博士論文
心肥大期から心不全期におけるヒストンのアセチル化修飾部位の変化を 介した転写制御機構
Transcriptional regulation mechanism via changes in histone acetylation domains during the transition from cardiac hypertrophy to heart failure
本論文は静岡県立大学大学院薬食生命科学総合学府博士論文である
2021 年 5 月
静岡県立大学大学院 薬食生命科学総合学府 薬科学専攻
分子病態学講座
船本 雅文
ii 略語表
本文中に用いた略語は下記のとおりである.
ACE-I : Angiotensin converting enzyme inhibitor ANF: Atrial natriuretic factor
ANOVA: Analysis of variance ARB : Angiotensin receptor blocker BNP: Brain natriuretic peptide
DMEM: Dulbecco's modified eagle medium DAB: Diaminobenzidine
DMSO: Dimethyl sulfoxide DR: Dahl salt-resistant DS: Dahl salt-sensitive
EDTA: Ethylenediaminetetraacetic acid ERK: Extracellular signal-regulated kinase FBS: Fetal bovine serum
FS: Fractional shortening
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase HAT: Histone acetyltransferase
HBSS: Hank's balanced salt solution HW / BW: Heart weigh t/ Body weight KD: Knockdown
KO: Knockout
LVIDd: Left ventricular internal dimension at end diastole LVIDs: Left ventricular internal dimension at end systole LVPWT: Left ventricular posterior wall thickness MAPK: Mitogen-activated protein kinase
MI: Myocardial infarction NAF: Sodium fluoride
NYHA : New york heart association PBS: Phosphate buffered saline PE: Phenylephrine
PMSF: Phenylmethylsulfonyl fluoride QOL: Quality of life
qRT-PCR: Quantitative reverse transcription polymerase chain reaction SDS: Sodium dodecyl sulfate
SFM: Serum-free media
SUMO: Small ubiquitin-related modifier
iii
TAC: Transverse aortic constriction
TBS: Tris buffered saline TG: Transgenic
WB: Western blotting WT: Wild type
β-MHC: β-myosin heavy chain
iv
目次
序論 ... 1
実験方法 ... 5
1.
使用した動物 ... 52.
初代新生仔ラット心筋細胞培養 ... 53.
心臓超音波検査 ... 54.
細胞刺激 ... 55.
核タンパク質、ヒストン画分回収 ... 66.
ウエスタンブロット... 67.
蛍光免疫染色 ... 78.
クロマチン免疫沈降法... 79.
心筋細胞面積測定... 810. mRNA
の抽出およびcDNA
の合成 ... 911. Quantiative real-time PCR (qRT-PCR) ... 9
12.
トランスフェクション... 1013.
組織片からの核タンパク質、ヒストン画分回収 ... 1014.
統計解析 ... 10目的・結果 ...11
1.
心筋細胞肥大時におけるヒストンのアセチル化の検討 ...112.
肥大反応遺伝子プロモーターにおけるヒストンのアセチル化の検討 ... 123. p300
ノックダウン処理によるヒストンのアセチル化の検討 ... 144.
クルクミン処理によるヒストンのアセチル化の検討 ... 185. p300
過剰発現によるヒストンのアセチル化の検討 in vitro ... 186. p300
過剰発現によるヒストンのアセチル化の検討 in vivo ... 187.
心肥大期・心不全期におけるヒストンのアセチル化の検討 ... 218.
心不全期におけるBRG1
の関与の検討 ... 24考察 ... 26
総括 ... 30
謝辞 ... 31
参考文献 ... 32
1
序論
日本において心疾患は昭和 50 年代に脳血管疾患にかわり死亡原因の中で悪性腫瘍 に次いで第 2 位となり、その後も死亡数・死亡率はともに上昇傾向にある [1]。心不 全は増加しつつある高血圧性心疾患や心筋梗塞、狭心症などの虚血性心疾患をはじめ としたあらゆる心疾患の最終像と位置付けられており、心疾患の中で最も多い死亡原 因となっている。高血圧などの様々な心肥大刺激によって心臓は心肥大を経て心不全 に至る。心不全とは心臓のポンプ機能が低下することで様々な臓器への血液供給が低 下した病態である [2]。心筋細胞は胎生期に盛んに分裂・増殖しているが、出生後は 分裂増殖能を失うため高血圧や心筋梗塞などのストレスがかかると、心臓はポンプ機 能を維持しようと個々の心筋細胞を肥大することで収縮力を増加させる。しかし、ス トレスが長時間にわたって加わることで、収縮力を維持していた心筋細胞は疲弊し、
その代償機構はやがて破綻する。その結果、心臓の収縮・拡張能の低下、間質の線維 化や心室腔の拡大が生じることによって最終的に心不全へと至る [3, 4]。したがって、
心不全の前段階で生じる心肥大の制御は結果的に心不全の予防・治療につながる重要 なターゲットである。さらに、超高齢社会の到来と共にますます増加する高血圧性心 疾患や虚血性心疾患による心不全に対して、その発症と進展を抑制するための新たな 治療法を確立することは社会的、医療経済的急務である。
心不全治療薬として前負荷を軽減するために利尿薬や強心作用をもつジギタリス 製剤が使用されてきた。さらにアンジオテンシン変換酵素阻害薬 (ACE-I) 、アンジオ テンシンⅡ受容体拮抗薬 (ARB) や 遮断薬などが標準的な治療薬として用いられて いる [5, 6]。これら治療薬の投与により 5 年生存率の改善が認められるが、心不全患 者の予後は悪く、ニューヨーク心臓協会が定めた心不全の症状の程度の分類 (NYHA 分類) Ⅲ度、Ⅳ度では 5 年生存率は 50 %程度のため、さらなる治療法の開発が期待さ れている [7]。慢性心不全の治療に用いられている ACE-I、ARB や遮断薬などは、
細胞膜受容体など心不全を引き起こすシグナル伝達経路の上流をターゲットとした ものであり、細胞内情報伝達経路の最終到達点である心筋細胞核内の転写経路を標的 とした治療薬は開発されていない。
心不全発症メカニズムとしてヒストン翻訳後修飾を含むエピジェネティクな制御
機構が注目されている [8, 9]。心不全発症におけるヒストンアセチル化酵素 (HAT) や
ヒストン脱アセチル化酵素の役割が盛んに研究されている [10-12]。主要な転写コア
クチベーターである p300 が心不全に関与する HAT として挙げられる。拡張型心筋症
や虚血性心不全患者の心臓は健常者の心臓と比較して p300 の発現量が有意に増大し
ていた [13]。また、心臓特異的に p300 を過剰発現したトランスジェニック (TG) マ
ウスでは心不全が引き起こされ、野生型 (WT) マウスと比べて心筋梗塞後の左室リモ
