444
2.7.6.13 第 I 相試験(190342-006 試験)[非日本人対象]
...
5.3.3.1-3
2.7.6.13.1 試験方法の概要
試験方法の概要を
表
2.7.6.13-1
に示した。
表
2.7.6.13-1 試験方法の概要
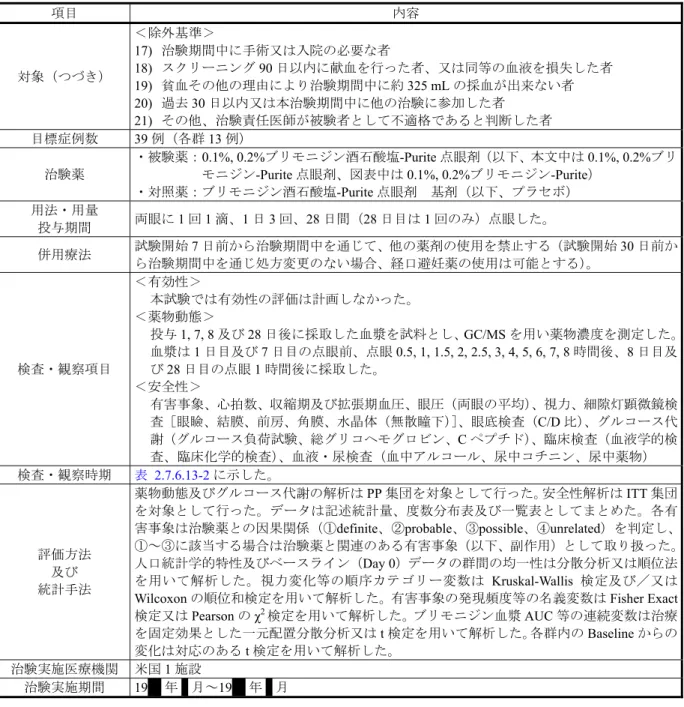
項目 内容 治験の表題 健康成人を対象とした1 日 3 回点眼時の安全性及び薬物動態の検討 治験の目的 健康成人男女を対象として0.1%又は 0.2%ブリモニジン酒石酸塩-Purite 点眼剤を 1 回 1 滴、 1 日 3 回、28 日間継続点眼による安全性及び薬物動態についてプラセボ(ブリモニジン酒 石酸塩-Purite 点眼剤基剤)を対照に比較検討した。 デザイン 二重盲検、無作為化、並行群間、プラセボ対照比較試験 対象 <選択基準> 1) 18 歳以上の健康な男女2) 標準身長/体重チャート(1996 Metropolitan Height and Weight Tables)で体重が±15% 以内の者 3) 文書による同意が得られた者 4) 医師の指示に従い、治験の全スケジュールを完了可能と思われる者 5) スクリーニング検査でグルコース代謝(空腹時血糖、グルコース負荷試験、C-ペプチ ド、総グリコヘモグロビン)が正常な者 6) 妊娠可能な女性の場合、Baseline(Day 0)において尿検査による妊娠検査の結果が陰性 の者 7) 血中アルコール反応、尿中アルコール又は中毒性の薬物反応(フェンシクリジン、ベ ンゾジアゼピン、カンナビノイド、アンフェタミン、バルビツール酸、コカイン、コ チニン及びアヘン等)が陰性の者 8) 試験開始 30 日前から治験期間中を通じて喫煙していない者 9) 両眼の眼圧が 12 mmHg 以上 21 mmHg 以下で、両眼の差が 5 mmHg 以下の者 10) 両眼の最高矯正視力(ETDRS)が Snellen 試視力表で 20/80 以上の者 11) 少なくとも 3 回、12 時間(午後 11 時から翌日の午前 11 時までの間)の絶食が可能な 者 <除外基準> 1) 妊婦、授乳婦、治験期間中に妊娠を希望する女性、妊娠の可能性があるが適切な避妊 手段をとることのできない女性。なお、両側の卵管を結索した者は参加できる。 2) 3 ヵ月以内に妊娠した者 3) 活動性の眼科疾患又は全身疾患を有する者 4) 糖尿病又はグルコース不耐性の既往歴又は家族歴(両親、兄弟姉妹)を有する者 5) 治験期間中コンタクトレンズの装用が予想される者。コンタクトレンズを装用してい る場合はSCL で 2 日間、RGP レンズで 7 日間の Washout 期間を設定した。 6) 3 ヵ月以内にレーザー手術、その他眼科的手術(屈折矯正手術を含む)、鼻涙管手術を 受けた者、眼瞼に外傷の既往を有する者 7) 治験薬成分のいずれかに過敏症がある者 8) 過去 6 ヵ月以内にアルコール及び薬物依存歴のある者 9) Day 1, 7, 8 及び 28 の来院日前 3 日間にアルコールを摂取した者 10) Day 1, 7, 8 及び 28 の来院日前 3 日間にキサンチン誘導体を含む飲料(茶、コーヒー、 チョコレート等)を摂取した者 11) 試験開始 30 日前から治験期間中を通じてニコチン(煙草、ガム、パッチ)を摂取した 者 12) 試験開始 7 日前から治験期間中を通じて経口避妊薬以外の何らかの薬剤(OTC 又はビ タミン剤を含む)を使用する必要がある者 13) 試験開始 3 日前から治験期間中を通じて生活習慣(運動又は食事等)の変更が予測さ れる者 14) 過去 60 日以内及び治験期間中に 10 ポンド以上体重が変動、あるいは変動が予測され る者 15) 血液学的検査、血液生化学的検査又は尿検査に異常が認められた者 16) HIV、B 型及び C 型肝炎の既往又は合併のある者
