創薬標的分子に対する選択性発現に関する 分子作用機序の解明
~カンナビノイド CB2受容体選択的アゴニストおよび Mps1 キナーゼ選択的阻害剤の創製~
日下部 兼一
1
目次
序論 ... 3
本論 ... 10
第1章 抗掻痒作用を有するカンナビノイドCB2受容体選択的アゴニストの創製と選択 性発現機構の解明 ... 10
第1節 ピリドン骨格を有するリード化合物の同定 ... 10
第1項 ピリドン骨格のデザインとシード化合物1-15aの同定 ... 10
第2項 シード化合物1-15aの最適化によるリード化合物1-39fの同定 ... 11
第3項 ピリドン誘導体の合成(1) ... 14
第2節 抗掻痒作用を有する縮環型ピリドン誘導体2-18eの創製 ... 17
第1項 リード化合物1-39fの最適化とCB2選択的アゴニスト2-18eの創製 ... 17
第2項 化合物2-18eと2-22cのin vivo評価 ... 21
第3項 ピリドン誘導体の合成(2) ... 22
第3節 CB2選択性に関する分子作用機序の解明 ... 24
第1項 リード化合物1-39fとCB2ホモロジーモデルとのドッキングモデル122) . 24 第2項 化合物2-18eのCB2選択性発現機序の解明 ... 27
第2章 Mps1キナーゼ選択的阻害剤の創製と分子作用機序の解明 ... 29
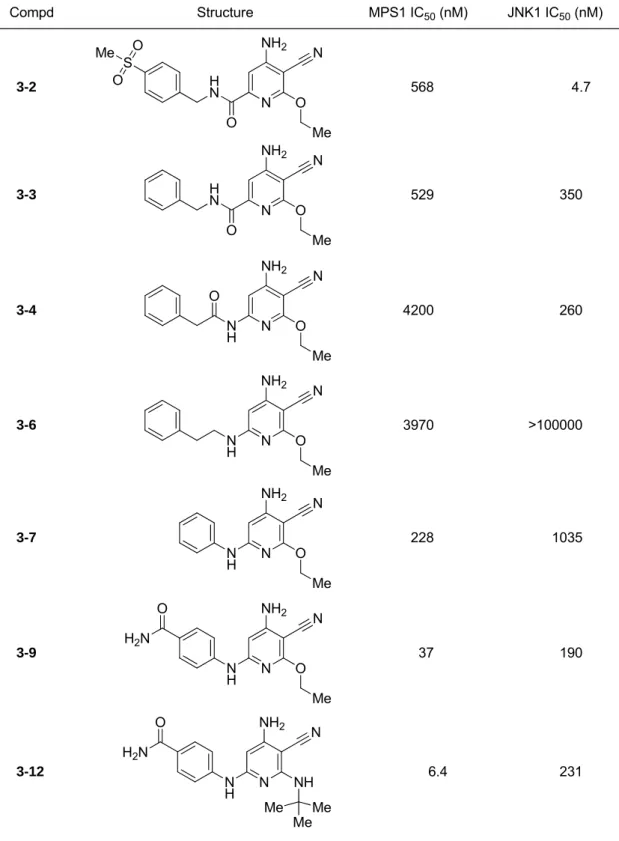
第1節 アミノピリジン骨格を有するMps1キナーゼ選択的阻害剤の創製124) ... 29
第1項 アミノピリジン骨格を有するリード化合物の同定 ... 29
第2項 Mps1キナーゼ選択的阻害剤3-12の同定 ... 30
第3項 化合物3-12のin vivo評価 ... 34
第4項 アミノピリジン誘導体の合成 ... 35
第2節 インダゾール骨格を有するMps1キナーゼ阻害剤の創製 ... 36
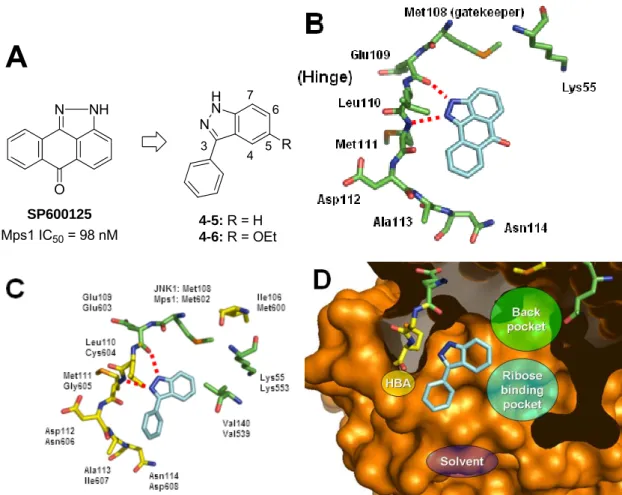
第1項 SP600125からインダゾール骨格を有するリード化合物4-6のデザイン . 36 第2項 リード化合物の最適化によるインダゾール4-32aと4-32bの同定 ... 38
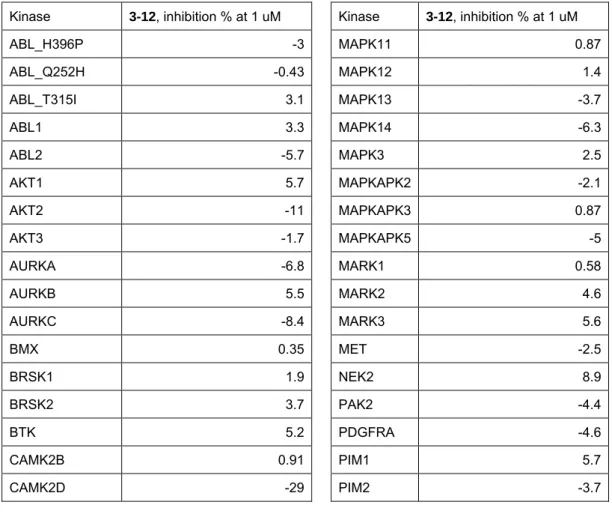
第3項 インダゾール化合物のキナーゼプロファイリング ... 44
第4項 インダゾール誘導体のPKプロファイル ... 47
第5項 インダゾール誘導体の合成 ... 48
2
第3節 Mps1 およびリガンド複合体 X 線結晶構造を用いた結合様式の解明とキナー
ゼ選択性発現に関する考察 ... 51
第1項 アミノピリジン3-9とMps1の複合体X線結晶構造解析 ... 51
第2項 インダゾール4-23dとMps1の複合体X線結晶構造解析 ... 54
第3項 アミノピリジンとインダゾール化合物を用いたキナーゼ選択性の考察 .... 56
総括 ... 62
謝辞 ... 66
実験の部 ... 67
第1章 実験の部 ... 67
第2章 実験の部 ... 92
引用文献 ... 116
3
序論
創薬標的分子に対して適正な選択性を有する医薬品を探索することは創薬研究におけ る重要な課題の一つである。医薬品分子は生体内で様々な物質と相互作用し得る。例えば、
タンパク質やDNA、RNA、脂質、代謝物、糖類、ペプチドなど多くの生体内分子が挙げら れるが、創薬標的分子以外のそれらとの相互作用は予期しない生体反応を引き起こし、重 篤な副作用を引き起こすこともある1)。最近、抗ヒスタミン薬・テルフェナジンやアステミ ゾールで心電図におけるQTc延長、それに続く心室性不整脈の発現(torsades de pointes)
による突然死が報告され、それらの薬剤が市場から撤退した2)。QTc延長の原因として、IKr
を形成するK+チャンネルサブユニットであるhERG(human ether-a-go-go-related gene)
が特定された3-5)。現在では医薬品のhERG阻害作用の回避は、安全性薬理試験で最も重要 な評価項目の一つとなっている6)。また、シトクロムP450(CYPs)は薬物代謝にかかわる 重要な酵素であるが、それらに対する阻害作用は、併用する薬物との相互作用により血中 濃度に大きな影響を与え、重大な副作用を引き起こすことがある7)。hERGチャンネルの阻 害と同様にCYP阻害作用の回避は、創薬の初期段階から重要な評価項目となっている8)。 さらに、創薬標的分子についてアミノ酸相同性の高いサブタイプが存在し、それらが望ま しくない薬理作用を引き起こす場合、それらとの選択性は創薬研究においてきわめて重要 な課題となる。一般的に、3次元構造が類似してアミノ酸相同性の高いそれらとの選択性を 獲得することは、CYP酵素やhERGチャンネルとの阻害作用の乖離よりも困難な課題とさ れる。特に G タンパク質共役型受容体(GPCR)やキナーゼ、プロテアーゼなどは創薬に おける重要なターゲットであり、3次元構造の類似したファミリーおよびアミノ酸相同性の 高いサブタイプが多く存在する。加えて、それらが独立した重要な役割を担っていること が多いことから、それらとの選択性の獲得は医薬品の探索研究において最も重要な課題の 一つとなっている1,9−12)。
