—Reviews—
ヒスタミン H
4受容体リガンドの開発とその現状
春 沢 信 哉
*, 荒 木 理 佐
Development of Histamine H
4Receptor Ligands and its Current Situation
Shinya H
ARUSAWAand Lisa A
RAKIOsaka University of Pharmaceutical Sciences, 4-20-1, Nasahara, Takatsuki, Osaka 569-1094, Japan
(Received September 26, 2008; Accepted November 4, 2008)
This paper looks at recent advances in the development of histamine H
4 receptor ligands and includes a discussion of the following items: 1) Discovery of histamine H
4 receptor (H4R) and difference between histamine H3 receptor (H3R) and H
4R. 2) Current situation with regard to GT-2331. 3) Binding affinities of known ligands in human (h) H3R and hH
4R. 4) Current situation with regard to selective hH4R agonists. 5) Current situation with regard to selective hH4R antagonists. 6) Selected patent compounds. 7) Key hH
4R ligands as pharmacological tools. Key words——histamine; histamine H3 receptor; histamine H4 receptor; agonist; antagonist
1. はじめに
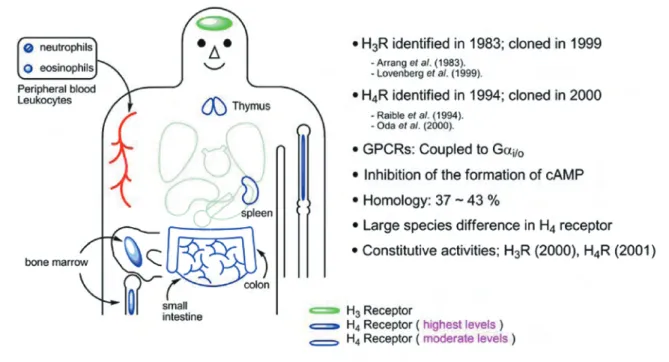
1999 〜 2001 年にかけて,ヒスタミン H3受容 体 (H3R) のクローニングの成功やヒスタミン H4受 容体 (H4R) の発見があり,さらに H3R と H4R はア ゴニストが存在しなくとも恒常的に活性化される 受容体(constitutively active receptor: 構成的活性 化受容体)であるとの報告により,ヒスタミンリ ガンド開発は大きな変革に見まわれた.その中で, 強力な H3R アゴニストとして報告された GT-2331 は,注意欠陥‐多動性障害(ADHD)の治療薬とし て臨床第 2 相試験まで進んだが,その後のヒトの H3R(hH3R)を用いた評価では,H3R アゴニスト であること等が報告され,GT-2331 の評価は混乱 した状況にある.最初に GT-2331 の現状について 整理した後,選択的 H4R リガンドの開発とその現 状について述べたい. H4R の生理学的役割や病態との係わりを解明す るには,選択的 H4R リガンドの開発が必須であ る.我々は幸い,初期の段階で H4R アゴニストの OUP-16 を見出したので,その経緯を紹介し, 続い て最近の選択的 H4R アゴニストである 4- メチル ヒスタミン(4-MeH) とクロザピンアナローグな どについて言及する.一方,Johnson & Johnson (J&J) 社は,高い選択性と結合親和性を持つ H4R アンタゴニスト JNJ7777120 とその誘導体を発表 している.これらの化合物は,ニュートラルアン タゴニストであり, H4R の選択的インバースアゴ ニストは未だ見出されていないと思われる. 2. H3R と H4R リガンド 2-1. H4R の発見と H3R との相違 H3R は,1983 年に Arrang ら 1 ) により発見された *大阪薬科大学 薬品合成化学研究室, e-mail: [email protected] 本総説は,第 11 回日本ヒスタミン学会(2007 年 12 月 14〜15 日,富山)のシンポジウムで講演したものを中心に記述したものである.