表
2.7.6.13-1 試験方法の概要(つづき)
項目 内容 対象(つづき) <除外基準> 17) 治験期間中に手術又は入院の必要な者 18) スクリーニング 90 日以内に献血を行った者、又は同等の血液を損失した者 19) 貧血その他の理由により治験期間中に約 325 mL の採血が出来ない者 20) 過去 30 日以内又は本治験期間中に他の治験に参加した者 21) その他、治験責任医師が被験者として不適格であると判断した者 目標症例数 39 例(各群 13 例) 治験薬 ・被験薬: 0.1%, 0.2%ブリモニジン酒石酸塩-Purite 点眼剤(以下、本文中は 0.1%, 0.2%ブリ モニジン-Purite 点眼剤、図表中は 0.1%, 0.2%ブリモニジン-Purite) ・対照薬:ブリモニジン酒石酸塩-Purite 点眼剤 基剤(以下、プラセボ) 用法・用量 投与期間 両眼に1 回 1 滴、1 日 3 回、28 日間(28 日目は 1 回のみ)点眼した。 併用療法 試験開始7 日前から治験期間中を通じて、他の薬剤の使用を禁止する(試験開始 30 日前か ら治験期間中を通じ処方変更のない場合、経口避妊薬の使用は可能とする)。 検査・観察項目 <有効性> 本試験では有効性の評価は計画しなかった。 <薬物動態> 投与1, 7, 8 及び 28 日後に採取した血漿を試料とし、GC/MS を用い薬物濃度を測定した。 血漿は1 日目及び 7 日目の点眼前、点眼 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8 時間後、8 日目及 び28 日目の点眼 1 時間後に採取した。 <安全性> 有害事象、心拍数、収縮期及び拡張期血圧、眼圧(両眼の平均)、視力、細隙灯顕微鏡検 査[眼瞼、結膜、前房、角膜、水晶体(無散瞳下)]、眼底検査(C/D 比)、グルコース代 謝(グルコース負荷試験、総グリコヘモグロビン、C ペプチド)、臨床検査(血液学的検 査、臨床化学的検査)、血液・尿検査(血中アルコール、尿中コチニン、尿中薬物) 検査・観察時期 表 2.7.6.13-2に示した。 評価方法 及び 統計手法 薬物動態及びグルコース代謝の解析はPP 集団を対象として行った。安全性解析は ITT 集団 を対象として行った。データは記述統計量、度数分布表及び一覧表としてまとめた。各有 害事象は治験薬との因果関係(①definite、②probable、③possible、④unrelated)を判定し、 ①~③に該当する場合は治験薬と関連のある有害事象(以下、副作用)として取り扱った。 人口統計学的特性及びベースライン(Day 0)データの群間の均一性は分散分析又は順位法 を用いて解析した。視力変化等の順序カテゴリー変数は Kruskal-Wallis 検定及び/又は Wilcoxon の順位和検定を用いて解析した。有害事象の発現頻度等の名義変数は Fisher Exact 検定又はPearson の χ2検定を用いて解析した。ブリモニジン血漿AUC 等の連続変数は治療 を固定効果とした一元配置分散分析又はt 検定を用いて解析した。各群内の Baseline からの 変化は対応のあるt 検定を用いて解析した。治験実施医療機関 米国 1 施設
446
表
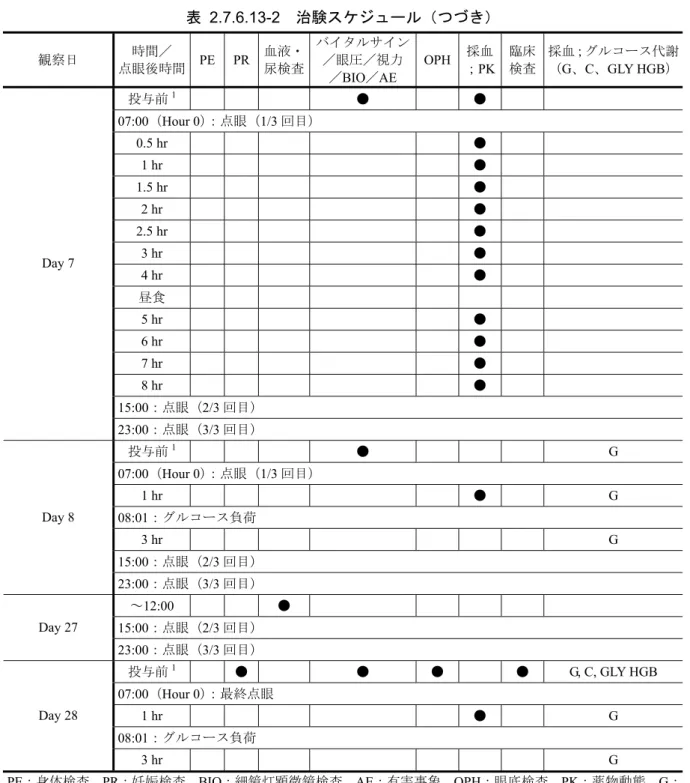
2.7.6.13-2 治験スケジュール
