─ ─261 ( ) ズラクトンによるアリル位アルキル化反応の報告例は無 い。 さらに,遷移金属錯体触媒アリル位アルキル化反応に 使用されるアリルエステル類としては様々なタイプのも のが使用可能であるが,置換基を一つだけ有する一置換 型アリルエステルの反応はその位置選択性の制御がやや 困難であることから,未だ発展途上の反応基質として言 われている。 以上の背景を基に,今回我々は一置換アリルアセター トとアズラクトンとのルテニム錯体触媒によるアリル位 アルキル化反応を行った。その結果,反応における位置 選択性を制御しつつ,生成物として混在する2 つのジア ステレオマーを連続した化学反応によって分離し,アズ ラクトン誘導体とオキサゾリノン誘導体をそれぞれ高エ ナンチオ選択的に得る事に成功したので,以下にその詳 細を報告する。 2.結 果 今回の反応検討は,光学活性なアリルアセタート1a (99% ee)を反応基質とし,求核剤をアズラクトン2とし 1.緒 言 遷移金属錯体触媒によるアリルエステル類のアリル位 アルキル化反応1)は炭素−炭素結合構築反応として,有 機合成化学分野において最も研究されている反応の一つ であるが,その報告例としてはパラジウム錯体を触媒と したものが大多数を占める。その一方で,近年になって イリジウム錯体など他の遷移金属錯体を触媒とした反応 例が活発に研究されるようになったが2),3),ルテニウム 触媒による反応例は未だ少なく4),更なる検討の余地が 残されている。 また,遷移金属錯体触媒によるアリルエステル類のア リル位アルキル化反応においては,求核剤としてマロン 酸エステルのアニオンが利用される場合が多い。しかし ながら,より実用的な反応系の構築を目指した場合には アミノ酸エステル誘導体などを求核剤とした触媒反応系 の構築が望まれる。そのような反応例の一つとして,ア ミノ酸誘導体であるアズラクトンを求核剤としたモリブ デン錯体触媒によるアリル位アルキル化反応が報告され ている5),6)。しかしながら,モリブデン錯体に比べてよ り扱いが容易であるルテニウム錯体などを触媒としたア
We investigated the ruthenium-catalyzed allylic alkylation of enantiomerically excess mono-substituted allylic ace-tates with azlactones, and obtained diastereomerically pure and enantiomerically excess azlactone derivetives and oxazoli-none derivatives. The reaction proceeded through the highly branch selective allylic alkylation, and sequential aza-Cope rearrangement. This protocol made easy to separate the two diastereomers, and we also confirmed that the both of azlac-tone derivetives and oxazolinone derivatives exhibit the high enantiomeric excess.
Keywords: ruthenium, allylic alkylation, azlactone, oxazolinone, regioselectivity, diastereomer, enantiomeric excess
アズラクトンを求核剤とした光学活性一置換アリルアセタートの
位置選択的なルテニウム触媒アリル位アルキル化反応と連続的
aza-Cope転位反応
Hiroaki TSUJI
*, Kenta UCHIDA
*, Yuki ISHIBASHI
*and Motoi KAWATSURA
* (Accepted November 30, 2017)* 日本大学文理学部化学科:
〒156-8550 東京都世田谷区桜上水3-25-40
* Department of Chemistry, College of Humanities and Sciences, Nihon
University:3-25-40, Sakurajosui, Setagaya-ku, Tokyo, 156-8550, Japan 日本大学文理学部自然科学研究所研究紀要
No.53 (2018) pp.261−264
1
裕章
*・内田健太
*・石橋勇輝
*・川面 基
*Ruthenium-Catalyzed Regioselective Allylic Alkylation of Chiral Mono-Substituted Allylic Acetates with
Azlactone, and Sequential aza-Cope Rearrangement
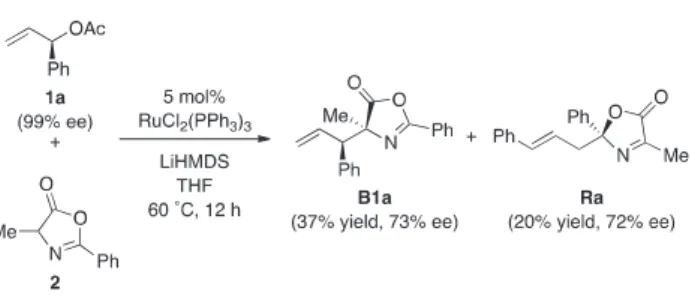
