FMO
計算に基づくマルチスケールシミュ レーション手法の開発と先導的応用
Development and Application of Multiscale Simulation Method Based on FMO Calculations
立教大学大学院 理学研究科 化学専攻 博士課程後期課程
奥脇弘次
i
目次
概要 ... 1
粗視化シミュレーションパラメータ算定スキームの構築 ... 4
緒言 ... 4
パラメータ算定法 ... 6
従来法 ... 6
新規手法 ... 8
信頼性の評価:テスト計算 ... 12
結果と考察 ... 14
Hexane-Nitrobenzene(溶媒-溶媒)系 ... 14
Polyisobuthylene-Diisobuthyl Ketone(ポリマー-溶媒系) ... 19
Polyisoprene –Polystyrene(ポリマー-ポリマー系) ... 20
汎用的システムの構築と公開 ... 22
FMO-DPD法を用いた応用計算 ... 24
緒言 ... 24
方法論 ... 24
DPDシミュレーション ... 24
Poisson-Boltzmann方程式による溶媒効果計算 ... 26
高分子電解質膜のパーコレーション解析 ... 28
Simulation details ... 30
結果と考察 ... 33
イオン系での検証 ... 46
脂質膜の解析 ... 50
Simulation details ... 50
結果と考察 ... 52
混合膜での検証 ... 57
リバースマップによる詳細解析 ... 60
緒言 ... 60
ii
リバースマップの実行スキーム ... 60
リバースマッププログラム ... 60
FMO用インプットの作成 ... 61
モデル設定 ... 61
結果 ... 63
パラメータ算定手法の改良の試み ... 66
機械学習手法を用いたパラメータ算定の効率化 ... 66
多体を用いたパラメータ算定 ... 68
パラメータ算定手順 ... 68
システム化 ... 69
実証計算 ... 70
結果 ... 71
同種粒子のパラメータの再定義 ... 71
小分子系での等温圧縮率の再現 ... 71
タンパク質の折り畳み ... 73
研究総括 ... 77
謝辞 ... 78
参考論文 ... 79
参考文献 ... 81
図目次 ... 94
表目次 ... 96
1
概要
本論文では、フラグメント分子軌道(FMO)法と粗視化シミュレーションを連 携した、マルチスケールシミュレーション手法の開発の試みについて報告する。
近年、材料開発や創薬の分野において、分子レベルで構造を制御した高度な製品 の開発が期待されており、開発の効率化のために、計算化学による物性の予測の 重要性が高まっている。そうした中、物性に関してメゾ領域の分子集団の分布が 大きく関わることが知られているために、散逸粒子動力学(DPD)といった、原子 集団を 1 つの粒子として粗視化する技術を用いたシミュレーションが注目を浴 びている。しかし、シミュレーションの際に必須となる、粒子間の相互作用を表 すパラメータ(χ)の精確な算定方法が確立しておらず、大きな課題となっていた。
そこで、FMO法による電子レベルの高精度計算を用いてパラメータを算出する ことで、精度の高いシミュレーション、解析手法(FMO-DPD 法)を確立した。
また、このワークフローをシステム化し、半自動でパラメータを算定できるプロ グラムとして無償で公開を行った。更に、構築した手法を用いた先導的な応用計 算では、高分子材料や生体分子について実験の物性値を多くの系で再現した他、
DPD の結果を再原子化し、FMO による解析を行うシステムを開発したことで、
ナノ(原子)-メゾ(粗視化)双方向の連携が可能となった。
本論文は6章の構成でなっている。本章では概要を述べ、第2章ではFMO法 を用いた新規パラメータ算出手法の詳細について説明する。第 3 章は確立した
FMO-DPD計算を用いた応用計算であり、高分子電解質膜、脂質膜を扱った。第
4 章では粗視化構造を再原子化するリバースマップ手法を用い、FMO により詳 細解析を行う手法の開発について報告する。第 5 章ではパラメータ算定手法の 更なる改良について触れ、機械学習を連携した算定効率化や、高精度計算に向け た手法の更なる改善のほか、DPD で用いるパラメータ自体の見直しや、タンパ ク質の折り畳みにも言及した。第6章は総括となっている。
パラメータ算定手法の改良では、高分子を構成する基本単位間の相互作用エ ネルギーから算出する手法1に着目し、相互作用エネルギーをFMO法で算定す ることで、汎用的かつ高精度の解析手法を確立した。更に系の異方性を評価する ことで、一連の評価法の精度を改良した。その結果、ヘキサンーニトロベンゼン といった単純な二成分混合系での相転移上部臨界温度が実験値と良好な一致を 示した。
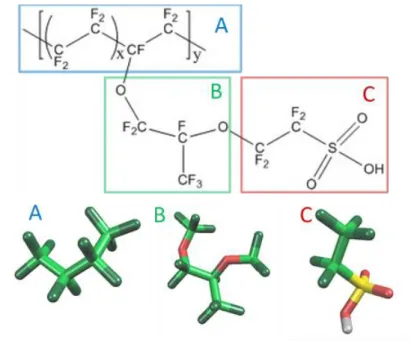
高分子電解質膜での応用計算では、フッ素系骨格の Nafionと芳香族炭化水素
骨格のSPEEKを対象に、電解質膜内の水クラスタの連結挙動についての解析を
2 行った。その結果、クラスタサイズ等の構造指標値について実験と良好な一致を 示した上、水クラスタの連結指標の傾向が H+ 伝導度の実測値と一致した。更