観察日 時間/ 点眼後時間 PE PR 血液・ 尿検査 バイタルサイン /眼圧/視力 /BIO/AE OPH ;採血PK 臨床検査 採血;グルコース代謝(G、C、GLY HGB) ~07:30 ● ● ● ● ● G, C, GLY HGB 08:00:グルコース負荷 08:30 G, C 09:00 G 09:30 G スクリーニング −28 日~−3 日 10:00 G Baseline (Day 0) ~12:00 ● ● 投与前1 ● ● 07:00(Hour 0):点眼(1/3 回目) 0.5 hr ● 1 hr ● 1.5 hr ● 2 hr ● 2.5 hr ● 3 hr ● 4 hr ● 昼食 5 hr ● 6 hr ● 7 hr ● 8 hr ● Day 1 15:00:点眼(2/3 回目) ~12:00 ● 15:00:点眼(2/3 回目) Day 6 23:00:点眼(3/3 回目) PE:身体検査、PR:妊娠検査、BIO:細鏡灯顕微鏡検査、AE:有害事象、OPH:眼底検査、PK:薬物動態、G: グルコース、C:C ペプチド、GLY HGB:総グリコヘモグロビン 1:点眼前 90 分以内に実施した。表
2.7.6.13-2 治験スケジュール(つづき)
観察日 時間/ 点眼後時間 PE PR 血液・ 尿検査 バイタルサイン /眼圧/視力 /BIO/AE OPH ;採血PK 臨床検査 採血;グルコース代謝(G、C、GLY HGB) 投与前1 ● ● 07:00(Hour 0):点眼(1/3 回目) 0.5 hr ● 1 hr ● 1.5 hr ● 2 hr ● 2.5 hr ● 3 hr ● 4 hr ● 昼食 5 hr ● 6 hr ● 7 hr ● 8 hr ● 15:00:点眼(2/3 回目) Day 7 23:00:点眼(3/3 回目) 投与前1 ● G 07:00(Hour 0):点眼(1/3 回目) 1 hr ● G 08:01:グルコース負荷 3 hr G 15:00:点眼(2/3 回目) Day 8 23:00:点眼(3/3 回目) ~12:00 ● 15:00:点眼(2/3 回目) Day 27 23:00:点眼(3/3 回目) 投与前1 ● ● ● ● G, C, GLY HGB 07:00(Hour 0):最終点眼 1 hr ● G 08:01:グルコース負荷 Day 28 3 hr G PE:身体検査、PR:妊娠検査、BIO:細鏡灯顕微鏡検査、AE:有害事象、OPH:眼底検査、PK:薬物動態、G: グルコース、C:C ペプチド、GLY HGB:総グリコヘモグロビン 1:点眼前 90 分以内に実施した。448
2.7.6.13.2 成績
2.7.6.13.2.1 対象
1) 症例の構成
本試験に
39 例が登録され、各群に 13 例が無作為に割付された。プラセボ群の 1 例が有害
事象発現(上室性期外収縮)のため
7 日目の最終投与後、また、プラセボ群の 1 例が自己都
合(静脈穿刺困難)による辞退により
6 日目の最終投与後、中止となった。37 例が試験を完
了した。
2) 解析対象集団の定義
ITT 集団は治験薬の投与を受け、1 回以上来院した被験者とし、安全性解析を行った。その
内訳は各群
13 例であった。PP 集団は重大な治験実施計画書違反がなく、1 回以上来院した被
験者とし、薬物動態及びグルコース代謝の解析を行った。その内訳は各群
13 例であった。
3) 人口統計学的及び他の基準値の特性
平均年齢は
33.4±10.11 歳(18~63 歳)、男性 21 例及び女性 18 例であった。平均身長は
69.29±3.727 インチ(約 176.0±9.5 cm)、平均体重は 160.87±23.524 ポンド(約 73.0±10.7 kg で
あり、標準身長/体重チャート(
1996 年の Metropolitan Life Height and Weight Tables)の±15%
