リクラスト点滴静注液
5mg に関する資料
本資料に記載された情報に係る権利及び内容についての責任は旭化成ファーマ
株式会社に帰属するものであり、当該情報を本薬剤の適正使用以外の営利目的
に利用することはできません。
リクラスト点滴静注液
5mg
ゾレドロン酸水和物
第
1 部(モジュール 1):
申請書等行政情報及び添付文書に関する情報
1.5 起原又は発見の経緯及び開発の経緯
旭化成ファーマ株式会社
略号一覧
略号 省略していない表現
AUC0-24 area under the plasma concentration−time curve from zero to 24 hours(投与後 24 時 間までの血漿中濃度−時間曲線下面積)
BMD bone mineral density(骨密度)
Cmax maximum plasma concentration(最高血漿中濃度) EMA European Medicines Agency(欧州医薬品庁) FDA Food and Drug Administration(米国食品医薬品局) NIH National Institutes of Health(米国国立衛生研究所) Novartis Novartis Pharma AG
NTX type I collagen cross-linked N-telopeptides(I 型コラーゲン架橋 N-テロペプチド) PMDA Pharmaceuticals and Medical Devices Agency(独立行政法人医薬品医療機器総合
機構)
PPK population pharmacokinetics(母集団薬物動態) QOL quality of life(生活の質)
RANKL receptor activator of nuclear factor kappa-B ligand RH relative humidity(相対湿度)
SERM selective estrogen receptor modulator(選択的エストロゲン受容体モジュレー ター)
WHO World Health Organization(世界保健機関)
ゾメタ® ゾメタ®点滴静注4mg/5mL およびゾメタ®点滴静注4mg/100mL ゾメタ®初回承 認時 2004 年の「悪性腫瘍による高カルシウム血症」に対する効能・効果でのゾメ タ®注射液4mg の承認時 本剤 Aclasta®/Reclast®、リクラスト点滴静注液5mg
目次
1. はじめに ... 4
1.1 骨粗鬆症の定義、背景、病態 ... 4 1.1.1 骨粗鬆症の発症機序、定義、診断、患者数 ... 4 1.1.2 骨粗鬆症の治療目的 ... 5 1.2 治療の現状 ... 5 1.2.1 骨粗鬆症の薬物治療 ... 5 1.2.2 ビスホスホネート製剤による治療の課題点 ... 52. 開発の経緯 ... 7
2.1 品質に関する開発の経緯 ... 8 2.2 非臨床試験に関する開発の経緯 ... 8 2.2.1 薬理試験結果の概略 ... 8 2.2.2 薬物動態試験結果の概略 ... 9 2.2.3 毒性試験結果の概略 ... 9 2.3 臨床試験に関する開発の経緯 ... 10 2.3.1 海外の開発 ... 10 2.3.2 国内の開発 ... 103. 特徴および有用性 ... 13
3.1 非臨床試験成績から見た特徴および有用性 ... 13 3.2 臨床試験成績から見た特徴および有用性 ... 134. 参考文献 ... 14
1. はじめに
ゾレドロン酸は、Novartis Pharma AG(以下、Novartis)が創製したビスホスホネート系の化 合物であり、破骨細胞の形成阻害、機能喪失およびアポトーシスを引き起こすことで骨吸収作 用を抑制する。 ゾレドロン酸の開発は、4 mg 点滴静注用製剤として「悪性腫瘍による高カルシウム血症」を 対象に海外で始まり、2000 年にカナダ、スイスで最初に承認され、2001 年には欧州諸国なら びに米国でも承認された(販売名:Zometa®)。その後、2002 年に欧州諸国ならびに米国で「多 発性骨髄腫及び各種固形癌の骨転移」の適応が追加承認された。 国内では2004 年に「悪性腫瘍による高カルシウム血症」を適応として承認され(承認時販 売名:ゾメタ®注射液4mg)、さらに 2006 年には「多発性骨髄腫による骨病変及び固形癌骨転 移による骨病変」の適応が追加承認された。 一方、5 mg 点滴静注用製剤としての開発は「骨パジェット病」を対象に海外で始まり、2005 年に欧州諸国で承認され、2007 年には米国で承認された(米国以外での海外販売名:Aclasta® /米国での販売名:Reclast®)。さらに、2007 年 8 月に米国で、同年 10 月に欧州諸国で「閉経 後骨粗鬆症」の適応で承認され、現在では海外115 ヵ国以上で承認されている。また、2009 年までに各国で「ステロイド性骨粗鬆症」、「男性骨粗鬆症」などの適応追加が承認されてい る。 国内では2010 年に、旭化成ファーマ株式会社が骨粗鬆症を適応症とする Aclasta®/Reclast®、 リクラスト点滴静注液5mg(以下、本剤)の独占的な開発・販売権のライセンス契約を Novartis と締結し開発に着手した。 1.1 骨粗鬆症の定義、背景、病態 1.1.1 骨粗鬆症の発症機序、定義、診断、患者数 骨粗鬆症は骨の代謝のバランスが崩れることにより発症する。骨の代謝は骨組織に存在する、 破骨細胞による骨吸収とそれに続く骨芽細胞による骨形成からなり、この骨吸収と骨形成の量 的バランスは、骨格の成長期には骨形成の割合が高く、成人では両者が均衡している。しかし ながら、閉経、骨代謝に影響を及ぼす内分泌疾患やステロイド剤投与などを要因として骨吸収 と骨形成のバランスが崩れ、相対的に骨吸収の割合が高まり骨量減少をきたすと骨粗鬆症が発 症する。その結果、骨粗鬆症では、骨量(骨密度)減少と骨微細構造の劣化をきたし、骨強度 が低下して骨折が発生しやすくなる。 骨粗鬆症は2000 年に開催された米国国立衛生研究所(以下、NIH)のコンセンサス会議にて 骨強度の低下を特徴とし、骨折のリスクが増大しやすくなる骨格疾患として定義され(NIH, 2001)、国内および海外では、この疾患定義が広く浸透している。 骨粗鬆症の診断は、国内では「原発性骨粗鬆症の診断基準(2012 年度改訂版)」(宗圓聰 他, 2013)が用いられ、国際的には骨粗鬆症の診断には世界保健機関(以下、WHO)が提唱した 診断基準が多くの国で用いられている。いずれも、脆弱性骨折、骨密度によって骨粗鬆症が診 断されており、国内外で大きな違いはない。 骨粗鬆症は高齢者ならびに閉経後の女性に多く発症することが知られている。国内の大規模 住民コホート研究で、日本骨代謝学会の診断基準の腰椎および大腿骨頸部の骨密度値から推定 した40 歳以上の骨粗鬆症患者数は 1,280 万人(男性 300 万人、女性 980 万人)と推計されてい
る(折茂肇 他, 2015a)。また、骨粗鬆症は年齢とともに有病率が増加する疾患である。総人 口に占める65 歳以上の割合は、2014 年の総務省統計局発表によると 25.9%と報告され年々増 加していることから、骨粗鬆症患者数は今後もさらに増加していくものと考えられる。 1.1.2 骨粗鬆症の治療目的 骨粗鬆症の治療目的は、骨折を防止し生活の質(以下、QOL)の維持改善を図ることである。 骨粗鬆症での骨折好発部位は、椎体、大腿骨近位部、橈骨、上腕骨であり、これらの骨折は、 連鎖的に発生すると言われている(萩野浩, 2014)、(Gehlbach S et al, 2012)。そのうち、最 も発生頻度の高いものは椎体骨折である。骨折した椎体は変形したまま治癒し、このような変 形椎体が複数発生することで、脊柱全体が変形し脊椎後弯を呈することとなる。脊椎後弯によ り内臓が圧迫されることで、逆流性食道炎(Yamaguchi T et al, 2005)や呼吸困難(Harrison RA
et al, 2007)等が生じるため、日常生活動作が低下し、QOL に影響を与えることとなる。 大腿骨近位部骨折は、2000 年以降の調査で欧州、北米など海外では発生率が下がる傾向にあ るが、長寿科学骨粗鬆症研究班の調査によると、国内では大腿骨近位部骨折患者の年間発生数 は2007 年には 148,100 人と推計され未だに増加傾向にある(折茂肇 他, 2015a)。また、大腿 骨近位部骨折の前後で、骨粗鬆症患者の日常生活動作の自立率が、87%から 50%に低下し、大 腿骨近位部骨折患者の10%が骨折後 1 年で死亡するとの報告がある(Sakamoto K et al, 2006)。 すなわち、大腿骨近位部骨折を予防することは死亡リスクを低下させることにつながると考え られる。 1.2 治療の現状 1.2.1 骨粗鬆症の薬物治療 「骨粗鬆症の予防と治療ガイドライン2015 年度版」(折茂肇 他, 2015b)では、骨粗鬆症の 治療は、食事指導、運動指導、理学療法、手術療法および薬物療法が推奨されている。 骨粗鬆症の薬物治療として、現在では、ビスホスホネート製剤、ヒト型抗RANKL モノクロー ナル抗体製剤、テリパラチド製剤、女性ホルモン製剤、選択的エストロゲン受容体モジュレー ター(SERM)製剤、活性型ビタミン D3製剤、ビタミンK2製剤、カルシトニン製剤、カルシ ウム製剤が使用されている。ビスホスホネート製剤は、骨に親和性が高く、破骨細胞に作用し 骨吸収を抑制することにより強力な骨量増加効果と骨折抑制効果を示し、国内外で標準的な骨 粗鬆症治療薬として使用されている(折茂肇 他, 2015b)、(Kanis JA et al, 2013)、(Cosman F et al, 2014)。 1.2.2 ビスホスホネート製剤による治療の課題点 ビスホスホネート製剤は化学構造上P-C-P を基本骨格とする骨粗鬆症治療薬である。ビス ホスホネートの作用により、破骨細胞の形成阻害、機能喪失およびアポトーシスを引き起こす ことで骨吸収作用を抑制する。ビスホスホネート製剤は、その構造から三世代に分類されてい る。第一世代は側鎖に窒素を含まない構造を有している。第二世代は側鎖に窒素を含むが、環 状構造を有さない。第三世代は側鎖に窒素を含み環状構造を有する。ビスホスホネート製剤は 側鎖の違いにより骨吸収抑制能に差が認められる。第一世代と比べ、第二、第三世代は1,000 ~1 万倍の骨吸収抑制能を有することが知られている(Fleisch H, 2000)。国内で市販されてい
