1.は
じ め に
化学という概念は広大であり,現実世界のあらゆる ものは化学から成るという見方もできる.当社に長く伝 えられている「化成萬物」という言葉は,その心をよく 表している.人類が土器を焼き始めたときそれは化学で あったし,現代の情報科学もまた化学の進歩なくしては あり得なかった.主役であれ脇役であれ,技術の進歩が あるときには必ず化学が伴っているともいえよう. したがって,化学と計算の間には古くからの関係があ るのも不思議ではない.産業化には予測と制御が必須で あり,これまでも幅広く計算が用いられてきた.特に大 規模化した製造では効率と安全を追求するために化学工 学を中心とした物質の移動と反応を取り扱う技術が,ま た研究開発では原理の解明と実験回数の削減のために量 子化学や分子動力学が使われてきている. しかし,化学の守備範囲はそれだけではない.例えば 医薬品や肥料などもまた化学である.統計学の祖の一人 であるフィッシャーが穀物と肥料を題材にしていたこと からも,化学と統計の関連の深さがうかがわれる.現代 の Evidence Based Medicine が統計を根拠としている ように,高度に複雑な系においては統計が主要な解析手 法である.広い意味での化学産業では,このように演繹 的な計算と帰納的な統計が長く使われてきた歴史を有す る. さて本号の特集であるマテリアルズインフォマティク ス(MI)は,これらの技術の延長であり発展形である と思われる.最終的に化学は実世界において価値をもつ が,その実現に至る期間を短縮し,あるいは新規なもの を生み出す手助けをしてくれる計算・統計という点では, これまで数十年の位置付けと大きな差はない.一方で興 味深いのは,これまでにないほど多くの,化学を専門と しない研究者・技術者の参入が見られることである.こ の点は,シリコンバレーで言うところの“Software is eating the world”のような野望とそれを支える知の民 主化や意思決定の速さ,産業界ではディジタルトランス フォーメーション(DX,詳細は付録を参照)と呼ばれ るインターネット経済の影響が大きいと思われる.著者 らの所属するような伝統的企業にとっては,これらの新 たな動きは脅威であると同時に,再び化学が大きな発展 をする絶好の機会でもあると捉えている.2.
化学における演繹と帰納
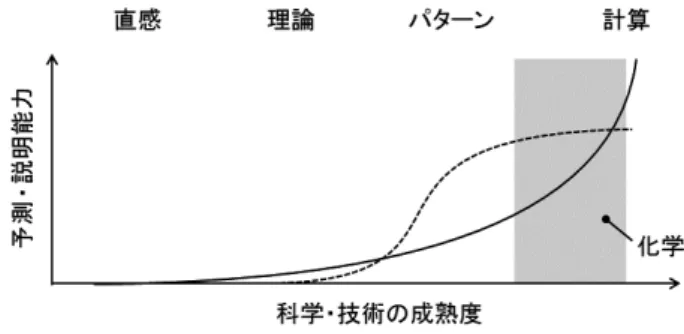
さて,化学における演繹的手法と帰納的手法の関係に ついて,著者らの考えを少し述べたい. 横軸に漠然とした科学・技術の成熟度のようなもの を,縦軸に説明能力あるいは予測能力のようなものをプ ロットすると,図 1 のようなイメージなのではないだろ うか.すなわち演繹的手法は基礎理論と計算速度の進展 とともに指数関数的な成長を示す一方で,帰納的な手法 はデータや情報の蓄積とともに垂直的に立ち上がるもの化学産業における分子デザイン
Molecular Design in Chemical Industry
磯村 哲
株式会社三菱ケミカルホールディングスTetsu Isomura Mitsubishi Chemical Holdings Corporation.
[email protected], https://www.mitsubishichem-hd.co.jp/
山下 博史
(同 上)Hiroshi Yamashita [email protected], https://www.mitsubishichem-hd.co.jp/
Keywords:
materials informatics, chemoinformatics, molecular design, human computation, Bayesian inference. 「マテリアルズインフォマティクス」図 1 演繹的手法と帰納的手法の関係.