2.7.6.13.2.2 安全性の結果
1) 薬物動態の解析
0.1%及び 0.2%ブリモニジン-Purite 点眼剤の反復投与 1 日目及び 7 日目の血漿中ブリモニジ
ン濃度の推移を
図
2.7.6.13-1
及び
図
2.7.6.13-2
に、薬物動態学的パラメータを
表
2.7.6.13-3
に
示した。
T
maxについて両群間の差及び
1 日目と 7 日目の差を比較した結果、いずれも統計学的に有
意な差は認められなかった。
0.1%及び 0.2%ブリモニジン-Purite 点眼剤の C
maxは
1 及び 7 日目
のいずれの時点においてもほぼ一定であった。
0.1%ブリモニジン-Purite 点眼剤に対する 0.2%
ブリモニジン
-Purite 点眼剤の C
max比は、
1 及び 7 日目においてそれぞれ 2.1 及び 2.2 であり、
ほぼ投与量に比例した血漿中濃度の上昇が認められた。
0.2%ブリモニジン-Purite 点眼剤の
AUC
(0-t)は
1 及び 7 日目でほぼ一定であった。0.1%ブリモニジン-Purite 点眼剤の AUC
(0-t)は
1
日目より
7 日目の方が有意に高かったが(P=0.0048)、被験者間に大きなバラツキが認められ
た。
0.1%ブリモニジン-Purite点眼剤に対する 0.2%ブリモニジン-Purite 点眼剤の AUC
(0-t)比は、
1 及び 7 日目においてそれぞれ 2.7 及び 1.9 であり、ほぼ投与量に比例した血漿中濃度の上昇
が認められた。同様に、
7 日目における 0.1%ブリモニジン-Purite 点眼剤に対する 0.2%ブリモ
ニジン
-Purite 点眼剤の AUC
(0-8)比及び
AUC
(0-∞)比は投与量に依存して増加した。
7 日目にお
ける
t
1/2はいずれの投与量においても
2 時間弱であり、投与群間に統計学的に有意な差は認め
られなかった。
以上の結果より、ブリモニジンは速やかに血漿中に移行すること、またすべての被験者の
血漿中濃度はすべての測定時点で
0.15 ng/mL 以下であり、全身循環への移行が少ないことが
示された。
図
2.7.6.13-1 1 日目の血漿中ブリモニジン濃度の推移
平均値±標準偏差(n=13) 0 20 40 60 80 100 0 1 2 3 4 5 6 7 8 9 投与後時間(h) 血漿 中ブ リモ ニジ ン 濃度 (pg/ m L ) 0.1%ブリモニジン-Purite 0.2%ブリモニジン-Purite450
図
2.7.6.13-2 7 日目の血漿中ブリモニジン濃度の推移
平均値±標準偏差(n=13)表
2.7.6.13-3 薬物動態学的パラメータ
薬剤 時点 Tmax (Hour) (pg/mL) Cmax AUC (0-t) (pg・hr/mL) AUC (0-8) (pg・hr/mL) AUC (0-∞) (pg・hr/mL) T1/2 (Hour) Day 1 1.54±0.660 (n=13) 23.3±14.07 (n=13) 79.3±47.78 (n=12) NA NA NA 0.1% ブリモニジン -Purite Day 7 1.50±0.677 (n=13) 30.0±17.78 (n=13) 127±86.8 (n=13) 136±84.8 (n=12) 153±96.9 (n=12) 1.88±0.809 (n=12) Day 1 1.77±0.599 (n=13) 48.4±35.10 (n=13) 211±147.0 (n=13) NA NA NA 0.2% ブリモニジン -Purite Day 7 1.35±0.944 (n=13) 64.7±37.76 (n=13) 245±124.1 (n=13) 245±123.8 (n=13) 268±129.8 (n=13) 1.95±0.628 (n=13) 検出限界値は2 pg/mL、値は平均値±標準偏差を示す。 