デリングが増悪する [12]。しかし、HAT 活性を欠如した p300 の心臓特異的過剰発現
TG マウスでは、心筋梗塞後の左室リモデリングの増悪は WT マウスと同程度にまで
抑えられた [11, 12]。反対に、p300 のホモ接合体のノックアウトマウスでは胎生致死
2
が引き起こされ、ヘテロ接合体のノックアウトマウスでは大動脈弓を結紮し大動脈に おける拍出量の減少から,左室内が高圧になる大動脈縮窄 (TAC) 手術により心肥大 が WT よりも抑制されることが報告されている [13, 14]。p300 の HAT 阻害作用を有 する天然物クルクミンを心筋梗塞後ラットや高食塩食による高血圧ラットに投与す ることで心不全の進展が抑制される [15]。さらに、p300 の HAT 阻害作用を有するア ナカルジン酸も TAC 手術を施したマウスにおいて心肥大並びに H3K9 のアセチル化 を抑制する [16]。また、 p300 の HAT 阻害剤の C646 や L002 はアンギオテンシンⅡに よる左室壁肥厚と心臓の線維化を抑制した [17]。心不全における p300 のシグナル伝 達経路として p300 / GATA4 経路がある。高血圧や心筋梗塞などのストレスにより交 感神経やレニン・アンジオテンシン系が作動し、様々なシグナル伝達経路を介して心 筋細胞核内の転写コアクチベーターである p300 が活性化する [18]。活性化した p300 は転写因子 GATA4 をアセチル化し、GATA4 の DNA 結合能を増加、GATA4 制御下の 心肥大反応因子である心房性ナトリウム利尿因子 (ANF)、脳性ナトリウム利尿ペプチ ド (BNP)、-ミオシン重鎖 (-MHC) などの発現を亢進させることで心筋細胞肥大を 引き起こし、心不全へと至る [19, 20] (Scheme. 1)。以上のことから、 p300-HAT 活性が 心不全治療の標的となることが示唆される。
Scheme. 1 心不全進展に関与する p300/GATA4 経路
p300 の HAT 活性は GATA4 などの転写因子だけでなくヒストンをアセチル化する
ことによって遺伝子の転写を制御している。ヒストンは真核生物でヌクレオソームを
構成する塩基性タンパク質であり、 長い DNA 鎖を核内に収納する役割を持ち DNA の
3
アクセシビリティの調節などによって遺伝子発現を調整する。ヒストンは H2A、 H2B、
H3、H4 という 4 つのヒストンコアタンパク質が 2 つずつのオクタマーの複合体を形
成している (Scheme. 2A)。このヒストンオクタマーに 145~147 bp の DNA が巻き付 きヌクレオソーム構造をとっている。クロマチンは、このヌクレオソームの繰り返し から構築されている [21]。ヒストンは、クロマチン形成のための構造的足場になり、
多様な翻訳後修飾を広範に受けることで転写制御に関与している [22]。ヒストンの翻 訳後修飾としてはメチル化、アセチル化、 SUMO 化、ユビキチン化およびリン酸化な どが報告されている [23]。その中でヒストンのアセチル化はヒストンと DNA との親 和性を低下したり、隣接するヌクレオソーム同士の距離を離したりすることで DNA が露出することにより、転写因子が DNA に結合しやすくなる開放的なクロマチンの 状態になる [24]。ヒストンのテールドメインのアセチル化に関する研究は盛んに行わ れており、特にヒストンのアセチル化修飾部位として H3 のリジン 9 (H3K9) に関し ての報告が多数ある [25-27]。ヒストンのテールドメインは、正電荷を持つアミノ酸 残基に富んでおり、負に帯電した DNA に巻き付くことで安定したヌクレオソーム構 造を形成していると考えられている。テールドメインである H3 の 9 番目のリジンが アセチル化されることによって電荷が変化し、ヒストンテールと DNA の相互作用が 減弱し、DNA への転写因子の結合が増加することで転写活性化が起こるといったメ カニズムが提唱されている。これらのヒストンのテールドメインは、 p300 によってア セチル化修飾され、転写活性化が起こることが報告されている [28-31] (Scheme. 3B)。
転写制御因子などは、ヒストンテールドメインのアセチル化を容易に認識し、集まる ことで DNA の露出や転写制御因子などとの相互作用により転写因子が DNA 上にリ クルートされやすくなる。これらのことから、ヒストンテールドメインはアセチル化 されることでクロマチンを転写不活性化状態のヘテロクロマチンから転写活性化状 態のユークロマチンに変化させ、転写を促進するクロマチンリモデリングの重要な場 所として役割を果たす [32]。一方で、ヒストン本体にある球状ドメインの翻訳後修飾 についてはほとんど研究されていない。近年、 p300 による新たなヒストンのアセチル 化修飾部位として H3 の 122 番目のリジンが報告された [33]。H3K122 は、ヒストン テールドメインの H3K9 とは異なりヒストンの球状ドメインに位置している。この
H3K122 は、ヒストンの 2 回転対称軸にあり、 DNA が巻き付く足場となる位置に存在
しているため、この場所でヒストンと DNA の結合は最長になる。H3K122 がアセチ
ル化修飾を受けるとヒストン-DNA の結合が減弱し、遺伝子の転写を活性化する [32,
33] (Scheme.3C)。すなわち、ヒストン球状ドメインはアセチル化されることでヒスト
ン-DNA との相互作用に直接影響を及ぼし、ヌクレオソームの構造が緩むことにより
DNA がむき出しになるヌクレオソームの構造変化を起こす重要な場所として役割を
果たしている。
4
Scheme. 2 p300 によるヒストンのアセチル化修飾部位 黄色 =H2A 、黄緑色= H2B 、青色= H3 、赤色= H4
ヒストン翻訳後修飾などのエピジェネティックな制御機構が生理機能におい て重要な役割を果たしているとともに、多くの疾患発症に関与していることも報告さ
れている [34-36] 。心不全においては、 TAC を施した心不全マウスの心臓においてヒ
ストンのアセチル化について ChIP-sequence を行った結果、 H3K9 並びに H3K27 のア
セチル化が遺伝子領域全体で亢進しており、そのアセチル化の亢進には p300 が関与
していることが報告されている [37] 。しかし、心不全におけるヒストンのアセチル化
についてはテールドメインである H3K9 や H3K27 のアセチル化に関しては報告があ
るものの、ヒストンの球状ドメインのアセチル化については全く知られていない。そ
こで本研究の目的は、高血圧誘導性心不全モデルであるダールラットの心肥大期から
心不全期への移行においてヒストンのテールドメインと球状ドメインのアセチル化
がどのように変化するのかを検討することである。
5
【実験方法】
1. 使用した動物
本動物実験は『静岡県立大学における動物実験に関する指針』に従い、静岡県立大学動物 保護委員会の承認を得たプロトコールに沿って行った。初代胎仔ラット心筋細胞培養のため の哺育
1-3
日齢胎児ラットは日本SLC
から購入した。心臓特異的p300
過剰発現 (-MHC-p300 TG)
マウスおよびWild Type (WT)
マウスを使用した [11]。高食塩食誘導性心不全モデルであるダールラットは、日本
SLC
から高食塩食感受性ダールラット (DS) と高食塩食抵抗 性ダールラット (DR) 購入した。ダールラットには、6週齢から8%NaCl (高塩分)
食を与え た。 動物は心肥大期 (12週齢) および心不全期 (21週齢) で心臓超音波検査後、サクリファ イスを行った。対照動物として正常血圧のDR
ラットの年齢を一致させ使用した。2. 初代新生仔ラット心筋細胞培養
哺乳
1
日齢の新生仔SD
ラット (10 匹) から心臓を摘出し、心臓以外のものをピンセット で剥ぎ取った。心臓をPBS (137 mM NaCl、 2.7 mM KCl、 10 mM Na
2HPO
4、1.8 mM KH
2PO
4)
で4
度洗浄後、をハサミでミンチ状になるまで細切れに刻み込み、Hanks Balanced Salt Solution
(HBSS,
ナカライテスク, Japan) で一度洗浄し、血球成分を除去した。次に37ºC