近年、X線結晶構造解析やNMRなどの創薬基盤技術の進展に伴い、医薬品分子とタン パク質などの高分子との相互作用における分子作用機序が明らかにされ、選択性発現が特 に困難とされるタンパク質ファミリー間やサブタイプ間の選択性に関しても分子レベルで 明らかになりつつある1,9,13)。一方で、結晶化が困難なGPCRおよび触媒部位のアミノ酸配 列と立体構造が高度に保存されているキナーゼにおいて、標的分子選択的医薬品を創製し、
4
選択性に関して分子レベルで解明することは現在でも困難な課題とされている1,10,12)。この ような背景から筆者は、カンナビノイドCB2受容体およびMps1キナーゼ選択的化合物の 探索研究を通して、これらの選択性に関する分子作用機序の解明に着手することとした。
カンナビノイドCB2受容体
Cannabis sativa(大麻)は数百年にわたり医薬品として広く使用されてきた。Δ9-テト ラヒドロカンナビノール(Δ9-THC, Figure 0-1)は大麻の主要有効成分であり、大麻に含ま れる化学物質はカンナビノイドと称され、これまで多くの化合物が特定されてきた 15,16)。 それらカンナビノイドは医薬品として優れた特徴を有しており、例えば痛み17−20)、嘔吐17,21)、
不安症17,22)、緑内障17)、不眠17)、食欲不振17)、がん17,23)、アルツハイマー病17)、てんか
ん 17)などへの効果が示唆されている。一方で、カタレプシー(強硬症)や依存性など中枢 性の副作用も有していることも広く認識されており、期待される薬効とそれらとの副作用 の乖離が長年の課題となっていた24)。1990年代にカンナビノイドが作用する2つの受容体 が同定された。それらはカンナビノイドCB1受容体25,26)とCB2受容体27)と称され、クラ
スA(ロドプシンファミリー)に属するGPCRであることが分かった。CB1受容体は主に
脳や脊髄など中枢神経系に存在し25,26)、一方でCB2受容体は脾臓や免疫細胞など末梢神経 系に主に発現し27)、最近ではグリア細胞にも存在していることが分かってきた28)。CB1・
CB2受容体は、サイクリック3'-5'-アデノシン一リン酸(cAMP)の産生を抑制するGi/o型 のGPCRであり、その内在性カンナビノイド受容体リガンド(エンドカンビノイド)とし て、アナンダミド(AEA)や2-アラキドノイルグリセロール(2-AG)などが同定され(Figure
0-1)、生体内でのカンナビノイド受容体の役割も明らかになりつつある29−30)。CB1とCB2
受容体のそれぞれに選択的・非選択的なアゴニスト、アンタゴニスト(例えば、非選択的 アゴニストCP-55,94031)やCB2選択的アゴニスト・WIN 55,212-232)、Figure 0-1)をツー ル化合物として用いた評価系も構築され、複数のカンナビノイドリガンドが同定された。
これらを用いた研究の結果、上述したカンナビノイドに由来する中枢性の副作用は、中枢 に存在するCB1受容体の活性化に起因することが分かってきた24)。これらの結果は、末梢 に主に作用するCB2受容体選択的なアゴニストが上述の副作用を回避した有用な薬剤にな り得ることを示唆している。
5
複数の研究グループよってカンナビノイドCB2受容体選択的アゴニスト(Figure 0-2)
が同定され、それらは動物モデルにおいて中枢性の副作用を回避して優れた鎮痛効果を発 揮している。例えばAM-1241はCB1受容体に対して80倍の選択性を有するCB2選択的 アゴニストであるが、中枢性の副作用を示すことなく、炎症性疼痛および神経障害性疼痛 モデルで有効であることを示した33)。CB1に対して1200倍の選択性を有するCB2選択的 アゴニスト・GSK554418Aは中枢性副作用を示すことなく、急性と慢性疼痛動物モデルの 両 方 で 優 れ た 薬 効 を 示 し た 34)。 他 に も 、HU-30835)や GW40583336)、A-79626037)、 GW842166X38)、CBS-055039)などCB2選択的アゴニストは中枢性副作用を示すことなく動 物モデルで薬理効果を示した。
6 N
N CF3 O
NH O NH
Cl Cl
N O
I
NO2
N Me
N O
Cl Cl
N O
AM-1241 L-768242 / GW405833
GW842166X
NMe O
N NH
Cl O
N O
N O Me
Me Me Me
A-796260
GSK554418A
N O
CBS-0550
N Me N
Me Me Me
F CF3 Figure 0-2.CB2 Agonists
OH
O HO
Me Me Me
HU-210
OMe
MeO HO
Me Me Me
HU-308
Me Me
アトピー性皮膚炎は「表皮なかでも角層の異常に起因する皮膚の乾燥とバリアー機能 異常という皮膚の生理学的異常を伴い、多彩な非特異的刺激反応および特異的アレルギー 反応が関与して生じる慢性に経過する炎症、および痒み(掻痒)をその病態とする湿疹・
