触媒的環化反応
有機金属化学第10回
2017年度
最初の例(量論反応)
Pauson-Khand反応 ([2+2+1]付加環化反応)
Pauson, P. L.; Khand, I. U., Ann. N. Y. Acad. Sci. 1977, 295, 2-14.
Co Co C C Ph H CO OC OC COCO CO (OC)4Co Co(CO)4 + H Ph –2CO H2C CH2 Ph O
初期の触媒反応の例
Rautenstrauch, V.; Mégard, P.; Conesa, J.; Küster, W.
Angew. Chem. Int. Ed. Engl. 1990, 29, 1413-1416.
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. Foreman, M. I., J. Chem. Soc., Perkin Trans. 1 1973, 977-981.
ホスフィン添加で効率向上&不斉化
Ts N Co2(CO)8, 20 mol% (S)-BINAP, 20 mol% 80 °C, 1 atm CO Ts N O H 54% yield 94% ee PPh2 PPh2 (S)-BINAPHiroi, K.; Watanabe, T.; Kawagishi, R.; Abe, I.
Tetrahedron Asym. 2000, 11, 797-808. N OAc 1) Co2(CO)8 2) NMO, H2O 3) Pd/C, H2 OAc N H H H O O N H H O (–)-dendrobine Cassayre, J.; Zard, S. Z., J. Am. Chem. Soc. 1999, 121, 6072-6073.
アルカロイド合成への応用
最初の例
アルキン三量化反応 ([2+2+2]付加環化反応)
OH OH HO Ni(CO)2(PPh3)2 HO OH OH + HO OH OH Reppe, W.; Schwecknediek, W. J.Justus Liebigs Ann. Chem. 1948, 560, 104-116.
nBu MeO MeO nBu Ni(cod)2/PPh3 nBu nBu MeO MeO
ヘリセン合成への応用
Teplý, F.; Stará, I. G.; Starý, I.; Kollárovič, A.; Šaman, D.;Rulíšek, L.; Fiedler, P., J. Am. Chem. Soc. 2002, 124, 9175-9180.
N Ts CO2Me N Ts [Rh(cod)]2BF4 (5 mol%) (S,S)-bdpp (5 mol%) (S,S)-bdpp 87%, 92% ee Ph2P PPh2
軸不斉シクロファン合成への応用
Araki, T.; Noguchi, K.; Tanaka, K.
カルベン錯体の反応
最初の報告
Rh触媒シクロプロパン化の反応機構
触媒的不斉シクロプロパン化
Simmons, H. E.; Smith, R. D., J. Am. Chem. Soc. 1958, 80, 5323-5324. CH2I2, Zn(Cu) R R’ I C H2 ZnI R R’ H2C I ZnI up to 70%
Fischer, E. O.; Dötz, K. H., Chem. Ber. 1970, 103, 1273-1278. (OC)5Cr C O Ph Me + Me CO2Me CO2Me Me PhOMe N
有機金属錯体を用いる最初の報告
O O R N2 CO2Me Ph + Rh cat. (1 mol%) CO2Me Ph 84% yield 84% ee –78 °C O O N H O O Rh Rh C12H25 4 O O R O O R O O R Rh RhDavies, H. M. L.; Bruzinski, P. R.; Lake, D. H.; Kong, N.; Fall, M. J., J. Am. Chem. Soc. 1996, 118, 6897-6907.
Nowlan, D. T.; Gregg, T. M.; Davies, H. M. L.; Singleton, D. A.
J. Am. Chem. Soc. 2003, 125, 15902-15911.
N EWG R N N EWG R N Rh N EWG R N Rh EWG R Rh R’ EWG R Rh R’ R’ EWG R Rh N N Cl Cl Cl Cl CuOTf, 10 mol% 11 mol% O + N I Ts Ph –78 °C O N Ts 75% yield 98% ee Li, Z.; Conser, K. R.; Jacobsen, E. N.
J. Am. Chem. Soc. 1993, 115, 5326-5327.
カルベン錯体の反応:オレフィンメタセシス
Banks, R. L.; Bailey, G. C.
Ind. Eng. Chem. Prod. Res. Dev. 1964, 3, 170-173.
H.S. Eleuterio , Ger. Pat. 1960, 1072811. MoO2/Al2O3/LiAlH4
AliBu3/MoO2/Al2O3 + + + isomers + isomers 42% 55% 2% + + isomers 1% “unsaturated polymer”
最初の報告
G. Natta, G. Dall'Asta, I. W. Bassi, G. Carella,
Makromol. Chem., 1966, 91, 87-106.
WCl6/EtAlCl2
n
P. J. L. Hérisson, Y. Chauvin, Makromol. Chem., 1971, 141, 161-176. J.-P. Soufflet, D. Commereuc, Y. Chauvin, C. R. Hebd.
Seances Acad. Sci. Série C, 1973, 276, 169-171.