に温めておいたコラゲナーゼ (0.5 mg/mL, Worthington, USA) / パンクレアチン (0.05%, Sigma, USA) 混合溶
液を
10 ml
加えた。10分間振盪し、パスツールピペットで上清を吸い取り、15 ml 遠沈チューブに移し
250 xg
で5
分間遠心、沈殿した細胞塊に10%FBS
入りDulbecco's Modified Eagle's
Medium (DMEM,
ナカライテスク) を5 ml
加えよく懸濁した後、37ºCにて保温した。残りの心臓残渣に再度コラゲナーゼ/パンクレアチン混合溶液を
10 ml
加え、10 分間振盪し、15 ml 遠沈チューブに上清を回収した。この操作を計8
回行った。回収した上清を250 xg
で10
分 間遠心した。上清を捨て、DMEM で1
度洗浄した後、沈殿した細胞塊にDMEM
を20 ml
加 えてよくピペッティングし、線維芽細胞を分離するために、10 cm
ディッシュ 2枚に播種し、CO
2incubator
で60
分間培養した。培養後、ディッシュの上清を50 mL
遠沈チューブに回収し、DMEM を
10 ml
加え、これを2
回繰り返した。細胞数をカウントし、DMEM で希釈し(1.5×10
5cell/mL)、必要に応じて、 100 mm dish (FALCON, USA)、 6-well plate (FALCON)、 12-well plate (FALCON)
と2-well slide chamber (Thermo scientific, USA)
に播種した。3. 心臓超音波検査
心機能は、10~12MHzのプローブと
sonos 5500
超音波システムを使用した心エコー検査に よって非侵襲的に評価した。左室収拡張期末期径 (left ventricular internal dimension, LVIDd)、左室収縮期末期径 (left ventricular internal dimension, LVIDs)、左室後壁厚 (posterior LV wall
thickness, PWT)
を測定し、左室内径短縮率 (fractional shortening, FS) は [LVIDd - LVIDs] /LVIDd
× 100 %から算出した。4. 細胞刺激
6
10 cm
ディッシュで心筋細胞を24
時間培養後、serum free のDMEM (SFM,
ナカライテスク) (10 ml) に
2
回交換し、血清を除去した。一晩培養後、再度SFM
に培地交換をしたのちに30 µM
フェニレフリン (PE, Wako, Japan) (最終濃度) にて刺激を行い、48時間 CO2incubator
にて培養した。クルクミン (Sigma) 処理を行う場合はPE
刺激を行う2
時間前に10 µM
クル クミン (最終濃度) となるように調整したSFM
に培地を交換した。コントロールは溶媒であ るDimethyl sulfoxide (DMSO, Wako)
を同量加えたSFM
を使用した。5. 核タンパク質、ヒストン画分回収
ディッシュの培地を廃棄し、
PBS
でWash
後にセルスクレーパーで細胞をかきとり、細胞を 新しいチューブに移した。805 xg、4 ºCで3
分間遠心し、上清を廃棄し冷PBS
を1 ml
加え懸 濁した。これらの作業を2
回繰り返した。遠心しPBS
を廃棄し低張バッファー (10 mM HEPES-KOH pH 8.0、1.5 mM MgCl
2、10 mM 2.5 M KCl、10 mM NAF、10 mM Sodium Butyrate、1%pepstatin、1 mM Na
3VO
4、1 mM PMSF、0.7% 2-mercaptoethanol) を細胞ペレットの3
倍量加え 懸濁し5
分間氷冷、さらに懸濁した。12,000 xg、4 ºCで5
分間遠心し上清を廃棄した。細胞 ペレットと等量の高張バッファー (1 M HEPES-KOH pH 8.0、1 M MgCl2、5 M NaCl、0.5 MEDTA、Glycerol 25 %、10 mM NAF、10 mM Sodium Butyrate、1% pepstatin、1 mM Na
3VO
4、1mM PMSF、0.7% 2-mercaptoethanol)
を加え懸濁し20
分間氷冷した。12,000 xg、4℃で5
分間 遠心し上清を核タンパク質として新しいチューブに回収し実験に使用した。その後、残渣に等量の
0.1 M HCl
を添加し、よく混合し5
分間氷冷、12,000 xg、4 ºCで5
分間遠心し、上清を新しいチューブに移した。次に全量の
1/5
量の1 M Tris buffer pH 8.0
を加え、中和混合した ものをヒストン画分として実験に使用した。核タンパク質とヒストン画分をProtein Assay Dye Reagent Concentrate (Bio-Rad, USA)
を用いてBradford
法にてタンパク質濃度を測定した。得 られたタンパク濃度をもとに、4x Sample Buffer (0.2 M Tris-HCl, 8% SDS, 40% glycerol, 0.004%bromophenol blue, 24% 2-mercaptoethanol)
を加えて、95ºC、5分間加熱してウエスタンブロッ トサンプルとした。6. ウエスタンブロット
7.5%または 18%ポリアクリルアミドゲルを用いて Laemmli
法に則り30 mA
でSDS-PAGE
電気泳動を行った。泳動後、ゲルからニトロセルロース膜 (Hybond-ECL、
GE healthcare, Japan)
に
200 mA
で1
時間膜転写を行った。その後、ニトロセルロース膜を1
時間 5% スキムミルク入り
PBS-T (0.05% Tween20 in PBS)
に浸し30
分間ブロッキングを行い、1次抗体を入れた1%
スキムミルク入りPBS-T
に封入し1
時間室温でインキュベートした。次にニトロセルロース膜を
PBS-T
で洗浄 (10分間 x 3回) し、2次抗体入りの1%
スキムミルク入りPBS-T
に封入し
1
時間室温でインキュベートした。PBS-Tで洗浄 (10分間 x 3回) 後、ECL, Primeま たはECL Select (GE Healthcare)
を用いて発色させ、LAS1000 (Fujifilm, Japan) で化学発光を検 出・撮影した。撮影した画像は、Multi Gauge V3.0 (Fujifilm)を用いて解析・シグナルの定量化 を行った。1
次 抗 体 は 、rabbit polyclonal anti-p300 antibody (Santa Cruz Biotechnology, USA), mouse
7
monoclonal anti--actin antibody (Sigma-Aldrich, USA), rabbit monoclonal anti-Histone-H3 antibody, rabbit polyclonal anti-acetyl-Histone-H3(K9) antibody (Cell signaling technology, USA), rabbit polyclonal anti-acetyl-Histone-H3(K122) antibody (Abcam, USA)を使用した。
2
次抗体は、goat anti-rabbit IgG–HRP (MBL, USA)
とsheep anti-mouse IgG-HRP (GE Healthcare)
を使用した。7. 蛍光免疫染色
心筋細胞を培養した
2 well slide chamber
をPBS
で洗浄し、15分間10%
中性ホルマリン緩 衝液で固定した。PBS
で洗浄後、1%BSA
と0.5%NP-40/ TBS-Ca
で 1時間ブロッキングした。次に、抗
Acetyl-Histone H3K9
抗体または抗Acetyl-Histone H3K122
抗体とともに抗myosin
heavy chain
抗体を添加し、湿潤箱で4 ºC
、一晩インキュベートした。PBS
で洗浄した後、抗rabbit Cy3
抗体及び抗mouse Dylight-649
抗体を添加し、湿潤箱で4 ºC
、2時間インキュベートした。その後、
Hoechst 33432 (ナカライテスク)
で核を染色し、PBS
で3