NA:not applicable 0 20 40 60 80 100 0 1 2 3 4 5 6 7 8 9 投与後時間(h) 血漿 中ブ リモ ニジ ン濃 度 ( pg/ m L ) 0.1%ブリモニジン-Purite 0.2%ブリモニジン-Purite2) 有害事象
(1) 全有害事象の表示及び分析
有害事象の発現例数及び発現頻度を
表
2.7.6.13-4
に示した。
有害事象は
0.1%ブリモニジン-Purite 点眼剤の 53.8%(7/13 例)、0.2%ブリモニジン-Purite
点眼剤の
69.2%(9/13 例)及びプラセボの 46.2%(6/13 例)に発現し、各群間の発現頻度
に差は認められなかった。発現頻度が高かった有害事象(いずれかの薬剤群で
3 例以上)
は結膜充血、鼻炎、頭痛であった。重症度別に検討したところ、すべて軽度と判定された。
452
表
2.7.6.13-4 有害事象の発現例数(発現頻度)
事 象 名 【MedDRA (ver.10.0) PT】 0.1%ブリモニジン -Purite (n=13) 0.2%ブリモニジン-Purite (n=13) プラセボ (n=13) 眼障害 結膜充血 4 30.8% 1 7.7% 1 7.7% 眼そう痒症 1 7.7% 1 7.7% 0 0.0% 眼瞼浮腫 1 7.7% 0 0.0% 0 0.0% 眼痛(LLT:眼痛) 1 7.7% 0 0.0% 0 0.0% 眼刺激(LLT:眼刺激) 1 7.7% 0 0.0% 0 0.0% 眼乾燥 0 0.0% 2 15.4% 0 0.0% 眼瞼紅斑 0 0.0% 1 7.7% 0 0.0% 眼刺激(LLT:眼の灼熱感) 0 0.0% 1 7.7% 0 0.0% 先天性、家族性及び遺伝性障害 先天性心臓疾患 0 0.0% 0 0.0% 1 7.7% 耳及び迷路障害 耳の障害 0 0.0% 1 7.7% 1 7.7% 胃腸障害 下痢 1 7.7% 0 0.0% 0 0.0% 歯肉増生 1 7.7% 0 0.0% 0 0.0% 肛門直腸障害 0 0.0% 1 7.7% 0 0.0% 悪心 0 0.0% 0 0.0% 1 7.7% 感染症及び寄生虫症 鼻炎 1 7.7% 4 30.8% 0 0.0% 咽頭炎 1 7.7% 2 15.4% 1 7.7% 副鼻腔炎(LLT:副鼻腔炎) 1 7.7% 1 7.7% 0 0.0% 筋骨格系及び結合組織障害 四肢痛(LLT:上肢痛) 0 0.0% 0 0.0% 1 7.7% 神経系障害 頭痛 3 23.1% 1 7.7% 1 7.7% 浮動性めまい 0 0.0% 1 7.7% 0 0.0% 生殖系及び乳房障害 性器分泌物 0 0.0% 1 7.7% 0 0.0% 呼吸器、胸郭及び縦隔障害 咳嗽 1 7.7% 0 0.0% 1 7.7% 鼻乾燥 0 0.0% 2 15.4% 0 0.0% 鼻出血 0 0.0% 1 7.7% 0 0.0% 皮膚及び皮下組織障害 紅斑 2 15.4% 1 7.7% 0 0.0% そう痒症 0 0.0% 1 7.7% 0 0.0%(2) 有害事象のその他の分析
a) 副作用
副作用の一覧を
表
2.7.6.13-5
に示した。
副作用の発現頻度は
0.1%ブリモニジン-Purite 点眼剤の 53.8%(7/13 例)、0.2%ブリモ
ニジン
-Purite 点眼剤の 69.2%(9/13 例)及びプラセボの 30.8%(4/13 例)であった。発現
頻度が高かった副作用(いずれかの薬剤群で