辻 裕章・内田健太・石橋勇輝・川面 基 ─ ─262 ( )2 供与性置換基であるMeO 基を有する1bや電子求引性基 置換基であるCF3基を有するもの1cでの反応において も良好な結果が得られる事が確認できた(Scheme 3)。 た組み合わせで行った (Scheme 1)。反応条件検討の初期 段階で,塩基をLiHMDSとして様々なルテニウム錯体の アリル位アルキル化反応に対する触媒活性を調査した。 具 体 的 に は,5 mol% の RuCl3, RuCl2(p-cymene)(PPh3)2, RuCl2(PPh3)3, Ru2Cl4(CO)6, Ru2Cl4(C10H16)2, RuCl2(C7H8), RuCl2(cod), Cp*RuCl(cod), [RuCp(CH3CN)3]PF6などによ る反応を検討した。その結果,その多くは目的の触媒活 性を示さなかったものの,RuCl2(PPh3)3を触媒とした時 のみ目的のアリル位アルキル化反応が進行する事を確認 した。また,その際,反応基質の消失と共に2 つの異な る生成物(B1aおよびRa)が得られている事が確認でき た。また,B1aおよび Raのエナンチオマー過剰率はそ れぞれ73% ee, 72% ee であった。この結果から,今回の アリル位アルキル化反応は極めて高い位置選択的に進行 しているものの,そのジアステレオ選択性は低く,生じ たジアステレオマーの一方が反応系中で分子内転位 (aza-Cope 転位)を起こし,転位体であるオキサゾリノ ン誘導体Raを与えたと予想できる。また,いずれの場 合にも反応基質の立体化学が生成物へ反映されているも のの,その光学純度は大きく低下している事が確認でき た。以上の反応結果を踏まえて,我々は反応条件の最適 化を行った。 最初に反応温度の最適化を行った。その結果,反応を 40 ˚C で 12 時間攪拌した後に 60 ˚C へと昇温することで, B1aの収率向上と共に,B1aとRa双方のエナンチオマー 過剰率を向上させることに成功した(Scheme 2)。更に 我々は反応溶媒の最適化を行った。 その結果,Table 1 に示したように,dioxaneを溶媒と して反応を行った時に,十分なエナンチオマー過剰率 (B1a: 97% ee, Ra: 89% ee)で,目的とするアズラクトン
誘導体B1a (65%)とオキサゾリノン誘導体Ra (34%)と
を合計収率99%で得る事に成功した(Table 1, run 5)。
また,今回見出した触媒反応条件は異なる置換基を有 する一置換アリルアセタートでの反応に対しても有効で あり,例えば置換基であるベンゼン環上のパラ位に電子
run solvent Yield (%) % ee B1a Ra B1a Ra 1 THF 55 26 92 86 2 dichloroethane 48 31 77 75 3 CH3CN 52 8 81 76 4 toluene 40 34 92 87 5 dioxane 65 34 97 89
Scheme 1. Ruthenium-catalyzed allylic alkylation of
1
with 2.
Scheme 2. Optimization of reaction conditions:
Reaction temperature.
Table 1. Optimization of reaction conditions: Solvent.
run solvent Yield (%) % ee
B1a Ra B1a Ra 1 THF 2 dichloroethane 3 CH3CN 4 toluene 5 dioxane 55 26 92 86 48 31 77 75 52 8 81 76 40 34 92 87 65 34 97 89
Scheme 3. Ruthenium-catalyzed reaction of
1b‒c with 2.
Scheme 4. Reaction pathway of aza-Cope
rearrangement.
Scheme 1. Ruthenium-catalyzed allylic alkylation of
1
with 2.
Scheme 2. Optimization of reaction conditions:
Reaction temperature.
Table 1. Optimization of reaction conditions: Solvent.
run solvent Yield (%) % ee
B1a Ra B1a Ra 1 THF 2 dichloroethane 3 CH3CN 4 toluene 5 dioxane 55 26 92 86 48 31 77 75 52 8 81 76 40 34 92 87 65 34 97 89
Scheme 3. Ruthenium-catalyzed reaction of
1b‒c with 2.
Scheme 4. Reaction pathway of aza-Cope
rearrangement.
Scheme 1. Ruthenium-catalyzed allylic alkylation of
1
with 2.
Scheme 2. Optimization of reaction conditions:
Reaction temperature.