R R1 R1 M R1 R1 M R1 R1 M R1 M
反応機構
Yves Chauvin Nobel Prize 2005Schrock, R. R., J. Am. Chem. Soc. 1974, 96, 6796-6797. Ta Cl Cl tBu tBu tBu 2 Li tBu H tBu – Ta tBu tBu tBu C tBu H Cl tBu Li Richard Schrock Nobel Prize 2005
構造の明確なアルキリデン錯体の合成
Murdzek, J. S.; Schrock, R. R., Organometallics 1987, 6, 1373-1374. Mo C Cl Cl O O Cl tBu NH iPr iPr Me3Si Mo C Cl Cl O O tBu N Dip H OLi CF3 CF3 Mo C O O tBu N Dip H F3C CF3 F3C CF3 Robert Grubbs Nobel Prize 2005
Schwab, P.; France, M. B.; Ziller, J. W.; Grubbs, R. H.
Angew. Chem. Int. Ed. Engl. 1995, 34, 2039-2041.
官能基許容性の高いRu錯体
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H.
Org. Lett. 1999, 1, 953-956. (Ph3P)2RuCl2 C N2 Ph H Ru PCy3 PCy3 C Cl Cl H Ph 1st generation 1) 2) PCy3 N N Mes Mes H OtBu Ru PCy3 C Cl Cl H Ph 2nd generation N N Mes Mes
カルベン錯体の反応:オレフィンメタセシスの応用
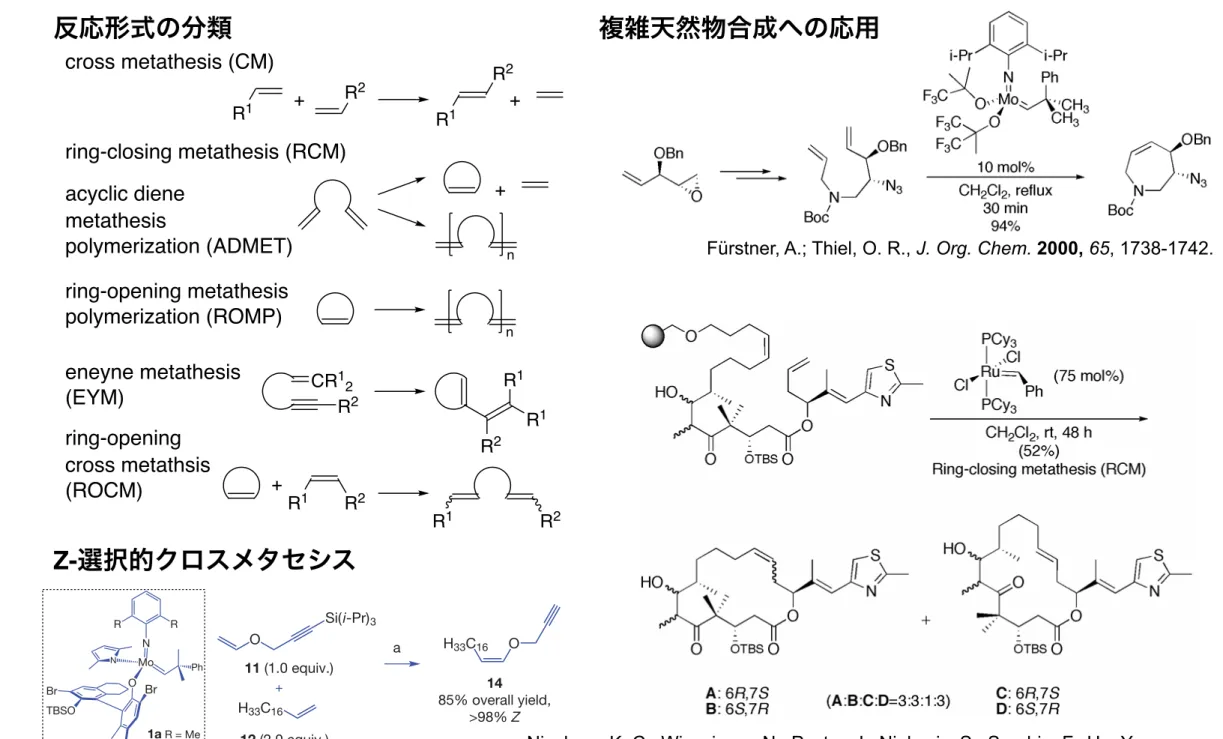
反応形式の分類
R1 R2 + R1 R2 + + n n R2 R1 R1 CR12 R2 + R1 R2 R1 R2 cross metathesis (CM) ring-closing metathesis (RCM) acyclic diene metathesis polymerization (ADMET) ring-opening metathesis polymerization (ROMP) eneyne metathesis (EYM) ring-opening cross metathsis (ROCM) 7 Scheme 6Scheme 7. The efficient RCM-based synthesis shown is due to Fürstner in his approach to balanol.10
Scheme 8. The example of the formation of a dicarba-analogue of a disulfide β-turn by RCM was disclosed by Grubbs.11
Fürstner, A.; Thiel, O. R., J. Org. Chem. 2000, 65, 1738-1742.