実線:演繹的手法(理論・シミュレーション), 破線:帰納的手法(データサイエンス)
の,データ量が十分になるにつれ飽和してシグモイド関 数のような曲線を示す. ここで横軸を四つのフェーズに分けて考えると,最 も左の領域は,演繹・帰納ともに役立たないため直感が 支配している.例えばアートが当てはまるだろう.次の フェーズはデータの蓄積は未熟であり,理論はあるが計 算には至らない.例えば経営のように断片的な理論が林 立するような領域がここに相当する.第 3 のフェーズで は,物理・化学ベースのシミュレーションで実問題を解 くには計算能力が不足しており,データからのパターン 認識でわかることのほうが多い.EC サイトにおける購 買行動予測などがこの典型である.最後のフェーズでは 圧倒的な計算パワーで理論から導かれる厳密解が得られ るため,理論的なバックボーンをもたないデータサイエ ンスは使われない.例えば流体などがここに相当する. なお,シミュレーションとデータサイエンスの融合であ るデータ同化をこの中に位置付けるなら,第 3 のフェー ズと第 4 のフェーズの境界領域に来るだろう. 材料科学におけるコンピュータサイエンスはこれまで 主に演繹的な手法が支配的である第 4 のフェーズを扱っ てきたが,データサイエンスを導入することで手前の第 3のフェーズを発見したのが MI であるといえよう.つ まり,実問題に対して理論や計算能力が不足している問 題こそ,MI の格好の標的となる.
3.
創薬における分子デザイン
さて,MI は産業界で今後どう発展していくのか.そ のヒントは,インフォマティクスで先行しているライフ サイエンスが参考になるのではないだろうか.特に創薬 化学における分子デザインは,現在 MI が置かれている 状況と非常に近いと感じる.その観点から,以降の章で は著者らの経験を中心に振り返ってみたい.読者にとっ て何らかの参考になれば幸いである. 創薬化学における計算機やインフォマティクスの利用 は Computer Aided Drug Design(CADD)と総称され, さらに分子動力学や量子化学などの演繹的・シミュレー ション中心の手法と定量的構造活性相関(Quantitative Structure-Activity Relationship:QSAR)などの帰納的・ 統計的な手法とに大別される.歴史的には統計的な手法 のほうが古く,1960 年代から検討が行われてきた.一 方で演繹的な手法は計算機のコスト低下に伴い飛躍的な 発展を遂げ,京コンピュータの存在もあって,つい数年 前までは CADD といえばシミュレーションを想起させ るほどの隆盛を誇った.この帰納─演繹の軸に加え,化 学構造から機能を予測する問題(ここでは“順問題”と 呼ぶ)と,機能を満たす構造を提示する問題(“逆問題”) との軸があり,四つの象限を形成している(表 1). 左上の象限は,いわば化学の基礎的な考え方をまっす ぐ計算機にもち込んだものであり,基本原理に則って系 の挙動を予測するものである.この手法は化学者にとっ て理解可能という利点が最も大きく,系の純粋な振舞い の予測に用いられる以外に,系の説明にも用いられる. 一方で複雑な系では巨大な計算資源を要するのが短所で ある. この象限をそのまま右に移動し逆問題を解こうとする と計算資源は絶望的に不足するため,何らかの計算を省 略する必要がある.このうち,探索領域を限定し特定の 化合物プールだけを対象とするのがバーチャルスクリー ニングであり,バーチャルスクリーニングをタンパク質 と化合物の立体構造の形状および静電的な相性で判定す るのがドッキングである. 右上の象限のもう一つの方法はラテン語で「最初から」 を意味する de novo 設計といわれており,タンパク質の ポケット内に置かれた薬剤の構造を元に,細かな改変を 施して機能を改善させる方法である.これは 1990 年代 に華々しく提案されて以降大きなインパクトを生んでい ないが,現代の MI と通ずるところが大きいのでご紹介 しよう. 3·1 低分子有機化合物の場合の数 de novo 設計ソフトウェアの一つである Ludi [Böhm 92]では,タンパク質内に結合しそうなフラグメントを 探索・連結することで,強固に結合し薬効が示唆される 化合物の構造を提案するという機能を有していた.