回洗浄後、Fluorescent mounting Medium S3203 (Dako, USA)で封入した。免疫蛍光は、レーザー顕微鏡 LSM510 (Carl Zeiss, Germany)
を用いて観察した。8. クロマチン免疫沈降法
10 cm
ディッシュで培養した心筋細胞に終濃度1%となるようにパラホルムアルデヒド溶
液 (Wako)を添加し、20分間反応させて
DNA /
タンパク質を架橋させた。次に0.5 M Glycine
(Wako)
溶液を1 ml
添加し5
分間置くことで架橋反応を停止させた。培地を廃棄し、冷PBS
で洗浄後、セルスクレーパーで細胞をかきとり、新しい
1.5 ml
チューブに移し805 xg, 4ºC
で5
分間遠心し上清を廃棄することで細胞を洗浄した。この操作を2
回行った後、SDS lysis buffer (50 mM Tris-HCl、140 mM NaCl、10 mM EDTA、1% SDS、1% Triton X-100)
を加え懸濁し5
分間氷冷した。次にバイオラプター (コスモ・バイオ, Japan) を用いて超音波破砕を行った。30
秒間破砕、30秒間インターバルの5
分間を計7
セット行った。12000 xg, 4ºCで 20分間遠 心し上清を回収した。これに10
倍量のDilution buffer (16.7 mM Tris-HCl、167 mM NaCl、1.2 mM EDTA、 1.1% Triton X-100、 100 mg/ml Salmon sperm DNA、 300 µg/ml BSA)
で希釈した後、抗
Acetyl-Histone H3K9
抗体、抗Acetyl-Histone H3K122
抗体およびコントロールとしてrabbit
IgG (Santa Cruz)
を添加し、一晩4ºC
にて転倒混和させた。また希釈したサンプルの一部 (免疫沈降に用いた
1/10
量) をInput DNA
とした。Dilution bufferで平衡化したDynabeads
®M-280
Sheep anti-Rabbit IgG
を30 µl
添加し、4ºCにて2
時間転倒混和させた。混和後、マグネットスタンドを用いて上清を廃棄し、
500 µl
のLow Salt Solution (20 mM Tris-HCl pH7.9、 150 mM NaCl、
2 mM EDTA、1% Triton X-100、0.1% SDS)
で1
回、High Salt Solution (20 mM Tris-HCl pH7.9、500 mM NaCl、2 mM EDTA、1% Triton X-100、0.1% SDS)
で1
回、LiCl Solution (10 mM Tris-HCl pH7.9、250 mM LiCl、1 mM EDTA、1% NP-40、1% NaDOC)
で6
回、マグネットビーズ を洗浄した。洗浄後、Elution Buffer (0.1 M NaHCO
3、1% SDS)
を250 µl
加え、5分間静置し、上清を新しいチューブに回収した。これに
5 M NaCl
を10 µl
加え、65ºCで6
時間加温するこ とで架橋構造を解離させた。このサンプルにRNase / ProteinaseK
溶液 (RNase 3 µl, ProteinaseK8
3 µl, 0.5 M EDTA pH 8 5 µl, 1 M Tris pH 6.8 10 µl)
を添加し65ºC
で1
時間反応することでRNA
およびタンパク質を分解させた。次にフェノールクロロホルム抽出およびエタノール沈殿を 行うことでDNA
オリゴを精製した。in vivoクロマチン免疫沈降法では心臓を1~3 mm
3程度 までスライスし、上記と同様にホルマリン固定後、Glycine (Wako)
溶液で架橋を停止させた。以後の操作は通常のクロマチン免疫沈降法と同様に行った。得られた
DNA
サンプルをテン プレートとして定量PCR
を行うことで肥大反応因子であるANF、BNP
や-MHCプロモータ ー上にあるヒストンのアセチル化の変化を測定した。プライマーは下記のFigure
のような⚫
ANF
プロモーターFw 5’- GATAACTTTAAAAGGGCATC -3’, Rv 5’- CTCTGGTTTCTCTCTCAGCC -3’
⚫
5’上流域の ANF
プロモーターFw 5’- TGTGTTTGCTTGTGCTAGGCCC -3’, Rv 5’- TAAGTGGGCTGGTATGTGCTTG -3’
⚫
BNP
プロモーターFw 5’- CTGCGGTTTATAGCCTCCAC -3’, Rv 5’- TGCCTCTGCCTTTTATCCTG -3’
⚫
5’上流域の BNP
プロモーターFw 5’- CACCAAGCCACACTCTGAAG -3’, Rv 5’- TGGCTGAAGATTGAATGCAG -3’
⚫ -MHCプロモーター
Fw 5’-TGGAAACAGGACCAGGCTAG -3’, Rv 5’-TGGCATGTTCAGGGTCTGAG -3’
⚫
5’上流域の-MHC
プロモーターFw 5’-GCAGTCTGGATCCCTGATGT -3’, Rv 5’-GACACTGGGGCACAGAGATT -3’
を使用した。以下の
ΔCt
法の計算式を用いてInput DNA
で補正し、コントロールを1
とする ことで相対値を算出した。ΔCt = Ct
Input– Ct
SamplePromoter occupation = 2
ΔCtScheme. 3 プライマーの標的領域
9. 心筋細胞面積測定9
心筋細胞を培養した
2 well slide chamber
をPBS
で洗浄し、15分間10%
中性ホルマリン緩 衝液で固定した。PBSで洗浄後、0.3% H2O
2in MeOH
で15
分間処理し内因性ペルオキシダー ゼを失活させた。PBS
で洗浄した。1
次抗体mouse monoclonal anti-myosin heavy chain IgG (Leica
Biosystems, USA)
を添加し、湿潤箱で4 ºC
、一晩インキュベートした。PBSで洗浄した後、2
次抗体sheep anti-mouse IgG-HRP (GE Healthcare)
を添加し4 ºC、30
分間インキュベートした 後、室温で30
分間インキュベートした。TBS-Ca (150 mM NaCl、50 mM Tris-HCl、 1mM CaCl
2)
に浸し、DAB溶液 (2mg/ml DAB錠 (Wako)、0.02% H2O
2in TBS-Ca)
で15
分間反応させた。蒸留水で
2
回洗浄後、PBSで洗浄し、蒸留水で洗浄した後、マイヤーヘマトキシリン (Wako) で核染色を8
分間行った。次に、15分間流水で洗浄し、90% Ethanol, 100% Ethanol、xyleneで 脱水した後、MGK-S (松浪ガラス工業, Japan) で封入した。-MHC 染色をした心筋細胞は顕 微鏡 (ECLIPSE 80i, NIKON, Japan) で形態を観察・写真撮影を行った。次に無作為に各群40
個の細胞を抽出してImage J 1.43
により細胞面積測定し、平均値を算出することで心筋細胞 の肥大を評価した。10. mRNAの抽出およびcDNAの合成
培養した心筋細胞を冷やした
PBS
で2
回洗浄後、TRI Reagent (ナカライテスク)
を添加し、室温で
10
分間静置し、新しい1.5 ml
チューブに移した。次に、Chloroform (Wako) 0.1 mLを 加え、15秒間激しく振り混ぜ、室温で5
分間静置した。その後、12,000 xg, 4 ºCで15
分間遠 心し、上清を回収した。回収後、2-Propanol (Wako) を0.25 µl
加えてvortex
し、室温で10
分 間静置した後、12,000 xg, 4ºC
で10
分間遠心した後、上清を取り除き、残ったペレットに75%
Ethanol (Wako) 500 µL
を加え、5,000 xg, 4 ºC
で5
分間遠心、上清を取り除いて10
分間風乾し、DNase / RNase free water (ナカライテスク)
を10 μl
を加え、55 ºCで15
分間インキュベートし て、RNA を溶解させた。抽出したmRNA
の濃度を吸光度測定器 (バイオフォトメーター,Eppendorf, USA)