複雑天然物合成への応用
Nicolaou, K. C.; Winssinger, N.; Pastor, J.; Ninkovic, S.; Sarabia, F.; He, Y.;
Vourloumis, D.; Yang, Z.; Li, T.; Giannakakou, P.; Hamel, E., Nature 1997, 387, 268.
8
Scheme 9. Solid phase synthesis used by Nicolaou in the syntheses epothilone A and its various derivatives. A RCM cyclization/cleavage strategy is used.12
Scheme 10. An efficient CM with allylic stereocontrol observed with a substituted allylic amine.13
Meek, S. J.; O’Brien, R. V.; Llaveria, J.; Schrock, R. R.; Hoveyda, A. H., Nature 2011, 471, 461.
varying amounts of 6 (see Supplementary Information for details). The latter studies established that, although fewer equivalents of 6 lead to reduced Z selectivity and competitive homocoupling, with 5 equiv. of the inexpensive and commercially available enol ether, 8a can be obtained in 93:7 Z:E selectivity and 71% yield (7% homo-coupled product). Excess enol ether 6 does not complicate product isolation, as this inexpensive reagent is volatile and can be easily removed in vacuo.
Z-Disubstituted enol ethers are obtained in 57–77% yield through exceptionally stereoselective (94% to .98% Z) CM with Mo alkylidene 1a (Fig. 2). Alkyl- (8) or aryl-substituted (10) Z enol ethers as well as those that bear a carboxylic ester (8c), a secondary amine (8e), a brom-ide (10b) or an alkyne (10c) are readily accessed. Reactions with the more electron-deficient enol ether 9 and the relatively electron-rich alkenes proceed with 2.0 equiv. of the aryl-substituted enol ether; in contrast, 10 equiv. of alkyl-substituted and easily removable 6 are required for similar efficiency. Such variations probably occur because when 9 is used there is a better electronic match3 between the
Mo-alkylidenes derived from the cross partners and either of the two alkenes, favouring CM versus homocoupling. Only 1.2 mol% 1a and 2.0 equiv. of the p-methoxyphenylenol ether (for example, 10a, 10b and 10d, Fig. 3c) are sufficient for an effective and exceptionally Z-selective CM to take place.
Synthesis of natural product C18 (plasm)-16:0 (PC) Next, we set out to demonstrate the utility of the catalytic CM process by a diastereo- and enantioselective synthesis of an anti-oxidant plasmalo-gen phospholipid, C18 (plasm)-16:0 (PC) (Fig. 2)16,17, the
correspond-ing E isomer of which has been shown to be less active17. This initiative
required addressing a challenge that is of general concern in catalytic CM: the inefficiency associated with the use of excess of one cross partner. The enol ether to be used (11) in the CM step is more valuable than the commercially available and inexpensive 1-octadecene (12 ), rendering utilization of excess amounts of the former unfavourable. Reducing the enol ether concentration diminishes efficiency and Z-selectivity, as detailed above and substantiated by the data in Table 2 (85% and 47% conversion with 5:1 and 1:1 11:12 ; entries 1 versus 2). Larger quantities of the less valuable 12 could improve yield and selectivity, as Mo-methylidene concentration is probably lowered through its reaction with excess alkene. However, increased amounts of an aliphatic alkene, unlike an enol ether, give rise to homocoupling and ethylene generation. Ethylene, in addition to being detrimental to the rate of CM (because it competes with the substrates for reaction with the available alkylidene), causes diminished stereoselectivity by increasing methylidene concentration, which promotes Z alkene isomeriza-tion (see above). We thus surmised that, if the negative effects of the generated ethylene were to be attenuated by performing the reaction
11 (1.0 equiv.) 12 (2.0 equiv.) H17C8 On-Bu 8b 76% conv., 68% yield, 98% Z On-Bu 8c 76% conv., 73% yield, 98% Z O
PhO (i-Pr)3SiO On-Bu
8d 86% conv., 77% yield, 94% Z On-Bu PhHN 8e 57% conv., 51% yield, >98% Z OPMP Br 10a 71% conv., 57% yield, >98% Z 10b 74% conv., 70% yield, >98% Z 10c 66% conv., 59% yield, >98% Z Cy OPMP 10d 81% conv., 75% yield, >98% Z Bn OPMP Me OPMP 3Si ( )6 ( )6 15 N Ph Ph S ON O O i-Pr i-Pr i-Pr a H33C16 O 14 85% overall yield, >98% Z H33C16 O Si(i-Pr)3 b H33C16 O OH OH 16 64% yield, 98:2 e.r., >98% Z H33C16 O O O C15H31 O C18 (plasm)-16:0 (PC) P O O O NMe3 Four steps 86% overall yield (see ref. 12) On-Bu 6 (10 equiv.) O 9 OMe or 1.2–5.0 mol% 1a C6H6, 22 °C, 2 h G G On-Bu or G O OMe 8 10 (2.0 or 10 equiv.)