皮膚炎群の一疾患」と定義されている 40)。本疾患は、苦痛軽減と耐え難い痒みに伴う掻破 行動により皮膚病態は悪化し、疾患は慢性化する。したがって、痒みを効果的に抑えるこ とが本疾患の治癒において重要な手段であるが、抗ヒスタミン薬が抗掻痒剤として使用さ れているものの、その抗掻痒効果は十分とはいえず、新たな薬剤の開発が期待されている
41)。
HU-210(Figure 0-2)を用いた試験の結果、カンナビノイドリガンドは上述のような 疼痛モデルのみでなく、ヒトにおいて痒みの抑制効果を有することが明らかとなった 42)。
7
一方でHU-210は、CB1とCB2に対する非選択的アゴニストであり、上述のような中枢性
CB1に起因する副作用が懸念された43)。筆者は、抗掻痒作用を有するCB2選択的アゴニス トがCB1に由来する副作用を軽減し、効果的な抗掻痒薬になり得ると考えて本研究に着手 した。
筆者は本論文中で、創製した CB2 アゴニストの CB2 選択性の分子作用機序を明らか とするために、2011年にKobilkaらが報告した活性型β2アドレナリン受容体X 線結晶構 造を鋳型としたCB2ホモロジーモデルを活用した。上述したように、膜タンパク質である GPCR の結晶化は極めて困難とされている。このことは、ゲノムに含まれる遺伝子の
20~30%が GPCR など膜タンパク質由来のもであるにもかかわらず、Protein Data Bank
(PDB)に登録されている膜タンパク質の構造は約1%程度に過ぎないことが明確に示して いる44)。一方で、膜タンパク質の構造解析は21世紀に入り加速度的に進展しており、PDB への登録件数も飛躍的に多くなっている。近年の目覚ましい発展の背景には、1) バキュロ ウイルスや動物細胞・酵母の発現系をはじめとした真核細胞を用いた大量発現系の方法論 が蓄積されてきたこと、2) 蛍光検出ゲルろ過クロマトグラフィー45)の確立により結晶化に 適したタンパク質を円滑に精製できるようになったこと、3) 脂質キュービックフェーズ法
(LCP 法)46)とよばれる生理的に近い脂質二重膜中での結晶化法が確立されてきたこと
47,48)、4) 抗体・融合タンパク質を利用して目的タンパク質を安定化させる方法論が見出さ
れたこと48,49)、が主に挙げられる44)。特に2007年には、3)のLCP法や4)の抗体融合タン
パク質を用いて不活性型のβ2アドレナリン受容体X線結晶構造が報告された48,50)。さらに
2011年には、Kobilkaらが不安定で結晶化が困難とされたβ2アドレナリン受容体とそのア
ゴニストとGsヘテロ 3量体との複合体(活性型β2アドレナリン受容体)の結晶構造解析 に成功した51)。この場合、T4 リゾチームを受容体 N末端に融合させ、さらにこの複合体 を抗原としてラクダ科のラマを用いて抗体を作製しGsの結合を安定化する抗体を選択して、
世界ではじめて活性型GPCRの結晶化を実現した。これまでアゴニストが結合する活性型 GPCRは、ロドプシンなどPDBに登録されている不活性型の結晶構造を基にして、NMR や分子動力学計算によって得られた知見からそれらを改変して使用する他なかったが52−55)、 この結晶構造の登場により活性型GPCRを鋳型としたホモロジーモデルの作成が可能とな った。筆者は、本結晶構造を鋳型として活性型CB2受容体ホモロジーモデルを作成し、CB1 との選択性に関する分子作用機序の解明に取り組んだ。
8 Mps1キナーゼ
がん細胞またはその前駆細胞は遺伝子的に不安定であり、その結果、遺伝子の正確な 複製が妨げられ、このことが正常細胞のがん化、さらにはがん細胞の悪性化につながって
いる56−59)。遺伝子の不安定化は遺伝子変異の蓄積により生じるとされ、がん細胞の特徴を
なすものであり、一般的にGenetic instability と称される。この遺伝子の不安定化は、ヌ クレオチドレベルで生じるマイクロサテライト不安定性(microsatellite instability, MIN)
と染色体レベルで生じる染色体不安定性(chromosomal instability, CIN)のいずれかから 生じるとされている 57)。前者について、ある細胞が遺伝子の損傷を修復できずに蓄積し、
さらにわずかな遺伝子変異が蓄積してMINへと至る57,60,61)。後者について、ある種の細胞 は、先天的な異常などいくつかの要因によって適切に染色体数を保つことができなくなり、
通常とは異なる数の染色体を有する細胞へと変質する。それらの細胞は異数性細胞
(aneuploidy cell)と称され、CINへと至る62,63)。なかでもCINが遺伝子不安定性の主要 な要因とされ、それに伴って生じる異数性細胞が、がん化の重要な要素とされた 64)。した がって、異数性細胞への変質の抑制、さらには異数性細胞の選択的増殖阻害作用を有する 化合物は有用な抗癌剤となり得るだろう65,66)。
Monopolar spindle 1(Mps1)はTTKとも称され、チロシン・セリン・スレオニンを