るビスホスホネート製剤は、エチドロン酸が第一世代、アレンドロン酸とイバンドロン酸が第 二世代、リセドロン酸、ミノドロン酸が第三世代に分類される(折茂肇 他, 2015c)。なお、 ゾレドロン酸は側鎖に窒素を含み環状構造を有するため、第三世代に分類される。 経口ビスホスホネート製剤は粘膜刺激による上部消化管障害を発現するため、嚥下困難、食 道炎、胃炎、十二指腸炎、または潰瘍等の上部消化管障害がある患者では使用しにくい。さら に、食道炎や胃炎のリスクが高まるため、服用後30 分以上、上体を起こしていることや立っ ていることのできない患者は禁忌とされている。このような服薬時の制限や留意点からコンプ ライアンス不良に陥りやすい。経口ビスホスホネート製剤の問題を改善するために、服薬率の 向上と治療の継続を目的として、投与間隔を広げる製剤の開発が行われてきた。具体的には、 経口連日投与製剤に始まり経口週1 回投与製剤、月 1 回投与製剤へと開発が進行してきた。し かし、経口製剤では月1 回投与であっても経口投与による問題点は解決できていない。 一方、既存の月1 回投与注射製剤では上記したような経口製剤による問題は生じないものの、 月1 回の通院が負担となる場合もあるため、投与間隔のより広い新たなビスホスホネート製剤 の開発が望まれている。
2. 開発の経緯
開発の経緯を以下に示す。 表 2-1 開発の経緯 評価 参考 品質に関する試験 製剤規格 製剤安定性試験 薬理試験 評価 評価 参考 参考 参考 参考 副次的薬理試験 参考 薬力学的薬物相互作用 参考 薬物動態試験 毒性試験 臨床試験 第I相 臨床試験 評価 第III相 臨床試験 評価 第III相 臨床試験 評価 第IIIa相 臨床試験 参考 第IIIb相 臨床試験 参考 第II相 臨床試験 参考 第IIIb相 臨床試験 参考 第IIIb相 臨床試験 参考 第IV相 臨床試験 参考 第IIIb相 臨床試験 参考 第III相 臨床試験 参考 第III相 臨床試験 参考 第III相 臨床試験 参考 第III相 臨床試験 参考 第III相 臨床試験 参考 第IIIb/IV相 臨床試験 参考 第IIIb/IV相 臨床試験 参考 治験相談 外国承認 FDA FDA ◆2007/8/17 (閉経後骨粗鬆症) EMA EMA ◆2007/10/3 ・ ◆ :実施期間および実施時期、 a) 文献著者名および発行年、 FDA:米国食品医薬品局、 EMA:欧州医薬品庁 試験項目 効力を裏付ける試験2.1 品質に関する開発の経緯 本剤は、ゾレドロン酸水和物を5mg/100mL の投与濃度に調製した点滴静注用製品として開 発した。添加剤の種類はゾメタ®点滴静注4mg/5mL およびゾメタ®点滴静注4mg/100mL(以下、 ゾメタ®)と同一である。 本剤は、ゾメタ®と有効成分が共通であることから、本申請のために新たに実施された原薬 の特性解析および安定性試験はなく、新たに設定した原薬の規格および試験方法はない。 本剤の国内における規格および試験方法には、ゾメタ®点滴静注4mg/100mL と同様のものを 設定した。また、本剤の安定性試験の結果、長期保存試験では25°C/60%RH で 60 ヵ月まで安 定、 °C/ - %RH で ヵ月まで安定、加速試験では40°C/75%RH で 6 ヵ月まで安定であっ た。したがって、有効期間は室温保存するとき36 ヵ月と設定した。 2.2 非臨床試験に関する開発の経緯 ゾレドロン酸の非臨床成績の多くは、2004 年の「悪性腫瘍による高カルシウム血症」に対す る効能・効果でのゾメタ®注射液4mg の承認時(以下、ゾメタ®初回承認時)に評価されている。 今回は新たに骨粗鬆症に対する効力を裏付ける薬理試験および薬力学的薬物相互作用試験 を追加実施した。なお、効力を裏付ける試験の一部および副次的薬理試験については、ゾメタ® 初回承認時の申請資料概要[1.13.1 既承認医薬品に係る資料]または公表論文をもとに検討し た。 本申請にあたり新たな毒性試験は実施しなかったものの、本剤の用法・用量はゾメタ®とは 異なることから、毒性試験結果についてゾメタ®初回承認時の申請資料概要[1.13.1 既承認医 薬品に係る資料]をもとに検討すると共に、本剤をヒトに投与したときの安全性について考察 した。 2.2.1 薬理試験結果の概略 ゾレドロン酸は、マウス頭蓋冠培養系において、骨吸収が亢進したマウス頭蓋冠からのカル シウム遊離を用量依存的に抑制したことから、骨吸収抑制作用を有すると考えられた。主な作 用機序として、破骨細胞の形成阻害、機能喪失およびアポトーシス誘導が示唆された。 ゾレドロン酸は、閉経後骨粗鬆症モデルである卵巣摘出ラットにおいて、単回静脈内投与後 32 週の骨密度の減少および骨微細構造の劣化を抑制するとともに、骨吸収マーカーを抑制し、 骨強度を増加させた。 ゾレドロン酸は、卵巣摘出ラットおよび卵巣摘出サルへの長期反復皮下投与(ラット52 週、 サル69 週)により、骨密度の減少および骨微細構造の劣化を抑制するとともに、骨吸収マー カーを抑制し、骨強度を増加させた。また、骨強度増加作用を示す用量で、骨石灰化の過剰な 抑制および骨組織の異常を示さなかった。 ゾレドロン酸は、ウサギ骨切延長術モデルにおいて、術後早期における骨延長部位の骨強度 を増加させ、術後長期における骨癒合を阻害しなかった。 ゾレドロン酸は、卵巣摘出ラットにおいて、骨形成促進薬との併用により、骨形成促進薬単 剤投与群に比べ骨密度を増加させた。
以上から、ゾレドロン酸は、骨粗鬆症患者に対する治療効果が期待できる薬剤と考えられた。 また、ゾレドロン酸は、骨石灰化を障害する可能性および骨折治癒過程に悪影響を及ぼす可能 性は低く、骨形成促進薬の骨密度増加作用を阻害しないと考えられた。 2.2.2 薬物動態試験結果の概略 新たな試験は実施していない。 2.2.3 毒性試験結果の概略 ゾレドロン酸の単回投与毒性は、ラットおよびイヌを用いた静脈内投与ならびにマウスを用 いた皮下投与により検討した。ラットおよびイヌの静脈内投与における概略の致死量は、それ ぞれ7.5 mg/kg および 10 mg/kg と考えられた。また、マウスの皮下投与における概略の致死量 は50 mg/kg と考えられた。 ゾレドロン酸の反復投与毒性は、イヌでは静脈内投与、ラットでは反復投与可能な皮下投与 により検討した。 ラットおよびイヌを用いたゾレドロン酸の反復投与毒性試験(連日投与)では、投与部位、 腎臓、肝臓、精巣、胃等に病理組織学的変化が認められた。これらの変化はビスホスホネート 製剤では既知のものであり、ゾレドロン酸に特異的な変化ではないと考えられた。1 年間隔で 投与する本剤では、これらの変化が臨床使用において問題となる可能性は低いと考えられた。 ラットに反復皮下投与したときの無毒性量は、1 ヵ月間、3 ヵ月間および 12 ヵ月間の連日投与 で、それぞれ0.013 mg/kg、0.01 mg/kg 未満および 0.001 mg/kg と考えられた。イヌに反復静脈 内投与したときの無毒性量は、4 週間、3 ヵ月間および 52 週間(それぞれ、連日、連日および 2 日または 3 日に 1 回)の投与で、それぞれ 0.013 mg/kg、0.01 mg/kg 未満および 0.005 mg/kg 未満と考えられた。 イヌを用いたゾレドロン酸の反復投与毒性試験(間欠投与)では、15 分間の静脈内投与を 3 週に1 回の投与間隔で 13 週間(計 5 回)および 6 ヵ月間(計 9 回)実施した。いずれの試験 においても、3 回投与 3 日後に剖検する中間検査を実施した。13 週間の試験では 0.23 mg/kg 以 上の群で投与部位、0.94 mg/kg 群で腎臓、6 ヵ月間の試験では 0.23 mg/kg 以上の群で腎臓に病 理組織学的変化が認められたものの、両試験とも0.23 mg/kg 群の 3 回投与後の中間検査では、 これらの変化が認められなかった。イヌに0.23 mg/kg を 15 分間静脈内投与したときの曝露量 (CmaxおよびAUC0-24)と、ヒトに本剤を臨床推奨用量で投与したときの曝露量との比較に基 づき算出した安全域は、Cmaxで2.0 倍、AUC0-24で2.1 倍であった。 以上の結果より、本剤の用法(1 年間隔の 1 回 15 分以上かけた点滴静脈内投与)を考慮する と、反復投与毒性試験でみられた毒性が臨床使用において問題となる可能性は低く、本剤の一 般毒性学的な安全性については大きな問題はないものと考えられた。ただし、本剤の安全域は 広くないことより、臨床使用時には用法・用量の遵守が必要と考えられた。 ラットおよびウサギを用いたゾレドロン酸の生殖発生毒性試験において、血中カルシウムの 低下に基づくと考えられる周産期の母動物の死亡が認められた。また、ラットでは着床前およ び着床後死亡率の増加、生存胎児数あるいは生存出生児数の減少がみられ、胎児の外表、内臓 および骨格検査において奇形が認められた。したがって、本剤は妊婦または妊娠している可能 性のある女性に対して禁忌にすべきと考えられた。