Figure 2 |Z-selective CM reactions of enol ethers with terminal alkenes and application to stereoselective synthesis of C18 (plasm)-16:0 (PC). Various Z enol ethers are synthesized with 1.2–5.0 mol% of Mo complex 1a, and typically require 2.0 equiv. (in the case of p-methoxyphenylvinyl ether) or 10.0 equiv. (with butylvinyl ether) of the terminal enol ether; excess butyl vinyl ether (6) is easily removed in vacuo. The desired Z alkenes are obtained in 51–77% yield and in 94% to .98% Z selectivity. Application to synthesis of C18 (plasm)-16:0 (PC) demonstrates the utility of the Z-selective Mo-catalysed CM, which is used in conjunction with a site- and enantioselective Cu-catalysed dihydroboration of the terminal alkyne in 14 (see Supplementary Information for details). All reactions shown were performed under N2atmosphere; catalysts were prepared
and used in situ. Conversions and Z selectivities were determined by analysis of 400 MHz1H NMR spectra of unpurified mixtures; the variance of selectivity values is estimated to be ,62%. Yields of isolated products are shown (65%). Reactions: for 8b–8e, we used 2.5 mol% 1a and 10 equiv. 6; for 10a, 10b, we used 1.2 mol% 1a and 2.0 equiv. 9 ; for 10c, 10d, we used 5.0 mol% 1a and 10 equiv. (10c) or 2.0 equiv. 9 (10d). See Supplementary Information for
experimental details. Conditions for synthesis of 16. Route a, step 1; 2.5 mol% 1a, C6H6, 22uC, 2.0 h, decalin, 1.0 torr: step 2; 5.0 equiv. (n-Bu)4NF, THF, 22uC, 2 h. Route b; 2.5 mol% 15 , 2.5 mol% CuCl, 20 mol% NaOt-Bu, 2.1 equiv. bis(pinacolato)diboron, 3.0 equiv. MeOH, THF, 0uC, 24 h; 30% H2O2, NaOH in aqueous THF, 1.0 h.
ARTICLE
RESEARCH
2 4 M A R C H 2 0 1 1 | V O L 4 7 1 | N A T U R E | 4 6 3
Macmillan Publishers Limited. All rights reserved
©2011
Z-Selective cross-metathesis of enol ethers
We began by evaluating the ability of stereogenic-at-Mo complexes to promote transformations of enol ethers, a class of substrates for which a CM reaction has not been previously reported (E-or Z-selective); the resulting products have proven to be of utility in chemical synthesis and can be found in biologically active molecules (see below). In the presence of 2.5 mol% 1a, CM between 6 and 7 (entry 1, Table 1) proceeds to 85% conversion to afford disubstituted enol ether 8a in 98% Z selectivity and 73% yield. With 1b, which bears a more sizeable 2,6-di-i-propyl-arylimido unit, the reaction is completely Z-selective (.98% Z) but 47% conversion is achieved within the same time span. When alkylidene 2 is used, CM proceeds to 37% conversion and
.98% Z-8a is generated; further transformation is not observed after six hours. Mo-based diolate 3 and Ru carbene 5 do not promote CM, and achiral Mo complex 4 catalyses a non-selective transformation (47.5% Z). Thus, stereogenic-at-Mo complexes prove to be effective in promoting enol ether CM, and although 1b or the less hindered 2 also afford exceptional stereoselectivity, neither delivers the efficiency of 1a. The 2,6-dimethylphenylimido 1a therefore offers the best balance between activity and stereoselectivity. Such performance variations may be observed because catalyst turnover is slower with the more sizeable 1b whereas the methylidene of the relatively unhindered 2 (compare IV, Fig. 1) might suffer from a shorter life span. Consistent with the above scheme, 82% 8a is formed when CM with 1b is allowed to continue for 16 hours; in contrast, conversion with 2 after 10 minutes or two hours is nearly identical (,38%).
