15 及び M16)がヒトの血漿中において認められた(2.7.2.2.2.2.3 マスバランス試験 [CL-007])。ミラベグロンの推定代謝経路を図 2.4-1 に示す。ミラベグロンの主な代謝経路は,1) 二級アミンの酸化(あるいは N-脱アルキル化),2) アミドの加水分解と生成したアミンへのアセ チル抱合,3) 二級水酸基へのグルクロン酸抱合,末端(チアゾール環の)一級アミンへのグルクロン酸抱合あるいは二級アミンへのカルバモイルグルクロン酸抱合,4) 二級水酸基のケトンへの 酸化,5) 上記 1),2),3)及び 4)の組み合わせであると推定された。 In vivo における代謝物の構造推定の結果,ヒト血漿中に認められた代謝物はすべて,少なくと もラットあるいはカニクイザルのいずれかの動物種においても認められ,両動物種を一般毒性試 験において使用することは妥当であると考えられた。 N S N H N H O OH NH2 N S N H O O O H NH2 N H O O H O N H N H O OH N H N H O O Gluc N S N H O O O H N H O N S N H N H O O NH2 Gluc N S N N H O O NH2 O O Gluc N S N N H O O H NH2 O O Gluc N S N H2 N H O NH2 N H NH2 OH O O OH O N H OH O N N H O OGluc O O N N H O O H O OGluc N S N H N H O O NH2 N S N H N H O OH N H Gluc N H NH2 OH O Gluc O Gluc N S N N H O NH2 OH Mirabegron M12 (YM-538858) H(P,U),R(P,B),Mk(P,U),Mo(P) M11 (YM-382984) H(P,U),R(P,B),Mk(P,U),Mo(P),Rb(P) M5 (YM-538852) H(P,U),R(P,U),Mk(P,U),Mo(P),Rb(P) M19 R(B) M8 (YM-538853) H(P,U),R( P, U,B),Mk(P,U),Mo(P) ,Rb(P) M20 (YM-345827) R(B) M6 (YM-538855) R(P, U) M16 (YM-208876) H(P,U),R(P,U),Mk(P),Mo(P),Rb(P) M1 R(U) M21 R(B) M22 R(B) M18 (YM-538856) R(U,B) M14 (YM-554028) H(P,U),R(P,B),Mk(P,U),Mo(P),Rb(P) M17 H(U) M15 (YM-9636324) H(P,U),Mk(P,U),Mo(P),Rb(P) M13 (YM-538859) H(P,U),R(P,B),Mk(P,U),Mo(P),Rb(P) M9 (YM-340790) H(U),R(U,B),Mk(P,U),Mo(P) M3 R(U)
Gluc: b-D-Glucuronosyl, H: human, R: rat, Mk: monkey, Mo: mouse, Rb: rabbit, P: plasma, U: urine, B: bile
図 2.4-1 ミラベグロンの推定代謝経路
2.4.3.5
排泄