に、Nafionの末端スルホン酸がイオン化したモデルを併せて扱い、シミュレーシ
ョンを行った。その際、正電荷を持つ水粒子モデルと、ニュートラルな水粒子モ デルの 2 種類を扱ったところ、正電荷水粒子が他の水粒子を囲む形で電離スル ホン酸と強く結びつく様子が観察された。また、Nafionの電離度を変更して複数 検討したところ、すべてのスルホン酸粒子が電離した際に、少ない含有量でクラ スタの連結が起こるという結果が得られた。
脂質膜については、代表的なリン脂質である POPC (1-palmitoyl-2-oleoyl-sn- glycero-3-phosphocholine)を対象に構造再現を試みたところ、脂質濃度に応じてベ シクル,二重膜構造が得られ、構造の膜面積,膜厚が実験値と良好な対応を示し た.また, ドラッグデリバリーシステム (DDS) へ向け、siRNAキャリアーとし てのリポソームを意図した正電荷脂質、リン脂質の混合膜についての構造検証 も行った。飽和リン脂質DPPCと, 不飽和正電荷脂質DOTAPについてパラメー タを算定し,混合・濃度の条件を変えながらDPDシミュレーションを行うこと で、膜が扁球状になるという知見が得られ、X 線小角散乱(SAXS)の結果と一致 した。
原子レベルの計算からボトムアップで粗視化シミュレーションにつなげるだ けでなく、DPD の結果に対し原子を割り当てるリバースマップの手法を用い、
FMOに繋げることで原子、電子レベルでの詳細解析を行うシステムを構築した。
脂質膜,高分子電解質膜について小規模のDPDモデルを作成し、実行したとこ ろ,スルホン酸-水,リン酸-水間の界面に強い水素結合が確認された。
更なる手法の改良として、まず機械学習を連携した算定効率化を試みた。単純 な分子系において回帰分析のモデルを組んだところ、距離情報から一定の精度 での相互作用の予測に成功した。精度方面の改良としては、2章の計算への多体 効果の盛り込みを行った。分子動力学(MD)を用いた多体配置構造から直接パ ラメータを算定するスキームを構築したところ、水-シクロヘキサン、水-ベンゼ ンの系において、界面張力を実験値と非常に近い値で再現した。また、DPD で 用いる同種粒子間パラメータの見直しにも着手し、セグメント間の相互作用か らパラメータの元となる等温圧縮率を現実的なオーダーで予測することができ た。更にこの試みをタンパク質に適用し、10 残基からなる最小のタンパク質で
あるChignolin の折り畳みを実行したところ、骨格がヘアピンカーブを描く構造
の再現に成功し、水素結合やCH/πといった、特徴的な残基の相互作用からなる 構造も確認された。
3 種々の応用計算が示すように、精度の高い分子軌道計算と粗視化シミュレー ション手法を連携する FMO-DPD 法は汎用的に物性を予測する設計ツールとし て大きな可能性を持っているといえる。
4
粗視化シミュレーションパラメータ 算定スキームの構築
緒言
粗視化シミュレーションは、分子動力学(MD)などの原子レベルのシミュレー ションと比較して、大規模で大きな時間スケールを処理することができるため、
大規模構造の解析に広く用いられる。粗視化手法の中でも、一般的な MD を粗 視化した概念である粗視化 MD と比べ、系内の成分の濃度の時間発展を計算す る動的平均場理論2,3や、流体の理論を取り込んだ散逸粒子動力学(DPD)4–9は、
相分離構造の予測に優れている。中でもDPDは粒子の扱いを保った上で相変化 挙動を低コストで追うことができるために、応用例は高分子電解質膜のメゾ構 造予測、両親媒性分子の小胞形成10,11や線状オリゴマー溶液の構造の解析12、両 親媒性二重層の平衡構造の解析 13やタンパク質や DNA の構造作成など多岐に 渡る。
平均場法やDPDシミュレーションでは、設定した粒子間の相互作用を記述す るパラメータが重要となり、これらは一般的にFlory-Huggins理論の成分間の親 和性を示すχパラメータに密接に関連している14,15。しかしながら、信頼できる χパラメータの評価は困難な作業であることがよく知られており、シミュレーシ ョンには実験値を元にした値や経験的な値が一般的に用いられていたため、適 用の限界が指摘されていた。
χ の予測方法の代表的なものは主に二つある。一つが、溶解度パラメータ SP 値からの予測である。SP値はHildebrand16によって考案され、原子団寄与モデル
17,18、bicerano19 の手法など様々な経験的な推算モデルが考案、検証されている。
単一分子の分子シミュレーションからSP値の予測する方法もある20。二つ目に、
異種分子間の相互作用からχ値を求める方法がある。代表的なものが、Fanらに よるセグメント間の接触エネルギーからの予測や1、凝集体モデルの凝集エネル ギーの差からの予測21である。本研究では、Fanの手法に着目した。この手法は 接触する粒子間の相互作用を直接分子論的に求めることができる利点がある。
Fan らの手法はセグメント対の網羅的な配座について相互作用計算を行った 後、メトロポリス法により温度ごとに取りうる状態を選択し、セグメント間の接 触エネルギーを求めている。しかし、古典力場(MM)のパラメータを対の接触 エネルギーの評価に使用していたために、相互作用の要因として電荷の移動が 本質的である系では、信頼性が低下することが知られていた。