There are several, mechanistically revealing, reasons for use of excess enol ether. CM generates a Mo-methylidene; this unhindered alkyli-dene can readily react with the Z-alkene product, reverse CM, cause equilibration and lower stereoselectivity. An enol ether reacts with a methylidene complex, circumventing diminution in Z selectivity. The more stable alkoxy-substituted alkylidene, generated from reaction of a methylidene complex and an enol ether (I in Fig. 1 with R15On -Bu), can undergo productive CM, giving rise to longer catalyst lifetime and improved turnover numbers. Furthermore, generation of the afore-mentioned alkoxy- or aryloxy-containing alkylidene means less of the alkyl-substituted derivative is formed and homocoupling of the aliphatic alkene is minimized. Owing to electronic factors, productive reaction between an enol ether-derived alkylidene and another O-substituted alkene is disfavored2. However, as use of excess enol ether is wasteful, we decided to examine the efficiency of the CM with
Z-Selective cross-metathesis of terminal alkenes
G1 G2 Catalyst G1 G2 G1 G2 G1 G1 G1 G2 G2 G2 Cross-metathesis
products Homocouplingproducts
Z alkenes higher in energy than corresponding E alkenes G2 G1
Catalyst-induced kinetic control is required for stereoselective Z-alkene synthesis (vs substrate-induced thermodynamic control)
Rotating and large monodentate aryloxide
ligand generates a significant steric presence
Mo O Br TBSO Br N N R1 R2 Mo O N R1 R2 Br TBSO Br N R1 1a R = Me 1b R = i-Pr Mo O Br TBSO Br N Ph N Mo O Br TBSO Br N Ph 2 Ru Oi-Pr NMes MesN Cl Cl i-Pr i-Pr CF3 F3C Mo N F3C O O F3C 4 5 3 Mo N Br TBSO Br N R2 R1 O R1 R2 N Mo O Br OTBS Br N I II III IV Mo N i-Pr i-Pr O O N R R Ph Ph Rotation around Mo–O bond R2
Figure 1|A catalytic CM reaction can afford as many as six alkenes, so the challenge is designing an efficient process that favours formation of the cross products. Particularly difficult is the development of a process that affords the higher-energy Z alkene predominantly. To accomplish a Z-selective CM, a variety of catalysts were considered, such as stereogenic-at-Mo complexes (1, 2 ) or other previously reported Mo- and Ru-based complexes
(3 –5 ). The structural flexibility of the stereogenic-at-metal complexes 1 and 2 can give rise to exceptional reactivity, and free rotation around the Mo–O bond of these alkylidenes might serve as the basis for development of highly Z-selective olefin metathesis reactions of terminal alkenes. The sphere represents an appropriate size imido substituent.
Table 1|Examination of various catalysts for CM with an enol ether
Entry no. Complex Time Conv. (%)* Yield (%){ Z:E*
1 1a 2 h 85 73 98:2 2 1b 2 h 47 ND .98:2 3 2 2 h 37 ND .98:2 4 3 2 h ,2 NA NA 5 4 10 min 80 ND 47.5:52.5 6 5 24 h ,2 NA NA
The reactions were carried out in purified benzene under an atmosphere of nitrogen gas; 10 equiv. of 6 was used (see Supplementary Information for details). NA, not available; ND, not determined. * Conversion (conv.) and Z:E ratios were measured by analysis of 400 MHz1H NMR spectra of unpurified mixtures; the variance of values is estimated to be ,62%.
{ Yield of isolated product after purification; the variance of values is estimated to be ,65%.
7 Ph 2.5 mol% Mo or Ru complex C6H6, 22 °C Bn On-Bu On-Bu 6 8a RESEARCH ARTICLE 4 6 2 | N A T U R E | V O L 4 7 1 | 2 4 M A R C H 2 0 1 1
Macmillan Publishers Limited. All rights reserved
©2011
触媒的C‒H結合官能基化
触媒的C–H結合官能基化
C–H結合官能基化の熱力学
C
–H結合切断メカニズム
C H C [M] C C C O C N C B C halogen C‒H 結合の活性化段階に金属 (M) が関与し, C‒M 結合を有する中間体を経由して進行する触媒反応. ∆G° (kcal/mol) in gas phase –16 –30 +5 –11 +10 –10 H H :C O O H :C O O + H SiMe3 H SiMe3 + H H H SiMe3 + H H H SiMe3 + O O C O O H +10 – HX – HX – HO2CR [M] H [M] H [M]H [M] H [M] X [M] H X [M] O O H R O [Ru] [Ru] O H [Ru] O H H Oxidative Addition (OA) M = Ru0, RhI, IrI, Ni0 Concerted Metallation Deprotonation (CMD) M = PdII, RuII Electrophilic Aromatic Substitution (SEAr) M = PdII; X = OC(O)R, halogen Nucleophilic Aromatic Substitution (SNAr) M = Ru0 σ-Bond Metathesis (SBM) M = RhIII, IrIII; X = B [M] H3C [M] H H 3C [M]H H3C [M] H X – HX H3C H – HX H3C [M] H X [M] O O H H3C R – HO2CR H3C [M] sp2炭素 sp3炭素 Oxidative Addition (OA) M = Ru0, RhI, IrI σ-Bond Metathesis (SBM) M = RhIII; X = B Electrophilic Substitution (SE) M = PtII; X = solvent Concerted Metallation Deprotonation (CMD) M = PdII触媒的C‒H結合官能基化
芳香族化合物の触媒的アルケニル化(S
EAr経由)
+ 90% cat. Pd(0/II) oxidantFujiwara, Y.; Moritani, I.; Danno, S.; Asano, R.; Teranishi, S.