3 例以上)は結膜充血、鼻炎、頭痛であっ
た。副作用はすべて治験終了までに、又は治験終了後すぐ(約
10 日以内)に回復した。
表
2.7.6.13-5 副作用の発現例数(発現頻度)
事 象 名【MedDRA (ver.10.0) PT】 0.1%ブリモニジン-Purite (n=13) 0.2%ブリモニジン-Purite (n=13)
プラセボ (n=13) 眼障害 結膜充血 4 30.8% 1 7.7% 1 7.7% 眼そう痒症 1 7.7% 1 7.7% 0 0.0% 眼瞼浮腫 1 7.7% 0 0.0% 0 0.0% 眼痛(LLT:眼痛) 1 7.7% 0 0.0% 0 0.0% 眼刺激(LLT:眼刺激) 1 7.7% 0 0.0% 0 0.0% 眼乾燥 0 0.0% 2 15.4% 0 0.0% 眼瞼紅斑 0 0.0% 1 7.7% 0 0.0% 眼刺激(LLT:眼の灼熱感) 0 0.0% 1 7.7% 0 0.0% 先天性、家族性及び遺伝性障害 先天性心臓疾患 0 0.0% 0 0.0% 1 7.7% 耳及び迷路障害 耳の障害 0 0.0% 1 7.7% 0 0.0% 胃腸障害 下痢 1 7.7% 0 0.0% 0 0.0% 感染症及び寄生虫症 鼻炎 1 7.7% 3 23.1% 0 0.0% 咽頭炎 0 0.0% 1 7.7% 1 7.7% 神経系障害 頭痛 3 23.1% 0 0.0% 1 7.7% 浮動性めまい 0 0.0% 1 7.7% 0 0.0% 呼吸器、胸郭及び縦隔障害 鼻乾燥 0 0.0% 2 15.4% 0 0.0% 鼻出血 0 0.0% 1 7.7% 0 0.0% 咳嗽 0 0.0% 0 0.0% 1 7.7% 皮膚及び皮下組織障害 紅斑 1 7.7% 1 7.7% 0 0.0%
454
b) 死亡、その他の重篤な有害事象
死亡、その他の重篤な有害事象はなかった。
c) 他の重要な有害事象(治験中止に至った有害事象)
プラセボの
1 例が有害事象発現(先天性心臓疾患)のため 7 日目の最終投与後治験中
止となった。
3) バイタルサイン、身体的所見及び安全性に関する他の観察項目
(1) 心拍数、収縮期血圧及び拡張期血圧
プラセボの
Day 8 において収縮期血圧が Baseline に比べ有意に低下したが(P=0.0401)、
0.1%及び 0.2%ブリモニジン-Purite 点眼剤では Baseline からの統計学的に有意な変化は認め
られなかった。また、いずれの時点においても薬剤群間に
Baseline からの統計学的に有意
な変化は認められなかった。
拡張期血圧に関しては、いずれの時点においても各薬剤群内及び薬剤群間に
Baseline か
らの統計学的に有意な変化は認められなかった。
心拍数に関しては、
0.2%ブリモニジン-Purite 点眼剤の Day 7 及び Day 28、プラセボの Day
7 において Baseline に比べ統計学的に有意な低下が認められた(P≤0.0125)が、0.2%ブリモ
ニジン
-Purite 点眼剤では Baseline からの統計学的に有意な変化は認められなかった。いず
れの時点においても薬剤群間に統計学的に有意な差はなく、臨床的に問題となる変化では
ないと判定された。
(2) 眼圧
プラセボの
Day 8 を除き、いずれの時点においても Baseline に比べ有意な眼圧下降が認
められた。
Day 8 及び Day 28 において、0.1%ブリモニジン-Purite 点眼剤及び 0.2%ブリモニ
ジン
-Purite 点眼剤はプラセボに比べ有意な眼圧下降を示した(P≤0.0347)。0.1%及び 0.2%
ブリモニジン
-Purite 点眼剤の Baseline からの眼圧下降幅は−3.4~−2.5 mmHg であり、健康