リン酸化する二重特異性キナーゼである67,68)。Mps1は細胞分裂期に活性化される分裂期キ ナーゼであり、中心体の複製、分裂期チェックポイント、さらにはCINの維持調節にかか わっている69−72)。Mps1はがん細胞に高発現していることが分かっており73−76)、その発現 の程度はがんの組織学的悪性度に比例している77)。高発現したMps1は、細胞分裂期にお いて異数性がん細胞の安定化と保護にかかわっていることが明らかとなった 77)。さらにそ れらのがん細胞において、Mps1の発現レベルをRNAiにより抑制することで異常な分裂状 態に導き、最終的にアポトーシスや細胞死を引き起こすことが示された。一方で Mps1 を 欠損した非がん化細胞において有意なアポトーシスの上昇は認められなかった 77)。これら の知見は、Mps1阻害が異数性がん細胞の選択的な増殖抑制につながることを示唆している。
これらの経緯から筆者は、Mps1が有望な抗癌剤のターゲットになり得ると考えた。
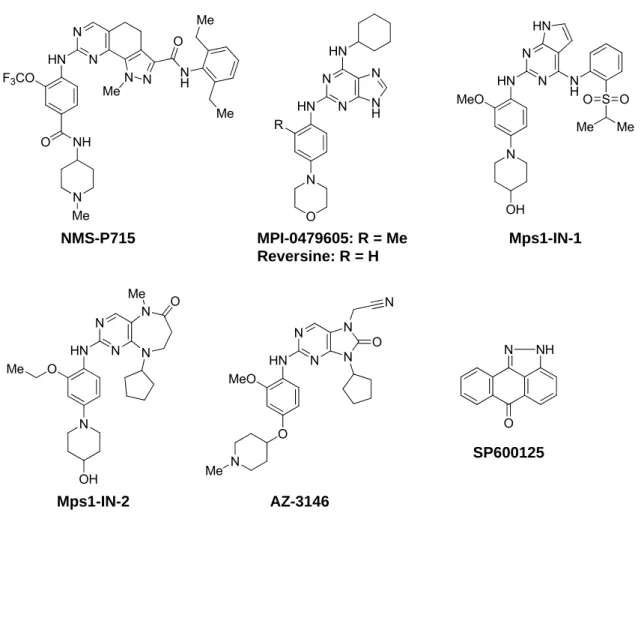
これまでに複数の Mps1阻害剤が報告されている(Figure 0-3)78)。特に、Nerviano Medical Science社とMyrexis社から報告されたNMS-P71579,80)とMPI-047960581)は、キ ナーゼ選択性とin vitro活性の良好な化合物で、マウスxenograftモデルで抗腫瘍活性を示
9
した。その他にもReversine(マウス筋芽細胞に対して特異的に脱分化を起こし、未分化な 細胞へ変化させる化合物として報告され、のちにMps1阻害を有することが分かった)82,83)、 Mps1-IN-184)、Mps1-IN-284)、AZ-314685)、SP60012572,86−88)がMps1選択的または非選択 的阻害剤として知られており、ツール化合物としても汎用されている。上述の阻害剤のな かでもNMS-P715とMPI-0479605が10 nM以下の酵素阻害IC50値を有し、その他の化 合物よりも強い活性を示す。しかしながら、がん細胞に対する増殖阻害活性がサブマイク ロモルオーダーで比較的弱く改善の余地があった79,81)。筆者らは、細胞系活性を改善し in vivoで有効性を示すMps1選択的阻害剤の創製を目指して探索研究に着手した。
N N
MeN N
NH O
Me
Me HN
NH O
N Me F3CO
NMS-P715 MPI-0479605: R = Me
Reversine: R = H
N
N N
H N HN
R
N O
HN N
N HN MeO
N HN
NH
OH
S
Me Me
O O
Mps1-IN-1
N N HN O
N
OH
N N
Me
Me O
Mps1-IN-2
N N HN MeO
O
AZ-3146
N N
O N
Me N
O
N NH
SP600125
Figure 0-3.Mps1 Kinase Inhibitors
10
本論
第1章 抗掻痒作用を有するカンナビノイド CB2 受容体選択的アゴニストの 創製と選択性発現機構の解明
第1節 ピリドン骨格を有するリード化合物の同定122)
第1項 ピリドン骨格のデザインとシード化合物1-15aの同定
ハイスループットスクリーニング(HTS)により見出されたヒット化合物 1-1 を基に して本研究を開始した(Figure 1-1)。ジヒドロイソキノロン骨格を有する化合物1-1は、
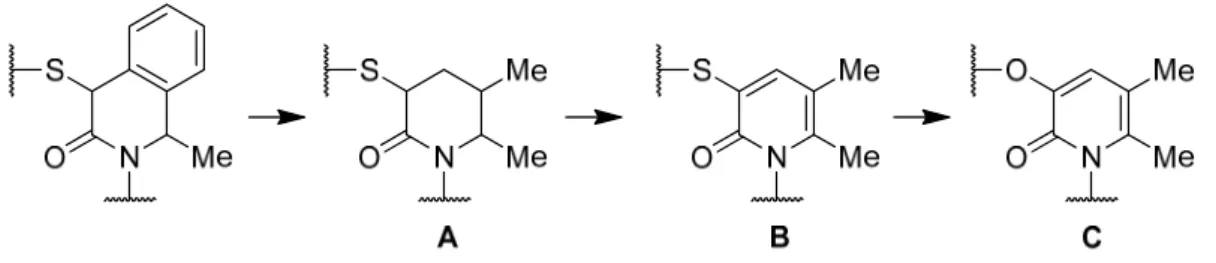
良好な活性(CB2親和性: CB2 Ki値)とCB1選択性を有するものの、2つの不斉点の制御 に伴って効率的な構造活性相関(Structure-activity relationship; SAR)を展開する困難さ があった。さらにイソキノロン骨格は、一般的に高い疎水性を伴うことから、ドラックラ イクネスの観点からも望ましいとはいえなかった 89)。そこで筆者は、不斉点と疎水性に起 因する課題を改善するために2-ピリドン骨格を有する化合物をデザインした(Figure 1-2)。
まず疎水性の低減を目的としてイソキノロン骨格のベンゼン環を除去して A に示す化 合物として、次に不斉点を除去するためにAを芳香化した2-ピリドン骨格Bをデザインし た。最後にチオエーテルに起因した代謝安定性を懸念して、エーテルリンカーに変換した ピリドンCをデザインした。実際に骨格Cを有する化合物1-15aを合成したところ(Table
1-1)、CB2に対して良好な活性を示した(CB2 Ki = 976 nM)。本化合物の活性はヒット化
合物と比較すると低下したものの、SAR の基点としては良好な活性と判断して、さらなる 構造変換を展開することとした。
11
第2項 シード化合物1-15aの最適化によるリード化合物1-39fの同定
ピリドン 1-15a のカルボニル基をチオカルボニル基に変換したところ(1-18a)、CB2