ラット及びカニクイザルに14C-ミラベグロン(10 mg/kg)を単回経口投与したとき,投与後 168 時間までの尿中放射能排泄率はそれぞれ 18.8%及び 46.8%であり,糞中放射能排泄率はそれぞれ 75.3%及び 54.2%であった(概要表 2.6.5.19 尿・糞中累積排泄)。ラット及びカニクイザルの尿試 料中の放射能成分をプロファイリングしたところ,ミラベグロンは尿中放射能のそれぞれ約 50% 及び約 10%を占めた(概要表 2.6.5.15 In vivo での代謝)。ラットに14C-ミラベグロン(10 mg/kg) を単回経口投与したとき,投与後 72 時間までの胆汁中放射能排泄率は 29.4%であった(概要表 2.6.5.20 尿・胆汁中累積排泄)。ラットの胆汁試料中の放射能成分をプロファイリングしたところ, ミラベグロンは胆汁中放射能の約 25%を占めた(概要表 2.6.5.15 In vivo での代謝)。これらの結果 より,ラットにおける放射能の主消失経路は胆汁中排泄を介した糞中排泄であり,カニクイザルにおける放射能の消失には尿中排泄と糞中排泄とがほぼ同等に寄与すると考えられた。ラットに おいてはミラベグロン由来成分の腸肝循環も認められた(概要表 2.6.5.21 腸肝循環)。これに対 して,ヒトにおける放射能の消失経路は,尿中排泄率が 55.0%,糞中排泄率が 34.2%であり,尿中 排泄の寄与の方が高かった(2.7.2.2.2.2.3 マスバランス試験[CL-007])。
2.4.3.6
ヒト代謝物に対する曝露量
ヒトに14C-ミラベグロンを 160 mg 単回経口投与したときに血漿中に認められたすべてのミラベ グロン代謝物 8 種(M5,M8,M11,M12,M13,M14,M15 及び M16)(2.7.2.2.2.2.3 マスバラン ス試験[CL-007])について,ヒト血漿中薬物濃度が測定された(2.7.2.2.2.3.1 性差及び高齢者試 験[CL-072])。その結果,ヒト血漿中で投与薬物に関連するすべての物質の合計曝露量(未変化 体及び代謝物の総和)の 10%を超える主代謝物は M11 及び M12 の 2 種であることが確認された。 M11 及び M12 は薬理活性が認められていない(2.6.2.2.5.1 ヒト β アドレナリン受容体に対する刺 激作用)グルクロン酸抱合体代謝物である。 実験動物の 15 日間反復経口投与試験において,最終投与後(投与 15 日目)における上述の 8 種のヒト血漿中代謝物に対する曝露量をヒトの定常状態(投与 7 日目)における代謝物に対する 曝露量と比較した。100 mg/kg/day を投与したラットは,臨床推奨用量(50 mg/day)のミラベグロ ンを投与したヒトと同等以上の濃度の代謝物 M5,M8,M11,M12,M13,M14 及び M16 に曝露 されていた(表 2.4-2)。30 mg/kg/day を投与したカニクイザルは,臨床推奨用量のミラベグロンを 投与したヒトと同等以上の濃度の代謝物 M5,M8,M11,M12,M13,M14,M15 及び M16 に曝 露されていた。100 mg/kg/day を投与したマウスは,臨床推奨用量のミラベグロンを投与したヒト と同等以上の濃度の代謝物 M5,M8,M11,M13,M14,M15 及び M16 に曝露されていた。また, 30 mg/kg/day を投与したウサギは,臨床推奨用量のミラベグロンを投与したヒトと同等以上の濃 度の代謝物 M5,M8,M11,M13,M14 及び M16 に曝露されていた。AUC24h(ng·h/mL) 種 マウス ラット ウサギ カニクイザル ヒト 投与量 100 mg/kg/day a, 15 日目 100 mg/kg/dayb, 15 日目 30 mg/kg/dayc, 15 日目 30 mg/kg/dayd, 15 日目 50 mg/daye(OCAS 錠), 7 日目 測定対象 雄 雌 雄 雌 雌 雄 雌 非高齢 男性 非高齢 女性 高齢 男性 高齢 女性 ミラベグロン 35007.17 23876.72 20472.89 18821.13 3219.66 5708.77 4646.55 413 471 341 512 M5 1303.97 1246.63 2085.39 1633.03 23556.28 102.18 174.29 