が,16 年後の 1999 年になって Lovenberg ら2 )が ようやくヒトゲノム情報に基づいて H3R のクロー ニングに成功した.翌 2000 年と 2001 年にかけ て, ほぼ同じ手法で日本の 2 つのグループを含む 6 つのグループから H4R のクローニングの成功 が相次いだ3-8).H4R については, すでに 1994 年に Raible ら9 )が H1R, H2R, H3R 以外のヒスタミン受容 体の存在を提示しており,2000 年にこれを同定 し,H4R と命名されたものである. H3R と H4R は,類似性と著しく異なる面を持っ ている1 0 ).H3R は,脳内に高密度で存在するのに対 して,H4R は骨髄, 末梢白血球(好酸球,好中球) に高いレベルで発現し,他に胸腺,膵臓,結腸, 小腸に見られる (Fig.1).H4R は免疫に係わる部位 での発現のため,発見当初から炎症やアレルギー 疾患の治療薬の標的と期待され,現在ではその方 向性にあると思われる11-20).H3R と H4R は,共に 7 回 膜貫通型の G タンパク質共役型受容体(GPCR) で, Gi を介した c-AMP の産生抑制により細胞内シグ ナル伝達が行われる.両受容体のアミノ酸配列の 相同性は, 全体で 40%,膜貫通部位で 60%と高 く,このため H3R リガンドの多くは H4R と親和性 を持つ.したがって H4R リガンドの開発において は,如何に H3R に弱く,H4R に対して強い親和性 と選択性を持つ H4R リガンドを開発するかに重点 が置かれてきた21).また H4R のアミノ酸配列は, 種 差が大きいため,リガンドの評価にはヒトの H4R (hH4R) とヒトの H3R (hH3R) を用いる必要がある. 2000 年には, H3R が構成的活性(constitutive activity)を示すことが正常動物(ラット)で最初 に確認された2 2 ).これは GPCR において内在性のリ ガンドが存在しなくとも細胞内にシグナル伝達が 生じるというものである23).このため,構成的活性 化受容体を活性化するものがアゴニストであり, 抑制するものがインバースアゴニストとなる.こ の発見により,それまで H3R アンタゴニストとさ れてきた多くの化合物は,実はインバースアゴニ ストであることが明らかとなった.2001 年には,
H4R が構成的活性化受容体であることも報告さ れた6 ).このように,2000 年前後にヒスタミン受 容体に関する新しい発見が続いたため, その後 のヒスタミン領域の研究は大きく進展し,隔世 の感すら覚える. H3R と H4R リガンド開発のための薬理学上の 評価方法は,それまでよく用いられた in vitro の モルモットの腸管を用いた方法から,遺伝子工 学的に強制発現させた hH3R と hH4R に変った. そのため,モルモットの腸管では H3R アンタゴ ニストと報告されていたものが,hH3R を導入 した細胞を用いるとアゴニストであるという報 告が見られるようになった.このような薬理学 上の評価法の進歩の中で悲運に見舞われたのは, ADHD の治療薬として期待された GT-2331 であ る. 2-2. GT-2331 の現状 GT-2331 (Cipralisant, PerceptinTM) は,1999 年に Gliatech 社が,モルモットの腸管を用いた 薬理評価から強力な H3R アンタゴニスト (pKi = 9.9) として発表したものである(Fig.2).2000 年24) には ADHD の第 2 相試験を実施し25),H3R を標的と した初めての医薬品の実現が期待された.しかし Gliatech 社は 2002 年に倒産し,その知的財産権 は Merck 社が引き継いだものの,2003 年には開 発を断念したという経緯がある2 6 ). この間の事情を学術論文で調べてみると,GT-2331 は,2002 年にラットと hH3R でアゴニスト と報告された2 7 ).驚いたことに, 2004 年に Abott 社 の研究陣は GT-2331 の効率合成と中間体の X 線 構造解析の結果から,絶対配置の間違いに気付い た28).その結果 GT-2331 は,それまでの 1R, 2R 配 置ではなく, 実はエナンチオマーの 1S, 2S 配置で あった.また Leurs らも hH3R では固有活性 α = 1 の完全アゴニストであることを報告した(200529)). なお,固有活性 (α) は,ヒスタミンが受容体に作 用することで惹起される最大応答を1としたもの である. 筆者が気になるのは, Abott 社の合成品の比旋光