J. Am. Chem. Soc. 1969, 91, 7166-7169.
[Pd+2] [Pd+2] R1 [Pd+2] EWG R1 [Pd0] EWG R1 EWG + HX [O]
R1 = (het)Ar, alkenyl, alkyl
X = halogen, OR3 [O] = oxidant CMD or SEAr R1–H H–X X X X X Pd(OAc)2 (2 mol %) BQ (1.0 equiv) TsOH (0.5 equiv) AcOH, 20 °C, 15 h H H N 91% + CO2Bu O Me H N O Me CO2Bu
Boele, M. D. K.; van Strijdonck, G. P. F.; de Vries, A. H. M.; Kamer, P. C. J.; de Vries, J. G.; van Leeuwen, P. W. N. M.,
J. Am. Chem. Soc. 2002, 124, 1586-1587.
応用例
RuH2(CO)(PPh3)3 (1 mol %) toluene, reflux, 0.5 h + H O Si(OEt)3 Si(OEt)3 O H 100 mmol 110 mmol 92–96%
芳香族化合物の触媒的アルキル化(OA or S
NAr経由)
Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.;
Kamatani, A.; Sonoda, M.; Chatani, N., Nature 1993, 366, 529-531.
H Br PdCl2 (5 mol %) AgOTf (1.0 equiv) DMF, 90 °C, 2 h + 56% CO2Me H N O H N O CO2Me
求電子剤とのカップリング
Zaitsev, V. G.; Daugulis, O., J. Am. Chem. Soc. 2005, 127, 4156-4157.
H I Cl Pd(OAc)2 (2.5 mol %) Cs2CO3 (2.0 equiv) DMF, MS4A, 110 °C, 21 h + 72% OH Ar OH
Satoh, T.; Kawamura, Y.; Miura, M.; Nomura, M.,
Angew. Chem. Int. Ed. Engl. 1997, 36, 1740-1742.
F H
Br
Pd(OAc)2 (5 mol %)
(t-Bu)2MeP•HBF4 (10 mol %)
K2CO3 (1.3 equiv) + 98% DMA,120 °C, 12 h Me F F F F F Ar F F F F
Lafrance, M.; Rowley, C. N.; Woo, T. K.; Fagnou, K.,
J. Am. Chem. Soc. 2006, 128, 8754-8756.
反応機構
触媒的C‒H結合官能基化
[Ir(OMe)(cod)]2 (1.5–5 mol %)dtbpy (3–10 mol %) alkane, rt–80 °C +
Ar H 1/2 (nip)B B(pin) Ar B(pin) + 1/2 H2
ベンゼン環C–Hホウ素化
B(pin) Cl I 82% B(pin) Cl MeO2C 80% B(pin) F 82% SiMe2H BF3K Cl 100% OH B(pin) Br NC 83% B(pin) Cl O SiMe2H via B(pin) Cl 82% Cl B(pin) Cl 82% Cl B(pin) (nip)B 97% B(pin) (nip)B (nip)B 83% B(pin) B(pin) 95%, o/m = 98:2 with [3,5-(F3C)2–C6H3]3P O OMe Ishiyama, Miyaura, Hartwig, et al. Angew. Chem. Int. Ed. 2002, 41, 3056;Marder, et al. Chem. Commun. 2005, 2172; Hartwig, et al. J. Am. Chem. Soc. 2008, 130, 7534. Ishiyama, Miyaura, et al. Chem. Commun. 2010, 159.
Ir N B B N B Ir N B B N B Ar H Ir N B H N B B–B or H–B H–B or H–H OA or SBM B = B(pin) Ar–H Ar–B
Ishiyama, Miyaura, Hartwig, et al. J. Am. Chem. Soc. 2002, 124, 390;
J. Am. Chem. Soc. 2005, 127, 14263; Sakaki, et al. J. Am. Chem. Soc. 2003, 125, 16114.
Cp*Rh(η4-C6Me6)3 (2.4 mol %) 150 °C
+ (nip)B B(pin)
R H R B(pin) + H B(pin)
or [Cp*RuCl2]2, [Cp*RuCl4] (5 mol %Ru)
B(pin) 49% B(pin) B(pin) + 73%, 5:1 O B(pin) 91% B(pin) O O 74% B(pin) F 83% n-C8F17 B(pin) 90% N B(pin) 55% Bu2N B(pin) 75%
C(sp
3)–Hホウ素化
(70-85%) that were similar to those of the catalytic process and with comparable or faster rates. The reaction of Cp*Rh(H)2(Bpin)2 or Cp*Rh(H)(Bpin)3 with benzene oc-curred at 80 °C in 82-85% yield, while the reaction with octane occurred at 125 °C, giving octylBpin in 70% and 72% yields, respectively. The reaction with octane formed 1-oc-tylBpin as the only alkylboronate ester product. Therefore, Cp*Rh(H)2(Bpin)2and Cp*Rh(H)(Bpin)3are chemically and kinetically competent to be intermediates in the borylation of arenes and alkanes.