69.2 85.4 79.8 112 M8 1238.51 1383.08 4044.34 2533.48 88.48 514.80 528.25 38.2f 57.3f 49.4f 104f M11 4153.10 2273.01 153.29 200.62 837.94 123581.30 131877.97 121 134 150 201 M12 0.00 0.00 133.64 137.40 0.00 284.27 321.09 115 94.5 82.9 98.5 M13 224.20 156.51 45.58 51.74 199.24 1466.19 1780.97 11.5 10.1 10.1 12.9 M14 821.44 581.71 496.78 517.07 889.99 122.08 117.58 52.2 61.3 68.5 75.0 M15 49.48 90.22 0.00 0.00 27.71 361.30 375.65 29.4 32.7 30.9 48.5 M16 1861.06g 1297.61g 157.42g 157.52g 10048.25g 59.88 70.78 40.2 47.8 41.5 69.5 a:マウスがん原性試験における最大用量(2.6.6.5.3 マウス 104 週間経口投与がん原性試験) b:ラット 26 週間反復経口投与毒性試験における最大用量(2.6.6.3.3 ラット 26 週間反復経口投与毒性試験) c:ウサギ生殖発生毒性試験における最大用量(2.6.6.6.7 ウサギ胚・胎児発生に関する試験) d:カニクイザル 52 週間反復経口投与毒性試験における最大用量(2.6.6.3.10 カニクイザル 52 週間反復経口投与毒性試験) e:ヒトにおける臨床推奨用量(2.7.2.2.2.3.1 性差及び高齢者試験[CL-072]) f:ヒトに 100 mg/day 投与したときのデータ(2.7.2.2.2.3.1 性差及び高齢者試験[CL-072]) g:他の測定対象とは別の試験において得られたデータ(概要表 2.6.5.17 反復経口投与後の血漿中代謝物濃度)
2.4.4
毒性試験
2.4.4.1
単回投与毒性試験
ラット単回経口投与毒性試験(300,500 及び 800 mg/kg)(2.6.6.2.1 ラット単回経口投与毒性試 験)において,300 mg/kg 以上の群の雌雄で自発運動の低下,流涎及び流涙,300 mg/kg の群の雌 雄で色素涙が認められた。500 mg/kg 以上の群の雌雄で腹臥及び一過性の体重減少が認められた。 800 mg/kg 群の雌雄で散瞳及び色素涙,雄で間代性痙攣,雌で横臥及び被毛の汚れが認められ,投 与後 24 時間以内に 5 例中 2 例の雄及び 5 例中 3 例の雌が死亡した。死亡動物 1 例の剖検において 肝臓の淡色化及び小葉明瞭化が認められ,病理組織学的検査において小葉周辺性に肝細胞の肥大 及び空胞変性が認められた。概略の致死量は雌雄とも 800 mg/kg と推定された。 イヌ単回経口投与毒性試験(0.3,3 及び 30 mg/kg)(2.6.6.2.2 イヌ単回経口投与毒性試験)にお いて,0.3 mg/kg 以上の群の雌雄で心拍数の増加及び皮膚の発赤が認められた。病理組織学的検査 において 0.3 mg/kg 群の雄及び 3 mg/kg 群の雌雄で頬骨腺腺房の巣状性拡張及び組織破壊が認めら れた。30 mg/kg 群では雌雄で嘔吐が認められ,雄は横臥,浅呼吸/喘ぎ呼吸を呈して投与後約 50 分に死亡した。病理組織学的検査において雌で頬骨腺の壊死が認められた。概略の致死量は雄が 30 mg/kg,雌が 30 mg/kg 以上と推定された。2.4.4.2
反復投与毒性試験