しか し,こうしたソフトウェアから出力される構造のバラエ ティには限界があり,また実際の合成に必ずしも向かな いものが多く,実利用は限定的であった. そこで,一度ステップバックして,そもそも化合物の 場合の数を実際に計算する研究が現れた.低分子有機化 合物の数学的に可能な集合(“化学空間”)はおよそ 10 の 60 乗と見積もられる [Bohacek 96].それに対し既知 の物質はわずか 10 の 8 乗程度しか存在しないため,化 学空間を把握するには十分ではなく,数学的な手法が必 要となる. 化学空間をより具体的に見積もるために,化合物をグ ラフで表現し,そのエッジラベル(単結合,二重結合な どの結合次数,正確には sp3,sp2などの結合様式)を固 定しノードラベル(C,N,O などの原子種)を網羅的 に置換することで,化合物をより正確に数え上げた研究 が存在する [Ogata 07].図 2 の左は,あるタンパク質の 表 1 CADD における四象限 順問題 逆問題 演 繹 分子力学分子動力学 量子化学 バーチャルスクリーニング (ドッキング) de novo 設計 (フラグメント付加) 帰 納 (QSAR)定量的構造活性相関 (リガンド法)バーチャルスクリーニング Inverse-QSARポケットに収まった薬剤化合物である.このタンパク質 ポケットに化合物が強く結合するとその機能が阻害され てより高い薬効が期待できるため,タンパク質ポケット に置かれた元々の薬剤化合物(図 2 左)のトポロジーを 保ったまま網羅的にノードラベル置換し,結合強度が最 大になるよう最適化した例が図 2 の右になる(注:一見 すると化合物らしいが,有機化学者からはかなり無理の ある構造であることが見て取れる). こういった変換パターンを種々のトポロジー構造に 対して計算機実験で試したところ,場合の数は驚くほど 法則性があることがわかった.図 3 の丸プロット Non-Filterでは,化合物中の水素を含まない原子数 N に対し, およそ 10 の N/2 乗程度と見積もれる.ただし,薬剤ら しさのフィルタを入れると場合の数が減ると同時に直線 性が崩れることがわかった.この関係式から MI で考慮 すべき化合物数の上限を見積もることができ,例えば原 子 30 個からなる化合物数は 10 の 15 乗程度と推定され る.なお,この研究 [Ogata 7] には続きがあり,単に化 合物を数え上げただけではない.実際に化合物を合成し, タンパク質の活性阻害試験と X 線構造解析を行った結 果,元化合物と同等の活性と結合位置であることが確認 されている(図 4)[Ogata 10]. この方向性の発展形の一つは,化合物グラフのトポ ロジーも変更する真の網羅的な数え上げである.例えば [猪口 15] ではグラフ理論と列挙アルゴリズムの技術を 応用し不適切な構造をもたない化合物グラフを高速に列 挙できるようにした(1 秒当たり約 77 000 個を列挙). 一方,組合せ最適化として別のアプローチも考えられ る.前述の [Ogata 07] では,化合物探索の際に組合せ 爆発を避けるため,化合物をフラグメントに分解し,フ ラグメントごとに構造の数え上げと結合強度の計算をし て,その情報から結合強度が最大となる全体構造を再構 築している.しかし近年の量子アニーラを用いれば,瞬 時に最適解が求まる可能性がある. ごく簡単なペプチドを標的タンパク質に,ベンゼンを 薬剤に見立てて検証を行った結果,確かに量子アニーラ で化学構造の最適化が行えることが確認された(図 5) [Sakaguchi 16].原子置換による結合強度(すなわち位 置エネルギー)の最適化はイジングモデルに自然に落ち るため当然かもしれないが,1 000 ビット程度で現実的 な有機化合物が扱えそうなことから,量子アニーラの実 用という面でも可能性を感じる結果である. 3·2 データ駆動型分子設計 表 1 左下の象限は QSAR として古くから検討されて おり,特に薬剤に共通の物性などデータが蓄積しやすい 系において日常的に用いられるようになった.しかし早 くから実業に根差した半面,一般的なデータサイエンス とは独自の進化を遂げる傾向にあった.近年のデータサ イエンスの隆盛で QSAR は深層学習などを取り込み久々 のイノベーション期にあるが,これまで分野特異的な知 識が詰め込まれたつくり込みを超えるのは容易ではな く,真の進歩が起きるかは定かではない. QSARでは化合物構造の特徴表現がまずもって重要で ある.