5 上記の問題を解決するためには、相互作用エネルギーの評価において、分極や 電荷移動を考慮した分子軌道(MO)計算や密度半関数(DFT)計算を用い、第 一原理計算の手続きを行う必要がある。これらの例として、Sepehr らは2015年 に、DFT法に基づいて、高分子電解質膜として代表的なNafionの水和モデルの DPD相互作用パラメータを評価した22。Nafionは電荷移動を伴い、古典的なMM の枠組みでは適切な描写が難しいとされており、この試みは非常に有効である。
しかし、セグメント対の分子サイズが大きくなった際に通常の第一原理計算を パラメータ評価に適用することは多くの計算コストがかかり、汎用的なスキー ムとして構築するのは難しい場合がある。
そこで、本章では、FMO法を用いてχパラメータを算定する新規手法を提案 する23。 FMO法は、高度に並列した実行によって、大規模分子(例えば、タン パク質および分子クラスター)に化学的信頼性を保った上で適用可能な効率的 な MO のフレームワークとして知られている。フラグメント間の相互作用エネ ルギーのリスト(inter-fragment interaction energy (IFIE) やpair interaction energy
(PIE))と呼ばれる)が、FMO計算と同時に得られる。FMOのこれらの特徴は、
後述するように、パラメータ算定にとって都合がよい。
本章では、2節でχパラメータの算出法の詳細に触れ、3, 4節ではこの新規手 法の信頼性の検証のために、Fan らの先行研究 1 で例示されている分子系三例 (Hexane-Nitrobenzene、Polyisobutylene-Diisobutyl Ketone、Polystyrene-Polyisoprene) に対し行ったテスト計算と結果ついて報告する。5節では、確立した手法のシス テム化と公開について説明する。
6 パラメータ算定法
従来法
Flory-Hugginsの格子理論において、2成分混合系の自由エネルギー変化ΔGは
式(1)で示される24。
∆𝐺 𝑅𝑇= 𝜑1
x1 ln𝜑1+𝜑2
x2ln𝜑2+ 𝜒𝜑1𝜑2 (1) ここで、φは体積分率、xi(i = 1,2)は鎖長である。 χパラメータは次のように 定義される。
𝜒 = 𝑍𝛥𝐸12
𝑅𝑇 (2)
𝑍はモデル格子の配位数であり、接触エネルギー𝛥𝐸12は次の式で与えられる。
∆𝐸12= 𝐸12−1
2(𝐸11+ 𝐸12) (3)
𝐸𝑖𝑗は成分iとjとの間の相互作用エネルギーであり、𝛥𝐸12は混合により得られる エネルギー量を表している。この関係は、メソスケールの事象をナノスケールに 落とし込むことを意味している。Fanらは分子シミュレーションに基づいて𝑍と
∆𝐸12を計算する手順を提案した 1。それにより、理論計算のみにより合理的な χ 値を計算することが可能となった。Fanらの手続き1の手順を以下に示す。
(i) 2分子接触構造の作成: パラメータを決めたいモノマーの小分子構造をセ
グメントとして作成する。これらの構造は、分子力場を用いて最適化を行う。
(ii) セグメントペアの配置の作成と相互作用エネルギーの計算: 各セグメン
ト対について、セグメント間の相互作用エネルギーを多数(数千単位の)配置に 対して計算する。配置生成の方法を以下に、イメージ図をFigure 1に示す。
(ii-1) 両セグメント分子の幾何中心を、Cartesian座標中心に配置する。
(ii-2) セグメント2の回転および並進方向を、3つのオイラー角および原点か
ら単位球の表面までのベクトルをランダムに選択することによって決定する
(セグメント1は固定する)。
(ii-3) セグメント2を、各分子のvan der Waals表面がちょうど互いに接触する
まで、ステップii-2により決定された角度、ベクトルに従って回転、移動する。
(ii-4) 上記の手続きで生成された配置の受け入れを、Metropolis MC 法に従い
判断する25。u𝑣𝑣′は、系が状態𝑣から、状態𝑣′に遷移する確率として定義すると、
u𝑣𝑣′は次のように与えられる。
u𝑣𝑣′ = exp (− ∆E𝑣𝑣′
𝑅𝑇 ) (4)
7 ここで、∆E𝑣𝑣′は、状態𝑣と𝑣′との間のエネルギー差である。Metropolis 法の手 続きにより採用された構造のエネルギーの平均値を、各セグメント対の相互作 用エネルギー (𝐸11、𝐸12、および𝐸22) として定義する。
(iii) 配位数𝑍の計算: Flory-Huggins理論では、式(2)に示すように、セグメン
トに隣接するセグメントの数(配位数𝑍)と、上記の相互作用エネルギー𝐸𝑖𝑗を掛 け合わせることでセグメントあたりの相互作用を見積もっており、配位数𝑍の値 が𝜒パラメータに大きな影響を与える。Fan らの先行研究 1では、上記の配座生 成ステップの (ii-2) と (ii-3) を繰り返すことにより、中心セグメントの周りに 配置可能なセグメントの数を求めている。分子ラベル 1 と 2 で示された二成分 混合の際、中心分子、周囲の分子にそれぞれセグメント 1, 2 であることが考え られるため、4つの異なる組み合わせが考えられる(1-1, 1-2, 2-1および2-2と記
載)。Flory-Huggins理論ではセグメントはすべて同じ大きさの球と仮定されてい
るため、4種類の配置の平均を配位数𝑍としている。生成されたセグメント構造 からのパラメータ計算のフローをFigure 2に示した。