ラット 2 週間反復経口投与毒性試験(10,30,100 及び 300 mg/kg/day)(2.6.6.3.1 ラット 2 週間 反復経口投与毒性試験)において,30 mg/kg/day 以上の群の雌雄でアラニンアミノトランスフェ ラーゼ(ALT)の増加,本薬の β3アドレナリン受容体刺激作用に起因すると考えられる白色脂肪 細胞の脂肪滴の減少,雄で中性脂肪の減少,肝臓及び腎臓の体重比重量の増加,雌でアルカリホ スファターゼ(ALP)の増加が認められた。100 mg/kg/day 以上の群の雌雄で血小板数の減少,摂 水量及び血漿中カリウムの増加,尿中ナトリウム及び塩素の総排泄量の増加,雄で体重増加量の 低値,ALP,総コレステロール,リン脂質及び尿中カリウムの総排泄量の増加,心臓の体重比重 量の増加,精巣及び精嚢の実重量あるいは体重比重量の減少,雌で肝臓及び腎臓の体重比重量の 増加,大動脈周囲の褐色脂肪細胞の脂肪滴の減少が認められた。300 mg/kg/day 群の雌雄で自発運 動の低下,体重増加抑制,摂餌量の減少,網赤血球率の減少,尿蛋白の増加,胸腺の実重量及び 体重比重量の減少が認められた。雄で尿量の増加,前立腺の実重量及び体重比重量の減少,大動 脈周囲の褐色脂肪細胞の脂肪滴の減少及び精嚢分泌液減少が認められた。雌で流涙,眼分泌物, 白血球数,リンパ球数,ヘマトクリット値及び平均赤血球容積(MCV)の減少,総コレステロー ル,リン脂質及び尿中カリウムの総排泄量の増加,心臓の実重量,脾臓,卵巣及び子宮の実重量 及び体重比重量の減少,子宮の萎縮,胸腺の萎縮及び骨髄造血低下が認められた。投与期間中に 認められた所見は休薬により回復性を示した。無毒性量は 10 mg/kg/day と推定された。ラット 13 週間反復経口投与毒性試験(10,30,100 及び 300 mg/kg/day)(2.6.6.3.2 ラット 13 週 間反復経口投与毒性試験)において,10 mg/kg/day 以上の群の雄で中性脂肪の減少,雌で白色脂 肪細胞の脂肪滴の減少が認められた。30 mg/kg/day 以上の群の雌雄で血小板数の減少,血漿中カ リウムの増加,クレアチニンの減少及び耳下腺のチモーゲン顆粒の減少が認められた。雄で流涎, 体重増加抑制,摂餌量及び摂水量の増加,リン脂質の減少及び ALT の増加,褐色脂肪細胞の脂肪 滴の減少が認められ,雌で流涙,尿中塩素の総排泄量の増加が認められた。100 mg/kg/day 以上の 群の雌雄で ALP の増加,尿 pH の低下,尿蛋白及びビリルビン陽性反応,肝臓のマクロファージ 及びクッパー細胞にリポフスチンの沈着が認められた。雄で流涙,アスパラギン酸アミノトラン スフェラーゼ(AST),尿中ナトリウム及び塩素排泄量の増加,a1-グロブリン分画比率の減少,白 色脂肪細胞の脂肪滴の減少及び骨髄のマクロファージにリポフスチンの沈着が認められた。雌で 流涎,腹部膨満,摂餌量の増加,アルブミン濃度の増加,肝臓及び腎臓の実重量あるいは体重比 重量の増加,子宮の実重量及び体重比重量の減少が認められた。300 mg/kg/day 群では雌 16 例中 2 例が死亡した。雌雄で下顎部の脱毛,アルブミン分画比率,アルブミン・グロブリン比及び総コ レステロールの増加,胸腺の実重量及び体重比重量の減少,顎下腺の顆粒管好酸性顆粒減少,肝 細胞腫脹,腎臓の尿細管上皮にリポフスチンの沈着,盲腸の粘膜固有層マクロファージにリポフ スチン及びヘモジデリンの沈着及び胸腺の萎縮が認められた。雄で下顎部の被毛の汚れ,耳介の 蒼白,散瞳,アルブミン濃度及び総ビリルビンの増加,白血球数,単球比率及び血糖の減少,膀 胱の結石,肝臓の小葉中心性肝細胞壊死及び線維化が認められた。雌で泌尿・生殖器付近の被毛 の汚れ,体重増加抑制,摂水量の増加,リンパ球比率,ALT 及びリン脂質の増加,好中球比率及 びa1-グロブリン分画比率の減少,下垂体の実重量及び体重比重量の減少,褐色脂肪細胞の脂肪滴 の減少,骨髄のマクロファージにリポフスチンの沈着及び子宮の萎縮が認められた。