を用いて260 / 280 nm
の波長を計測することで決定した。次にReverTra Ace
®qPCR RT Master Mix (TOYOBO, Japan)
を用い、標準プロトコールにそって抽出したmRNA
から
cDNA
を合成した。Total RNA
量は1 sample
あたり250 ng
になるよう添加し、DNase / RNase
free water
でTotal 10 μl
になるよう調製した。穏やかに撹拌させた後、37 ºCで60
分インキュベートし、95 ºCで
5
分インキュベートとし、5倍希釈したものをcDNA
のテンプレートとし た。11. Quantitative real-time PCR (qRT-PCR)
定量的
PCR
はKOD SYB
®qPCR Mix (TOYOBO)
を用いて、その標準プロトコールに従ってを行った。プライマーは
⚫ ANF (Fw 5’- AGGCCATATTGGAGCAAATC -3’, Rv 5’- CATCTTCTCCTCCAGGTGGT -3’)
⚫ BNP (Fw 5’-ATCTGCCCTCTTGAAAAGCA -3’, Rv 5’- TCGAGCAGATTTGGCTGTTA -3’)
⚫ -MHC (Fw 5’-ATCACCAACAACCCCTACGA -3’, Rv 5’-GCGCCTGTCAGCTTGTAAAT -3’)
⚫ GAPDH (Fw 5’-TGGTGAAGGTCGGTGTGAAC -3’ , Rv 5’-GTTGAACTTGCCGTGGGTAG -3’) を用いた。反応後、得られた増幅曲線の適当な箇所に閾値を設定し、増幅曲線との交点のサ
10
イクル数を
Ct
値とした。以下のΔCt
法の計算式を用いてrat GAPDH
で補正し、コントロー ルを1
とすることでANF, BNP, -MHC
のmRNA
量の相対値を算出した。ΔCt = Ct
GAPDH– Ct
SamplemRNA
量 = 2ΔCt12. トランスフェクション
培養心筋細胞への
siRNA
のトランスフェクションはLipofectamine RNAiMAX (Invitrogen,
USA)
を用い、標準プロトコールに則って行った。播種24
時間後の培養心筋細胞の培地を二度
SFM
に交換した。Opti-MEM (Invtirogen) にsiRNA-p300 (si-p300)
及びコントロールsiRNA (si-control)
を溶解、同時に別チューブにOpti-MEM
にLipofectamine RNAiMAX
を溶解させた 後、この二つを混合、15
分間静置し、Lipofectamine / siRNA
複合体を形成させた。続いてOpti- MEM
を加え希釈した後 (終濃度50 nM)、心筋細胞の培地と入れ替え、 2
時間CO
2インキュベ ーター内で培養した。次にSFM
培地に交換し更に2
時間培養を行った後、PE 刺激 (30 µM) を与え、48時間培養した。培養心筋細胞への
p300
の過剰発現には、作成したpCMV-p300HA
及びコントロールとして
pcDNA-null
を用いて行った [12]。プラスミドのトランスフェクションはLipofectamine
及び
Plus Reagent (Inviteogen)
を用い、標準プロトコールに則って行った。播種24
時間後の培養心筋細胞の培地を二度
SFM
に交換した。Opti-MEM (Invtirogen) にpCMV-p300HA
またはpcDNA-null
とPlus Reagent
を溶解、同時に別チューブにOpti-MEM
にLipofectamine
を溶解さ せた後、この二つを混合、15分静置し、Lipofectamine / プラスミド複合体を形成させた。続いて
Opti-MEM
を加え希釈した後、心筋細胞の培地と入れ替え、2時間CO
2インキュベーター内で培養した。次に
SFM
培地に交換し更に48
時間培養した。13. 組織片からの核タンパク質、ヒストン画分抽出
液体窒素で凍結させた心臓組織片をすり鉢で擂り潰し、ダウンスガラスホモジナイザーに 移した。三倍量の低調バッファーを加えて、ホモジナイズし
10
分間放置した。805 xg、 4℃で 5
分間遠心し上清を廃棄した。残渣に等量の高張バッファーを添加し混合後、12,000 xg、4℃で
5
分間遠心し上清を核タンパク質として回収した。残渣に等量の0.1 M HCl
を添加し、よ く混合し5
分間氷冷、12,000 xg、4 ºCで5
分間遠心し、上清を新しいチューブに移した。次に全量の
1/5
量の10%SDS
と1 M Tris buffer pH 8.0
を加え、中和混合したものをヒストン画分として実験に使用した。
14. 統計解析
結果は平均±SEとして表した。実験群間の統計的比較は、一元配置分散分析または二元配 置分散分析に続いて、Tukey testと対応のない
t
検定を使用して実行した。p値は0.05
未満を 有意であるとした。11
【結果】
1. 心筋細胞肥大時にH3K9とH3K122のアセチル化は亢進した
心筋細胞肥大時にヒストンの
H3K122
のアセチル化が増加するかどうかは不明である。そ こで初代培養心筋細胞にフェニレフリン (PE) 刺激を行い、心筋細胞肥大を誘導し、H3K9
とH3K122
のアセチル化を検出した。免疫蛍光染色の結果、PE 刺激時に心筋細胞核内でH3K9
並びに
H3K122
のアセチル化が確認された (Figure. 1A)。次にウエスタンブロット (WB) 法の結果、H3K9と
H3K122
のアセチル化レベルがPE
刺激で増加した (Figure. 1B)。Figure 1. PE刺激は心筋細胞肥大を誘導し、ヒストンのアセチル化を亢進した
(A) 初代培養心筋細胞にPE刺激 (30 μM) を与え心筋細胞肥大を誘導した。刺激48時間後に抗 MHC 抗体、Hoechst、抗 Acetyl-Histone H3K9抗体及び抗 Acetyl-Histone H3K122抗体で蛍光免疫染色を行い、
細胞の写真を撮影した。(B) 刺激48時間後にヒストン画分を回収し、抗 Acetyl-Histone H3K9抗体、抗
Acetyl-Histone H3K122抗体及び抗 Histone H3抗体を用いてヒストンのアセチル化をWBで検出した。
12
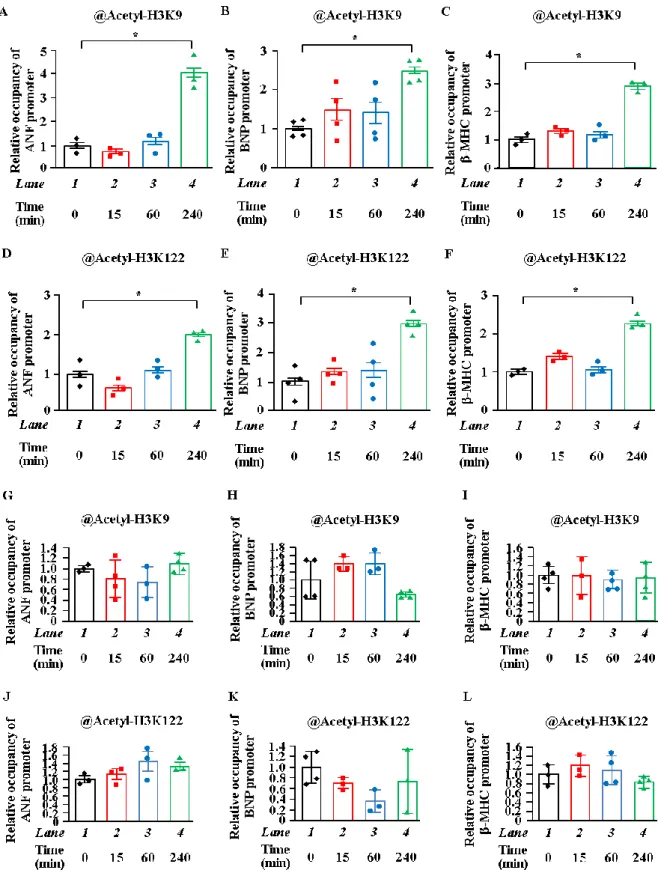
2. H3K9とH3K122のアセチル化は肥大反応因子プロモーター近辺で亢進した
H3K9
およびH3K122
のアセチル化がANF、BNP、-MHC
などの肥大応答因子遺伝子プロモーターで増加するかどうかを調べるために、クロマチン免疫沈降 (ChIP) アッセイを行っ た。 PE刺激に応答して
H3K9
およびH3K122
のアセチル化レベルは、PE刺激で240
分後にANF、BNP
および-MHCプロモーターで有意に増加した (Figure. 2A~F)。これらのプロモーターの上流領域における
H3K9
並びにH3K122
のアセチル化レベルは、PE
処理で240
分後に おいても変化しなかった (Figure. 2G~L)。まとめると、ヒストンのテールドメイン (H3K9) および球状ドメイン (H3K122) のアセチ ル化が心筋細胞肥大時に肥大応答遺伝子プロモーター近辺で誘導されることを示している。
13
Figure 2. PE刺激は心筋細胞肥大を誘導し、肥大反応因子プロモーター近辺でヒストンのア
セチル化を亢進した
(A~L) 初代培養心筋細胞にPE刺激0分、15分、60分、240分後、抗 Acetyl-H3K9抗体および抗 Acetyl-
H3K122 抗体にて ChIP アッセイを行った。(A~F) 肥大反応遺伝子プロモーターや (G~L) 肥大反応遺
伝子プロモーターの上流領域におけるヌクレオチドのレベルをqPCR法にて定量化した。
14
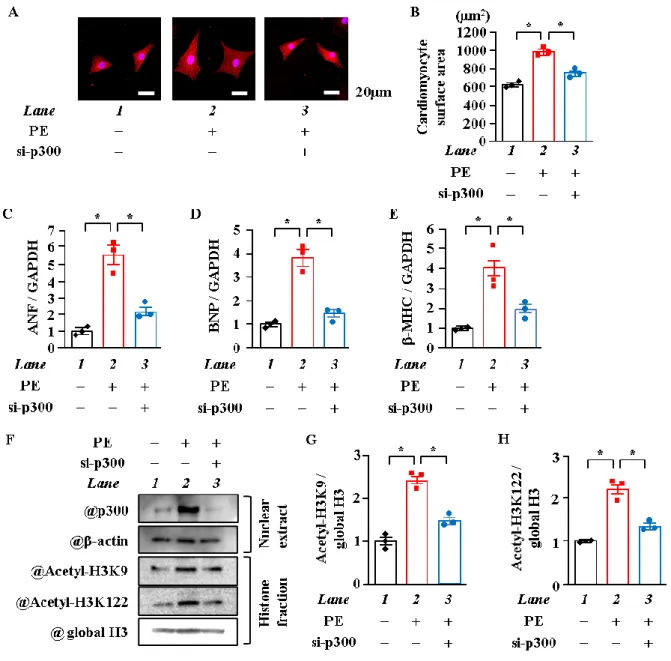
3. p300のノックダウンはヒストンのアセチル化及び心筋細胞肥大反応を抑制した
心筋細胞肥大時においてヒストンのテールドメインだけでなく球状ドメインのアセチル化 が亢進していた。しかし、心筋細胞肥大時にこれらのヒストンのアセチル化が
p300
によって 引き起こされているかどうかは不明である。心筋細胞核内においてp300