Table 1. Optimization of reaction conditions: Solvent.
run solvent B1a Yield (%) Ra B1a % ee Ra
1 THF 2 dichloroethane 3 CH3CN 4 toluene 5 dioxane 55 26 92 86 48 31 77 75 52 8 81 76 40 34 92 87 65 34 97 89
Scheme 3. Ruthenium-catalyzed reaction of
1b‒c with 2.
Scheme 4. Reaction pathway of aza-Cope
rearrangement.
Table 1 Optimization of reaction conditions: Solvent.
Scheme 1. Ruthenium-catalyzed allylic alkylation of
1
with 2.
Scheme 2. Optimization of reaction conditions:
Reaction temperature.
Table 1. Optimization of reaction conditions: Solvent.
run solvent Yield (%) % ee
B1a Ra B1a Ra 1 THF 2 dichloroethane 3 CH3CN 4 toluene 5 dioxane 55 26 92 86 48 31 77 75 52 8 81 76 40 34 92 87 65 34 97 89
Scheme 3. Ruthenium-catalyzed reaction of
1b‒c with 2.
Scheme 4. Reaction pathway of aza-Cope
rearrangement.
Scheme 1 Ruthenium-catalyzed allylic alkylation of 1 with 2.
Scheme 2 Optimization of reaction conditions: Reaction temperature.
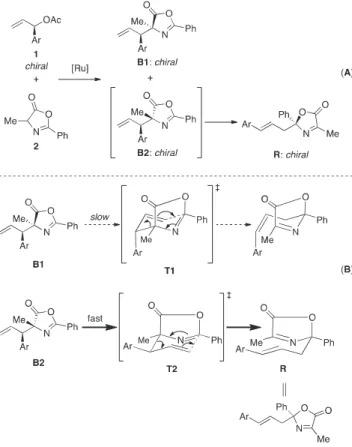
─ ─263 ( ) アズラクトンを求核剤とした光学活性一置換アリルアセタートの位置選択的なルテニウム触媒アリル位アルキル化反応と連続的aza-Cope転位反応 3 エチルで抽出を行った。続いてロータリーエバポレー ターで溶媒を留去して残渣を得,それをシリカゲルクロ マト−グラフィー(ヘキサン/ 酢酸エチル = 4 /1)にて 精製し,対応するアズラクトン誘導体B1a (0.182 mmol) およびオキサゾリノン誘導体Ra (0.095 mmol) をそれぞ れ無色透明の液体として得た。また,エナンチオマー過 剰率はキラルカラムによるHPLC 分析によって決定し た。 (S)-4-Methyl-2-phenyl-4-((R)-1-phenylallyl)oxazol-5(4H )-one ((4S, 1’R)-B1a): Colorless oil. 1H NMR (600 MHz, CDCl3) δ 1.35 (s, 3H), 3.69 (d, J = 9.5 Hz, 1H), 5.09 (dd, J = 1.3, 10.1 Hz, 1H), 5.19 (d, J = 16.9 Hz, 1H), 6.01 (ddd, J = 9.5, 10.1, 16.9 Hz, 1H), 7.26 (d, J = 14.7 Hz, 1H), 7.33 (t, J = 7.7 Hz, 2H), 7.45–7.50 (m, 4H), 7.57 (t, J = 7.7 Hz, 1H), 8.01 (dd, J = 1.4, 8.2 Hz, 2H). 13C NMR (151 MHz, CDCl 3) δ 22.5, 57.6, 73.6, 118.9, 125.9, 127.4, 128.0, 128.5, 128.8, 129.2, 132.7, 135.6, 138.4, 159.9, 180.7. IR (neat) 3405, 2980, 1815, 1739, 1653, 1581, 1493, 1241, 1003, 927, 703. HRMS (ESI): m/z: calcd for C19H18NO2+ [M+H]+ 292.1338, found 292.1328. [α]D26-43.2 (c 3.2, CHCl3) (86% ee). Enantiomer ratio was determined by HPLC using a Daicel CHIRALCEL OJ-H (hexane/2-propanol = 25/1, flow: 1.0 mL/min, 254 nm, 35 ˚C, tR 19.1 min (minor); tR 23.7 min 3.考 察 今回行った反応はScheme 4 に示す反応経路で進行し ていると考えられる(Scheme 4-A)。