投与期間中 に認められたほとんどの所見は休薬により回復性を示した。無毒性量は 10 mg/kg/day と推定され た。 ラット 26 週間反復経口投与毒性試験(3,10,30 及び 100 mg/kg/day)(2.6.6.3.3 ラット 26 週間 反復経口投与毒性試験)において,3 mg/kg/day 以上の群の雌で本薬のbアドレナリン受容体刺激 作用に起因すると考えられる流涙が認められた。10 mg/kg/day 以上の群の雌雄でクレアチニンの 減少,雄で腹臥,流涙及び摂餌量の増加,血小板数の減少,血漿中カリウムの増加,尿中カリウ ム及び塩素の総排泄量の増加,雌で白色脂肪細胞の脂肪滴の減少が認められた。30 mg/kg/day 以 上の群の雌雄で流涎,肝細胞の好酸性化及び褐色脂肪細胞の脂肪滴減少,雄で体重増加抑制,摂 水量の増加,ヘマトクリット値,ヘモグロビン量,MCV 及び平均赤血球血色素量(MCH)の増 加,中性脂肪の減少,ALT の増加,尿浸透圧上昇,pH 低下,ビリルビン増加,胸腺の実重量の減 少,白色脂肪細胞の脂肪滴の減少,脾臓にヘモジデリンの沈着の増加,雌で腹臥,摂餌量の増加, 血漿中カリウムの増加,尿中塩素の総排泄量の増加,肝臓の実重量あるいは体重比重量の増加, 耳下腺のチモーゲン顆粒の減少が認められた。100 mg/kg/day 群の雌雄で尿蛋白の増加,雄で総コ レステロール,ALP,無機リン,総蛋白,アルブミン濃度,b-及びg-グロブリン濃度の増加,a1 -グロブリン(濃度及び分画比率)の減少,尿中ナトリウムの総排泄量の増加,尿量の減少,尿の
黄褐色化,脳及び脾臓の実重量の減少,耳下腺のチモーゲン顆粒の減少が認められた。雌で摂水 量の増加,MCV の増加,血小板数,中性脂肪及び血漿中塩素の減少,尿 pH の低下及びビリルビ ンの増加,胸腺の実重量及び体重比重量の減少が認められた。投与期間中に認められたほとんど の所見は休薬により回復性を示した。無毒性量は 3 mg/kg/day と推定された。 ラット 2 週間反復静脈内投与毒性試験(1,3 及び 10 mg/kg/day)(2.6.6.3.5 ラット 2 週間反復静 脈内投与毒性試験)において,3 mg/kg/day 以上の群の雌雄で白色脂肪細胞の脂肪滴の減少が認め られた。10 mg/kg/day 群の雌雄で散瞳,自発運動の低下及び腹臥,雌で摂餌量の増加が認められ た。無毒性量は 3 mg/kg/day と推定された。 イヌ 2 週間反復経口投与毒性試験(1,3,10 及び 20 mg/kg/day)(2.6.6.3.6 イヌ 2 週間反復経口 投与毒性試験)において,1 mg/kg 以上の群の雌雄で皮膚の発赤,頬骨腺の変性及び炎症が認めら れた。1 mg/kg/day 群の雌で眼周囲の腫脹が認められ,眼,眼瞼及び鼻口部の腫脹,眼球突出が認 められた 3 mg/kg/day 群の雌 1 例が投与 6 日に剖検された。3 mg/kg/day 以上の群の雌雄で流涎, 雌で嘔吐及び眼分泌物が認められた。3 mg/kg 群の雌雄で強膜の充血が認められた。10 mg/kg/day 以上の群の雌雄で心拍数の増加,P 波及び QRS 間隔の延長,T 波の増高,雄で嘔吐及び眼分泌物 が認められた。20 mg/kg/day 群の雌雄で眼周囲の腫脹及び心室頻拍,雌で眼の炎症が認められ,5 例中 1 例の雌が死亡し,投与 7 日までに全例が剖検された。病理組織学的検査において左心室心 内膜の出血,左心室心筋変性及び肝臓の門脈周囲に空胞形成が認められた。本試験において無毒 性量は特定できなかった。 イヌ 3 日間反復経口投与唾液腺毒性確認試験(20 mg/kg/day)(2.6.6.3.7 イヌ 3 日間反復経口投 与唾液腺毒性試験)において,皮膚,眼結膜及び口腔粘膜の潮紅,嘔吐,自発運動の低下,潜血 便,流涎,眼結膜蒼白,摂餌量の減少,ALP 及び ALT の軽度な増加が認められた。