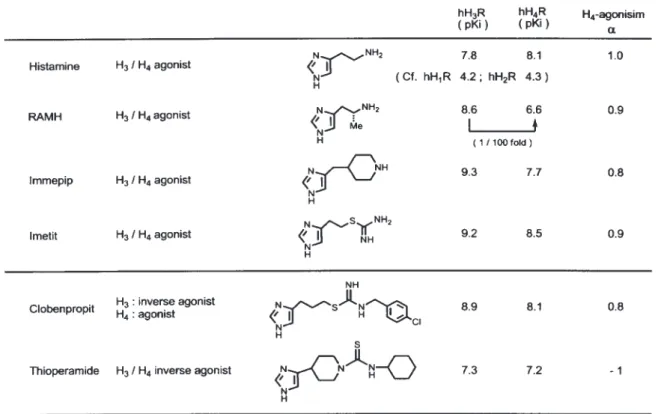
度が [α]D: +277°に対して元の Gliatech 社のもの は +140°と低く鏡像体過剰率は 50% e.e. に過ぎ ない. したがって Gliatech 社の GT-2331 は,(1S, 2S)-eutomer と(1R, 2R)-distomer の 3:1 混 合 物 であったと思われる.現在,中国の会社から GT-2331 とされるものが販売されているようである が,これを薬理実験に使用する場合は, 事前に比 旋光度等の物理恒数をチェックするなど慎重を期 す必要がある. 3. 既存の hH3R と hH4R リガンドの結合親 和性 多くの H3R リガンドは,ヒスタミンのイミダ ゾールをコアーとして開発されてきた5,19,29,30). 先に述 べたように,H3R リガンドの多くは, H3R のみな らず H4R とも親和性を持つ.Table 1 に,ヒスタ ミン及び既存の hH3R リガンドの hH3R と hH4R に 対する結合親和性とα 値についてまとめた. ヒスタミンは,H1R 〜 H4R の共通の内因性リガ ンドであるが,これらの GPCR に対する親和性は それぞれ異なっている.hH1R と hH2R は,ヒス タミンに対する pKi 値はそれぞれ 4.2 と 4.3 と弱 いが,hH3R と hH4R では 7.8 と 8.1 と高親和性受 容体で,hH4R はヒスタミンに対して最も高い結 合性を示す.R-α-メチルヒスタミン (RAMH) は, hH3R で pKi 値 は 8.6 で あ る が,hH4R で は 100 分の 1 弱い親和性のパーシャルアゴニスト (pKi = 6.6, α = 0.9) である.このように H3R リガンドは, 通常 H4R では親和性が弱くなる.H3R アゴニスト であるイメピップ,イメティットも H4R では同様 に作用を弱めたパーシャルアゴニストである. クロベンプロピットとチオペラミドは,従来 H3R アンタゴニストのプロトタイプとされていた が,H3R の構成的活性の確認後,共に H3R ではイ ンバースアゴニストであることが分った.その内 クロベンプロピットは,hH4R では逆にアゴニス ト (α = 0.8) として作用する.一方,チオペラミド
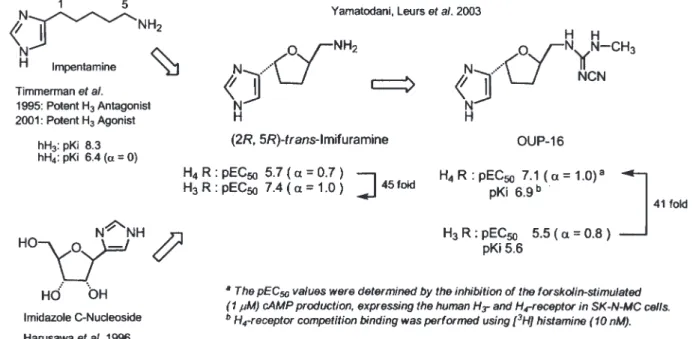
は,hH3R と hH4R の両方のインバースアゴニス ト (α = –1) である. 4. hH4R アゴニスト 4-1. 最初の選択的 hH4R アゴニスト OUP-16 1999 年以前は, H3R のクローン技術はなく, モルモットの腸管を用いて H3R リガンドの薬 理評価が行われる場合が多かった.1995 年に Timmerman らは,ヒスタミンの中間鎖を C2か ら C5に延長したイムペンタミンは,強力なアン タゴニスト作用を示すと報告したが (Fig. 331)),後 に同グループは,hH3R を用いて評価した場合, イムペンタミンはアゴニストであると発表した (200132,33)). 一方, 我々はイミダゾールを塩基として持つ C- ヌクレオシドの合成法及びその応用研究を 行っていたので34,35),イムペンタミンをテトラヒド ロフラン環に組み込むことで,H3R に対して作 用を示す立体配置を知ることが出来るのではな いかと考えた.そこで, 二つのキラル中心を持つ 5- アミノメチルテトラヒドロフランをデザイン し,その 4 異性体すべてを合成した36,37).それらは生 きたラットの脳を用いるマイクロダイアリシスで 調べると,2R,5R 配置を持つイミフラミンにの み脳内ヒスタミン遊離をイメピップと同程度まで 減少させる H3R アゴニスト活性を見出した 36) .さら に, 我々と Leurs ら(アムステルダム自由大学)は, 共同してイミフラミン及びその誘導体の hH3R と hH4R に対する結合親和性 (binding affinity) と機 能性評価 (functional assay) を行ったところ, イミ フラミンは, hH3R アゴニストで, hH4R より 45 倍 強いものであった38).一方, 興味深いことに, その シアノグアニジン誘導体 OUP-16 は,逆に hH3R (pEC50: 5.5; α = 0.8) よ り hH4R (pEC50: 7.1; α = 1.0) に 41 倍の強さを持つ完全アゴニストである ことを明らかとした38).我々の発見した OUP-16 は hH4R に選択性を持つ最初のアゴニストであった ため,さらに OUP-16 の効率的合成法も別に開発 した39).