その他、ゾレドロン酸の遺伝毒性、がん原性、局所刺激性および抗原性ならびに類縁物質に 関する毒性の各試験において問題となる結果は認められなかった。 2.3 臨床試験に関する開発の経緯 2.3.1 海外の開発 Novartis は骨粗鬆症を新たな効能としてゾレドロン酸の開発を進めることとし、まず、骨粗 鬆症に対する至適用法・用量を検討する目的で、閉経後骨粗鬆症を対象に用量反応試験(0041 試験)を実施した。その結果、至適用量は明確にできなかったものの、ゾレドロン酸4 mg を 12 ヵ月に 1 回投与することで、骨粗鬆症患者の骨量を増加させることを確認した。一方、骨吸 収マーカーは、ゾレドロン酸4 mg の投与により投与 1 ヵ月後に低下したものの、その後回復 する傾向にあった。骨吸収抑制剤の効果発現には骨吸収マーカーを低下させ、その後低値を維 持することが重要であり、そのためには4 mg では用量が不足している可能性が考えられた。 そこで、骨吸収マーカーを1 年間持続的に低下させることを意図し、第 III 相試験(H2301 試 験)の投与量を4 mg から 5 mg に変更することについて、米国食品医薬品局(FDA)と協議し、 合意した。 この合意に従い、第III 相試験はゾレドロン酸 5 mg を年 1 回点滴静注する用法・用量で、閉 経後骨粗鬆症を対象としたプラセボ対照二重盲検並行群間比較試験(H2301 試験)を実施した。 本試験結果から、ゾレドロン酸5 mg の骨折抑制効果および骨量増加効果が検証され、また忍 容性も認められた。この結果に基づき、2007 年 8 月に米国で「閉経後骨粗鬆症」の適応で承認 され、同年10 月に欧州諸国で承認され、現在では海外 115 ヵ国以上で承認されている(米国 での販売名:Reclast®/米国以外での海外販売名:Aclasta®)。また、2009 年までに各国で「ス テロイド性骨粗鬆症」、「男性骨粗鬆症」などの適応追加が承認されている。 2.3.2 国内の開発 2.3.2.1 国内開発戦略 [1] 申請データパッケージの検討 本剤の国内開発を進めるにあたり、海外では既に骨粗鬆症に対する製造販売承認を取得 していたことから、海外臨床試験結果を活用したブリッジング戦略を検討した。しかし、 海外の用量反応試験である0041 試験では明確な用量反応性が認められなかったため、用量 反応性を国内外で比較することは不可能であった。また、海外の骨粗鬆症患者におけるゾ レドロン酸5 mg の薬物動態データが存在しないため、骨粗鬆症患者での国内外の薬物動 態を直接比較することができず、一般的なブリッジング戦略を用いることは困難な状況に あった。 一方、「悪性腫瘍による高カルシウム血症」等を適応としたゾレドロン酸4 mg 製剤は、 既に日本人と外国人との間で薬物動態の類似性が確認されていた。さらに、日本人と外国 人で有効性・安全性についても臨床上の差異は認められていないことから、国内外で同一 の用法・用量で承認されており、本剤は民族(人種)差の影響を受けにくいものと考えら れた。なお、骨粗鬆症を適応とした本剤は、東アジアを含む海外115 ヵ国以上で 5 mg を 1 年間隔で点滴静脈内投与する同一の用法・用量で承認されている。
以上から、本剤の骨粗鬆症患者における薬物動態は、人種差がないことが類推でき、骨 折抑制試験で本剤の有効性・安全性が国内外で類似していることを示すことができれば、 本剤の承認申請に既存の海外臨床試験結果が利用できるものと考えた。 そこで、臨床薬理試験(AK156-I-1 試験)および骨折抑制試験(AK156-III-1)を新たに 国内で実施し、下記に示した考え方に沿って、ゾレドロン酸5 mg 製剤の日本人骨粗鬆症患 者での有効性を検証し安全性を検討することを計画した。 (1) 臨床薬理試験(AK156-I-1 試験)では、日本人の原発性骨粗鬆症患者でのゾレドロン酸 4 mg および 5 mg 製剤の薬物動態データを取得し、日本人の悪性腫瘍骨転移患者を対象 に実施した臨床薬理試験(1101 試験)の薬物動態と比較することにより、疾患差が本剤 の薬物動態に与える影響を検討することとした。また、AK156-I-1 試験成績に加え、悪 性腫瘍骨転移患者を対象に実施した国内外の臨床試験(J001、1101、503 および 506 試 験)成績も含めた母集団薬物動態(以下、PPK)解析を実施することを計画した。これ らの検討により、骨粗鬆症で本剤の薬物動態に人種差のないことを類推することとした。 (2) 臨床薬理試験(AK156-I-1 試験)の結果から、骨粗鬆症患者における本剤の薬物動態に 人種差がないことを類推した後、海外で実施したH2301 試験と同様のデザインで日本人 骨粗鬆症患者を対象とした骨折抑制試験(AK156-III-1 試験)を実施し、ゾレドロン酸 5 mg 年 1 回投与の骨折抑制率および安全性が海外試験結果と類似していることを示すこ ととした。 新たに国内で実施する試験と比較検討する試験の構成を以下に示す(図 2-1)。 図 2-1 新たに国内で実施する試験と比較検討する試験の構成
[2] 独立行政法人医薬品医療機器総合機構(以下、PMDA)との対面助言 国内開発計画の妥当性を協議するために、 年 月 日にPMDA との対面助言を実 施した。協議の結果、国内で実施する試験と比較対応する試験の構成および実施すべき国 内試験の骨子などの助言を受け、国内の開発を開始した。 2.3.2.2 国内臨床試験の結果 [1] 臨床薬理試験(AK156-I-1 試験)および PPK 解析 日本人骨粗鬆症患者にゾレドロン酸4 mg を投与した際の薬物動態を、既存の日本人悪性 腫瘍骨転移患者での結果と比較した結果、疾患間で大きな違いはみられなかった。さらに PPK 解析の結果、疾患(悪性腫瘍骨転移、骨粗鬆症)および人種差(日本人、外国人)は 薬物動態に影響を及ぼす要因にはならないと考察した。以上から、日本人および外国人骨 粗鬆症患者にゾレドロン酸5 mg を点滴静注したときの薬物動態に顕著な人種差はないと 類推できると考察した。 [2] 第 III 相試験(AK156-III-1 試験) 上記の結果を踏まえ、日本人骨粗鬆症患者を対象として、ゾレドロン酸5 mg 年 1 回投与 の有効性、安全性を検討する骨折抑制試験(AK156-III-1 試験)を実施した。その結果、主 要評価項目である新規椎体骨折抑制率では投与期間2 年間でゾレドロン酸 5 mg のプラセボ に対する優越性が示され、海外のピボタル試験である骨折抑制試験(H2301 試験)の結果 と類似していた。また、安全性プロファイルは海外試験の結果と特段の違いは認められな かった。 以上、新たに実施した国内臨床試験で想定どおりの結果が得られたため、本剤の国内での製 造販売承認申請が可能と判断した。
3. 特徴および有用性
3.1 非臨床試験成績から見た特徴および有用性 ゾレドロン酸の製剤学的特徴、非臨床試験から確認された特徴および示唆された有用性を以 下に示す。 [1] 長期間にわたり骨組織に特異的に分布し、骨吸収を抑制する作用を持つ。 [2] 骨粗鬆症モデル動物において、骨量の減少を抑制し、骨強度を増加させる。 [3] 骨折の治癒過程に悪影響を及ぼさない。 3.2 臨床試験成績から見た特徴および有用性 ゾレドロン酸の臨床試験から確認された特徴および有用性を以下に示す。 [1] 1 年間隔の投与で効果を発揮する。 [2] 様々なタイプの骨粗鬆症患者に対し高い有効性を示す。 [3] 静脈内への直接投与のため、確実に血中に投与され、また、経口ビスホスホネート製剤に 起こりうる上部消化管障害発現の可能性が低い。 本剤は注射製剤であり投与間隔が1 年と長いことから、経口ビスホスホネート製剤の服用ご とに発現する消化器系の副作用を減少させ、かつ飲み忘れや通院間隔の遅延による服薬コンプ ライアンスの低下を回避でき、長期間の投与継続を可能とすることが期待される。さらに定期 的な来院が困難な患者、認知症を合併する患者への骨粗鬆症治療の面および通院負担の軽減の 面で有益である。以上を踏まえ本剤は、医療現場に新たな選択肢を提供するものである。4. 参考文献
Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al.The National Osteoporosis Foundation. Clinician's Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int 2014; 25(10): 2359-81.