化合物の構造的特徴はあらかじめ定義した部分構 造セットにわたってその有無を調べた結果(0/1)をフィ 図 2 同一ジオミトリの原子置換. (左)タンパク質ポケットに収まった元化合物と(右)結合 強度が最適になるようノードラベル置換した化合物 図 5 量子アニーラによる化合物の原子置換. (a)原子タイプの情報を消した初期構造 ,(b)原子タイプ を割り振った最安定構造 ,(c)2 番目に安定な構造 図 3 化合物をノードラベル置換したときの場合の数. 横軸は化合物の水素を除いた原子数,縦軸はノードラ ベル置換の場合の数(対数).●:数学的に可能なすべ ての場合 ▲:“薬剤らしさ”という知識に照らして不 適切な構造を除いた場合 図 4 原子置換前後の X 線構造解析結果. タンパク質 JNK3 に結合した化合物の(a)元構造と(b) 原子置換後の構造
ンガープリントとして表すところから始まり [Durant 02],化学構造に含まれる部分構造をそのつど認識して ハッシュ値に変換するものへと進化した [Rogers 10]. これまでに,3 000 以上の構造表現方法が開発された [Todeschini 09].また,機械学習コミュニティでは,特 徴量の次元を無限に取れるグラフカーネルが多く考案さ れた [Kashima 03, Mahé 09, Yamashita 14].グラフカー ネルは構造情報ロスが少ないという利点があったもの の,予測に重要な部分構造がその他多くに埋もれてしま うという課題があった.そして近年,化合物グラフ(入 力)から特性値(出力)まで一気通貫で学習できるグラ フ畳込みニューラルネットワーク [Wu 19, Xu 18] が出現 し,データから予測への寄与が高い部分構造の認識を踏 まえた化合物構造の特徴量作成が自動化された.しかし, ニューラルネットワークの学習では大量にデータがある ことが前提であるため,データが少ない MI では,蓄積 があるデータからの転移学習,シミュレーションによる データ生成,少ないデータからの学習を可能にする少数 ショット学習が重要となる. 表 1 右下の象限のうち,バーチャルスクリーニング(リ ガンド法)では,データから学習した相関関係を因果関係 の代わりに使って入手可能なあらゆる化合物について順問 題を解く.結果の成否は探索対象にした化合物空間の偏り に左右される.この偏りを緩和する試みとして,反応と 試薬の情報を使って網羅的に生成した大量の仮想化合物 (10 の 9 乗個程度)を利用する研究がある [長谷川 15]. 一方,帰納ベースで分子を組み立てるアプローチはこ れまで盛り上がったとは言いづらい.Inverse-QSAR と 呼ばれる研究自体は昔からあるが,構造ベースの陰に隠 れていた印象である.ここまで背景が整いながら,本象 限は MI のほうが先に着目されているというのは複雑な ものである. 図 6 は近年の研究成果であり,ベイズ推定による分 子設計の考え方でもある [Ikebata 17].前述したよう に [Ogata 07] から本来対象とすべき化合物空間は途方 もなく広大であるため,ここでは化合物の構造発生器を 用いた目的地周辺からのランダムサンプリングを用いて いる.ここで,S が化合物(原因),Y が特性(結果), Uが望ましい特性のセットを表す.ベイズの反転公式 p(S|Y ∈U)∝p(Y∈U|S)p(S)を使うアプローチで は,化合物 S の実現性についての信念の分布 p(S)を データという証拠からつくられる化合物が満たすべき特 性をもつ確率の分布 p(Y∈U|S)で更新して,満たす べき特性をもつ化合物の分布 p(S|Y∈ U)を手にする ことができる.p(Y∈U|S)は予測特性値と理想特性 値のずれの大きさから定義することができる.p(S)は 化学構造の文字列表記(SMILES)のパターンを既存化 合物データから学習した確率的言語モデルであり,構造 発生器として用いる.そして,モンテカルロ法によって p(S|Y ∈U)からサンプリングすることで具体的な設 計化合物が得られる.妥当な構造を出すには,ルール化 したケミストの知識で p(S)に制約を入れることが肝要 である. 3·3 合成可能性の取扱い さて,以降は逆問題の鬼門である合成可能性について 触れたいと思う. 逆問題では,構造生成と構造評価の二つの機能が本質 的である.