病理組織学的 検査において唾液腺(顎下腺,大舌下腺,小舌下腺,耳下腺及び頬骨腺)の腺房細胞の萎縮及び 壊死,導管の拡張,導管上皮の壊死,剥離,増生及び鉱質沈着,間質の水腫,出血,血栓及び細 胞浸潤が認められ,これらの変化は頬骨腺で顕著であった。4 週あるいは 13 週間の休薬後には腺 房細胞萎縮,導管の増生を伴う線維化及び鉱質沈着が認められたが,所見の程度は軽減していた。 肝臓で小葉周辺性の肝細胞肥大,小葉中心性及び周辺性の脂肪滴沈着,小葉中心性のグリコーゲ ン顆粒の増加が認められたが,13 週間の休薬後にはこれらの所見は認められなかった。 カニクイザル 2 週間反復経口投与毒性試験(10,30 及び 60 mg/kg/day)(2.6.6.3.8 カニクイザル 2 週間反復経口投与毒性試験)において,30 mg/kg/day 群の雌で投与初日に眼瞼下垂及び口粘膜の 蒼白化が認められた。60 mg/kg/day 群の雌雄で眼瞼下垂及び口粘膜の蒼白化,横臥,腹臥,自発 運動の低下,心室頻拍,PR 及び QRS 間隔の延長が認められた。無毒性量は 10 mg/kg/day と推定 された。 カニクイザル 13 週間反復経口投与毒性試験(3,10 及び 30 mg/kg/day)(2.6.6.3.9 カニクイザル 13 週間反復経口投与毒性試験)において,10 mg/kg/day 以上の群の雌雄で PR 間隔の延長傾向あ るいは延長,白色脂肪組織で細胞質内に多数の小脂肪滴を有する小型脂肪細胞が認められた。
30 mg/kg/day の雄で QRS 間隔の延長傾向及び心室頻拍が認められた。投与期間中に認められた所 見は休薬により回復性を示した。無毒性量は 3 mg/kg/day と推定された。 カニクイザル 52 週間反復経口投与毒性試験(3,10 及び 30 mg/kg/day)(2.6.6.3.10 カニクイザ ル 52 週間反復経口投与毒性試験)において,3 mg/kg/day 群の雌雄及び 30 mg/kg/day 群の雄の白 色脂肪組織で多房性細胞質を有する小型脂肪細胞が認められた。30 mg/kg/day 群の雌雄で投与初 期に眼瞼下垂,自発運動の低下,よろめき歩行及び口粘膜の蒼白化,PR,QRS 及び QTc 間隔の延 長あるいは延長傾向が認められた。本試験における無毒性量は 10 mg/kg/day と推定された。 カニクイザル 2 週間反復静脈内投与試験(0.3,1 及び 3 mg/kg/day)(2.6.6.3.12 カニクイザル 2 週間反復静脈内投与毒性試験)において 1 mg/kg/day 群の雄で PR 間隔の延長及び心室頻拍が認め られた。3 mg/kg/day 群の雌雄で PR 及び QRS 間隔の延長,心室頻拍が認められた。3 例中 1 例の 雄で昏睡が認められ,雌で尿素窒素の軽度な増加が認められた。無毒性量は 0.3 mg/kg/day と推定 された。
2.4.4.3
遺伝毒性試験
ネズミチフス菌及び大腸菌を用いた復帰突然変異試験(2.6.6.4.1 細菌を用いる復帰突然変異試 験)(1.6~5000 µg/plate)において,代謝活性化系の存在下及び非存在下に遺伝子突然変異誘発性 は認められなかった。 ヒト末梢血リンパ球を用いた染色体異常試験(2.6.6.4.2 ヒト末梢血リンパ球を用いる染色体異 常試験)(110~1280 µg/mL)において,代謝活性化系の存在下及び非存在下に 3 時間処理した場 合に染色体異常を持つ細胞数の軽度な増加が認められたが,いずれも強い細胞毒性を示す濃度(細 胞生存率 40%未満)における変化であった。 ラットを用いた小核試験(100,200 及び 400 mg/kg/day)(2.6.6.4.3 ラットを用いる小核試験) において,小核誘発性は認められなかった。2.4.4.4