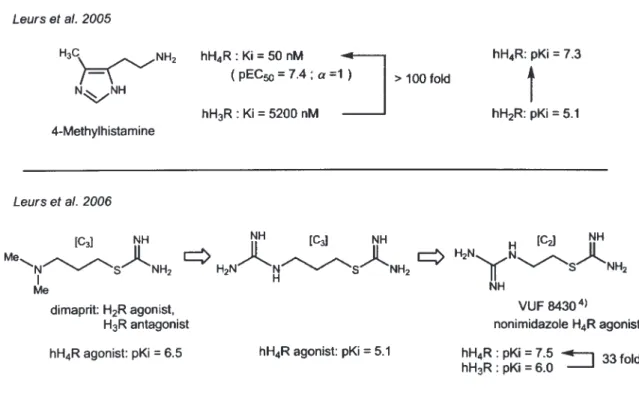
ここで Leurs らが用いた hH3R と hH4R は,小 児の癌細胞由来の SK-N-MC 細胞に hH3R と hH4R を遺伝子工学的に強制発現させたものである.こ の手法は J&J 社が開発し,特許を持つもので, Leurs らは研究目的でのみその使用が認められて いるものであった.このため, SK-N-MC 細胞を用 いる評価法を用いることの出来る J&J 社と Leurs らが, その後の hH4R リガンド開発のほとんどを 独占的に進めることとなった. 4-2. 4-MHA と VUF 8430 ヒスタミンの特定の部位にメチル基を導入する と H1R 〜 H3R に選択性を持つアゴニストを作り分 けることができる.2-MHA は, 選択的 H1R アゴ ニストであり,アミノ基の α 位炭素にメチル基を 入れた RAMH は, H3R アゴニストのプロトタイプ である.4-MHA は, 弱い H2R アゴニスト(hH2R: pKi = 5.1) とされていたが, Leurs らは, 4-MHA の hH3R と hH4R に対する結合親和性と機能性評価を 先に述べた SK-N-MC 細胞を用いる手法で調べたと ころ,4-MHA は hH4R の選択的アゴニストである ことを明らかにした.この場合,4-MHA は hH3R (Ki = 5200 nM) より hH4R (Ki = 50 nM) で 100 倍 以上の親和性を示すことが明らかとなった40).また Tocris 社は,2006 年より 4-MHA・2HCl を発売 している. Leurs らは, さらにこの研究の中で H2R アゴニ スト,ジマプリットが hH4R で緩和ながらもアゴ ニスト活性 (pKi = 6.5) を示すことから,さらに構 造活性相関を進めた.ジマプリットのジメチルア ミノ基をさらに塩基性の強いグアニジル基に置換 した場合,アゴニスト活性は弱くなるものの (pKi
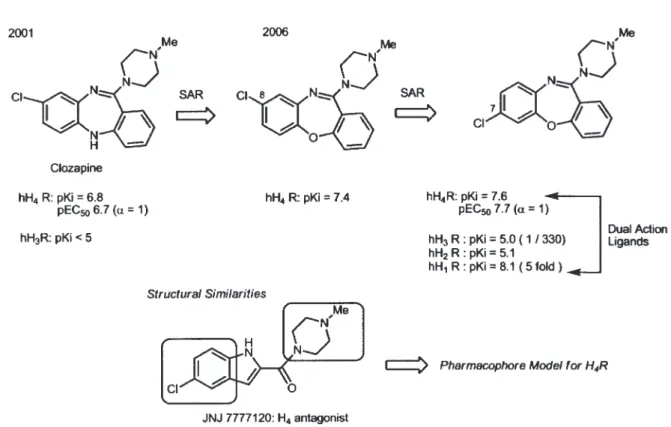
= 5.1),中間炭素鎖を一つ短くした S-(2- グアニジ ルエチル ) イソチオウレア (VUF8430) は,hH3R (pKi = 6.0) より hH4R (pKi = 7.5) で 33 倍の選択性 を示した41).VUF8430 は,最初の非イミダゾール hH4R アゴニストである. 4-3. クロザピンアナローグ ベンゾジアゼピン誘導体である抗精神薬クロザ ピンは,H4R 発見の初期の段階で H3R に親和性が なく,弱いながらも H4R 選択的アゴニストとして 知られていた3-5).Leurs らは, クロザピンが, 大抵の GPCR に対してアンタゴニストとして作用するこ とに興味を持ち, hH4R に対して緩和なアゴニスト (pKi = 6.8; pEC50 = 6.7; α = 1) であることをあらた めて明らかとした40).そこで,クロザピンのジアゼ ピン環の N の一つを O に置換したオキザゼピン では,そのアゴニスト活性は上昇した. さら に,塩素を 8 位から 7 位に置換した 7- クロロ ジベンゾオキザゼピン体は,hH4R アゴニスト (pKi = 7.6; pEC50 = 7.7; α = 1) で あ り,hH3R (pKi = 5.0) より 330 倍の選択性を示した42).こ のジベンゾオキザゼピンは,hH1R (pKi = 8.1) に対して, hH4R よりさらに 5 倍の親和性を合 わせ持つ.H1R と H4R に同時に作用するとい うことは,hH1R と hH4R のダブルブロックに よる新しいタイプの抗アレルギー剤の開発に繋 がる可能性があり興味深い.このジベンゾオキ ザゼピンは, 次に述べる hH4R アンタゴニスト の JNJ7777120 と比べると N-メチルピペラジ ンとクロロベンゼン部分が共通であり, H4R の ファーマコフォアモデルに対する示唆を与えて いる.