つまり,アリルア セタート1とアズラクトン2のルテニウム錯体触媒によ る ア リ ル 位 ア ル キ ル 化 反 応 は 位 置 選 択 的 に 進 行 し branch 体のみを与えるものの,そのジアステレオ選択 性は低く,B1およびB2の双方が生じていると考えられ る。しかし,そこで反応温度を高くすることにより一方 のジアステレオマーであるB2が優先的にaza-Cope 転位 を起こしてオキサゾリノン誘導体Rを与える(Scheme 4-A)。なお,アリル位アルキル化反応および aza-Cope 転位いずれの反応段階においても,反応基質であるアリ ルアセタート1の立体化学は保持され,その結果として キラルなアリル位置換体であるアズラクトン誘導体と転 位体であるオキサゾリノン誘導体をそれぞれ高いエナン チオマー過剰率で与えていると考えられる。なお,2つ のアリル位アルキル化体B1および B2のうち,B2が優 先的にaza-Cope 転位を起こす理由は以下のように説明 できる。Scheme 4-Bに示すように,アリル位アルキル化 体はaza-Cope 転位反応を引き起こすに当たって六員環 遷移状態を経由する必要がある。しかしながら,遷移状 態T1はT2に比べてAr基がアキシャル位を占めるために 不利である。そのため,遷移状態T2を経由するB2が優 先的に転位すると説明できる。また,本反応で得られる 2つのタイプの生成物は,いずれも不斉四置換炭素を有 するアミノ酸誘導体である事から,有機合成化学分野に おいて極めて価値ある反応であると言える。 以上,我々はルテニウム錯体触媒による光学活性アリ ルアセタートとアズラクトンとのアリル位置換反応を行 い,アズラクトン誘導体とオキサゾリノン誘導体をそれ ぞれ十分な収率かつエナンチオマー過剰率で得る事に成 功した。 実験の部 代表的な実験例 窒素気流下で,セプタム付きスクリューキャップにア リルアセタート1a (0.28 mmol),アズラクトン 2 (0.42
mmol) および RuCl2(PPh3)3 (5 mol%) を入れ,室温にて dioxane (0.5 mL) に溶解させた。この反応溶液へ 0 ˚C に てLiHMDS (1M in THF) (0.39 mmol) を滴下した後,40 ˚Cにて12時間攪拌した。その後,反応溶液を60 ˚Cへと 昇温し,さらに12 時間加熱攪拌した。アリルアセター ト1aの消失を TLC にて確認した後,反応溶液に飽和炭 酸水素ナトリウム水溶液を加えて反応を停止させ,酢酸
Scheme 1. Ruthenium-catalyzed allylic alkylation of
1
with 2.
Scheme 2. Optimization of reaction conditions:
Reaction temperature.
Table 1. Optimization of reaction conditions: Solvent.
run solvent Yield (%) % ee
B1a Ra B1a Ra 1 THF 2 dichloroethane 3 CH3CN 4 toluene 5 dioxane 55 26 92 86 48 31 77 75 52 8 81 76 40 34 92 87 65 34 97 89
Scheme 3. Ruthenium-catalyzed reaction of
1b‒c with 2.
Scheme 4. Reaction pathway of aza-Cope
rearrangement.
辻 裕章・内田健太・石橋勇輝・川面 基
─ ─264 ( )4
1) (a) Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395-422. (b) Hayashi, T. J. Organomet. Chem. 1999, 576, 195-202. (c) Helmchen, G.; Pfaltz, A. Acc. Chem. Res. 2000, 33, 336-345. (d) Dai, L.-X.; Tu, T.; You, S.-L.; Deng, W.-P.; Hou, X.-L. Acc. Chem. Res. 2003, 35, 659-667. (e) Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921-2943. (f) Trost, B. M. J. Org. Chem. 2004, 69, 5813-5837. (g) Trost, B. M.; Machacek, M. R.; Aponick, A. Acc. Chem. Res. 2006, 39, 747-760. (h) Lu, Z.; Ma, S. Angew. Chem. Int. Ed. 2008, 47, 258-297. (i) Diéguez, M.; Pàmies, O. Acc. Chem. Res. 2010, 43, 312-322.