Fleisch H. Bisphosphonates in Bone Disease, Fourth Edition: From the Laboratory to the Patient. Academic Press 2000; p. 34-55.
Gehlbach S, Saag KG, Adachi JD, Hooven FH, Flahive J, Boonen S, et al. Previous fractures at multiple sites increase the risk for subsequent fractures: the Global Longitudinal Study of Osteoporosis in Women. J Bone Miner Res 2012; 27: 645-53.
Harrison RA, Siminoski K, Vethanayagam D, Majumdar SR. Osteoporosis-related kyphosis and impairments in pulmonary function: a systematic review. J Bone Miner Res 2007; 22: 447-57.
Kanis JA, McCloskey EV, Johansson H, Cooper C, Rizzoli R, Reginster JY. Scientific Advisory Board of the European Society for Clinical and Economic Aspects of Osteoporosis and
Osteoarthritis (ESCEO) and the Committee of Scientific Advisors of the International Osteoporosis Foundation (IOF). European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int 2013; 24(1): 23-57.
Kanis JA. WHO Study Group. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. Osteoporos Int 1994; 4: 368-81.
NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285(6): 785-95.
Prevention and management of osteoporosis. WHO Technical Report. World Health Organization 2003; 31-52.
Sakamoto K, Nakamura T, Hagino H, Endo N, Mori S, Muto Y, et al. Report on the Japanese Orthopaedic Association's 3-year project observing hip fractures at fixed-point hospitals. J Orthop Sci 2006; 11: 127-34.
Yamaguchi T, Sugimoto T, Yamauchi M, Matsumori Y, Tsutsumi M, Chihara K. Multiple vertebral fractures are associated with refractory reflux esophagitis in postmenopausal women. J Bone Miner Metab 2005; 23: 36-40.
折茂肇 他. 骨粗鬆症の予防と治療ガイドライン作成委員会. 骨粗鬆症の予防と治療ガイ ドライン 2015 年版. ライフサイエンス出版; 2015a. p. 4-5.
折茂肇 他. 骨粗鬆症の予防と治療ガイドライン作成委員会. 骨粗鬆症の予防と治療ガイ ドライン 2015 年版. ライフサイエンス出版; 2015b. p. 78-85. 折茂肇 他. 骨粗鬆症の予防と治療ガイドライン作成委員会. 骨粗鬆症の予防と治療ガイ ドライン 2015 年版. ライフサイエンス出版; 2015c. p. 96-97. 宗圓聰, 福永仁夫, 杉本利嗣, 曽根照喜, 藤原佐枝子, 遠藤直人,他. 日本骨代謝学会, 日本 骨粗鬆症学会合同 原発性骨粗鬆症診断基準改訂検討委員会. 原発性骨粗鬆症の診断基準 (2012 年度改訂版). Osteoporosis Japan 2013; 21(1); 9-21. 萩野浩. 骨折の連鎖を断つための新たな活動. 臨床整形外科 2014; 49(9): 775-80.
リクラスト点滴静注液
5mg
ゾレドロン酸水和物
第
1 部(モジュール 1):
申請書等行政情報及び添付文書に関する情報
1.6 外国における使用状況等に関する資料
旭化成ファーマ株式会社
略号一覧
略号 省略していない表現
目次
1. 外国における使用状況 ... 4
2. 主たる承認国における添付文書等 ... 7
2.1 米国における添付文書 ... 7 2.2 欧州における添付文書 ... 76 2.3 企業中核データシート ... 1121. 外国における使用状況
ゾレドロン酸は、骨パジェット病や骨粗鬆症領域を適応症として米国ではRECLAST®、米国 以外ではACLASTA®の販売名で承認されている。2016 年 6 月時点で、本剤は 115 以上の国ま たは地域で販売承認を得ている。本剤の主な承認国、初回承認日の一覧を表 1に、欧州および 米国における効能・効果別の承認日を表 2に示す。また、本剤の欧州および米国における承認 内容を 表 3に示す。 なお、ゾレドロン酸を有効成分とする製剤は、腫瘍領域でZOMETA®としても承認されてい る。ZOMETA®の欧州および米国における承認内容を 表 3に示す。 表 1 主な承認取得国または地域 地域区分 国または地域 初回承認日 欧州 フランスa) 2005 年 4 月 15 日 ドイツa) 2005 年 4 月 15 日 英国a) 2005 年 4 月 15 日 北中南米 カナダ 2005 年 6 月 30 日 ブラジル 2005 年 8 月 1 日 米国b) 2007 年 4 月 16 日 アジア インド 2005 年 8 月 17 日 韓国c) 2005 年 11 月 9 日 香港 2006 年 2 月 26 日 中国 2007 年 4 月 26 日 台湾 2007 年 9 月 10 日 オセアニア ニュージーランド 2007 年 8 月 30 日 オーストラリア 2008 年 6 月 23 日 中近東 カタール 2006 年 1 月 19 日 アラブ首長国連邦 2006 年 3 月 6 日 サウジアラビア 2006 年 12 月 10 日 アフリカ ケニア 2006 年 2 月 10 日 エジプト 2007 年 7 月 12 日 南アフリカ 2007 年 11 月 30 日 a) 欧州の中央審査方式で承認された b) 販売名:RECLAST®c) 2012 年 12 月 26 日に Novartis から Sandoz へ製造販売承認を移管(現在の商標名は「Sandoz Zoledronic acid」)
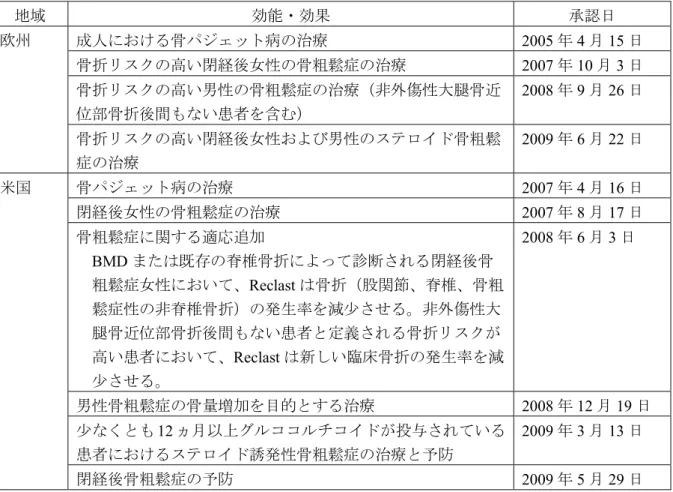
表 2 欧州および米国における効能・効果別の承認日 地域 効能・効果 承認日 欧州 成人における骨パジェット病の治療 2005 年 4 月 15 日 骨折リスクの高い閉経後女性の骨粗鬆症の治療 2007 年 10 月 3 日 骨折リスクの高い男性の骨粗鬆症の治療(非外傷性大腿骨近 位部骨折後間もない患者を含む) 2008 年 9 月 26 日 骨折リスクの高い閉経後女性および男性のステロイド骨粗鬆 症の治療 2009 年 6 月 22 日 米国 骨パジェット病の治療 2007 年 4 月 16 日 閉経後女性の骨粗鬆症の治療 2007 年 8 月 17 日 骨粗鬆症に関する適応追加 BMD または既存の脊椎骨折によって診断される閉経後骨 粗鬆症女性において、Reclast は骨折(股関節、脊椎、骨粗 鬆症性の非脊椎骨折)の発生率を減少させる。非外傷性大 腿骨近位部骨折後間もない患者と定義される骨折リスクが 高い患者において、Reclast は新しい臨床骨折の発生率を減 少させる。 2008 年 6 月 3 日 男性骨粗鬆症の骨量増加を目的とする治療 2008 年 12 月 19 日 少なくとも12 ヵ月以上グルココルチコイドが投与されている 患者におけるステロイド誘発性骨粗鬆症の治療と予防 2009 年 3 月 13 日 閉経後骨粗鬆症の予防 2009 年 5 月 29 日