Figure 1. 構造生成のイメージ図。青、赤色の球はそれぞれセグメント1, セグメ
ント2を示す。
8
Figure 2. 生成されたセグメントの構成からのパラメータ計算のフロー。青、赤
色の球はそれぞれ、セグメント 1 およびセグメント 2 を表す。平均相互作用エ ネルギー𝑬𝒊𝒋は生成された構成に対して Metropolis MC 法を用いて計算される。
〇および×の記号は MC サンプリングにおける採用、不採用を示す。得られた 平均エネルギーに配位数𝒁𝒊𝒋が乗算される。
新規手法
本研究で提案する新規手法では、式(3)で示されるペアエネルギー𝐸𝑖𝑗を、FMO
法23,26によって評価している。手続きの詳細は以下の通りである。
(i) FMOに基づく相互作用エネルギー: 一般的なFMO計算は以下のように要
約される23。任意の対象分子系を、モノマーと呼ばれる適切なフラグメントに分
割し、 Hartree-Fock(HF)の手順を使用して、単量体段階で環境静電ポテンシャ
ル(ESP)下の各フラグメントのMO計算を行う。フラグメント単量体の大反復 は、self-consistent-charge(SCC)が満足されるまで繰り返される。フラグメント 内処理は並列化され、モノマーリストの手続きも並列処理(または 2 層並列処 理)される。単量体のSCCの反復が完了すると、単量体の場合と同様の並列性 を有する単量体 SCC の ESP 下で、ダイマー(隣接したモノマーからからなる)
の一連のHF計算が実施される。分極および電荷移動は、モノマー段階およびダ イマー段階でそれぞれ考慮され、全電子エネルギーEは、以下の簡単な式によっ て得られる。
9 𝐸 = ∑ 𝐸𝐼𝐽− (𝑁f− 2)
𝐼>𝐽
∑ 𝐸𝐼
𝐼
(5) ここで、𝐸𝐼はモノマーIのエネルギー(𝑁𝑓はモノマーの数)であり、𝐸𝐼𝐽はダイマ ーIJのエネルギーである。 2次のMoller-Plesset摂動論(MP2)などの電子相関 によるエネルギー補正を加算的に組み込むことができる。また、式(5)は、フラグ メント相互作用エネルギー(IFIE)の合計で書き直すことができる。
𝐸 = ∑ ∆𝐸̃𝐼𝐽+
𝐼>𝐽
∑ 𝐸𝐼′
𝐼
(6) 𝐸𝐼′ はESPの寄与を除いた架空のモノマーエネルギーである。IFIE値は、計算対 象系におけるフラグメント相互作用の性質を分析するのに有用な値 23 であり、
本研究の手法における式(3)のセグメント対の相互作用エネルギーとして使用す ることができる。
IFIEは詳細解析のために、PIEDA (Pair Interaction Energy Decomposition Analysis)
27,28を用いて、静電相互作用(ES)、交換反発(EX)、分散寄与(DI)、電荷移動
他(CT +mix) に分解することができる。ES、EXおよびCT +mix項はHFレベ ルで評価され、DI項は通常MP2計算によって得られる。 PIEDAは古典的なMM とFMOとの間のエネルギー評価の違いを解析するために有益である。
(ii) 非結合原子間の距離の再評価: Fanらは、対象となるセグメント対の配置
の作成の際、原子のvan der Waals半径のセットを使用した。しかしながら、水 素結合のような、より密接な接触が必要である特定の相互作用が描写できない といった問題が生じる。そこで本研究では、各原子対における非結合原子間距離 パラメータを、一連の量子化学計算に基づいて再評価した。
(iii) 周囲の環境の評価: 本手法では、Metropolis 法による採用した配置のエ
ネルギーの平均値をセグメント対当たりの平均相互作用としている。次いで、対 応する相互作用エネルギーにそれぞれの配位数を乗じることによって、周囲の 場との相互作用エネルギーの合計が計算される。配位数は空間充填モデルから のみ計算される。セグメント対の特定の部分が強い安定化を示す場合、メトロポ リスアルゴリズムにおいてサンプリングの潜在的な偏りが生じる。そのため、周 囲の場との相互作用エネルギーの合計を過大評価する傾向がある。そのような 過大評価を検証するために、中心セグメントの重心から周囲の空間をFigure 3に 示すような6つの領域に分割し、モンテカルロ計算の結果から、各領域 i(= 1- 6)の重みを以下の式で計算した。
𝑑𝑖 = 𝐴𝑐𝑖
𝐴𝑐𝑚𝑎𝑥 (7)
10 ここで、𝐴𝑐𝑖は領域iでの採用数、𝐴𝑐𝑚𝑎𝑥は採用数が最大の領域での採用数である。
採用数の差は以下のように自由エネルギーの差に相当する。
−𝑙𝑛 ( 𝐴𝑐𝑖
𝐴𝑐𝑚𝑎𝑥) ≅ ∆𝐺 (8)
しかし、Flory-Huggins 理論では、セグメント分子は等方的であり、この方程式 の ΔG はゼロでなければならない。そこで、系の等方性を表すパラメータとし て、以下のSを採用した。
𝑆 = ∑𝑑𝑖
𝑖 𝑁
(9)
ここで、𝑁は領域の数である。もし、系が完全に等方的であれば、𝑑𝑖はすべての 領域において同じ数になり、𝑆は 1 であり、異方的であるほど値は小さくなる。
このことから、S値を等方性の指標として扱った。S値も空間の分割に依存する ため、空間の分割を変え100種類S 値を計算し、平均した値をScaling factorと して後の手続きに使用した。
(iv) 𝐸𝑖𝑗, 𝑍の計算とχの算出: Metropolis法では、サンプル数が少ないと信頼