がん原性試験
マウス 104 週間投与がん原性試験(25,50 及び 100 mg/kg/day)(2.6.6.5.3 マウス 104 週間経口 投与がん原性試験)において,すべてのミラベグロン投与群で投与約 3 カ月まで対照群と比較し て体重の高値が散見されたが,その後は体重増加抑制が認められた。すべてのミラベグロン投与 群で投与期間を通して摂餌量の増加が認められた。いずれのミラベグロン投与群においても腫瘍 の発生頻度の増加は認められなかった。 ラット 104 週間投与がん原性試験(雄 12.5,25 及び 50 mg/kg/day,雌 25,50 及び 100 mg/kg/day) (2.6.6.5.4 ラット 104 週間経口投与がん原性試験)において,100 mg/kg/day 群の雌で死亡の増加 が認められた。すべてのミラベグロン投与群の雌雄で投与期間を通して摂餌量の増加が認められ, 雄で用量依存的な体重増加抑制が認められた。いずれのミラベグロン投与群においても腫瘍の発 生頻度の増加は認められなかった。2.4.4.5
生殖発生毒性試験
雄ラット授胎能及び着床までの初期胚発生に関する試験(30,100 及び 300 mg/kg/day)(2.6.6.6.1 雄ラット授胎能及び着床までの初期胚発生に関する試験)において,すべてのミラベグロン投与 群で体重増加抑制及び摂餌量の減少が認められた。30 及び 100 mg/kg/day 群では投与後期に摂餌 量の増加が認められた。300 mg/kg/day 群では振戦及び自発運動の低下が認められ,20 例中 14 例 が死亡した。雄ラットの授胎能及び着床までの初期胚発生に対する無毒性量は 100 mg/kg/day と推 定された。 雌ラット受胎能及び着床までの初期胚発生に関する試験(30,100 及び 300 mg/kg/day)(2.6.6.6.2 雌ラット受胎能及び着床までの初期胚発生に関する試験)において,100 mg/kg/day 群で摂餌量の 増加が認められた。300 mg/kg/day 群では自発運動の低下,被毛の汚れ,流涙,振戦,体重及び摂 餌量の減少が認められ,20 例中 2 例が死亡し,1 例が瀕死屠殺された。発情休止期の延長,黄体 数,着床数及び生存胎児数の減少が認められたが,交尾までに要した日数,交尾率,受胎率,着 床前死亡率及び着床後死亡率に影響は認められなかった。雌ラットの生殖機能に対する無毒性量 は 100 mg/kg/day,初期胚発生に対する無毒性量は 300 mg/kg/day と推定された。 ラット胚・胎児発生に関する試験(10,30,100 及び 300 mg/kg/day)(2.6.6.6.4 ラット胚・胎児 発生に関する試験)において,100 mg/kg/day 以上の群で母動物の体重増加抑制及び摂餌量の減少, 胎児の波状肋骨の増加及び中足骨骨化数の低値が認められた。300 mg/kg/day 群では 3 例の母動物 の死亡が認められ,胎児体重の低下,肩甲骨及び前腕骨の屈曲,胸骨分節及び仙尾椎骨の骨化数 の低値が認められた。100 mg/kg/day 群で実施した生後 4 日の出生児の骨格検査において,波状肋 骨の増加が認められた。胚・胎児発生に対する無毒性量は 30 mg/kg/day と推定された。 ラット胎児で認められた波状肋骨の回復性試験(100 mg/kg/day)(2.6.6.6.5 ラット胚・胎児発生 に関する試験(波状肋骨の回復性に関する試験))において,母動物で体重増加抑制及び摂餌量の 減少が認められた。妊娠末期胎児で波状肋骨の増加及び胸骨分節及び左右中手骨の骨化数の低値 が認められたが,生後 4 日及び 63 日の出生児では認められなかったことから,これらの変化は発 育に伴い回復するものと考えられた。 