に対する親和性が向上した(Table 1-1, CB2 Ki = 101 nM)。次にピリドン環の窒素原子上 の置換基を検証した。その結果、n-ブチル基が適したサイズであり、鎖長を短くした場合
(1-18b, 1-18c)や長くした場合(1-18d, 1-18e)のいずれも活性が減弱した。これらの結 果は、この部位が疎水性の領域で、ポケットのサイズもある程度制限されていることを示 唆している。
N X O
R Me O
N
Me
Compd 1-15a
X R hCB2 Ki (nM)
1-18a 1-18b 1-18c 1-18d 1-18e
O S S S S S
n-Bu n-Bu Et n-Pr n-Pentyl n-Hexyl
976 101 3855 276 214 609
Table 1-1.Binding affinity of pyridones1-15aand1-18a-e
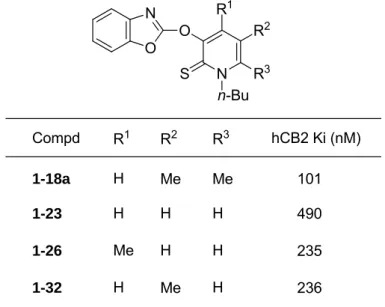
ピリドン環上の置換基の影響を検証した(Table 1-2)。R1にメチル基を導入した化合
12
物1-26とR2に置換基を導入した化合物1-32は、無置換体の化合物1-23と比較して活性 が向上した。これらの中で最も強い活性を示したものは R2と R3の両方にメチル基を有す
る化合物1-18aであり、対応する無置換体1-23と比較して5倍程度強い活性を示した。こ
れらの結果は、ピリドン環上への疎水性置換基の導入が活性向上において重要であること を示唆している。
N O S
n-Bu N
O
R1 R2 R3
Table 1-2.Binding Affinity of Thiopyridones1-18a,1-13,1-26, and1-32
Compd R1 R2 hCB2 Ki (nM)
H Me
1-18a 1-23 1-26 1-32
H Me H
101 490 235 236 R3
Me
Me
H H
H H
H
上記で見出した最適なピリドン環上の部分構造を用いて、ベンゾオキサゾールオキシ 基のバイオアイソスターであるアミド基への変換を試みた。本変換に際して、共有結合能 などの懸念のあるチオカルボニル基 90)からカルボニル基に変換して構造活性相関を実施し た(Table 1-3)。ベンゾオキサゾールオキシ基をアセトアミド基に変換した化合物1-39aは 大幅に活性が減弱したものの、フェニルアセトアミド基に変換した化合物 1-39b は、対応 するベンゾオキサゾールを有する 1-15a と同程度の親和性を示した。さらにフェニルアセ トアミド基をベンズアセトアミド基に置換した化合物1-39cは15倍程度活性が向上しCB2
Ki = 89 nMを示した。さらなる活性向上を目指してベンズアセトアミド基のフェニル基上
の置換基効果を検証した。2-フルオロ基を導入した化合物1-39dは無置換と同程度の活性で あったが、2-クロロ基を導入した化合物1-39eは活性が向上して16 nMのKi値を示した。
2-クロロ基を対応するメチル基に置換した化合物1-39fも同程度の強い活性を有し(14 nM)、
CB1に対して29倍の選択性を示した。2-メチル基(オルト位)と比較して、メタ位(1-39g)
13
とパラ位(1-39h)にメチル基を有する化合物はいずれも活性が減弱した。1-ナフチル基を
有する1-39iは、1-メチル基を有する1-39fと同程度の活性を示したものの、CB1に対する
選択性が大きく低下した。また2-ナフチル基を有する1-39jはCB2活性が大幅に低下した。
N O
n-Bu
HN Me
O Me R
Compd R hCB2 Ki (nM)
Me 1-39a
1-39b 1-39c 1-39d
Benzyl Phenyl 2-F-phenyl
>5000 1310 89 88 1-39e
1-39f 1-39g 1-39h 1-39i 1-39j
2-Cl-phenyl 2-Me-phenyl 3-Me-phenyl 4-Me-phenyl 1-Naphthyl 2-Naphthyl
hCB1 Ki (nM)
16 14 42 72 12 768
>5000
>5000 1376 617 391 390 343 854 19
>5000 Table 1-3.Binding affinity of pyridones39a-j
活性とCB1に対する選択性に優れたピリドン1-39fについて、cAMPを指標としたCB2 機能活性を測定した。ヒトCB2受容体を過剰発現させたCHO細胞に被験物質を投与し、
フォルスコリンで惹起したサイクリック 3'-5'-アデノシン一リン酸(cAMP)産生の抑制効 果を確認した。フォルスコリン刺激による cAMP 産生をフォルスコリン無刺激に対して 100%とし、50%のcAMP抑制作用を示す濃度をcAMP IC50とした。その結果、化合物1-39f
のcAMP IC50値は13 nMで、強いアゴニスト活性を示した。以上のように、CB2受容体
に対して強い親和性と機能活性を示し、CB1に対して良好な選択性を示すCB2アゴニスト 1-39fを同定した。この化合物は、適切な分子量(312)、tPSA(45)91)、CLogP(3.6)91)、
14