After having identified likely reaction intermediates, the reactions of methane with CpRh(H)2(BO2C2H4)2 and CpRh(H)(BO2C2H4)3were studied by Harwig, Hall, and co-workers as models for the reactions of alkanes with Cp*Rh(H)2(Bpin)2and Cp*Rh(H)(Bpin)3(Scheme 12). Novel conclusions were drawn from these studies.8 The empty p-orbital of the boryl ligand in the CpRh(BO2C2H4)2 inter-mediate was found to be involved in the C-H borylation process in several ways.
Reaction of the bisboryl intermediate with methane was calculated to occur by coordination of the alkane, followed by conversion of the alkane complex to a borane complex through a single transition state. This process occurs without formation of a discrete Rh(V) intermediate, much like the pathway lacking a high-valent intermediate calculated for the stoichiometric reactions of the iron and tungsten boryl complexes. Coupling of the alkyl group with the second boryl ligand is calculated then to form the final products (part (a) of Scheme 12).
Reaction of the boryl hydride intermediate with methane was calculated to occur by a related but distinct mechanism. After coordination of the alkane, simultaneous cleavage of the alkane C-H bond and formation of a borane B-H bond is calculated to form an alkyl complex containing a coor-dinated borane. Coupling of the boryl moiety with the alkyl group would then form the final products (part (b) of Scheme 12).
Miyamoto and co-workers69 performed computational studies on the reaction of BH3with CH4catalyzed by CpRh. This set of calculations, like those conducted by Hall, Hartwig, and co-workers, indicated that the rate-determining step in the functionalization of alkanes catalyzed by Cp*Rh is C-H bond cleavage and that the reductive elimination of a C-B bond is rapid. However, these authors proposed that a pathway involving oxidative addition and reductive elimi-nation through a high-valent rhodium intermediate is fol-lowed (Scheme 13).
The difference between the two sets of computational conclusions can be explained by some simplifications made by Miyamoto and co-workers. For example, HBpin and B2pin2are the only reagents, so far, that undergo the
rhodium-catalyzed borylation of alkanes and arenes. The alkoxy groups on HBpin modulate the strength of B-H interactions, which are critical to the catalytic process. Thus, BH3 might be a poor computational model for HBpin and B2pin2in these reactions. Second, the intermediates in the catalytic process are bisboryl dihydride and trisboryl monohydride complexes, not the monoboryl trihydride complexes studied by Miyamoto.
5.4. Rh-Catalyzed Borylation of Benzylic C
-H
Bonds
Rhodium complexes also catalyze the borylation of ben-zylic C-H bonds. After the observation of benben-zylic bory-lation as a side product from the reaction of B2pin2 with m-xylene reported by Smith63 when using the Cp*Rh-based catalyst developed by Hartwig, Marder and co-workers described, in 2001, the benzylic borylation of toluene, p-xylene, and mesitylene with HBpin catalyzed by [Rh(Cl)(N2)(PiPr3)2].65 [Rh(Cl)(N2)(PiPr3)2] (3 mol %) and 1 M HBpin were allowed to react with toluene at 140 °C, and the major product was 2-benzyl-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (Scheme 14). Products resulting from the borylation of arene C-H bonds or from diborylation of the benzylic position were produced in low yields. The quantity of PhCH(Bpin)2produced indicated that borylation of the benzylic C-H bonds in toluene further activated one of the remaining benzylic hydrogens to C-H borylation. para-Xylene was borylated at the benzylic position in preference to the aromatic position (98:2). The reaction of mesitylene with HBpin catalyzed by [Rh(Cl)(N2)(PiPr3)2] yielded 2-(3,5-dimethyl-benzyl)-4,4,5,5-tetramethyl-[1,3,2]di-oxaborolane, resulting from the borylation of a benzylic C-H bond, as the sole monoborylation product, along with smaller amounts of bisborylation products and traces of trisborylation products.
Scheme 12. Mechanism of C-H Bond Cleavage and C-B Bond Formation Supported by Density Functional Theory (DFT) Calculations for Hydridoboryl (a) and bis(Boryl) (b) CpRh Complexes
Scheme 13. Mechanism of the Borylation of Alkanes Catalyzed by Cp*Rh Proposed by Miyamoto and Co-workers
898 Chemical Reviews, 2010, Vol. 110, No. 2 Mkhalid et al.
(70-85%) that were similar to those of the catalytic process and with comparable or faster rates. The reaction of
Cp*Rh(H)2(Bpin)2 or Cp*Rh(H)(Bpin)3 with benzene
oc-curred at 80 °C in 82-85% yield, while the reaction with octane occurred at 125 °C, giving octylBpin in 70% and 72% yields, respectively. The reaction with octane formed 1-oc-tylBpin as the only alkylboronate ester product. Therefore,
Cp*Rh(H)2(Bpin)2and Cp*Rh(H)(Bpin)3are chemically and
kinetically competent to be intermediates in the borylation of arenes and alkanes.