しかし実際には,構造生成においてグラフを 組み立てる技術は数学的に閉じた問題であるのに対し, 現実に合成できるかどうかは数学の外側であるだけでな く,有機合成自体の技術革新によってラベルが変わり得 る問題であり,さらには収率や原料・精製コストなどの 状況,専門家どうしの見解の相違でもラベルが変化する. したがって,グラフ組立てと合成可能性という二つの問 題は切り分けて考えるのが,長期的視座に立てば重要で ある. 合成可能性を情報科学に取り込むにはいくつかのアプ ローチがある.例えば化学反応データと深層学習や強化 学習を組み合わせて逆合成経路を探索する方法 [Liu 17, Segler 18],過去に存在する化合物データ中に現れる部 分構造の出現頻度とグラフ複雑度から合成可能性スコア を算出する方法 [Ertl 09],専門家の直観を取り込む方 法 [Baba 18] などである.バーチャルスクリーニングで は合成が困難な天然物由来の構造などを除外できればよ かったので,どの方法でも大して問題はなかった.しか し逆問題では,これまでにない構造でありながら化学的 に妥当であるという非常に困難な問題設定を強いられ る.一般のデータベースに登録されている構造のほとん どが合成された,すなわち合成可能な化合物からなるこ とを考えればなおさらである. これに対する一つの切り口は,ヒューマンコンピュ テーションで専門家の能力を手続きに取り込むことであ 図 6 ベイズ推定による分子設計
る(図 7).化学者は専門家のみならず,ある程度の専 門性を有する技術者の意見をうまく集約することで一定 水準の回答を維持できることが示されており [Baba 18], 未知な問題に対してインフォマティクス単独で取り組む よりも大きな可能性が見えている.
4.MI の
定着と発展に向けて
MIが定着するためには,データの管理,統計モデリ ング,モデルの運用の三つすべてが回る必要があるが, データ管理と運用は実験化学者の近くで行うべきであ り,可能なら実験化学者自身が携わるべきである.その 理由は心理学的でいう保有効果が大きいと考えている. つまり,自らが使うために自分で準備したデータであれ ば積極的に標準化や保存を行うし,自らが予測した化学 構造は実験で検証するモチベーションが上がる.モデリ ングの部分はデータの標準化と経験の増加に伴い,自動 化できる部分が増えてデータサイエンティストの負担は 下がるだろう.これが定着への道筋と考えられる.もち ろんトップダウンでルール化する選択肢もあるが,イノ ベーティブな風土を維持促進するためには内発的動機付 けがベターではないだろうか. 図 8 は社内で開発した,合成化学者が実験を構想す る際のアシストツールであり,マウスを用いて構造を描 画するとリアルタイムで機械学習による予測物性が表示 される.当然ながらプロジェクトごとに要求物性は異な るため,予測モデルの構築はデータサイエンティストが 行ったうえでデプロイし,実験化学者はモデリングやシ ステムを意識しないで使えるようになっている. 一方でデータサイエンティストと計算科学者や実験 チームとのコミュニケーションを密にすることも定着と レベル向上に肝要である.そのためには背景知識やキャ リアパスの多様なチームを形成し,本質的な理解を深め ることが重要だろう. MIのデータサイエンスとしての側面を考えると,一 般的にはデータ活用に三つの段階があると考えている (表 2).Level 0 はデータを活用していない段階であり, 数年前までの材料科学の研究開発はここに相当するだろ う.Level 1 はデータとデータサイエンスを利用してい るが,新規技術としての興味であって,業務フローや意 思決定の根幹に関わる部分は従来と同様である.Level 2 ではデータが起点となって種々の状況判断や意思決定が 行われている.近年のデータサイエンスブームの文脈で は当然のように Level 2 が推奨されているが,実は基礎 科学が十分に発達している場合には Level 1 のほうが質 の高い判断ができるケースも往々にして存在する.重要 なのは結論の正確さとそこに至る速さであって,データ サイエンスはその手段であることを失念してはいけない. さて最後に,MI がさらなる発展を遂げるための制約 をあげたいと思う.