高いリガンド効率(ligand efficiency, LE)(0.47)92)を有しており、優れたリード化合物で あるといえる。
第3項 ピリドン誘導体の合成(1)
ピリドン1-15aと1-18a–eはScheme 1-1とScheme 1-2に従って合成した。エナミン 1-8はβケトエステル1-7とベンジルアミンとの縮合により合成し、メトキシアセチル化し た後に、ナトリウム存在下エタノール中で加熱することでピリドン1-10を得た93)。この化 合物とクロロフェニルテトラゾールを反応させて化合物1-11に変換し、中圧条件で水素添 加反応を実施することで鍵中間体1-12を合成した。この鍵中間体に対してヨウ化ブチルを 作用させて 1-13a とし、メトキシ基を脱保護し、水素化ナトリウム存在下クロロベンゾオ キサゾールと反応させることでピリドン1-15aを合成した(Scheme 1-1)。
Scheme 1-2に示すように、鍵中間体1-12に対して各種アルキル化剤を用いて化合物
1-13a–eとし、Lawesson試薬によりチオピリドン1-16a–eに変換し、それらをScheme1-1 と同様の方法で脱保護、ベンゾオキサゾール付加させることでチオピリドン 1-18a–e を合
15 成した。
チオピリドン1-23と1-26はScheme 1-3に従って合成した。チオピリドン1-23は化 合物1-19を用いてチオピリドン1-18a と同様に合成した。チオピリドン1-26は、化合物 1-22をテトラメチルメチレンジアミンで化合物1-24に変換し、メチル化、トリフェニルホ スフィンの付加、加水分解、ベンゾオキサゾール付加反応により合成した。
N MeO
X
a b c
N HO
S
n-Bu NMe2
N HO
S
n-Bu Me N
HO S
n-Bu N
O S
n-Bu N
O
N O S
n-Bu N
O
Me e
f g
1-19:R1= H, X = O 1-20:R1=n-Bu, X = O 1-21:R1=n-Bu, X = S
1-22 1-23
1-24 1-25 1-26
R1
d
Scheme 1-3. Reagents and conditions: a) n-BuI, NaH, DMF; b) Lawesson's reagent, toluene, reflux; c) pyridinium chloride, 200 °C; d) 2-chlorobenzo[d]oxazole, NaH, DMF, rt. e) N,N,N',N'- tetramethylmethylenediamine, EtOH-H2O, 75 °C; f) i) MeI, DCM, rt, ii)PPh3, EtOH, 75 °C, iii) aq.
NaOH, MeOH, 60 °C; g) 2-chlorobenzo[d]oxazole, NaH, DMF, rt.
16
5-メチルチオピリドン1-32は、3-メトキシピリドン1-19をテトラメチルメチレンジア
ミンと反応させて化合物1-27に変換した後に、Scheme 1-3に記載の化合物1-23と同様の 方法で合成した(Scheme 1-4)。
アミド基を有するピリドン誘導体1-39a–jはScheme 1-5に従って合成した。エステル
1-33をn-ブチルアミンと反応させて対応するアミド1-34に変換した後に、塩基性条件下で
環化させることで 3-シアノピリドン 1-35 とした。シアノ基を加水分解によりカルボン酸 1-36に変換し、Crutius転位反応によりCbz保護したアミンとした後に、脱保護を行うこ
とで3-アミノピリドン1-38を合成した。化合物1-38を各種酸クロライドと反応させるこ
とで対応するアミド誘導体1-39a–jを合成した。
17 N
R1 O
a b
N O
n-Bu R2
c d e Me
Me f
N O
n-Bu
HN Me
O Me R3
1-33:R1= OEt
1-34:R1= NHn-Bu 1-35:R2= CN 1-36:R2= CO2H 1-37:R2= NHCbz 1-38:R2= NH2
1-39a-j 1-39a:R3= Me 1-39b:R3= Benzyl 1-39c:R3= Phenyl 1-39d:R3= 2-F-phenyl 1-39e:R3= 2-Cl-phenyl 1-39f:R3= 2-Me-phenyl 1-39g:R3= 3-Me-phenyl 1-39h:R3= 4-Me-phenyl 1-39i:R3= 1-Naphthyl 1-39j:R3= 2-Naphthyl Scheme 1-5.Reagents and conditions: a) n-BuNH2, rt; b) 2-methyl-3-oxobutanal sodium salt, piperidine, AcOH, DMF, 135 °C; c) KOH, 80% aq. EtOH, reflux, 85%; d) DPPA, Et3N, BnOH, dioxane, 110 °C; e) 10% Pd/C, MeOH, rt; f) R3COCl, pyridine, THF, rt.