After having identified likely reaction intermediates,
the reactions of methane with CpRh(H)2(BO2C2H4)2 and
CpRh(H)(BO2C2H4)3were studied by Harwig, Hall, and
co-workers as models for the reactions of alkanes with
Cp*Rh(H)2(Bpin)2 and Cp*Rh(H)(Bpin)3 (Scheme 12). Novel
conclusions were drawn from these studies.8 The empty
p-orbital of the boryl ligand in the CpRh(BO2C2H4)2
inter-mediate was found to be involved in the C-H borylation process in several ways.
Reaction of the bisboryl intermediate with methane was calculated to occur by coordination of the alkane, followed by conversion of the alkane complex to a borane complex through a single transition state. This process occurs without formation of a discrete Rh(V) intermediate, much like the pathway lacking a high-valent intermediate calculated for the stoichiometric reactions of the iron and tungsten boryl complexes. Coupling of the alkyl group with the second boryl ligand is calculated then to form the final products (part (a) of Scheme 12).
Reaction of the boryl hydride intermediate with methane was calculated to occur by a related but distinct mechanism. After coordination of the alkane, simultaneous cleavage of the alkane C-H bond and formation of a borane B-H bond is calculated to form an alkyl complex containing a coor-dinated borane. Coupling of the boryl moiety with the alkyl group would then form the final products (part (b) of Scheme 12).
Miyamoto and co-workers69 performed computational
studies on the reaction of BH3with CH4catalyzed by CpRh.
This set of calculations, like those conducted by Hall, Hartwig, and co-workers, indicated that the rate-determining step in the functionalization of alkanes catalyzed by Cp*Rh is C-H bond cleavage and that the reductive elimination of a C-B bond is rapid. However, these authors proposed that a pathway involving oxidative addition and reductive elimi-nation through a high-valent rhodium intermediate is fol-lowed (Scheme 13).
The difference between the two sets of computational conclusions can be explained by some simplifications made by Miyamoto and co-workers. For example, HBpin and
B2pin2are the only reagents, so far, that undergo the
rhodium-catalyzed borylation of alkanes and arenes. The alkoxy groups on HBpin modulate the strength of B-H interactions,
which are critical to the catalytic process. Thus, BH3 might
be a poor computational model for HBpin and B2pin2in these
reactions. Second, the intermediates in the catalytic process are bisboryl dihydride and trisboryl monohydride complexes, not the monoboryl trihydride complexes studied by Miyamoto.
5.4. Rh-Catalyzed Borylation of Benzylic C
-H
Bonds
Rhodium complexes also catalyze the borylation of ben-zylic C-H bonds. After the observation of benben-zylic
bory-lation as a side product from the reaction of B2pin2 with
m-xylene reported by Smith63 when using the Cp*Rh-based
catalyst developed by Hartwig, Marder and co-workers described, in 2001, the benzylic borylation of toluene,
p-xylene, and mesitylene with HBpin catalyzed by
[Rh(Cl)(N2)(PiPr3)2].65 [Rh(Cl)(N2)(PiPr3)2] (3 mol %) and
1 M HBpin were allowed to react with toluene at 140 °C, and the major product was 2-benzyl-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (Scheme 14). Products resulting from the borylation of arene C-H bonds or from diborylation of the benzylic position were produced in low yields. The
quantity of PhCH(Bpin)2 produced indicated that borylation
of the benzylic C-H bonds in toluene further activated one of the remaining benzylic hydrogens to C-H borylation.
para-Xylene was borylated at the benzylic position in
preference to the aromatic position (98:2). The reaction of
mesitylene with HBpin catalyzed by [Rh(Cl)(N2)(PiPr3)2]
yielded 2-(3,5-dimethyl-benzyl)-4,4,5,5-tetramethyl-[1,3,2]di-oxaborolane, resulting from the borylation of a benzylic C-H bond, as the sole monoborylation product, along with smaller amounts of bisborylation products and traces of trisborylation products.
Scheme 12. Mechanism of C-H Bond Cleavage and C-B Bond Formation Supported by Density Functional Theory (DFT)
Calculations for Hydridoboryl (a) and bis(Boryl) (b) CpRh Complexes
Scheme 13. Mechanism of the Borylation of Alkanes Catalyzed by Cp*Rh Proposed by Miyamoto and Co-workers
898 Chemical Reviews, 2010, Vol. 110, No. 2 Mkhalid et al.
反応機構
反応機構
Hartwig, Hall, et al. J. Am. Chem. Soc. 2005, 127, 2538. Hartwig, et al. Science 2000, 287, 1995; J. Am. Chem. Soc. 2003, 125, 858;
J. Am. Chem. Soc. 2004, 126, 15334; J. Am. Chem. Soc. 2006, 128, 13684.