一般に科学技術の発展にはアカデミ アが大きな役割を果たすが,ことデータサイエンスに関 しては,質と難易度の高いオープンデータを生み出すと いう点において実験科学系ラボの果たす役割は大きい. しかし,アカデミアにおける材料科学,特に高分子系の 研究室は減少する一方であり,あるいはかなりライフサ イエンスに寄ることで予算を得ていることが多く,産業 的に有用なデータが生み出されるような研究が幅広く行 われているとは言いがたい状況である.この状況を打破 し,材料科学に夢を与えることが最も重要であり,アカ デミアと産業界が共に盛り上げる必要がある.MI がそ の牽引役になることを期待している. 図 7 候補構造の妥当性を化学者に問うための UI. 回答者は提示された構造を見て,文献などを調べることな く直観で化学構造の妥当性を 5 段階で回答する 図 8 合成化学者のための物性予測 GUI. 化学者が構造を描画・編集すると,あらかじめつくられた 予測モデルを通じて物性値がリアルタイムに推算される 表 2 データ利用に関する三つの段階 Level 2: データ駆動 データが起点となった状況判断や意思決定 Level 1: データ利用 データ解析を利用しているが,位置付けは既存の方法のサポート Level 0: データ未活用 データは存在しないか,データを生かした意思決定をしていない《付録》ディジタルトランスフォーメーション
近年ビジネス界でバズワードのように使われている「ディジタ ルトランスフォーメーション(DX)」という語がある.これはディ ジタを用いてビジネス変革を行うことを指すが,当社では MI を DXの一環と捉えており,著者らも数多くの DX プロジェクトに関 わっているため,その考え方を紹介しておきたい. DXはディジタル技術とその思想を用いて企業内のあらゆる要素 やシステムを変革することであるが,中心的な概念は取引コスト の減少,ネットワーク効果,知の民主化,データ駆動,サイバーフィ ジカルシステム(CPS)などインターネット革命と重なるところ が大きい.このため価値観の背景が伝統的な大企業と異なり,内 部人材のみでは対応できないことから,当社ではチーフデジタル オフィサー(CDO)をはじめ多くの人材を外部から登用し,内部 人材との積極的な交流を図っている. DX プロジェクトも従来の慎重かつ大規模な投資と異なり,リー ンスタートアップのように小さく始め,有用性が見えてきた段階 で規模を拡大するという手順を踏んでいる.プロジェクト推進に はさまざまな知識とスキルが状況に応じて必要になるため,DX 専 門家は本社でチームを形成し,有機的にさまざまなプロジェクト に出入りする構造となっている. MI も同様であり,材料科学の素人であるデータサイエンティス トと内部で長く材料開発に携わってきた化学者が同じプロジェク トに入ることで,これまでにないアプローチが迅速な成果に結び付 いている.また,知の民主化にならい解析プログラムや UI は社内 で広く利用できるようなシステムを提供している.これらを通じ ディジタルの思想が研究開発から広まることも期待の一つである.◇ 参 考 文 献 ◇
[Baba 18] Baba, Y., Isomura, T. and Kashima, H.: Wisdom of crowds for synthetic accessibility evaluation, J. Mol. Graph
Model., Vol. 80, pp. 217-223(2018)
[Böhm 92] Böhm, H. J.: The computer program LUDI: A new method for the de novo design of enzyme inhibitors, J. Comput.
Aided Mol. Des., Vol. 6, No. 1, pp. 61-78(1992)
[Bohacek 96] Bohacek, R. S., McMartin, C. and Guida, W. C.: The art and practice of structure-based drug design: A molecular modeling perspective, Med. Res. Rev., Vol. 16, No. 1, pp. 3-50 (1996)
[Durant 02] Durant, J. L., Leland, B. A., Henry, D. R. and Nourse, J. G.: Reoptimization of MDL keys for use in drug discovery, J.