がこれらのヒストン をアセチル化していることを証明するために、初代培養心筋細胞でp300
のノックダウンを試 みた。心筋細胞面積を測定した結果、p300 のノックダウンはPE
刺激による心筋細胞面積の 増加を有意に抑制した (p < 0.05) (Figure. 3A, B)。RT-qPCRにて肥大反応因子のmRNA
を測定 したところ、PE刺激によって増加したANF、BNP
並びに-MHCのmRNA
レベルはp300
の ノックダウンによって有意に抑制された (p < 0.05) (Figure. 3C~E)。WBの結果、PE刺激によ りp300
のタンパク質レベルは増加するが、p300のノックダウンによりp300
のタンパク質レ ベルは有意に減少した。また、PE
刺激で亢進したヒストンのテールドメインの H3K9だけで なく球状ドメインのH3K122
のアセチル化は、p300 ノックダウンにより抑制された (Figure.3F~H)。
以上の結果から、p300をノックダウンすることで心筋細胞肥大反応の抑制とともに、ヒス トンのアセチル化が有意に減少した。
15
Figure 3. p300のノックダウンはPE刺激による心筋細胞肥大とヒストンのアセチル化を抑
制した
(A, B) 培養心筋細胞にsiRNA (各々50 nM) を導入し、2時間後にPE刺激 (30 μM) を与えた。刺激48 時間後に抗MHC抗体で免疫染色を行い、細胞の写真を撮影し無作為に選んだ細胞50 個の面積を
Image J 1.43 にて測定した。代表的な心筋細胞の写真 (A) と定量化したグラフ (B) を示す。(C~E)
PE刺激48時間後にmRNAを抽出し、ANF、BNP並びにβ-MHCのmRNAレベルをqPCRにより定 量化した。(F~H) PE刺激48時間後に核タンパク質及びヒストン画分を抽出し、抗 p300抗体、抗 - actin抗体、抗 Acetyl-Histone H3K9抗体、抗 Acetyl-Histone H3K122抗体及び抗 Histone H3抗体を用 いて検出した。代表的な図 (F)と定量化したグラフ (G, H) を示す。
16
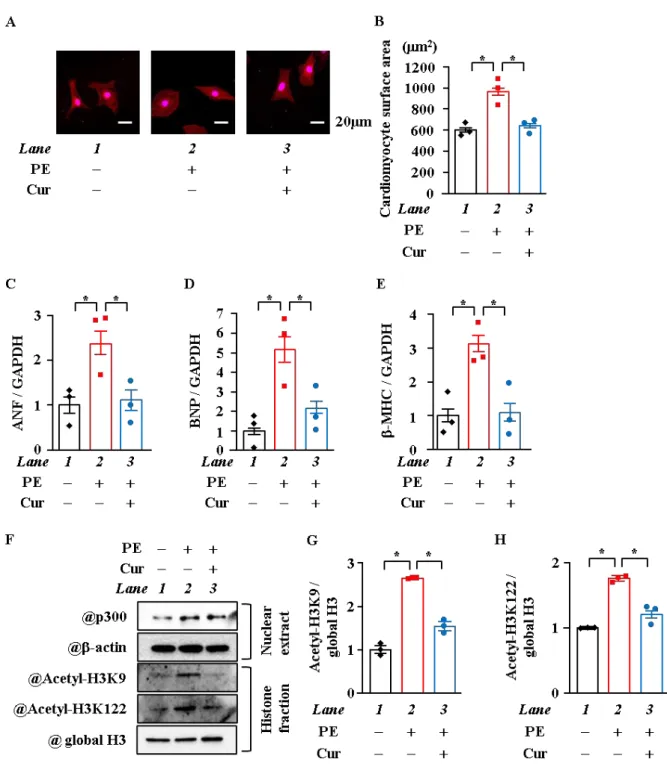
4. クルクミン処理はヒストンのアセチル化及び心筋細胞肥大反応を抑制した
p300
のHAT
阻害剤であるクルクミンを用いて、心筋細胞肥大時に起きているヒストンの アセチル化がp300
のHAT
活性によるものなのかを検討した。心筋細胞面積を測定した結 果、クルクミン処理はPE
刺激による心筋細胞面積を有意に抑制した (p < 0.05) (Figure.4A,B)。また、肥大反応因子の mRNA
をRT-qPCR
法にて測定した。クルクミン処理はPE
刺激によって増加した
ANF、BNP
並びに-MHCを抑制した (p < 0.05) (Figure. 4C~E)。ヒストン のアセチル化をWB
にて検討した結果、PE刺激によりp300
のタンパク質レベルの増加がみ られるが、クルクミン処理を加えても、そのp300
のタンパク質レベルの増加に変化は見ら れなかった。PE刺激はテールドメインのH3K9
と球状ドメインのH3K122
のアセチル化を 亢進するが、クルクミン処理を加えるとこれらのアセチル化は全て抑制された (Figure.4F~H)。
以上の結果から、クルクミンは
p300
のHAT
活性の阻害を介して心筋細胞肥大反応を抑制 するだけでなく、ヒストンのテールドメインや球状ドメインのアセチル化を阻害しているこ とが示唆された。17
Figure 4. クルクミン処理はPE刺激による心筋細胞肥大とヒストンのアセチル化を抑制した
(A, B) 培養心筋細胞にクルクミン (10 μM) 処理し、2時間後にPE刺激 (30 μM) を与えた。刺激48
時間後に抗β-MHC抗体で免疫染色を行い、細胞の写真を撮影し、無作為に選んだ細胞50 個の面積
をImage J 1.43にて測定した。代表的な写真 (A)と定量化したグラフ (B) を示す。(C~E) PE刺激48
時間後にmRNAを抽出し、ANF、BNP、β-MHCのmRNAレベルを測定した。(F~H) PE刺激48時間 後に核タンパク質とヒストン画分を抽出し、抗 p300抗体、抗 -actin抗体、抗 Acetyl-Histone H3K9 抗体、抗 Acetyl-Histone H3K122抗体及び抗 Histone H3抗体を用いて検出した。代表的な図 (F)と定 量化したグラフ (G, H) を示す。
18
5. 心筋細胞にp300を過剰発現させるとヒストンのアセチル化は亢進した
心筋細胞に
p300
を一過性に過剰発現させて、ヒストンのアセチル化の変化を検討した。初 代培養心筋細胞にpCMV-p300HA [12]
及びコントロールとしてpcDNA-null
をトランスフェ クションし、48時間培養した。WB
の結果、p300発現プラスミドの導入により、p300
の過剰 発現を確認し、p300の過剰発現はヒストンのテールドメインのH3K9
とともに球状ドメイン のH3K122
のアセチル化を亢進させた (Figure. 5A~C)。Figure 5. p300を心筋細胞に過剰発現するとヒストンのアセチル化は亢進した
(A~C) 初代培養心筋細胞に pCMV-p300HA および pcDNA-null をトランスフェクションし、培養 48
時間後に核タンパク質およびヒストン画分を抽出し、抗 p300抗体、抗 -actin抗体、抗 Acetyl-Histone H3K9 抗体、抗 Acetyl-Histone H3K122 抗体及び抗 Histone H3 抗体を用いてヒストンのアセチル化を WBで検出した。代表的な図 (A) と定量化したグラフ (B, C) を示す。
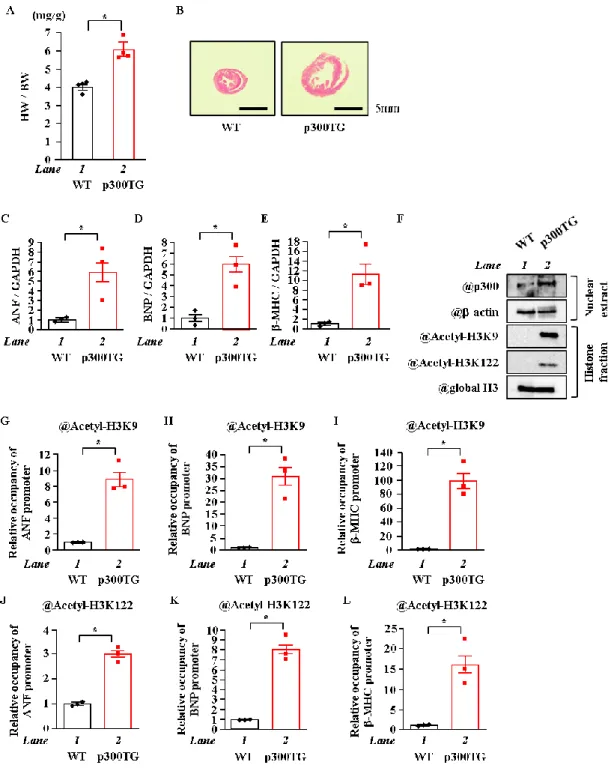
6. 心臓特異的p300過剰発現マウスにおけるヒストンのアセチル化の検討
初代培養心筋細胞を用いた
p300
のノックダウンや過剰発現によってH3K9
並びにH3K122
のアセチル化が変動したことから、これらのヒストンのアセチル化はp300
によって引き起 こされていることが示唆された。そこで、in vivoにおいてもp300
がこれらのヒストンのH3K9
とH3K122
のアセチル化を起こしているのかどうか検討を行った。26週齢のWT
マウス (N=4) 及び
p300-TG
マウス (N=4) の体重及び心臓重量を測定し、心体重比 (HW / BW,Heart weight per body weight)
の算出を行った。その結果、p300-TGマウスはWT
マウスと比 較して有意に心体重比が増加した (Figure. 6A)。また心臓を輪切りにし、ヘマトキシリン-エ オシン染色して心臓組織を観察したところ、p300-TGマウスではWT
マウスと比べて左室壁 の肥大及び左室腔の拡張を認めた (Figure. 6B)。次に摘出したこれらの心臓からANF、BNP
並びに-MHCといった肥大反応因子のmRNA
レベルをqPCR
にて測定した。その結果、WT
マウスと比べてp300-TG
マウスで有意に上昇した (p < 0.05) (Figure. 6C~E)。p300-TGマ ウスはWT
マウスと比べてp300
の発現量が増加し、それに伴いヒストンのテールドメイン のH3K9
や球状ドメインのH3K122
のアセチル化が大きく亢進していることが判明した(Figure. 6F)
。In vivo ChIPにより肥大反応遺伝子プロモーター上のH3K9
及びH3K122
のア19
セチル化を解析したところ、WTマウスと比べて
p300-TG
マウスでANF、BNP
や-MHCの プロモーター上のヒストンのテールドメインのH3K9
と球状ドメインのH3K122
のアセチル 化レベルがともに有意に亢進した (p < 0.05) (Figure. 6G~L)。以上の結果から、in vivoにおいても
p300
は肥大反応因子プロモーター上のヒストンのテー ルドメインや球状ドメインをアセチル化することが示唆された。20
Figure 6. 心臓特異的p300過剰発現はヒストンのアセチル化を亢進させた
(A, B) 26週齢の心臓特異的p300過剰発現 (p300TG) マウスおよびwild type (WT) マウス から心臓を 摘出し、重量を計測することで心体重比 (HW / BW) を算出した。摘出した心臓を輪切りにし、HE染 色で細胞質等を染色して細胞の写真を撮影した。(C~E) 各心臓からmRNAを抽出し、ANF、BNP並