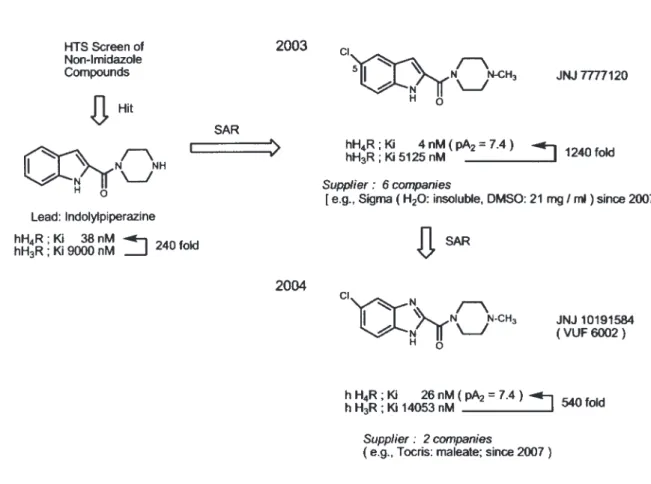
5. hH4R アンタゴニスト 5-1. JNJ7777120 と JNJ10191584 最 初 の 強 力 な 選 択 的 hH4R ア ン タ ゴ ニ ス ト JNJ7777120 は, 2003 年に J&J 社の研究陣が発 表した43).彼らは,H3R に親和性の高いイミダゾー ル含有化合物では,H4R の高選択性は期待できな いという判断から, 自社の化合物ライブラリーの 中から非イミダゾール化合物を選び,ハイスルー プットスクリーニングで, リード化合物となるイ ンドリールピペラジンを見つけた.この化合物 は,hH4R で Ki = 38 nM, hH3R で Ki = 9000 nM の結合親和性を示し,240 倍の hH4R 選択性を持 つ.このリードの SAR から 5 位に塩素を加え, さらに N- メチルピペリジンとした JNJ7777120 (hH4R: Ki = 4 nM; hH3R: Ki = 5152 nM ) は, hH4R と hH3R の差が 1240 倍の高選択的 hH4R アンタ ゴニストであった.さらに, インドールをベン ズイミダゾールにした JNJ10191584 を発表し た44,45).JNJ10191584 (hH4R: Ki = 26 nM; hH3R: Ki = 14053 nM ) は, hH4R と hH3R の選択性は 540 倍 と JNJ7777120 より劣るが, 分子内にイミダゾー ル部位を持つため二つの N 間で H が移動する互 変異性体を生じ,極性が生まれる.J&J 社が開発 したこれらは 2007 年より試薬として発売されて いる.JNJ7777120 は,Sigma 社を含む 6 社から 購入でき, 水に不溶で DMSO に溶解する.また, JNJ10191584 は, Tocris 社ともう一社よりマレ イン酸塩として発売されている.
一方, Leurs らは,JNJ10191584 と同一化合物 を VUF6002 として同時期に報告した45).彼らはさ らにこれらの hH4R アンタゴニストの詳細な薬理 実験を行ない,これらはニュートラルアンタゴニ ストであることを明らかとした13,45). 5-2. チエノピロールピペリジンカルボキシアミド と 2- アリールベンツイミダゾール J&J 社 は, JNJ7777120 の ベ ン ゼ ン 環 を チ オ フェンに置換した hH4R アンタゴニスト,チエノ ピロールピペリジンカルボキシアミドを報告した (200546)).チオフェンの S とピロールの N が同じ方 向を向く head-to-head 型 (hH4R: Ki = 25 nM) と, 逆向きのものを head-to-tail 型 (hH4R: Ki = 3 nM) は, ともに hH4R の高親和性物質である. 彼らは, さらに 2-アリールベンツイミダゾール類を見出 し47),中でも末端に N-メチルホモピペラジン持つも のは hH4R で Ki = 1 nM という高い値を示した. 5-3. シクロプロパン骨格に基づく H4R アンタゴ ニスト 周東らは,2- アミノエチルシクロプロピルイ ミダゾール [(1S, 2S)-cis-AECI (Ki = 1.31 nM)] が,
選択的 H3R アゴニストであることを見出し
47) ,そ
れを基にシクロプロパン環の立体配置と側鎖疎水 性部位の検討を行った.その結果, N-クロルベン ジル基を持つ (1R, 2S)-trans-CAIC が, hH4R (Ki = 8.4 nM) と hH3R (Ki = 7.6 nM) の両方でアンタゴ ニスト活性を示すこと,さらに 1R, 2R 配置を持 ち側鎖を 1 炭素減らした (1R, 2R)-trans-CAIC は, hH4R アンタゴニストとして hH3R (Ki = > 1000 nM) より hH4R (Ki = 118 nM) で 8.5 倍の結合親和 性を示すことを報告した48). 6. 特許化合物 特許審査中のものから H4R アンタゴニストを調 べると,Bayer 社と Pfizer 社から新しい構造を持 つものとして 2- アミノピリジン49,50) と 4- アルキルア ミノピリジン類51) が見られる また,JNJ 化合物由 来のベンズイミダゾール52) やチエノイミダゾール類53) がある.また, 重原子に富む Janssen 社のトリア