第2節 抗掻痒作用を有する縮環型ピリドン誘導体2-18eの創製123)
第1項 リード化合物1-39fの最適化とCB2選択的アゴニスト2-18eの創製
上述のように、良好な活性とCB2選択性を示すCB2アゴニスト1-39fを同定した。しか しながら、本化合物は経口吸収性が低く、それに伴いin vivo薬効を示さなかった。そこで
筆者は、in vivo薬効を有するCB2選択的アゴニストの同定を目的として、化合物1-39fを
リード化合物として更なる最適化研究を行った。ピリドン環 3 位のアミド基について、各 種官能基への変換を試みたところ、スルホンアミド2-10やウレア2-11への変換はいずれも 大幅に活性が低下した(Table 1-4)。一方で、逆のアミド基を有する2-13は、対応する1-39c と比較して若干活性が向上した。逆アミド2-13は合成面でも1-39cのアミドタイプよりも 有利であることから、以降のSARでは逆アミド基を用いて実施した。
18 N O
Me Me
Me R
O
Ph H
N
Ph S H N
O O
O HN H Ph N
NH Ph
O 1-39c
2-10
2-11
2-13
Compd R hCB2 Ki (nM) hCB1 Ki (nM)a
aNT = not tested.
89
>5000
>5000
66
1376
NT
NT
606 Table 1-4.SAR of the 3-position on the pyridone ring
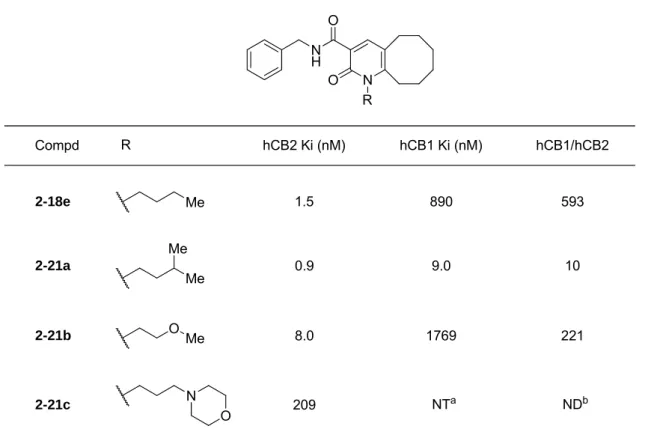
更なる活性と選択性の改善を目指して、化合物2-13のピリドン環5位と6位のメチル 基に相当する部分について構造変換を実施した(Table 1-5)。6位のメチル基をややサイズ の大きいエチル基に変換したところ、CB2に対する親和性が3倍程度向上した(2-18a)。
ピリドン環5位と6位をシクロペンタン環で縮環した化合物2-18bは、化合物2-18aと同 程度の活性を示した。一方で、化合物2-18aと2-18bのCB1に対するCB2選択性はそれ ぞれ16 倍と8.9倍であり、ジメチル基を有する化合物2-13と同様に低かった。次にシク ロヘキシル基を有する化合物2-18cを合成したところ、CB1 に対する選択性が大幅に改善 し(44倍)、CB2活性も維持した(CB2 Ki = 17 nM)。加えて、cAMP IC50は17 nMを示 し、アゴニスト活性も維持していることが分かった。さらに各種縮環化合物を合成して検 証したところ、環サイズが大きくなるとともに活性が向上する傾向があり、シクロヘプタ
19
ン環、シクロデカン環ではそれぞれ6.0 nM、2.5 nMのKi値を示した(本データはTable には示していない)。最終的にシクロオクタン環を有する化合物2-18eが最も優れた活性と CB2選択性を有し、合わせて優れたアゴニスト活性も示した(CB2 Ki = 1.5 nM, CB1/CB2
= 593)。一方で、化合物2-18dのようにシクロヘキサン環上に酸素原子を導入した場合、
CB2 に対する親和性が大幅に低下した。このことから縮環部位における疎水性相互作用は CB2親和性向上に重要であることが示唆された。
N O NH
Me O
A
Compd A hCB2 Ki (nM)
Me Me
20 2-18a
2-18b
2-18c
2-18d
2-18e
O
hCB2 cAMP IC50(nM)a
hCB1 Ki (nM)a hCB1/hCB2b
NT 322 16
16 NT 143 8.9
17
204
1.5
17
NT
1.4
746
NT
890
44
ND
593 Table 1-5.SAR of the A ring on the bicyclic pyridone ring
aNT = not tested.bND = not determined.