びにβ-MHCのmRNAレベルを定量化した。(F) 心臓から核タンパク質およびヒストン画分を抽出
し、抗 p300抗体、抗 -actin抗体、抗 Acetyl-Histone H3K9抗体、抗 Acetyl-Histone H3K122抗体及 び抗 Histone H3抗体を用いてヒストンのアセチル化をWBで検出した。(G~L) 抗 Acetyl- Histone H3K9抗体および抗 Acetyl- Histone H3K122抗体にてin vivo クロマチン免疫沈降を行った。肥大反応 遺伝子プロモーター内のヌクレオチドのレベルをqPCR法にて定量化した。
21
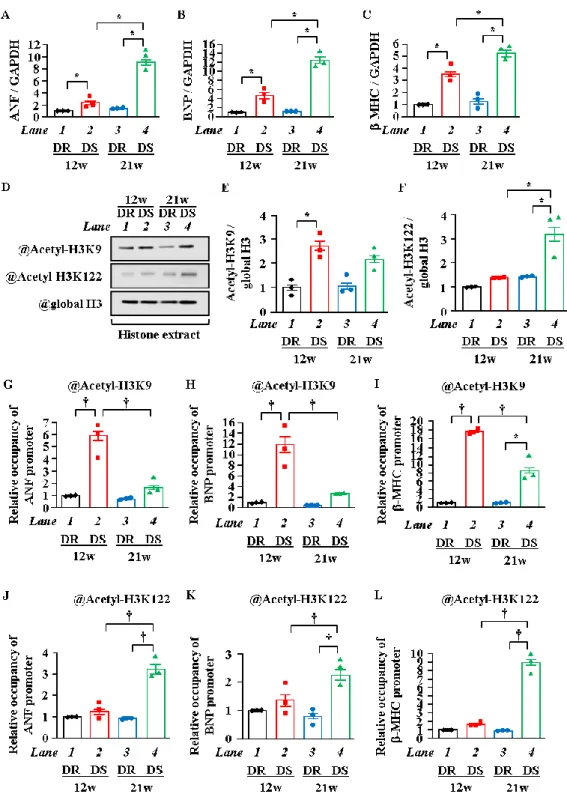
7. 心肥大期から心不全期にヒストンのアセチル化修飾部位が変遷した
ヒストンの
H3K9
とH3K122
は心筋細胞肥大時にp300
によってアセチル化されることがinvitroとin vivoの実験から明らかとなった。これらのヒストンのアセチル化によるエピジェネ
ティックな心筋細胞肥大反応の制御が実際の心肥大から心不全の移行における変化が見られ るのかどうか調べるため、高血圧心不全モデルラットであるダールラットを用いた。食塩感 受性ダールラット (DS, Dahl salt sensitive rat) 及び食塩抵抗性ダールラット (DR, Dahl salt
resistant rat)
で検討を行った。このDS
は6
週齢時から8%の高食塩を含む食餌を与えること
で著明な高血圧に至り、
12
週齢で心機能は維持されているが有意に心臓が肥大した心肥大期、21
週齢で左室収縮力の低下を伴った心不全期へと移行する。コントロールとして高食塩食を 与えても高血圧にならず心不全に至らないDR
を用いた。心臓超音波検査の結果、DS
は心肥 大期、心不全期で左室後壁厚の肥大がみられた。左室収縮能の指標であるFS (Fractional Shortening)
は、DS
の心不全期で低下していた (DS: 34.2 ±5.2%、DR: 57.1±4.0 %,
p < 0.05)。DS
は12
週齢で心肥大期、21週齢で心不全期あることを確認した (Table. 1)。Stage LVH HF
Parameter DR 12w DS 12w DR 21w DS 21w
LVPWT (mm) 2.2 ± 0.3 3.2 ± 0.3
*2.4 ± 0.2 3.4 ± 0.3
†LVIDd (mm) 6.3 ± 0.4 5.7 ± 0.6 7.1 ± 0.2 6.9 ± 0.5 LVIDs (mm) 2.7 ± 0.4 2.2 ± 0.6 3.1 ± 0.2 4.6 ± 0.4
†‡FS (%) 57.8 ± 5.5 62.9 ± 6.9 57.1 ± 4.0 34.2 ± 5.2
†‡Table1. ダールラットの心エコーのデータ
数値は平均±SEで表している。LVH: left ventricular hypertrophy, HF: heart failure, LVPWT: left ventricular posterior wall thickness, LVIDd: left ventricular internal dimension at end diastole, LVIDs: left ventricular internal diameter at end systole, FS: fractional shortening,N=3; two-way ANOVA followed by Tukey test. * p < 0.05 DR 12w vs DS 12w, † p < 0.05 DR 21w vs DS 21w, ‡ p < 0.05 DS 12w vs DS 21w.
これらの心臓を用いて肥大反応遺伝子である
ANF、BNP
及び-MHCのmRNA
レベルを測 定した結果、DR
と比べてDS
では心肥大期並びに心不全期で有意に増加した。さらに、DS
同 士においても心肥大期から心不全期にかけて有意に増加した (Figure. 7A~C)。WB
によりヒス トンのアセチル化を検討したところ、DS
ではDR
と比べてヒストンのテールドメインのH3K9
のアセチル化が心肥大期、球状ドメインのH3K122
のアセチル化が心不全期で亢進している ことが判明した (Figure. 7D~F)。次に、肥大反応因子のプロモーター上におけるヒストンのア セチル化レベルを検討するため、in vivo ChIPアッセイを行った。その結果、心肥大期ではDR
と比べて
DS
でANF、BNP
並びに-MHC のプロモーター上でヒストンのテールドメインである
H3K9
のアセチル化が有意に高値であった (p < 0.05)。ヒストンのテールドメインのH3K9
のアセチル化も認められるものの、心肥大期ほど高値ではなかった(Figure. 7G~I)。一方で、22
DR
と比べてDS
で心肥大期ではヒストンの球状ドメインのH3K122
のアセチル化は低値であ った。心不全期では、これらプロモーター上のヒストンの球状ドメインであるH3K122
のア セチル化が有意に高値であった(p < 0.05) (Figure. 7J~L)。以上の結果から、H3K9のアセチル化は心肥大期に著明に亢進し、
H3K122
のアセチル化は 心不全期に亢進し、心肥大期から心不全期にかけてヒストンのアセチル化修飾部位が変化す ることが示唆された。23
Figure 7. 高血圧性心不全モデルであるダールラットは肥大反応因子プロモーター上で
H3K9とH3K122のアセチル化レベルが亢進していた
(A~C) 各心臓からmRNAを抽出し、ANF、BNP並びにβ-MHCのmRNAレベルを定量化した。
(D~F) 核タンパク質およびヒストン画分は、12週齢または21週齢のDRおよびDSの心臓から抽出
した。 代表的な図 (D)と定量化したグラフ (E, F) を示す。(G~L) 抗 Acetyl-Histone H3K9抗体およ び抗 Acetyl-Histone H3K122抗体にてin vivo クロマチン免疫沈降を行った。ANF、BNP及び-MHC のプロモーターのヌクレオチドのレベルをqPCR法にて定量化した。N=3, * p <0.05, †p <0.01
24
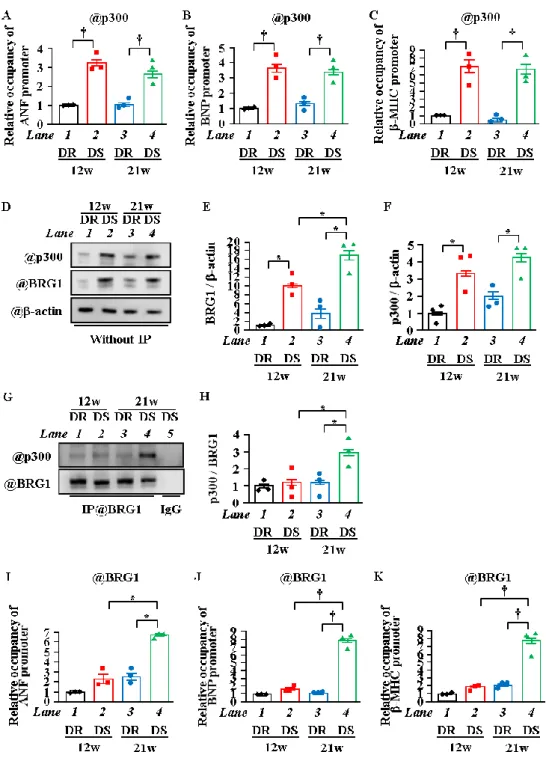
8. p300とBRG1の結合およびBRG1の肥大反応因子プロモーターへのリクルートは心不全期に増 加した
これまでの結果から心肥大期から心不全期への移行に伴って起こる肥大反応遺伝子の転写 の亢進が、ヒストンのアセチル化修飾部位の変化に関連していることが示された。肥大反応 遺伝子プロモーターへの
p300
のリクルートの変化がヒストンアセチル化ドメインの変化に 影響を与えるかどうかを検討するために、in vivo ChIPアッセイを実施した。その結果、ANF、BNP、および-MHC
プロモーターにリクルートされたp300
の量がそれぞれ12w
および
21w
のDR
と比較して、12wおよび21w
のDS
で有意に増加していることを示した。しか し、リクルートされたp300
の量は、12wのDS
と21w
のDS
で同程度であった (Figure.8A~C)。すなわち、心肥大期と心不全期の間でヒストンのアセチル化部位の変化が肥大反応
遺伝子プロモーターへのp300
のリクルートに依存していないことを示唆している。したが って、このヒストンのアセチル化修飾部位の変化はp300
に結合する他の因子によって調節 されている可能性が考えられた。そこで関与する可能性のある1
つの因子としてクロマチン リモデリング因子のBRG1
に着目した。BRG1はSWI / SNF
クロマチンリモデリング複合体 の触媒サブユニットであり、転写のアップレギュレーション中にヒストンのアセチル化(H3K27)
においてp300
と協調的に働くことが報告されている [38]。そこで心不全期にBRG1
とp300
の協調的に働いているかどうかを調べるために、心肥大期と心不全期の心臓を用いて
IP-WB
を実施した。その結果、Without IPではDR
と比較してDS