表 3 欧州および米国の承認内容 国 販売名 剤型・含量 効能・効果 用法・用量 欧 州 ACLASTA® 注射剤・5 mg 骨折リスクの高い、以下の男女にお ける骨粗鬆症の治療 - 閉経後女性 - 成人男性(非外傷性大腿骨近位部 骨折後間もない患者を含む) 骨折リスクの高い、以下の男女にお けるステロイド性骨粗鬆症の治療 - 閉経後女性 - 成人男性 成人における骨パジェット病の治療 15 分以上かけて点滴静注する。 閉経後骨粗鬆症、男性骨粗鬆症、 ステロイド性骨粗鬆症:5 mg を 年1 回 骨パジェット病:5 mg を単回 ZOMETA® 注射剤・4 mg 成人悪性腫瘍骨転移患者における骨 関連事象(病的骨折、脊髄圧迫、骨 に対する放射線治療あるいは外科手 術、腫瘍による高カルシウム血症) の予防 成人における腫瘍による高カルシウ ム血症の治療 15 分以上かけて点滴静注する。 悪性腫瘍骨転移患者における骨 関連事象の予防:4 mg を 3~4 週間に1 回 腫瘍による高カルシウム血症の 治療:4 mg を単回 米 国 RECLAST® 注射剤・5 mg 閉経後骨粗鬆症の治療および予防 男性骨粗鬆症の骨量増加を目的とす る治療 ステロイド性骨粗鬆症の治療および 予防 骨パジェット病の治療 15 分以上かけて点滴静注する。 閉経後骨粗鬆症の治療、男性骨 粗鬆症の骨量増加を目的とする 治療、ステロイド性骨粗鬆症の 治療および予防:5 mg を年 1 回 閉経後骨粗鬆症の予防:5 mg を 2 年に 1 回 骨パジェット病の治療:5 mg を 単回 ZOMETA® 注射剤・4 mg 悪性腫瘍による高カルシウム血症 多発性骨髄腫および固形癌骨転移 15 分以上かけて点滴静注する。 悪性腫瘍による高カルシウム血 症:4 mg を単回 多発性骨髄腫および固形癌骨転 移:4 mg を 3~4 週間に 1 回
2. 主たる承認国における添付文書等
2.1 米国における添付文書 米国における添付文書の和訳および原文を以下に示す。【米国添付文書の和訳】
添付文書の概要 概要には、RECLAST®を安全かつ有効に使用するために必要な情報がすべて含まれているわけで はない。本剤の添付文書(全文)を参照すること。 RECLAST®点滴静注 米国での承認取得:2001 年 効能・効果 本剤は以下の適応を有するビスホスホネートである: • 閉経後骨粗鬆症の治療および予防(1.1、1.2) • 男性骨粗鬆症の骨量増加を目的とする治療(1.3) • ステロイド性骨粗鬆症の治療および予防(1.4) • 骨パジェット病の治療(1.5) 使用の制限 至適使用期間は確立されていない。骨折リスクの低い患者では、3~5 年間の使用後に投与中止を 検討すること(1.6) 用法・用量 15 分以上かけて点滴静注する。 • 閉経後骨粗鬆症の治療(2.2)、男性骨粗鬆症の骨量増加を目的とする治療(2.4)、ステロイ ド性骨粗鬆症の治療および予防(2.5):5 mg を年 1 回 • 閉経後骨粗鬆症の予防:5 mg を 2 年に 1 回(2.3) • 骨パジェット病の治療:5 mg を単回点滴静注。カルシウム 1500 mg/日およびビタミン D 800 IU/日を補給すること(2.6) 剤形および含量 100 mL 中にゾレドロン酸として 5 mg を含有する点滴静注液(3) 禁忌 • 低カルシウム血症患者(4) • クレアチニンクリアランスが35 mL/分未満の患者および急性腎機能障害の所見を有する患者 (4、5.3) • 本剤の成分に対して過敏症を有する患者(4、6.2) 警告および使用上の注意• 同じ有効成分を含有する医薬品:ZOMETA®を使用している患者には本剤を投与しないこと (5.1) • 低カルシウム血症:治療中に低カルシウム血症が悪化する場合がある。カルシウムおよびビ タミンD を十分に補うこと(5.2) • 腎機能障害:1 回の投与量は 5 mg 以下とし、点滴静注時間は 15 分以上とすること。投与前 から存在する腎機能障害、高齢、脱水等の危険因子を有する患者では、腎毒性が強まる可能 性がある。各投与前にクレアチニンクリアランスを測定すること(2.7、5.3) • 顎骨壊死:顎骨壊死の発現が報告されている。本剤の投与前に必ず歯科検査を行うこと(5.4) • 非定型大腿骨骨折:大腿骨の非定型骨折が報告されている。大腿部痛または鼡径部痛を有す る患者には適切な診察・検査を行い、大腿骨骨折の可能性を除外すること(5.5) • 妊娠:本剤は胎児に影響を与える恐れがある。妊娠の可能性がある女性には、本剤投与中の 妊娠に関して説明すること(5.6、8.1) • 重度の骨痛、関節痛および筋痛:これらの疼痛が生じる可能性がある。重度の疼痛が発生し た場合は、以降の本剤投与を中止すること(5.7) 副作用 頻度の高い副作用(>10%)は発熱、筋痛、頭痛、関節痛および四肢痛であった(6.1)。他の重 要な副作用は、インフルエンザ様症状、悪心、嘔吐、下痢(6.2)および眼の炎症(6.1)であった。 副作用が疑われる症状を報告する場合は、ノバルティスファーマ株式会社(1-888-669-6682)また は米国食品医薬品局(FDA)(1-800-FDA-1088 または www.fda.gov/medwatch.)に連絡すること 薬物間相互作用 • アミノグリコシド系抗生物質:長期間にわたって血清カルシウム値が低下する可能性がある (7.1) • ループ利尿剤:低カルシウム血症のリスクが増大する可能性がある(7.2) • 腎毒性を有する薬剤:慎重に使用すること(7.3) • 主に腎排泄される薬剤:腎機能障害に伴って曝露が増加する可能性がある。このようなリス クを有する患者では血清クレアチニン値を測定すること(7.4) 特殊な集団における使用 授乳婦:授乳婦には投与しないこと(8.3) 小児への投与:小児での使用は適応外である(8.4) 高齢者への投与:腎機能に留意し、慎重に投与すること(8.5) 患者向け情報と患者向け医薬品ガイドについては17 項を参照すること 改訂:2016 年 4 月
添付文書(全文):目次* 1 効能・効果 1.1 閉経後骨粗鬆症の治療 1.2 閉経後骨粗鬆症の予防 1.3 男性骨粗鬆症 1.4 ステロイド性骨粗鬆症 1.5 骨パジェット病 1.6 重要な使用制限 2 用法・用量 2.1 全般的な注意 2.2 閉経後骨粗鬆症の治療 2.3 閉経後骨粗鬆症の予防 2.4 男性骨粗鬆症 2.5 ステロイド性骨粗鬆症の治療および予防 2.6 骨パジェット病の治療 2.7 投与前の臨床検査および歯科検査 2.8 カルシウムおよびビタミン D の補給 2.9 投与方法 3 剤形および含量 4 禁忌 5 警告および使用上の注意 5.1 同一有効成分を含有する薬剤 5.2 低カルシウム血症およびミネラル代謝障害 5.3 腎機能障害 5.4 顎骨壊死 5.5 非定型大腿骨転子下骨折および大腿骨骨幹部骨折 5.6 妊娠 5.7 筋骨格痛 5.8 喘息患者 6 副作用 6.1 臨床試験 6.2 市販後の使用経験 7 薬物間相互作用 7.1 アミノグリコシド系抗生物質 7.2 ループ利尿剤 7.3 腎毒性を有する薬剤 7.4 主に腎排泄される薬剤 8 特殊な集団における使用 8.1 妊娠 8.3 授乳婦 8.4 小児への投与
8.5 高齢者への投与 8.6 腎機能障害患者 8.7 肝機能障害患者 10 過量投与 11 性状 12 薬効薬理 12.1 作用機序 12.2 薬力学 12.3 薬物動態 13 非臨床毒性 13.1 がん原性、変異原性、生殖能障害 13.2 動物薬理学 13.3 生殖発生毒性 14 臨床成績 14.1 閉経後骨粗鬆症の治療 14.2 閉経後骨粗鬆症の予防 14.3 男性骨粗鬆症 14.4 ステロイド性骨粗鬆症の治療および予防 14.5 骨パジェット病の治療 16 包装・保存および取扱い 17 患者向け情報 *添付文書(全文)から省略されたセクションまたはサブセクションは記載していない