性が十分ではない。しかし、力場計算と比較してFMO計算を実行するには、多 くの計算時間がかかる。そこで、本研究では、セグメントペアごとに2,000個の 配置を生成し、Metropolis法を使用して10,000回の採用を実施した。その上で、
MC シミュレーションにおける平均エネルギーを相互作用エネルギー𝐸𝑖𝑗として 用いた。配位数の評価は、各セグメントの組み合わせ(セグメント1-1、セグメ
ント1-2、セグメント2-1、およびセグメント2-2)に適用し、結果を配位数𝑍𝑖𝑗と
して使用した。中心セグメントがポリマーである場合には、連結部分の排除体積 を考慮する必要がある。Fanらの先行研究に倣い 1、中心分子がポリマーセグメ ントであるとき、元の計算値から2を引く形で反映させた29。 次に、以下の式 を用いてχパラメータを算出した。
𝜒 =(𝐸12𝑍12𝑆12+𝐸21𝑍21𝑆21) − (𝐸11𝑍11𝑆11+ 𝐸22𝑍22𝑆22)
𝑅𝑇 (10)
ここで、𝑍𝑖𝑗は中心セグメントiの周囲にセグメントjを配置した際の配位数、𝑆𝑖𝑗 は中心セグメントiの周囲にセグメントjを配置した際のScaling factorである。
本章のテスト計算では、𝑆12と𝑆21を同じ値として扱った。一連の手続きをまとめ たフローをFigure 4に示す。
(v) 相転移上部臨界温度の評価: 𝜒パラメータの温度依存性から二成分混合物 の上限臨界温度(Tc)を計算することが可能である。Flory-Huggins理論では、臨
11 界点の𝜒の理論値 (𝜒𝑐) が、各成分の重合度のみに依存して以下の式によって得 られる。
𝜒𝑐 = ( 1
√𝑛𝑎+ 1
√𝑛𝑏)
2
2 (11)
ここで𝑛𝑎, 𝑛𝑏は各成分の重合度で、ポリマー分子量(Mw)とセグメント分子量の 商である。 セグメントが溶媒である場合、この値は1である。本研究の手法に より得られた温度ごとの𝜒値と𝜒𝑐を比較し、一致する際の温度が臨界温度の計算 値Tcに相当する。
Figure 3. 接触配向の算出のための中央セグメントの周りの空間の分割。6 つの
領域は、立方体に従って定義される。青色および赤色の球体は、それぞれセグメ ント1およびセグメント2を表す。
Figure 4. FMO計算を用いたパラメータ算定のワークフロー
12 信頼性の評価:テスト計算
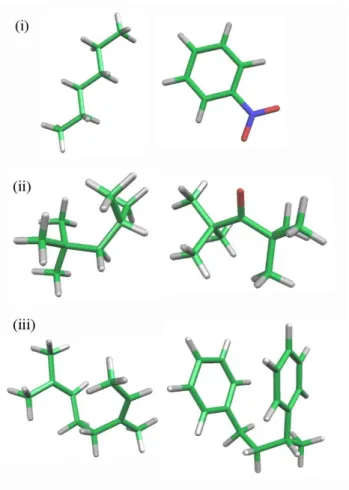
本手法の有用性と信頼性を検証するために、χパラメータセットを以下の3種 の基礎的な二成分混合系について計算した30,31。分子構造をFigure 5に示す。
(i) Hexane-Nitrobenzene(溶媒-溶媒)
(ii) Polyisobuthylene-Diisobuthyl Ketone(ポリマー-溶媒)
(iii) Polyisoprene-Polystyrene(ポリマー-ポリマー)
この検証計算において、セグメント対の生成のための非結合原子間の距離を 再評価した (2.2.2を参照)。Table 1にまとめたように、この再評価にはいくつか の小分子を用いた。原子間の最適な距離を決定するために、GAUSSIAN 09プロ グラム32を用いて、B97D33密度汎関数、6-31G(d)34基底にて1組の小分子対を最 適化した。再評価された距離をTable 2に示す。
各セグメント構造は、GAUSSIAN0932を用いて B97D33/6-31G(d)34 レベルで最 適化した。本章では、J-OCTA35のシステムを用いて配置の生成および配位数𝑍の 計算を行った。構造の生成はRef.1に倣い、再評価した非結合原子距離を使用し た。次に、セグメントの各組み合わせについて、ABINIT-MP36を用いて FMO2-
MP2 / 6-31G(d)レベルで2000配座の相互作用計算を行った。溶媒分子(Hexane,
Nitrobenzene, Diisobuthyl Ketone) は1分子をセグメント構造として扱った。ポリ マー分子(Polyisobutylene, Polyisoprene, Polystyrene)は2モノマーを1 つのセグメ ント構造として扱い、計算ではモノマーごとに 2 つのフラグメントに分割して 扱った。これらのポリマーセグメント間の相互作用エネルギーは、異なるセグメ ントに属するフラグメント間の相互作用を合計することによって計算した。例 えば、1つのセグメントがフラグメント1および2から成り、他のセグメントが フラグメント3および 4からなる場合、2 つのセグメント間の相互作用∆𝐸̃𝑠𝑒𝑔は 以下の式によって計算される。
∆𝐸̃𝑠𝑒𝑔 = ∆𝐸̃13+ ∆𝐸̃14+ ∆𝐸̃23+ ∆𝐸̃24 (12)
∆𝐸̃𝑖𝑗はフラグメントiとjの間の相互作用である。
生成した各配置の相互作用の計算が終わると、平均相互作用エネルギー𝐸𝑖𝑗と
Scaling factor 𝑆𝑖𝑗を算出し、配位数𝑍𝑖𝑗の計算の後、𝜒パラメータを式(10)を用いて
評価した。その後、相転移臨界温度 Tcの計算値を実験データと比較した。いく つかのケースにおいて力場計算と結果と比較を行ったが、この際、general AMBER force field (GAFF)とDREIDING 力場 37の結果を参照データとして用い た。更に、ペア相互作用エネルギーの詳細解析として、Hexane-Nitrobenzene系に
おいてPIEDA27,28による詳細解析を行った。