の12w
と21w
でp300
とBRG1
のタンパク質レベルは増加した (Figure. 8D~F)。p300とBRG1
の結合は21w
のDR
と比較して21w
のDS
で増加したが、12wのDS
では増加しなかった (Figure. 8G,H)。次に心肥大期から心不全期への移行中に、BRG1
がヒストンのテールドメインから球状ドメインへとアセチル化修飾部位の変化に関連するかどうかを検討するために、in vivo ChIP アッセイを実施した。その結果、ANF、BNP、および-MHCプロモーターへの
BRG1
のリ クルートは21w
のDR
と比較して21w
のDS
で増加したが、12wのDS
では増加しなかった(Figure. 8I~K)。
以上の結果から、BRG1と
p300
の結合およびBRG1
の肥大反応遺伝子上へのリクルート は心不全期に亢進することが示された。25
Figure 8. 心不全期にBRG1の肥大反応遺伝子プロモーターへのリクルートは増加した
(A-C) In vivo ChIPアッセイは、12週齢または21週齢のDRおよびDSの心臓から抽出し、抗p300抗 体にて行った。ANF、BNP並びに-MHCのプロモーターのヌクレオチドのレベルをqPCR法にて定量
化した。(D~F) 核タンパク質は12週齢または21週齢のDRまたはDSの心臓から抽出した。代表的な
図 (D)と定量化したグラフ (E, F) を示す。(G, H) ダールラット心臓溶解物を、対照ウサギIgGまたは 抗BRG1抗体のいずれかを免疫沈降に使用した。 p300およびBRG1に対してWBを行った。代表的 な図 (G) と定量化したグラフ (H) を示す。(I~K) 抗 BRG1抗体にてin vivo ChIPアッセイを行った。
ANF、BNPおよび-MHCのプロモーターのヌクレオチドのレベルをqPCR法にて定量化した。N=3、
* p <0.05、†p <0.01
26
【考察】
心不全発症に関するエピジェネティックな制御機構については盛んに研究されている [39-
41]。本研究では高血圧性心不全モデルであるダールラットを用いた検討において、ヒストン
のテールドメインであるH3K9
のアセチル化は心肥大期から起きていたがヒストンの球状ド メインであるH3K122
のアセチル化は心不全期にはじめて起きることを見出した。すなわち、心肥大から心不全への移行にかけて起きるヒストンのテールドメインと球状ドメインの段階 的アセチル化修飾による転写制御機構という新たなモデルを提唱する。
心不全発症に関するエピジェネティックな制御機構として以下の仮説を考える。まず、高 血圧などの心臓への負荷により様々な情報伝達経路を介して
p300
が活性化し、クロマチンへ とリクルートされヒストンのテールドメインがアセチル化される。その結果、クロマチンリ モデリングが起き肥大反応因子の転写が亢進し心肥大期へと至る。次に、活性化したp300
とBRG1
の結合が増加し、BRG1 がDNA
へリクルートされる。その結果、リクルートされたBRG1
によりp300
のHAT
部位がヒストンとDNA
との間に入り込みH3K122
がアセチル化さ れる。このH3K122
のアセチル化によりDNA
とヒストンとの親和性が減弱しヌクレオソーム 構造が緩むことでヒストンがDNA
上を移動しやすくなり、DNAがより露出することで転写 因子を含む巨大な複合体がDNA
へリクルートされやすくなる。このヌクレオソーム構造の 変化によって心不全発症に関連する遺伝子の転写が心肥大期よりも著明に亢進したと考えら れる (Figure. 9)。Figure 9. 心肥大から心不全へのヒストンのアセチル化状態を示すモデル
赤
Ac:H3K9ac、青 Ac:H3K122ac
ヒストンの翻訳後修飾は、転写、複製および
DNA
修復などの細胞プロセスに関与してお り、これらの細胞プロセスはクロマチンリモデリングなどのDNA
へのアクセス性の向上を 伴う [42]。すなわち、これまでに報告された翻訳後修飾のほとんどは、ヒストンのテールド メインに位置し、転写制御因子などをヒストンへ誘導することによってクロマチン構造に間 接的に影響を及ぼしている。H3K122
は、ヒストンの球状ドメインに見られ、ヌクレオソーム 構造中でヒストンのDNA
結合表面に特異的に位置する。Schneider らの研究によりH3K122
がp300
によりアセチル化されることでDNA
とヒストンとの親和性が低下し、ヌクレオソー27
ムの構造変化が起きることが明らかとなっている [33]。この
H3K122
のアセチル化はヌクレ オソーム構造の変化に対する直接的な影響を示唆する。すなわち、ヒストンのテールドメイ ンと球状ドメインにおける翻訳後修飾が異なる調節機構によって転写を制御している [32,33, 43]。Fenley
らはヒストンの球状ドメインにおける一つのリジン残基がアセチル化されるだけでも
DNA
とヒストンとの相互作用の減少に大きな影響もたらすことを示している [44]。一方、ヒストンのテールドメインのアセチル化はヌクレオソーム構造の変化に影響を与えな いと考えられている [32]。さらに、H3K122 のアセチル化は発現が亢進している遺伝子プロ モーターおよびエンハンサー領域において頻繁に起きており、これらの領域における
H3K122
のアセチル化レベルは遺伝子発現のレベルに正比例することも報告されている [33]。本研究において
ANF、BNP
や-MHC のmRNA
が心肥大期よりも心不全期で有意に増加していた。これらの遺伝子プロモーター領域において
H3K122
のアセチル化が生じることで爆発的な転 写亢進が起き、これらの遺伝子発現レベルの上昇に関与していることが示唆される。ヒスト ンのアセチル化は心不全の遺伝子発現変化における情報伝達経路において最終の共通経路で あると考えられ、本研究の結果からH3K122
のアセチル化が心不全発症の重要なトリガーの 一つであることが示唆された。BRG1
は、クロマチンリモデリング因子でありSWI / SNF
複合体の主要なタンパク質でATP
依存的にヌクレオソーム構造を緩め、遺伝子の転写を制御している。BRG1
はGATA4、 Nkx2- 5、及び TBX5
などの心臓特異的転写因子と相互作用することで心筋細胞の発生・分化を調節 している [45]。このBRG1
は胎仔期に高発現されているが新生仔の心臓ではダウンレギュレ ーションされており、ヒト心筋症においてBRG1
並びに-MHC の高発現が確認されている。また、BRG1コンディショナルノックアウトマウスでは
TAC
手術による心不全が抑制されて いる [46]。さらに胚性幹細胞においてBRG1
とp300
はブロモドメインタンパク質のBrd4
を介して
H3K27
とH3K56
をアセチル化し、転写を調節することが報告されている [38]。しかし、心肥大期や心不全期における
BRG1
とp300
の直接結合や協調性に関しては全く知られて いない。本研究によりBRG1
とp300
の結合は心肥大期から心不全期に増加すること、BRG1 の肥大反応遺伝子プロモーター上へのリクルートも同様に心肥大期から心不全期で増加する ことが見出された。心筋細胞においてp300
は自己アセチル化を起こしプロテオソーム分解に 対して耐性が増加することで安定化し、結果的にHAT
活性が上昇し [47]、心不全時にBRG1
のブロモドメインがp300
の自己アセチル化部位を認識することで直接的に結合する可能性 が考えられる。また、p300とBRG1
は様々なタンパク質と結合し巨大な複合体を形成するこ と [48, 49]、BRG1とp300
はBrd4
を介して協調的に働くことでヒストンH3
のアセチル化や クロマチンリモデリングを促進し、転写を亢進させることも報告されていることから[38]、心 不全時にBRG1
とp300
はBrd4
などの別のタンパク質を介して間接的に結合することで転写 を調整している可能性も考えられる。ヒストンの翻訳後修飾には、アセチル化以外にもリン酸化、ユビキチン化やメチル化等が 挙げられる[50-52]。これらの翻訳後修飾がヒストンの球状ドメインのアセチル化を制御して いる可能性も考えられる。なかでも、ヒストンのメチル化については、リジンやアルギニン といった残基がモノメチル化、ジメチル化やトリメチル化されることが知られている [53, 54]。