ウサギ胚・胎児発生に関する試験(3,10,及び 30 mg/kg/day)(2.6.6.6.7 ウサギ胚・胎児発生 に関する試験)において,10 mg/kg/day 以上の群で母動物の摂餌量の減少及び胎児体重の低値が 認められた。30 mg/kg/day 群で母動物の体重減少あるいは増加抑制が認められ,1 例が横臥及び呼 吸困難を呈して死亡した。胎児で着床後死亡胚数の増加,大動脈拡張,巨心及び肺副葉欠損の増 加,胸骨分節癒合の増加,中手骨及び前後肢の中節骨の数の低値が認められた。胚・胎児発生に 対する無毒性量は 3 mg/kg/day と推定された。 ウサギ胎児で認められた心血管系の異常に対するb1アドレナリン受容体遮断薬の影響に関する 試験(30 mg/kg/day)(2.6.6.6.9 ウサギ胚・胎児発生に関する試験で認められた胎児所見に関する 基礎的検討試験(b1アドレナリン受容体遮断薬の影響))において,ミラベグロン投与群で母動物 の体重増加抑制及び摂餌量の減少,メトプロロール(3 mg/kg/day)併用投与群で母動物の摂餌量の減少が認められた。ミラベグロン投与群で認められた胎児における大動脈拡張及び巨心の発現 頻度の増加は,メトプロロールを併用することにより減少したことから,ウサギ胎児で発現した 大動脈の拡張及び巨心はミラベグロンのb1アドレナリン受容体刺激作用に起因している可能性が 示唆された。 ラット出生前及び出生後の発生並びに母体の機能に関する試験(10,30 及び 100 mg/kg/day) (2.6.6.6.10 ラット出生前及び出生後の発生並びに母体の機能に関する試験)において, 100 mg/kg/day 群で母動物 2 例の死亡及び体重増加抑制が認められ,出生児で 4 日生存率の低値及 び体重増加抑制が認められた。母動物の生殖機能に対する無毒性量は 100 mg/kg/day,出生児に対 する無毒性量は 30 mg/kg/day と推定された。
2.4.4.6
局所刺激性試験
ウサギ皮膚刺激性試験(2.6.6.7.1 ウサギ皮膚刺激性試験)において,皮膚刺激性は認められな かった。 ウサギ眼粘膜刺激性試験(2.6.6.7.2 ウサギ眼粘膜刺激性試験)において,軽微な眼粘膜刺激性 が認められたが,直ちに洗浄することにより眼粘膜刺激性は軽減された。 ウサギ血管局所刺激性試験(2.6.6.7.3 ウサギ血管局所刺激性試験)において,静脈内投与によ り注射部位とその下流に限局性浮腫が認められたが,投与後 8 時間までに消失した。静脈周囲投 与により投与部位周囲に浮腫及び紅斑が投与後 24 時間まで認められ,投与後 48~96 時間には紫 斑が認められた。病理組織学的検査において皮下組織に炎症性反応が認められた。2.4.4.7
その他の毒性試験
モルモット皮膚感作性試験(Adjuvant and Patch 法及び Buehler 法)[2.6.6.8.1 モルモット皮膚感 作性試験(Adjuvant and Patch 法)及び 2.6.6.8.2 モルモット皮膚感作性試験(Buehler 法)]におい て,中等度の皮膚感作性が認められた。 約 1%(実測値:1.51%)の不純物(YM-181687)を含むミラベグロン原薬を用いたラット 2 週 間反復経口投与毒性試験(3 及び 10 mg/kg/day)(2.6.6.8.3 高レベルの不純物を含む原薬を用いた ラット 2 週間反復経口投与毒性試験)において,3 mg/kg/day 以上の群の雌で腸間膜リンパ節周囲 の白色脂肪細胞の小型化が認められた。10 mg/kg/day 群の雌雄で摂餌量の増加,雄でフィブリノー ゲン及び β グロブリン比の増加,雌で AST 及び ALT の増加,中性脂肪の減少が認められた。無 毒性量は 3 mg/kg/day と推定された。 溶血性試験(2.6.6.8.4 溶血性試験)において,ヒト血液に対して溶血性は認められなかった。