13 Figure 5. テスト計算に使用した分子構造。(i) Hexane-Nitrobenzene(溶媒-溶媒)
(ii) Polyisobuthylene-Diisobuthyl Ketone( ポ リ マ ー-溶 媒 )(iii) Polyisoprene-
Polystyrene(ポリマー-ポリマー)。
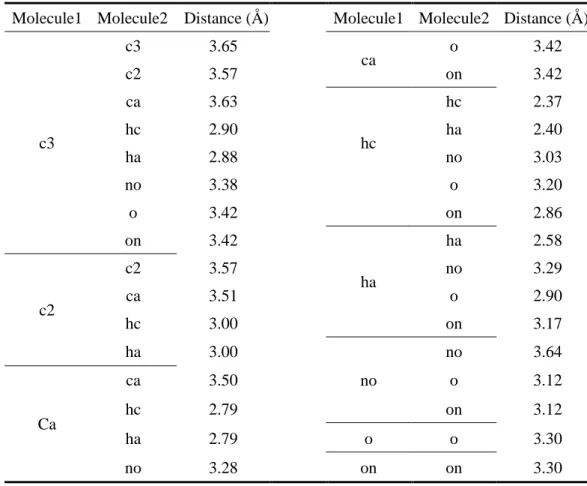
Table 1. 非結合接触距離の再評価に用いた小分子モデルの設定
Atom Type Definition Molecule
c3 sp3 carbon C2H6
c2 sp2 carbon C2H4
ca aromatic carbon C6H6
hc Hydrogen bonded to aliphatic carbon C2H6
ha Hydrogen bonded to aromatic carbon C6H6
no Nitrogen of the nitro group CH3NO2
o Oxygen with one atom CH3CHO
on Oxygen of the nitro group CH3NO2
14
Table 2. 再評価した非結合原子間の距離情報
Molecule1 Molecule2 Distance (Å) Molecule1 Molecule2 Distance (Å)
c3
c3 3.65
ca o 3.42
c2 3.57 on 3.42
ca 3.63
hc
hc 2.37
hc 2.90 ha 2.40
ha 2.88 no 3.03
no 3.38 o 3.20
o 3.42 on 2.86
on 3.42
ha
ha 2.58
c2
c2 3.57 no 3.29
ca 3.51 o 2.90
hc 3.00 on 3.17
ha 3.00
no
no 3.64
Ca
ca 3.50 o 3.12
hc 2.79 on 3.12
ha 2.79 o o 3.30
no 3.28 on on 3.30
結果と考察
Hexane-Nitrobenzene (溶媒-溶媒) 系
𝜒の結果について議論する前に、再評価した距離情報の影響について述べる。
ニトロベンゼンのペアについて従来の距離情報(van der Waals 半径由来の距離 情報)と、量子計算により算出した距離情報を用いてパラメータ算定を行った場 合について、Metropolis 法により採用された配置の動径分布関数を比較した。
Figure 6からわかるように、再評価したパラメータでは、近い距離での採用配座
が増加し、サンプリングの問題が改善していることがわかる。
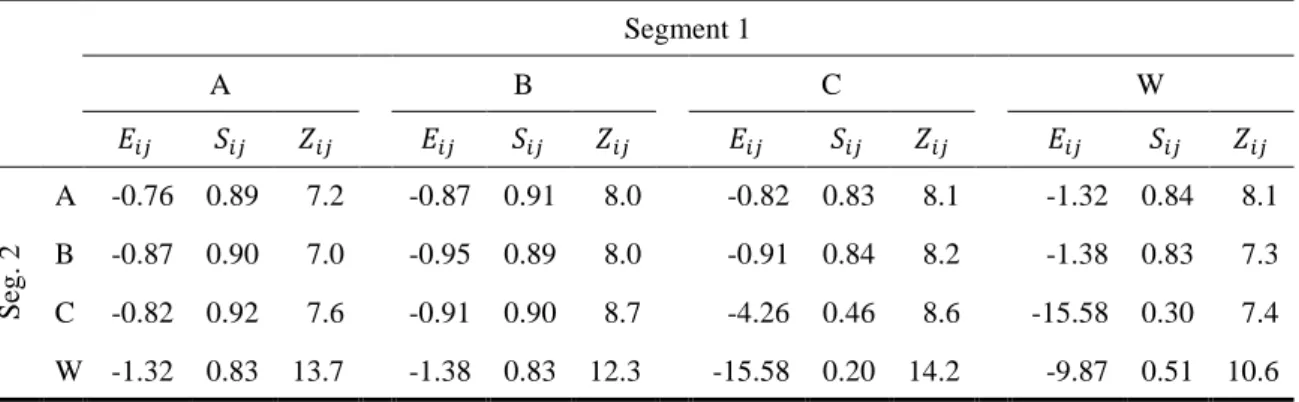
溶媒-溶媒系として扱ったヘキサン-ニトロベンゼンについて、Table 3に300K での3ペアでの相互作用値、配位数、採用配座の偏りから算出したScaling factor を示した。ヘキサンのような n-アルカンでは相互作用値が小さいが、Scaling
factorは0.88である。これは系が等方的であり、複数の配置で均等に近い状態で
相互作用している様子が示されている。一方、ニトロベンゼンはニトロ基による
15 静電相互作用やフェニル基による π 電子の相互作用により大きな安定化を示す ことがわかる。しかし、Scaling factorは0.75であり、系が異方的で、配置による 相互作用の差が大きいことがわかる。また、各温度での結果から得られた温度ご とのχ値の結果をFigure 7に示した。溶媒-溶媒系は重合度が1なため、式(11)か らこの系でのχc の理論値は 2.0である。臨界温度の実験値と本計算から得られ
た値をTable 3に示した。実験値は293K30に対し計算値は286Kであり、実験値
を10%程度の誤差で再現した。
相互作用が最も大きい配座 (A)、相互作用が最も小さい配座 (B)、DREIDING による計算結果との差が大きい配座 (C) に対し、PIEDAによる詳細解析を行っ
た結果をTable 4、Figure 8に示す。どのペアにおいても、骨格が平行に並んでい
る配座で安定となっている。ヘキサンペアは、分散力によってのみ安定化を示し ている。一方、ニトロベンゼンでは、分散力の寄与だけでなく、静電相互作用、
電荷移動により大きな相互作用を示している。また、3ペア全てにおいて、電荷 移動量が大きい配座で、FMOとDREIDING計算の差が大きくなっていることが わかる。このことは電子状態を考慮することの重要性を示しているといえる。ニ トロベンゼンペアの安定配座(A)の電荷移動量が小さく出ているのは、両セグメ ントから電子の行き来が起きているためと考えられる。
Figure 6. Nitrobenzene-Nitrobenzene対のMCサンプリングにおいて採用された配 置 の 分 子 間 か ら 得 ら れ た 動 径 分 布 関 数 。 各 ピ ー ク は 1 に 統 一 さ れ て い
る。”Original”ラベルは Fan の手法による van der Waals 接触ベースの結果を表
し、”Present”ラベルは MO ベースの構造最適化により非結合原子間距離を再評
価した結果に対応する。
0 0.2 0.4 0.6 0.8 1
3 4 5 6 7 8 9
RDF
Distance (Å)
Original Present