リールイミダゾール54) と Astrazeneca 社のアシルピ ペリジン化合物55) などが見られた. 7. おわりに ここで,薬理学ツールとして有用であり,且つ 購入できるリガンドをまとめると,H4R アゴニス トとしては 4-MHA,ニュートラルアンタゴニスト としては,JNJ7777120 と JNJ10191584,hH4R と hH3R 共通のインバースアゴニストとしてチオ ペラミドをあげることができる (Table 2).なお, hH4R 選択的インバースアゴニストは,今回の調 査ではまだ確認できなかった.J&J 社の Venable と Thurmond は,優れた総説を出し, 2005 年ぐ らいまでがよくまとめられている56).本稿では,こ れを踏まえ 2005 年以降の報告分と我々が得た知 見を加えた. H4R は,2000 年に確認されて以来 8 年になろ うとしているが,既存の H1R アンタゴニストに対 して H4R アンタゴニストが新しい作用機序を持つ Fig. 9. Patent Compounds
抗アレルギー剤となる可能性が次第に明らかにさ れている.そうした中,有機合成化学者と薬理学 者が協力することで,我が国から優れた H4R リガ ンドが創製されることを期待するものである.こ の小文が, hH4R リガンドの研究の発展に寄与する ことが出来るなら筆者のこれに勝る喜びはない. 謝辞 本稿をまとめるにあたって,大阪大学 大学院医学系研究科保健学専攻生体情報科学講座 の大和谷厚教授と波多野浩太修士にご指導を賜り 深く御礼申し上げます.香川大学医学部の橋本剛 博士には, ご助言をいただき御礼申し上げます. また, アルフレッサファーマの坂本靖彦博士と下 田綾子氏には, 助言及び特許調査などでご協力を いただき深謝いたします.さらに大阪薬科大学の 栗原拓史元学長からは終始全面的な協力をしてい ただき深く御礼申し上げます.最後に, 機会ある ごとに暖かく励ましていただいた名古屋市立大学 名誉教授, 塩入孝之先生に深く感謝の意を表しま す. REFERENCES
1) Arrang J.-M., Garbarg M., Schwartz J.-C., Nature, 302, 832-837 (1983).
2) Lovenberg T. W., Roland, B. L., Wilson S. J., Jiang X., Pyati J., Huvar A., Jackson M. R., Erlander M. G., Mol. Pharmacol. 55, 1101-1107 (1999).
3) Oda T., Morikawa N., Saito Y., Masuho Y., Matsumoto S., J. Biol. Chem., 275, 36781-36786 (2000).
4) Nakamura T., Itadani H., Hidaka Y., Ohta M., Tanaka K., Biochem. Biophys. Res. Commun., 279, 615-620 (2000).
5) Liu C., Ma X. -J., Jiang X., Wilson S. L., Hofstra C. L., Blevitt K., Li X., Chai, W., Carruthers, N, Lovenberg T. W., Mol. Pharmacol., 59, 420-426 (2001).
6) Morse K. L., Behan J., Laz, T. M., West R. E., Jr., Greenfender S. A., Anthes J. C., Umland S., Wan Y., Hipkin R. W., Gonsiorek W., Shin N., Gustafson E. L., Qiao X., Wang S., Hendrick J. A., Green J., Bayne M., Monsma F. J., Jr., J. Pharmacol. Exp. Ther., 296, 1058-1066 (2001).
7) Nguyen T., Shapiro D. A., George S. R., Setola V., Lee D. K., Cheng R., Rauser L., Lee S. P., Lynch K. R., Roth B. L., O’Dowd B. F., Mol. Pharmacol., 59, 427-433 (2001).
8) Zhu Y., Michalovich D., Wu H. -L., Tan K. B., Dytko, G. M., Mannan I. J., Boyce R., Alston J., Tierney L. A., Li X., Herrity N. C., Vawter L., Sarau H. M., Ames R. S., Davenport C. M., Hieble, J. P., Wilson, S., Bergsma D. J., Fitzgerald L. R., Mol. Pharmacol., 59, 434-444 (2001).