添付文書(全文) 1 効能・効果 1.1 閉経後骨粗鬆症の治療 本剤は、閉経後骨粗鬆症の治療に適応される。本剤は、骨密度(BMD)または既存椎体骨折に基 づいて閉経後骨粗鬆症と診断された患者で骨折(大腿骨近位部骨折、椎体骨折および非椎体骨折) の発生率を低下させる。骨折リスクの高い患者(非外傷性大腿骨近位部骨折後間もない患者)で は、新規臨床骨折の発生率を低下させる[「臨床成績」(14.1)参照]。 1.2 閉経後骨粗鬆症の予防 本剤は、閉経後骨粗鬆症の予防に適応される[「臨床成績」(14.2)参照]。 1.3 男性骨粗鬆症 本剤は、男性骨粗鬆症の骨量増加を目的とする治療に適応される[「臨床成績」(14.3)参照]。 1.4 ステロイド性骨粗鬆症 プレドニゾロン換算で7.5 mg/日以上のステロイド剤の全身投与を開始または継続しており、ステ ロイド剤の使用を12 ヵ月間以上継続することが予想される患者において、本剤は性別を問わずス テロイド性骨粗鬆症の治療および予防に適応される[「臨床成績」(14.4)参照]。 1.5 骨パジェット病 本剤は、性別を問わず骨パジェット病の治療に適応される。骨パジェット病患者のうち、血清ア ルカリホスファターゼ値が年齢別基準範囲上限より2 倍以上高い患者、症状を有する患者、合併 症のリスクを有する患者に用いられる[「臨床成績」(14.5)参照]。 1.6 重要な使用制限 骨粗鬆症治療薬としての本剤の安全性および有効性は、3 年間の臨床成績に基づいている。至適 使用期間は確立していない。ビスホスホネート製剤を投与している患者では、治療継続の必要性 を定期的に再検討すること。骨折リスクが低い患者では、3~5 年間の使用後に投与中止を検討す ること。投与中止後は定期的に骨折リスクを再評価すること。 2 用法・用量 2.1 全般的な注意 本剤は15 分以上かけて点滴静注する。 • 本剤の投与前に十分な水分補給を行うこと[「警告および使用上の注意」(5.3)参照]。 • 可能な限り、薬液に異物や変色がないことを目視により確認してから使用すること。 • 点滴静注後、静脈ラインを10 mL の生理食塩水でフラッシュすること。 • 本剤の投与後にアセトアミノフェンを投与すると、急性期反応の症状発現を抑制できること がある。
2.2 閉経後骨粗鬆症の治療 通常、5 mg を 15 分以上かけて年 1 回点滴静注する。 2.3 閉経後骨粗鬆症の予防 通常、5 mg を 15 分以上かけて 2 年に 1 回点滴静注する。 2.4 男性骨粗鬆症 通常、5 mg を 15 分以上かけて年 1 回点滴静注する。 2.5 ステロイド性骨粗鬆症の治療および予防 通常、5 mg を 15 分以上かけて年 1 回点滴静注する。 2.6 骨パジェット病の治療 通常、5 mg を一定の速度で 15 分以上かけて点滴静注する。 骨パジェット病での再投与 骨パジェット病患者で、本剤の初回単回投与による寛解期間の長期持続が認められている。再投 与に関するデータはないが、血清アルカリホスファターゼ値の上昇に基づき再発と診断された患 者や、血清アルカリホスファターゼ値が正常化しなかった患者、症状が確認された患者では、本 剤の再投与を考慮してもよい。 2.7 投与前の臨床検査および歯科検査 • 本剤の各投与前に血清クレアチニン値を測定し、体重を基にCockcroft-Gault 式を用いてクレ アチニンクリアランスを算出すること。本剤は、クレアチニンクリアランスが35 mL/分未満 の患者および急性腎機能低下の患者には禁忌である。クレアチニンクリアランスが35 mL/分 以上の患者では、本剤5 mg の静脈内投与が推奨される。腎機能に基づく用量調節の必要性を 裏付ける安全性データおよび有効性のデータはない。したがって、クレアチニンクリアラン スが35 mL/分以上の患者で投与量の調節は必要ない[「禁忌」(4)および「警告および使用 上の注意」(5.3)参照]。 • 本剤の投与開始前に歯科検査を行うこと[「警告および使用上の注意」(5.4)参照]。 2.8 カルシウムおよびビタミン D の補給 • 骨パジェット病の治療を受けている患者に対しては、カルシウムおよびビタミンD の補給の 重要性ならびに低カルシウム血症の症状を説明すること。本剤投与後2 週間は、カルシウム 1,500 mg/日(750 mg を 1 日 2 回または 500 mg を 1 日 3 回)およびビタミン D 800 IU/日を補 給すること[「警告および使用上の注意」(5.2)参照]。 • 骨粗鬆症の治療を受けている患者に対しては、食事によるカルシウムおよびビタミンD 摂取 が不十分な場合、カルシウムおよびビタミンD を補うよう指示すること。最低 1,200 mg/日の カルシウムおよび800~1,000 IU/日のビタミン D の補給が推奨される。
2.9 投与方法 本剤は一定の速度で15 分以上かけて点滴静注する。点滴静注後は 10 mL の生理食塩水で静脈ライ ンをフラッシュすること。 カルシウム等の2 価陽イオンを含有する溶液と混合しないこと。他の薬剤とは別のベント式点滴 ラインから単回点滴静注液として投与すること。 冷蔵保存した場合は、室温に戻してから使用すること。開封後の薬液は2~8℃(36~46°F)で 24 時間安定である[「包装・保管および取扱い」(16)参照]。 3 剤形および含量 100 mL 中にゾレドロン酸として 5 mg を含有する点滴静注液 4 禁忌 以下の患者には禁忌である。 • 低カルシウム血症患者[「警告および使用上の注意」(5.2)参照] • クレアチニンクリアランスが35 mL/分未満の患者および急性腎機能障害の所見を有する患者 (腎不全リスクが高くなるため)[「警告および使用上の注意」(5.3)参照] • ゾレドロン酸または本剤の成分に対し、過敏症の既往歴のある患者(蕁麻疹、血管浮腫、ア ナフィラキシー反応・ショック等の過敏症が報告されている)[「市販後の使用経験」(6.2) 参照] 5 警告および使用上の注意 5.1 同一有効成分を含有する薬剤 本剤には、癌治療に使用されるZOMETA®と同一の有効成分が含まれるため、ZOMETA®を使用 している患者には本剤を投与しないこと。 5.2 低カルシウム血症とミネラル代謝障害 低カルシウム血症やミネラル代謝障害(副甲状腺機能低下症、甲状腺手術、副甲状腺手術、吸収 不良症候群、小腸切除等)が存在する場合は、本剤の投与開始前に有効な治療を行うこと。これ らの患者では、カルシウム値、リン値およびマグネシウム値の測定が推奨される[「禁忌」(4) 参照]。 本剤投与後の低カルシウム血症は、骨パジェット病患者にとって重大なリスクである。骨パジェッ ト病患者に対しては、低カルシウム血症の症状ならびに血清カルシウム値の維持におけるカルシ ウムおよびビタミンD の補給の重要性を説明すること[「用法・用量」(2.8)、「副作用」(6.1)、 「患者向け情報」(17)参照]。 骨粗鬆症患者に対しては、血清カルシウム値の維持におけるカルシウムおよびビタミンD の補給 の重要性を説明すること[「用法・用量」(2.8)、「副作用」(6.1)、「患者向け情報」(17) 参照]。 5.3 腎機能障害 1 回の投与量は 5 mg 以下とし、15 分以上かけて投与すること[「用量・用法」(2)参照]。