16 Table 3. Hexane (label 1)-Nitrobenzene (label 2) ペアにおける300Kの平均相互作 用エネルギー(𝑬𝒊𝒋), scale factor(𝑺), 配位数(𝒁𝒊𝒋)と臨界温度
Pair (i-j) 𝐸𝑖𝑗 (kcal/mol) 𝑆𝑖𝑗 𝑍𝑖𝑗 Tc (K)
𝜒𝑐 exptl. FMO
1-1 -0.77 0.88 10.6
2.0 293 286
1-2
-1.78 0.83 10.4
2-1 10.7
2-2 -3.28 0.75 10.6
Figure 7. Hexane-Nitrobenzeneペアの温度ごとの𝛘値。正方形、十字、三角の記号 は、スケーリング(S)付きのFMO、スケーリングなしのFMO、GAFFの結果に それぞれ対応している。点線の横線はTable 3の𝝌𝒄を示す。正方形のカーブ(ス ケーリング付きFMO)は、𝝌 = −𝟐. 𝟐𝟑 + 𝟏. 𝟐𝟏𝟐 × 𝟏𝟎𝟑/𝑻でFittingしている。
-1.0 0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0
270 320 370 420 470 520 570
χ
Temperature(T)
FMO (with S) GAFF
FMO (without S) χc
17 Figure 8. Hexane-Nitrobenzeneペアの特徴的な配置。(A)最も安定した配置(B)
最も不安定な配置(C)IFIEとFF値との間のエネルギー差が最も大きい配置。
18 Table 4. Hexane (label 1)-Nitrobenzene(label 2)系のPIEDAの結果。ES、EX、CT + mixおよびDI項はkcal / molで、q(Mulliken charge transfer)はeで与えられる。
DREIDINGの結果も併せて記載している。ラベルA、B、およびCはFigure 8に
対応する。
Pair (i-j) IFIE
PIEDA
Dreiding
Difference between IFIE and Dreiding ES EX CT + mix DI (MP2) q (I=>J)
1-1
A -2.12 -0.13 0.20 -0.38 -1.81 -0.0001 -2.41 0.29
B -0.19 0.06 0.03 -0.05 -0.23 -0.0002 -0.29 0.10
C -1.44 0.18 0.13 -0.29 -1.45 0.0001 -1.99 0.55
1-2
A -4.12 -0.71 0.33 -1.06 -2.68 -0.0076 -3.09 1.04
B -0.01 0.16 0.02 -0.05 -0.14 0.0005 -0.34 0.33
C -2.99 -1.14 0.15 -0.70 -1.31 -0.0049 -1.13 1.86
2-2
A -7.13 -2.09 0.94 -1.20 -4.79 0.0013 -6.06 1.07
B 2.36 2.84 0.14 -0.29 -0.32 0.0001 2.47 0.10
C -4.68 -2.53 0.10 -0.81 -1.43 0.0070 -2.59 2.09
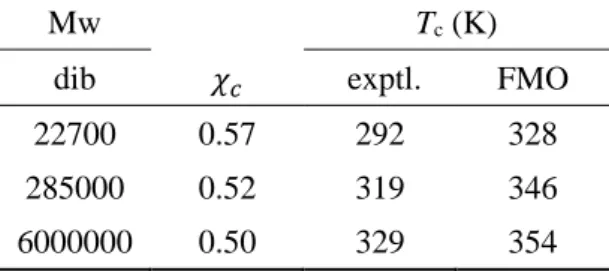
19 Polyisobuthylene-Diisobuthyl Ketone (ポリマー-溶媒) 系
ポリマー-溶媒系としては、Polyisobutylene - Diisobutyl Ketoneを扱った。Table 5に溶媒系と同様、300Kでの 3ペアでの相互作用値、配位数と、採用配座の偏 りから算出した Scaling factor を表に示した。この系には Nitrobenzene のような 強い相互作用を持たず、配置による偏りも少ないことがわかる。各温度での結果 から得られた温度ごとのχ値の結果をFigure 9に示した。臨界温度については、
先行研究に習い、Polyisobuthyleneについて実験値のある 3種の重合度の結果に ついて比較を行った結果をTable 6に示した。それぞれの重合度での実験値は292,
319, 329K29に対し計算値は328, 346, 354Kであり、いずれの結果においてもFMO
計算から得られたχ値は実験値を10%程度の誤差で再現した。
Table 5. Polyisobuthylene (label 1) - Diisobuthyl Ketone(label 2)系の300Kでの相 互作用エネルギー(𝑬𝒊𝒋), scale factor(𝑺), 配位数(𝒁𝒊𝒋)
Pair (i-j) 𝐸𝑖𝑗 (kcal/mol) 𝑆𝑖𝑗 𝑍𝑖𝑗
1-1 -0.95 0.92 7.6
1-2 -1.13 0.92 6.9
2-1 9.7
2-2 -1.38 0.88 9.1
Table 6. Polyisobuthylene-Diisobuthyl Ketone(dib)系の臨界温度
Mw Tc (K)
dib 𝜒𝑐 exptl. FMO
22700 0.57 292 328
285000 0.52 319 346
6000000 0.50 329 354
20 Figure 9. Polyisobuthylene- Diisobuthyl Ketone系の温度ごとのχ値。正方形、十字、