Chem. Inf. Comput. Sci., Vol. 42, No. 6, pp. 1273-1280(2002) [Ertl 09] Ertl, P. and Schuffenhauer, A.: Estimation of synthetic
accessibility score of drug-like molecules based on molecular complexity and fragment contributions, J. Cheminformatics, Vol. 1, No. 8(2009)
[長谷川 15] 長谷川亜樹,藤原康広,森本元太郎,平野秀典,沖本憲明, 泰地真弘人,船津公人:創薬に向けた大規模仮想化合物ライブ ラリの開発と高度化,第 38 回ケモインフォマティクス討論会東 京,pp. 50-51(2015)
[Ikebata 17] Ikebata, H., Hongo, K., Isomura, T., Maezono, R. and Yoshida, R.: Bayesian molecular design with a chemical language model, J. Comput. Aided Mol. Des., Vol. 31, No. 4, pp. 379-391(2017)
[猪口 15] 猪口明博,磯村 哲:グラフコーディングを用いたスーパー グラフ検索の効率化,第 105 回人工知能学会知識ベースシステ ム研究会,pp. 26-33(2015)
[Kashima 03] Kashima, H., Tsuda, K. and Inokuchi, A.: Marginalized kernels between labeled graphs, Proc. 20th Int.
Conf. on Machine Learning(ICML 2003), Washington, DC, U.S.A., August 21-24, 2003, Fawcett, T. and Mishra, N., Eds., pp. 321- 328, AAAI Press: Chicago, IL, U.S.A.(2003)
[Liu 17] Liu, B., Ramsundar, B., Kawthekar, P., Shi, J., Gomes, J., Nguyen, Q. L., Ho, S., Sloane, J., Wender, P. and Pande, V.: Retrosynthetic reaction prediction using neural sequence-to-sequence models, ACS Cent. Sci., Vol. 3, No. 10, pp. 1103-1113 (2017)
[Mahé 09] Mahé, P. and Vert, J.-P.: Graph kernels based on tree patterns for molecules, Mach. Learn., Vol. 75, pp. 3-35(2009) [Ogata 07] Ogata, K., Isomura, T., Yamashita, H. and Kubodera,
H.: A quantitative approach to the estimation of chemical space from a given geometry by the combination of atomic species, QSAR Comb. Sci., Vol. 26, No. 5, pp. 596-607(2007) [Ogata 10] Ogata, K., Isomura, T., Kawata, S., Yamashita,
H., Kubodera, H. and Wodak, S. J.: Lead generation and optimization based on protein-ligand complementarity,
Molecules, Vol. 15, No. 6, pp. 4382-4400(2010)
[Rogers 10] Rogers, D. and Hahn, M.: Extended-connectivity fingerprints, J. Chem. Inf. Model., Vol. 50, No. 5, pp. 742-754 (2010)
[Sakaguchi 16] Sakaguchi, H., Ogata, K., Isomura, T., Utsunomiya, S., Yamamoto, Y. and Aihara, K.: Boltzmann sampling by degenerate optical parametric oscillator network for structure-based virtual screening, Entropy, Vol. 18, No. 10, pp. 365-375(2016)
[Segler 18] Segler, M. H. S., Preuss, M. and Waller, M. P.: Planning chemical syntheses with deep neural networks and symbolic AI, Nature, Vol. 555, No. 7698, pp. 604-610(2018) [Todeschini 09] Todeschini, R., Consonni, V.: Molecular
Descriptors for Chemoinformatics(2 volumes), Wiley-VCH: Weinheim(2009)
[Wu 19] Wu, Z., Pan, S., Chen, F., Long, G., Zhang, C. and Yu, P.: A comprehensive survey on graph neural networks, arXiv preprint arXiv:1901.00596(2019)
[Xu 18] Xu, K., Hu, W., Leskovec, J. and Jegelka, S.: How powerful are graph neural networks, arXiv preprint arXiv:1810.00826(2018)
[Yamashita 14] Yamashita, H., Higuchi, T. and Yoshida, R.: Atom environment kernels on molecules, J. Chem. Inf. Model., Vol. 54, No. 5, pp. 1289-1300(2014) 2019年 3 月 6 日 受理