本剤は、クレアチニンクリアランスが35 mL/分未満の患者および急性腎機能障害の所見を有する 患者には禁忌である[「禁忌」(4)参照]。病歴または診察所見から脱水が示唆される場合は、 脱水が回復するまで本剤を投与しないこと[「市販後の使用経験」(6.2)参照]。 慢性腎機能障害患者では慎重に使用すること。特に投与前から存在する腎障害、高齢、腎毒性を 有する薬剤の併用、利尿剤の併用、本剤の投与前後の重度脱水等の危険因子を有する患者におい て、ゾレドロン酸投与後に、腎不全を含む急性腎機能障害が認められている。単回投与後の患者 では急性腎不全が認められている。投与前から中等度または重度の腎機能障害が存在する患者お よび上記の危険因子のいずれかを有する患者では、入院・透析あるいは致死的転帰に至った報告 がある[「市販後の使用経験」(6.2)参照]。腎機能障害患者では、主に腎排泄される併用薬や その代謝物の曝露が増える場合がある[「薬物間相互作用」(7.4)参照]。 本剤の各投与前に、体重を基にCockcroft-Gault 式を用いてクレアチニンクリアランスを算出する こと。腎機能障害患者では、投与後の血清クレアチニン値の一過性上昇度が大きくなる可能性が あるので、このようなリスクを有する患者では、投与後も必要に応じてクレアチニンクリアラン スを確認すること。高齢患者や利尿剤の投与を受けている患者では急性腎不全のリスクが増大す る。これらの患者では、本剤の投与前に体液量の状態を確認し、十分な水分補給を行うこと。腎 毒性を有する薬剤と併用する場合は慎重に使用すること[「薬物間相互作用」(7.3)参照]。主 に腎排泄される薬剤を併用しており、急性腎不全のリスクを有する患者では、クレアチニンクリ アランスのモニタリングを検討すること[「薬物間相互作用」(7.4)参照]。 5.4 顎骨壊死 ゾレドロン酸製剤を含むビスホスホネート製剤が投与された患者において顎骨壊死が報告されて いる。報告された症例の多くは、歯科処置中にビスホスホネート剤の静脈内投与を受けた癌患者 であるが、ビスホスホネート製剤の経口投与または静脈内投与を受けた閉経後骨粗鬆症患者でも 発生している。したがって、ビスホスホネート剤の投与開始前に、主治医は口腔内検査を行うこ と。顎骨壊死の危険因子(悪性腫瘍、化学療法、血管新生阻害剤、放射線治療、ステロイド剤の 使用、口腔の不衛生、既存の歯科疾患または感染症、貧血、血液凝固異常等)を有する患者では、 ビスホスホネート剤の投与前の歯科検査および適切な歯科予防処置を検討すること。顎骨壊死を 発症するリスクは、ビスホスホネート剤への曝露期間に応じて増大する可能性がある。顎骨壊死 と関連付けられている薬剤の併用は、顎骨壊死の発症リスクを高める可能性がある。 顎骨壊死の危険因子を有する患者では、ビスホスホネート製剤の投与中、侵襲的な歯科処置を可 能な限り回避すること。投与中に顎骨壊死が発現した場合、歯科手術により病態が悪化する可能 性がある。歯科処置を要する患者がビスホスホネート製剤を休薬した場合の顎骨壊死リスク軽減 の有無を示すデータはない。主治医は臨床的判断のもと、患者ごとにベネフィット・リスク評価 に基づいて治療計画を立てること[「副作用」(6.1)参照]。 5.5 大腿骨転子下および大腿骨骨幹部の非定型骨折 ビスホスホネート剤の投与を受けた患者で、大腿骨骨幹部の非外傷性非定型骨折が報告されてい る。非定型骨折は、粉砕の所見を伴わない横骨折または短斜骨折で、大腿骨の小転子直下から顆 上部までどの部位でも起こりうる。非定型大腿骨骨折は、ビスホスホネート剤の投与を受けてい ない骨粗鬆症患者にも発生することがあるため、ビスホスホネート製剤との因果関係は不明であ る。
非定型大腿骨骨折は軽微な外傷後に、または外傷がなくても発生する。両側性の場合もあり、多 くの患者は大腿骨の完全骨折が発生する数週間から数ヵ月間前に、大腿部の鈍痛等の前駆痛を訴 える。骨折時にステロイド剤(プレドニゾン等)を併用していたという報告も多い。 ビスホスホネート剤の使用歴を有する患者が大腿部痛または鼡径部痛を訴えた場合は、非定型大 腿骨骨折の発生を疑い、大腿骨の不完全骨折の有無を確認すること。非定型大腿骨骨折が発生し た患者では、対側の大腿骨骨折の症候の有無も確認すること。リスク・ベネフィット評価の結論 が出るまでは、ビスホスホネート製剤の休薬を検討すること。 5.6 妊娠 妊娠中は本剤を投与しないこと。妊娠中の女性に本剤を投与すると、胎児に悪影響を及ぼす恐れ がある。本剤の投与中に妊娠した場合は、胎児に悪影響を及ぼす可能性があることを患者に説明 すること。妊娠の可能性がある女性には、本剤の投与中は避妊するよう指導すること[「特殊な 集団における使用」(8.1)参照]。 5.7 筋骨格痛 市販後に、本剤を含むビスホスホネート製剤の投与を受けている患者で、日常生活に支障を来す ほど重度の骨痛、関節痛および筋痛がまれに報告されている。疼痛発現までの期間は、投与開始 後1 日から数ヵ月まで幅があった。重度の疼痛が発現した場合は、以降の投与中止を検討するこ と。患者の大半は投与中止後に疼痛が軽減したが、同じビスホスホネート製剤または他のビスホ スホネートを再投与したところ、疼痛が再発した患者も認められた[「副作用」(6.2)参照]。 5.8 喘息患者 本剤の臨床試験では認められていないが、ビスホスホネート製剤の投与を受けているアスピリン 感受性患者において気管支収縮の報告が複数ある。アスピリン感受性患者に本剤を投与する場合 は、慎重に投与すること。 6 副作用 6.1 臨床試験 臨床試験は試験ごとに条件が異なるため、ある薬剤の臨床試験で認められた副作用発現率と、他 の薬剤の臨床試験における副作用発現率とを直接比較することはできない。また、臨床試験の副 作用発現率は、日常診療での副作用発現率を反映していない場合もある。 閉経後骨粗鬆症の治療 骨密度または既存椎体骨折の有無に基づいて診断された閉経後骨粗鬆症患者7,736 名(65~89 歳) を対象とした大規模な無作為化、二重盲検、プラセボ対照、国際共同試験(試験1)において、 閉経後骨粗鬆症の治療における本剤の安全性を検討した。試験期間は3 年間で、3,862 名に本剤を、 3,852 名にプラセボを投与した。本剤は、1 回 5 mg/100 mL を 15 分以上かけて年 1 回、計 3 回点 滴投与した。すべての患者にカルシウム1,000~1,500 mg/日およびビタミン D 400~1,200 IU/日を 補給した。
全死亡率は、本剤群で3.4%、プラセボ群で 2.9%であり、両群で同程度であった。重篤な有害事象 の発現率は本剤群で29.2%、プラセボ群で 30.1%であった。有害事象の発現により試験中止に至っ た患者の割合は、本剤群で5.4%、プラセボ群で 4.8%であった。 50~95 歳の男女 2,127 名を対象とした無作為化、二重盲検、プラセボ対照、国際共同、エンドポ イント評価試験(試験2)で、非外傷性大腿骨近位部骨折後間もない(90 日以内)骨粗鬆症患者 の治療における本剤の安全性を検討した。1,065 名が本剤群に、1,062 名がプラセボ群に無作為に 割り付けられた。本剤は、1 回 5 mg/100 mL を 15 分以上かけて年 1 回点滴投与した。平均約 2 年 間の追跡調査を行った試験対象集団おいて、臨床骨折が211 名以上の患者に確認されるまで試験 を継続した。ビタミンD 値は定期的に測定しなかったが、投与開始 14 日以上前に、初回負荷投 与量のビタミンD(50,000~125,000 IU)を経口投与または筋肉内投与した。また、カルシウム 1,000 ~1,500 mg/日およびビタミン D 800~1,200 IU/日を補給した。 全死亡率は本剤群で9.6%、プラセボ群で 13.3%であった。重篤な有害事象の発現率は本剤群で 38.3%、プラセボ群で 41.3%であった。有害事象の発現により試験中止に至った患者の割合は、本 剤群で5.3%、プラセボ群で 4.7%であった。 上記のいずれかの試験で骨粗鬆症患者の2%以上に認められ、プラセボ群より本剤群で発現率が高 かった副作用を表1 に示す。 表1 骨粗鬆症患者の 2%以上に認められ、プラセボ群より発現率が高かった副作用 Study 1 Study 2 器官別大分類 本剤5 mg 年1 回点滴 静注 % (N=3862) プラセボ 年1 回投与 % (N=3852) 本剤5 mg 年1 回点滴 静注 % (N=1054) プラセボ 年1 回投与 % (N=1057) 血液およびリンパ系障害 貧血 4.4 3.6 5.3 5.2 代謝および栄養障害 脱水 0.6 0.6 2.5 2.3 食欲不振 2.0 1.1 1.0 1.0 神経系障害 頭痛 12.4 8.1 3.9 2.5 浮動性めまい 7.6 6.7 2.0 4.0 耳および迷路障害 回転性めまい 4.3 4.0 1.3 1.7 心臓障害 心房細動 2.4 1.9 2.8 2.6 血管障害 高血圧 12.7 12.4 6.8 5.4 胃腸障害 悪心 8.5 5.2 4.5 4.5 下痢 6.0 5.6 5.2 4.7 嘔吐 4.6 3.2 3.4 3.4 上腹部痛 4.6 3.1 0.9 1.5 消化不良 4.3 4.0 1.7 1.6 筋骨格系および結合組織障害 関節痛 23.8 20.4 17.9 18.3 筋肉痛 11.7 3.7 4.9 2.7