三角の記号は、スケーリング(S)付きのFMO、スケーリングなしのFMO、GAFF の結果にそれぞれ対応している。3つの点線の横線はTable 6 の𝝌𝒄を示す。正方 形のカーブ(スケーリング付きFMO)は、𝛘 = −𝟎. 𝟑𝟗 + 𝟑. 𝟏𝟑𝟖 × 𝟏𝟎𝟐/𝐓でFitting することができる。
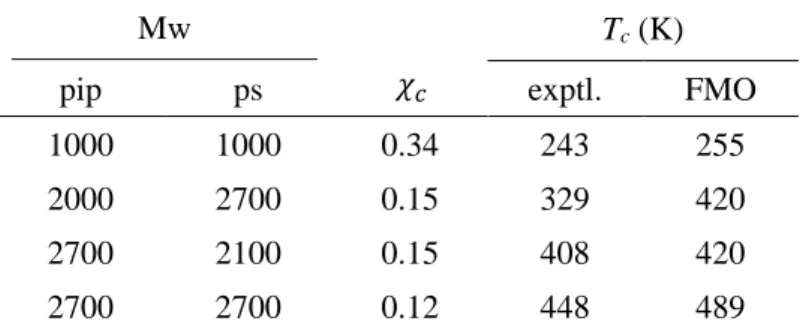
Polyisoprene –Polystyrene (ポリマー-ポリマー) 系
ポリマー-ポリマー系として、Polyisoprene –Polystyreneを扱った。上記二例と 同様、300Kでの3ペアでの相互作用値、配位数と、採用配座の偏りから算出し たScaling factorをTable 7に示した。Polyisopreneは強い相互作用を持つような 官能基を持たないため配置による偏りがすくないが、PolystyreneではPh基の存 在により相互作用が強くなっている。
各温度での結果から得られた温度ごとのχ値の結果をFigure 10に示した。臨 海温度については、それぞれについて重合度の異なる 4 種類の結果について比 較を行い、結果をTable 8 38–40に示した。こういった系では重合度によってχが 大きく異なるため,重合度による臨界温度の差が大きい。上記の二例とくらべて
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
270 320 370 420 470 520 570
χ
Temperature(T)
FMO (with S) GAFF
FMO (without S) χc
21 綺麗な一致を示すことはできなかったが、基本的に 10%程度の良好な一致を示 した。
Table 7. Polyisoprene (label 1)-Polystyrene (label 2)系の300Kでの相互作用エネル ギー(𝑬𝒊𝒋), scale factor(𝑺), 配位数(𝒁𝒊𝒋)
Pair (i-j) 𝐸𝑖𝑗 (kcal/mol) 𝑆𝑖𝑗 𝑍𝑖𝑗
1-1 -1.16 0.84 8.0
1-2 -1.78 0.80 7.0
2-1 9.0
2-2 -2.59 0.76 7.8
Table 8. 300KにおけるPolyisoprene (pip)-Polystyrene (ps)系の臨界温度 (式(11)に 従うとpip-psのMwが2000-2700, 2700-2100のとき、𝝌𝒄は同じ値(0.15)となる。
そのため、FMOのTcも同じ値となる)。
Mw Tc (K)
pip ps exptl. FMO
1000 1000 0.34 243 255
2000 2700 0.15 329 420
2700 2100 0.15 408 420
2700 2700 0.12 448 489
𝜒𝑐
22 Figure 10. Polyisoprene -Polystyrene 系の温度ごとのχ値。正方形、十字、三角の 記号は、スケーリング(S)付きのFMO、スケーリングなしのFMO、GAFFの結 果にそれぞれ対応している。3つの点線の横線はTable 8 の𝝌𝒄を示す。正方形の カーブ(スケーリング付きFMO)は、𝝌 = 𝟎. 𝟒𝟕 × 𝟏𝟎−𝟏+ 𝟏. 𝟗𝟏𝟖 × 𝟏𝟎𝟑/𝑻𝟐でFitting されている。
汎用的システムの構築と公開
本章で開発したフレームワークでは非経験的MO法から直接DPDシミュレー ションを行うパラメータを生成しているため,幅広い分子に対して適用可能で あるという利点がある.このようなソフトウェアは高い実用性が求められる領 域にこそふさわしいと考えられるため、任意の分子間の χ パラメータを算定で きるシステムを開発した。FMO-based Chi parameter Evaluation Workflow System (FCEWS) という名称で、計算工学ナビ(http://www.cenav.org/fcews_ver1_rev2/)に て公開している。プログラムは Python (3 系)と Fortran 90 からなっており、
ABINIT-MP が使用可能であることが前提となっている。本章での実行では、配
0.0 0.5 1.0 1.5 2.0
270 320 370 420 470 520 570
χ
Temperature(T)
FMO (with S) GAFF
FMO (without S) χc
23 置の作成にJ-OCTAの配置生成機能を用いたが、独立した機能として開発した。
ユーザーはパラメータを作成したいセグメントの xyz ファイルを作成するのみ で、構造の作成、ABINIT-MPによる計算、χパラメータの算出を一括で行うこと ができる。計算コストの観点から、Linuxでの運用を想定しているが、Windows での使用も可能である。各種ジョブ管理システム(PBS, Torque, LSF, Lava)のほ か、FOCUSや京といったスーパーコンピュータへのジョブ投入にも対応してい る。
接触原子間の距離に関して、本章では量子化学計算を元に修正したが、FCEWS では汎用性を維持するために、Bondiらのvdw半径41をデフォルトの設定とし、
水素結合に関わる距離情報のみ変更している。対応していない原子がある場合 はUFF力場の値を設定できるようにしている42。
なお、このプログラムはJ-OCTA ver. 4.1以降に同梱されている。