活性とCB2選択性に優れたピリドン2-18eを基にして窒素原子上のブチル基の構造活 性相関を検証した(Table 1-6)。n-ブチル基からイソペンチル基(2-21a)に変換するとCB1 親和性が強くなり、選択性が大幅に低下した。ブチル基上に酸素原子を導入したメトキシ
20
エチル基2-21bはやや活性が減弱し、さらにプロピルモルホリノ基(2-21c)を導入した場
合は大幅に活性が低下した。これらの結果は、ピリドン環上と同様にNアルキル部位にお ける疎水性相互作用の重要性を示唆するものである。以上の結果からピリドン環窒素原子 上の置換基はn-ブチル基が最適であることが分かった。
N O
R NH
O
Compd R hCB2 Ki (nM)
2-18e
hCB1 Ki (nM) hCB1/hCB2
1.5 890 593
Table 1-6.SAR of N-1 position on the bicyclic piridone
Me
Me Me
O Me
N O
0.9 9.0 10
8.0 1769 221
209 NTa NDb 2-21a
2-21b
2-21c
aNT = not tested.bND = not determined.
次にアミド側鎖の構造活性相関を検証した(Table 1-7)。その結果、疎水性の向上に伴 いCB1 とCB2 の両方の親和性が向上した。例えばベンジル基を水素に置換した無置換の
アミド2-22aは、CB2に対する活性が10倍以上低下しCB1活性は消失した。一方で疎水
性のイソプロピル基を導入した化合物2-22bは、無置換の2-22aと比較して10倍程度CB2 活性が向上した。さらに疎水性を付与したシクロヘキシル基を有する2-22cは、イソプロピ ル基と比較して20倍程度活性が向上し0.2 nMのKi値を示した。この化合物はCB1親和 性がやや強いものの、CB1に対して95倍の選択性を示した。また2-18eのベンジル位にジ メチル基を導入した2-22eは、対応するベンジル基を有する2-18eと比較して15倍程度活
21
性が向上し0.1 nMのKi値を示したが、CB1に対する親和性も増強しCB1 Ki値は1.0 nM を示した。フェネチル基を有する2-22fは、ベンジル基を有する2-18eと同程度の活性と選 択性を示した。これらの結果から、CB2 活性・選択性に優れたベンジル基を有する 2-18e とシクロヘキシル基を有する2-22cをin vivo評価化合物として選択して次項で評価した。
N O NH
Me O
R
Compd R hCB2 Ki (nM)
2-18e
hCB2 cAMP IC50(nM)a
hCB1 Ki (nM)a hCB1/hCB2b
1.5 1.4 890 593 Table 1-7.SAR of C-3 amide derivatives
Benzyl
2-22a 40 >5000 >125
2-22b i-Pr 4.0 2.5 591 148
2-22c Cyclohexyl 0.2 <0.2 19 95
H
2-22d
2-22e
2-22f
Me N
Me Me
2-Phenethyl
147
0.1 0.2 1.0 1.0
1.6 1.7 908 568 NT
NT NT ND
aNT = not tested.bND = not determined
第2項 化合物2-18eと2-22cのin vivo評価
CB2アゴニスト2-18eおよび2-22cのマウス薬物動態(Pharmacokinetics, PK)試験 を実施した(Table 1-8)。化合物1-22cを10 mg/kgで経口投与したところ、48%の生体内 利用率(Bioavailability, BA)と 6917 ng·h/mL の薬物血中濃度-時間曲線下面積(area under the blood concentration time curve, AUC)を示した。加えて静脈内投与において 35 mL/min/kg の良好なクリアランスを示した。一方で化合物 2-18e は 25%の BA と 96
mL/min/kg のクリアランスを示しており、2-22c と比較して薬物動態プロファイルはやや
22 劣る結果であった。
化合物 2-18eと2-22c をマウス薬効モデルで評価する前に、これら2化合物のマウス
におけるCB1とCB2のKi値を測定した。その結果、これらの2化合物はCB2に対して ヒトと同程度の親和性を示したが、一方で化合物2-22cはCB1に対する親和性が強くなり、
選択性が低下することが分かった(mCB2 Ki = 12 nM)。これらの結果から、化合物2-18e
をin vivo薬効評価化合物として選択した。
マウス痒みモデルにより化合物 2-18e の抗掻痒効果を評価した。この試験は Inagaki らが報告した方法 94)を改良して実施した。雌性 ICR マウスの予め剃毛した背部に
compound 48/90を皮下注射して痒みを惹起した。痒みは注射後すぐに惹起され、引っ掻き
回数は30分間カウントした。抗掻痒効果は化合物投与群とビークル群を比較することで算 出した。化合物2-18eを100 mg/kg経口投与したところ、compound 48/80で惹起された
痒みを81%阻害することが分かった。さらに本マウス薬効試験において、CB1に由来する
と思われる中枢性の副作用は確認されなかった。
第3項 ピリドン誘導体の合成(2)
ピリドン2-10と2-11の合成をScheme 1-6に示した。アミン2-9をベンゼンスルホニ ルクロリドと反応させることで化合物2-10を得た。またウレア2-11は、アミン2-9とフェ ニルイソシアネートを反応させることにより合成した。アミド 2-13 は、カルボン酸 2-12 とベンジルアミンを縮合させることで得た(Scheme 1-7)。
23 N O
Me Me
Me H2N
N O
Me Me
Me HN
Ph S N O O
O
Me Me
Me HN
HN O
Ph a
2-9 2-10
2-11
Scheme 1-6.Reagents and conditions: (a) benzenesulfonyl chloride, pyridine, THF, 0 °C (70%); (b) phenyl isocyanate, THF, rt (94%).
b
a N
O HO
O
Me Me
Me
N O NH
O
Me Me
Me Ph
2-12 2-13
Scheme 1-7.Reagents and conditions: (a) DCC, HOBt, benzylamine, THF, rt (27%).
ピリドン2-18aと縮環型ピリドン18b–eはScheme 3に記載の方法に従って合成した。
ケトン2-14a–eをn-ブチルアミンと縮合することにより対応するケチミンとし、さらにマ
ロン酸エステル 2-15 と反応させることでピリドンエステル 2-16a–e を得た。エステル
2-16a–eを加水分解し、酸クロライドに変換した後に、ベンジルアミンと反応させることで
ピリドン2-18a–eを合成した。