四肢痛 11.3 9.9 5.9 4.8 肩痛 6.9 5.6 0.0 0.0 骨痛 5.8 2.3 3.2 1.0 頚部痛 4.4 3.8 1.4 1.1 筋痙縮 3.7 3.4 1.5 1.7 変形性関節症 9.1 9.7 5.7 4.5 筋骨格痛 0.4 0.3 3.1 1.2 一般・全身障害および投与部位の状態 発熱 17.9 4.6 8.7 3.1 インフルエンザ様症状 8.8 2.7 0.8 0.4 疲労 5.4 3.5 2.1 1.2 悪寒 5.4 1.0 1.5 0.5 無力症 5.3 2.9 3.2 3.0 末梢性浮腫 4.6 4.2 5.5 5.3 疼痛 3.3 1.3 1.5 0.5 倦怠感 2.0 1.0 1.1 0.5 高熱 0.3 <0.1 2.3 0.3 胸痛 1.3 1.1 2.4 1.8 臨床検査 腎クレアチニンクリアランス減少 2.0 2.4 2.1 1.7 腎機能障害 ゾレドロン酸製剤を含むビスホスホネート製剤の静脈内投与後に、腎機能低下(血清クレアチニ ン値の上昇)およびまれに急性腎不全として発現する腎機能障害が認められている。閉経後骨粗 鬆症を対象とした臨床試験では、ベースラインのクレアチニンクリアランス(体重に基づく)が 30 mL/分未満の患者、尿試験紙において蛋白が 2+以上の患者、スクリーニング期間中に血清クレ アチニン値が0.5 mg/dL を超えた患者を除外した。3 年間にわたって、クレアチニンクリアランス (各投与前に年1 回測定)の変動および腎不全・腎機能障害の発現率は、ベースラインのクレア チニンクリアランスが30~60 mL/分の患者を含め、本剤群とプラセボ群で同程度であった。本剤 群の1.8%の患者およびプラセボ群の 0.8%の患者で、投与後 10 日以内に血清クレアチニン値が一 過性に上昇したが、特に治療を行うことなく自然に回復した[「警告および使用上の注意」(5.3) 参照]。 急性期反応 試験1 では、本剤の点滴静注後に発熱(18%)、筋痛(9%)、インフルエンザ様症状(8%)、頭 痛(7%)、および関節痛(7%)等の急性期反応が発現した。これらの症状の大半は本剤投与後 3 日以内に発現し、多くは発現後3 日以内に回復したが、回復に 7~14 日要することもあった。試 験2 では、アセトアミノフェンが禁忌ではない患者に対して、点滴静注時に標準投与量の経口ア セトアミノフェンを提供し、点滴静注後72 時間は自宅で追加のアセトアミノフェンを頓服するよ う指導した。この試験では、本剤による一過性の急性期反応の発現が少なかった(発熱7%および 関節痛3%)。以降の本剤投与では急性期反応の発現率が低下した。 臨床検査所見
試験1 では、閉経後骨粗鬆症患者の約 0.2%で、本剤投与後に血清カルシウム値の顕著な低下(7.5 mg/dL 未満)が認められた。症候性の低カルシウム血症はみられなかった。試験 2 では、ビタミ ンD による前治療後、本剤投与中に血清カルシウム値が 7.5 mg/dL 未満に低下した患者はいなかっ た。 注射部位反応 骨粗鬆症を対象とした試験では、点滴投与部位における局所反応(そう痒感、発赤、疼痛等)が、 本剤投与患者の0~0.7%、プラセボ投与患者の 0~0.5%で認められている。 顎骨壊死 閉経後骨粗鬆症患者7,736 名を対象とした試験 1 では、投与開始後にプラセボ群の 1 名と本剤群 の1 名で、顎骨壊死と合致する症状が発現した。いずれも適切な治療後に回復した[「警告と使 用上の注意(5.4)」参照]。試験 2 では、いずれの投与群においても顎骨壊死の報告はなかった。 心房細動 閉経後骨粗鬆症を対象とした試験1 では、重度の有害事象と判定された心房細動の発現率は、本 剤群で1.3%(3,862 名中 50 名)、プラセボ群で 0.4%(3,852 名中 17 名)であった。全心房細動の 発現率は、本剤群で2.5%(3,862 名中 96 名)、プラセボ群で 1.9%(3,852 名中 75 名)であった。 いずれの投与群においても、心房細動の関連事象の90%以上は、投与後 1 ヵ月以上経過してから 発現した。心電図サブスタディでは、投与前と投与9~11 日後に 559 名の部分集団で心電図測定 を行った。投与群間で心房細動の発現率に大差はなく、心房細動が点滴静注後の急性期反応とは 無関係であることが示唆された。試験2 では、重篤な有害事象と判定された心房細動の発現率が 本剤群で1.0%(1,054 名中 11 名)、プラセボ群で 1.2%(1,057 名中 13 名)で、群間差がないこと が示された。 眼障害 ゾレドロン酸製剤を含むビスホスホネート製剤の投与を受けた患者において虹彩炎、ブドウ膜炎、 上強膜炎、結膜炎が報告されている。骨粗鬆症を対象とした試験では、本剤群の1 名(0.1%未満) ~9 名(0.2%)およびプラセボ群の 0 名(0%)~1 名(0.1%未満)で虹彩炎、ブドウ膜炎および 上強膜炎が認められた。 閉経後骨粗鬆症の予防 45 歳以上の閉経後女性 581 名を対象とした 2 年間の多施設共同、無作為化、二重盲検、プラセボ 対照試験において、閉経後骨減少症(低骨量)患者における本剤の安全性を検討した。患者を以 下の3 群のいずれかに無作為に割り付けた。 ・本剤群:無作為化時および12 ヵ月後に本剤を投与(n=198) ・本剤/プラセボ群:無作為化時に本剤を、12 ヵ月後にプラセボを投与(n=181) ・プラセボ群:無作為化時および12 ヵ月後にプラセボを投与(n=202) 本剤は、1 回 5 mg/100 mL を 15 分以上かけて点滴投与した。すべての患者にカルシウム 500~1,200 mg/日およびビタミン D 400~800 IU/日を補給した。
重篤な有害事象の発現率は、本剤群が10.6%、本剤/プラセボ群が 9.4%、プラセボ群が 11.4%で、 いずれの投与群も同程度であった。有害事象の発現により試験中止に至った患者の割合は、本剤 群で7.1%、本剤/プラセボ群で 7.2%、プラセボ群で 3.0%であった。骨減少症患者の 2%以上に認 められ、プラセボ群より本剤群で発現率が高かった副作用を表2 に示す。 表2 骨減少症患者の 2%以上に認められ、プラセボ群より発現率が高かった副作用 器官別大分類 本剤5 mg 年1 回点滴静注 % (n=198) 本剤5 mg 単回点滴静注 % (n=181) プラセボ 年1 回投与 % (n=202) 代謝および栄養障害 食欲不振 2.0 0.6 0.0 神経系障害 頭痛 14.6 20.4 11.4 浮動性めまい 7.6 6.1 3.5 感覚鈍麻 5.6 2.2 2.0 耳および迷路障害 回転性めまい 2.0 1.7 1.0 血管障害 高血圧 5.1 8.3 6.9 胃腸障害 悪心 17.7 11.6 7.9 下痢 8.1 6.6 7.9 嘔吐 7.6 5.0 4.5 消化不良 7.1 6.6 5.0 腹痛* 8.6 6.6 7.9 便秘 6.6 7.2 6.9 腹部不快感 2.0 1.1 0.5 腹部膨満 2.0 0.6 0.0 皮膚および皮下組織障害 発疹 3.0 2.2 2.5 筋骨格系および結合組織障害 関節痛 27.3 18.8 19.3 筋肉痛 19.2 22.7 6.9 背部痛 18.2 16.6 11.9 四肢痛 11.1 16.0 9.9 筋痙縮 5.6 2.8 5.0 筋骨格痛** 8.1 7.2 7.9 骨痛 5.1 3.3 1.0 頚部痛 5.1 6.6 5.0 関節炎 4.0 2.2 1.5 関節硬直 3.5 1. 1 2.0 関節腫脹 3.0 0.6 0.0 側腹部痛 2.0 0.6 0.0 顎痛 2.0 3.9 2.5 一般・全身障害および投与部位の状態 疼痛 24.2 14.9 3.5 発熱 21.7 21.0 4.5