9) Raible D. G., Lenahan T., Fayvilevich Y., Kosinski, R., Schulman E. S., Am. J. Respir. Crit. Care Med., 149, 1506-1511 (1994).
10) Hough L. B., Mol. Pharmacol., 59, 415-419 (2001) and references therein.
11) Thurmond R. L., Desai P. J., Dunford P. J., Fung-Leung W.-P., Hofstra C. L., Jiang W., Nguyen S., Riley J. P., Sun. S., Williams K. N., Edwards J. P., Karlsson L., J. Pharmacol. Exp. Ther., 309, 404-413 (2004).
12) Ling P., Ngo K., Nguyen S., Thurmond R. L., Edwards J. P., Karlsson L., Fung-Leung W.-P., Br. J. Pharmaco.,
142, 161-171 (2004).
13) de Esch I. J. P., Thurmond R. L., Jongejan A., Leurs R., Trends Pharmacol. Sci., 26, 462-469 (2005).
14) Daugherty B. L., Br. J. Pharmacol., 142, 5-7 (2004). 15) Jablonowski J. A., Carruthers N. I., Thurmond R. L. J.
A., Mini Rev. Med. Chem., 4, 993-1000 (2004). 16) B el l J. K ., McQ u een D . S ., Rees J . L. , B r. J.
Pharmacol., 142, 374-380 (2004).
17) Fung-Leung W. P., Thurmond R. L., Ling P., Karlsson L., Curr. Opin. Investig. Drugs, 5, 1174-1183 (2004). 18) Buckland K. F., Williams T. J., Conroy D. M., Br. J.
Pharmacol., 140, 1117-1127 (2003).
19) Lim H. D., van Rijin R. M., Ling, P., Bakker, R. A., Thurmond R. L., Leurs R., J. Pharmacol. Exp. Ther.,
314, 1310-1321 (2005).
20) Celanire, S., Wijtmans, M., Talaga, P., Leurs R., de Esch I. J. P., Drug Discovery Today, 10, 1613-1627 (2005).
21) Leurs R., Bakker R. A., Timmerman H., de Esch I. J. P., Nature Reviews Drug Discovery, 4, 107-120 (2005). 22) Morisset S., Rouleau A., Ligneau X., Gbahou F.,
Tardivel-Lacombe J., Stark H., Schunack W., Ganellin C. R., Schwartz J.-C., Arrang J.-M., Nature, 408, 860-864 (2000).
23) Bakker R. A., Leurs R., “Constitutively Active Histamine Receptors, Part 13, G Protein-Coupled Receptors,” eds. by Seifert R., Wieland T., WILEY-VCH Verlag GmbH & Co. kGaA, Weinheim, 2005, pp. 195-222.
24) Ali S. M., Tedford C. E., Gregory R., Handley M. K., Yates S. L., Hirth W. W., Phillips J. G., J. Med. Chem.,
42, 903-909 (1999).
25) Tedford C. E., Mant T. G. K.,Mah M., Shaffer L. M., Program & Abstract for International Sendai Histamine Symposium: Nov. 22-25; O-12, p 53 (2000).
26) From Wikipedia, USA (2007).
27) Wulff B. S., Hastrup S., Rimvall K., Eur. J. Pharmacol.,
453, 33-41 (2002).
28) Liu H., Kerdesky F. A., Black L. A., Fitzgerald M., Henry R., Esbenshade T. A., Hancock A. A., Bennani, Y. L., J. Org. Chem., 69, 192-194 (2004).
29) Kitbunnadaj R., Hoffmann M., Fratantoni, S. A., Bongers G., Bakker R. A., Wieland K., Jilali A. el, De Esch I. J. P.; Menge W. M. P. B., Timmerman H., Leurs R., Bioorg. & Med. Chem., 13, 6309-6323 (2005). 30) Kitbunnadaj R., Zuiderveld O. P, Christophe B.,
Hulscher S., Menge W. M. P. B., Gelens E., Snip E., Bakker R. A., Celanire S., Gillard M., Talaga P., Timmerman H., Leurs R., J. Med. Chem., 47, 2414-2417 (2004).
31) Vollinger R. C., Menge W. M. P. B., Leurs R., Timmerman H., J. Med. Chem., 38, 266-271 (1995). 32) Wieland K., Bongers G., Yamamoto Y., Hashimoto T.,
Yamatodani A., Menge W. M. P. B., Timmerman H., Lovenberg T. W., Leurs R., J. Pharmacol. Exp. Ther.,
299, 908-914 (2001).
33) Govoni M., Lim H. D., El-Atmioui D., Menge W. M. P. B., Timmerman H., Bakker R. A., Leurs R., De Esch I. J. P., J. Med. Chem., 49, 2549-2557 (2006).