成人において臨床的に意味のある眼圧下降が認められた。安全性の観点からは危険性はな
いと治験責任医師は判断した。
(3) 視力
Baseline と比較して最終観察時に 2 ライン以上の視力悪化がみられたものは、プラセボの
1/13 例(7.7%)のみであった。最終観察時における Baseline との変化は薬剤群間に統計学
的に有意な差はなく、臨床的に問題となる変化ではないと判定された。
(4) 細隙灯顕微鏡検査
Baseline と比較して 1 段階以上の悪化がみられたものは、0.1%ブリモニジン-Purite 点眼
剤の水晶体(白内障)
3 例であった。いずれもスクリーニング時及び最終観察時に軽度の
白内障の所見が認められたが、
Day 1、Day 7 及び Day 8 においては所見なしと報告されて
おり、誤って変化ありと記録されたと考えられた。
(5) 眼底検査
C/D 比に関して、いずれの薬剤も群内及び薬剤群間に Baseline からの統計学的に有意な
変化は認められず、臨床上問題となる所見は認められなかった。
(6) グルコース代謝
グルコース負荷試験及び総グリコヘモグロビンは男女別に解析した。グルコース負荷試
験に関して、男性はプラセボの
Day 28 の投与前において、女性は 0.1%ブリモニジン-Purite
点眼剤の
Day 28 の Hour 1、0.2%ブリモニジン-Purite 点眼剤の Day 28 の投与前及び Day 28
の
Hour 1、プラセボの Day 28 の投与前においてスクリーニング時に比べ統計学的に有意な
低下が認められたが(
P≤0.0429)、いずれの時点においても薬剤群間に統計学的に有意な差
は認められず、臨床上問題となる変化は認められなかった。総グリコヘモグロビンは薬剤
群内及び薬剤群間にスクリーニング時からの統計学的に有意な変化は認められなかった。
(7) 臨床検査
臨床化学的検査のすべての項目において薬剤群間に統計学的に有意な差は認められなか
った。スクリーニング時に比べ統計学的に有意な変化が認められた項目を以下に示す。い
ずれの項目においても臨床上問題となる変化は認められなかった。
・
0.1%ブリモニジン-Purite 点眼剤:アルブミン(減少)、ALT(減少)、LDH(減少)、
K(減少)、尿酸(減少)
・
0.2%ブリモニジン-Purite 点眼剤:アルブミン(減少)、Fe(減少)、クレアチニン
(増加)
・ プラセボ:
ALK(減少)、AST(減少)、LDH(減少)、尿酸(減少)、クレアチニ
ン(増加)
血液学的検査に関して、スクリーニング時の
MCH 及び MCV は、0.2%ブリモニジン
-Purite 点眼剤及びプラセボに比べ 0.1%ブリモニジン-Purite 点眼剤で有意に低い値を示し
た。
Day 28 において、白血球数は 0.1%ブリモニジン-Purite 点眼剤及びプラセボに比べ 0.2%
ブリモニジン
-Purite 点眼剤は Baseline に比べ有意に高値へ変動した。
スクリーニング時に比べ統計学的に有意な変化が認められた項目を以下に示す。いずれ
の項目においても臨床上問題となる変化は認められなかった。
・
0.1%ブリモニジン-Purite 点眼剤:好酸球%(増加)、単球%(増加)、ヘマトクリ
ット(減少)
、ヘモグロビン(減少)
、赤血球数(減少)
・
0.2%ブリモニジン-Purite 点眼剤:単球数(増加)、ヘマトクリット(減少)、ヘモ
グロビン(減少)、赤血球数(減少)
・ プラセボ:好酸球
%(増加)、Lymph%(増加)、MCH(増加)、MCV(減少)、好
中球
%(減少)、好中球数(減少)、白血球数(減少)
456