34) Harusawa S., Murai Y., Moriyama H., Imazu T., Ohishi H., Yoneda R., Kurihara T., J. Org. Chem., 61, 4405-4411 (1996).
35) 総説:春沢信哉,荒木理佐,栗原拓史,有合化,
61, 682-693 (2003).
36) Harusawa S., Imazu T., Takashima S., Araki L., Ohishi H., Kurihara T., Yamamoto Y., Yamatodani A., Tetrahedron Lett., 40, 2561-2564 (1999).
37) Harusawa S., Imazu T., Takashima S., Araki L., Ohishi H., Kurihara T., Sakamoto Y., Yamamoto Y., Yamatodani A., J. Org. Chem., 64, 8608-8615 (1999). 38) Hashimoto T., Harusawa S., Araki L., Zuiderveld O.
P., Smit M. J., Imazu T., Takashima S., Yamamoto Y., Sakamoto Y., Kurihara T., Leurs R., Bakker R. A., Yamatodani A., J. Med. Chem., 46, 3162-3165 (2003). 39) Harusawa S., Araki L., Terashima H., Kawamura M.,
Takashima S., Sakamoto Y., Hashimoto T., Yamamoto Y., Yamatodani A., Kurihara T., Chem. Pharm. Bull.,
51, 832-837 (2003).
40) Lim H. D., van Rijin R. M., Ling P., Bakker R. A., Thurmond R. L., Leurs R., J. Pharmacol. Exp. Ther.,
314, 1310-1321 (2005).
41) Lim H. D., Smits R. A., Bakker R. A., van Dam C. M. E., de Esch I. J. P., Leurs R., J. Med. Chem., 49, 6650-6651 (2006).
42) Smits R. A., Lim H. D., Stegink B., Bakker R. A., de Esch I. J. P., Leurs R., J. Med. Chem., 49, 4512-4516 (2006).
43) Jablonowski J. A., Grice C. A., Chai W., Dyorak C. A., Venable J. D., Kwok A. K., Ly K. S., Wei J., Baker S. M., Desai P. J., Jiang W., Wilson S. J., Thurmond R. L., Karlsson L., Edwards J. P., Lovenberg T. W., Carruthers N. I., J. Med. Chem., 46, 3957-3960 (2003). 44) Venable J. D., Cai H., Chai W., Dvorak C. A., Grice C.
A., Jablonowski J. A., Shah C. R., Kwok A. K., Ly K. S., Pio B., Wei J., Desai P. J., Jiang W., Nguyen S., Ling P., Wilson S. J., Dunford P. J., Thurmond R. L., Lovenberg T. W., Karlsson L., Carruthers N. I., Edwards J. P., J. Med. Chem., 48, 8289-8298 (2005).
45) Terzioglu N., van Rijn R. M., Bakker R. A., de Esch I. J. P., Leurs R., Bioorg. & Med. Chem. Lett., 14, 5251-5256 (2004).
46) L.-Dutra A., Arienti K. L., Buzard D. J., Hack M. D., Khatuya H., Desai, P. J., Nguyen S., Thurmond R. L., Karlsson L., Edwards J. P., Breitenbucher J. G., Bioorg. & Med. Chem. Lett., 16, 6043-6048 (2006).
47) Kazuta Y., Hirano K., Natsume K., Yamada S., Kimura R., Matsumoto S., Furuichi K., Matsuda A., Shuto S., J. Med. Chem., 46, 1980-1988 (2003).
48) Watanabe M., Kazuta Y., Hayashi, H., Yamada S., Matsuda A., Shuto S., J. Med. Chem., 49, 5587-5596 (2006).
49) Sato H., Fukushima K., Shimazaki M., Urbahns K., Sakai K., Gantner F., Bacon K., Eur. Pat. Appl. 1505064, Feb. 9, 2005.
50) Sato H., Tanaka K., Shimazaki M., Urbahns K., Sakai K., Gantner F., Bacon K., PCT Int. Appl. WO 2005054239, Jun. 16, 2005.
51) Bell A. S., Lane C. A. L., Mowbray C. E., Selby M. D., Swain N. A., Williams D. H., PCT Int. Appl. WO 2007072163, Jun. 28, 2007.
52) Lane C. A. L., Price D. A., U. S. Pat. Appl. 2006111416, May 25, 2006.
53) Cai H., Carruthers N. I., Dvorak C. A., Edwards J. P., Kwok A. K., U. S. Pat. Appl. 2004048878, Mar. 11, 2004.
54) Buzard D. J., Edwards J. P., Kindrachuk D. E., Venable J. D., PCT Int. Appl. WO 2005092066, Jan. 6, 2005. 55) Burns S., Hamley P., PCT Int. Appl. WO 2005014579,
Feb. 17, 2005.
56) Venable J. D., Thurmond R. L., Inflamm. & Anti-Allergy Agents in Med. Chem., 5, 307-322 (2006).