2) (a) Jiang, X.; Beiger, J. J.; Hartwig, J. F. J. Am. Chem. Soc. 2017, 139, 87-90. (b) Rossler, S. L.; Krautwald, S.; Carreira, E. M. J. Am. Chem. Soc. 2017, 139, 3603-3606. (c) Wu, Q.-F.; He, H.; Liu, W.-B.; You, S.-L. J. Am. Chem. Soc. 2010, 132, 11418-11419. (d) Giacomina, F.; Riat, D.; Alexakis, A. Org. Lett. 2010, 12, 1156-1159. (e) Onodera, G.; Watabe, K.; Matsubara, M.; Oda, K.; Kezuka, S.; Takeuchi, R. Adv. Synth. Catal. 2008, 350, 2725-2732. (f) Nemoto, T.; Sakamoto, T.; Fukuyama, T.; Hamada, Y. Tetrahedron Lett. 2007, 48, 4977-4981. (g) Gnamm, C.; Förster, S.; Miller, N.; Brödner, K.; Helmchen, G. Synlett 2007, 790-794. (h) Alexakis, A.; Polet, D. Org. Lett. 2004, 6, 3529-3532. (i) Kinoshita, N.; Marx, K. H.; Tanaka, K.; Tsubaki, K.; Kawabata, T.; Yoshikai, N.; Nakamura, E.; Fuji, K. J. Org. Chem. 2004, 69, 7960-7964.
3) Kawatsura, M.; Tsuji, H.; Uchida, K.; Itoh, T. Tetrahedron 2011, 67, 7686-7691.
4) (a) Isobe, S.; Terasaki, S.; Hanakawa, T.; Mizuno, S.; Kawatsura, M. Org. Biomol. Chem. 2017, 15, 2938-2946. (b) Mizuno, S.; Shinozawa, T.; Terasaki, S.; Kawatsura, M. Org. Lett. 2017, 19, 504-507. (c) Shinozawa, T.; Terasaki, S.; Mizuno, S.; Kawatsura, M. J. Org. Chem. 2016, 81, 5766-5774. (d) Kawatsura, M.; Uchida, K.; Terasaki, S.; Tsuji, H.; Minakawa, M.; Itoh, T. Org. Lett. 2014, 16, 1470-1473. (e) Kawatsura, M.; Sato, M.; Tsuji, H.; Ata, F.; Itoh, T. J. Org. Chem. 2011, 76, 5485-5488. (f) Kawatsura, M.; Ata, F.; Hirakawa, T.; Hayase, S.; Itoh, T. Tetrahedron Lett. 2008, 49, 4873-4875. (g) Kawatsura, M.; Ata, F.; Hayase, S.; Itoh, T. Chem. Commun. 2007, 4283-4285. (h) Kawatsura, M.; Ata, F.; Wada, S.; Hayase, S.; Uno, H.; Itoh, T. Chem. Commun. 2007, 298-300.
5) (a) Trost, B. M.; Dogra, K. J. Am. Chem. Soc. 2002, 124, 7256-7257. (b) Trost, B. M.; Lee, C. J. Am. Chem. Soc. 2001, 123, 12191-12201. (c) Trost, B. M.; Ariza, X. J. Am. Chem. Soc. 1999, 121, 10727-10737. (d) Trost, B. M.; Heinemann, C.; Ariza, X.; Weigand, S. J. Am. Chem. Soc. 1999, 121, 8667-8668. (e) Trost, B. M.; Lee, C. B. J. Am. Chem. Soc. 1998, 120, 6818-6819. (f) Trost, B. M.; Ariza, X. Angew. Chem. Int. Ed. Engl. 1997, 36, 2635-2637. 6) (a) Frébault, F.; Luparia, M.; Oliveria, M. T.; Goddard, R.;
Maulide, N. Angew. Chem. Int. Ed. 2010, 49, 5672-5676. (b) Weber, M.; Jautze, S.; Frey, W.; Peters, R. J. Am. Chem. Soc. 2010, 132, 12222-12225. (c) Kawatsura, M.; Ikeda, D.; Ishii, T.; Komatsu, Y.; Uenishi, J. Synlett 2006, 2435-2438.
引用文献 (major)).
(S)-2-(3-Phenyl-2-propenyl)-4-methyl-2-phenyloxazol-5(2H)-one ((S)-Ra): Colorless oil. 1H NMR (600 MHz, CDCl3) δ 2.27 (s, 3H), 2.99 (ddd, J = 1.1, 7.5, 14.2 Hz, 1H), 3.05 (ddd, J = 1.1, 7.5, 14.2 Hz, 1H), 5.90 (dt, J = 7.5, 15.8 Hz, 1H), 6.39 (d, J = 15.8 Hz, 1H), 7.22–7.29 (m, 5H), 7.36– 7.41 (m, 3H), 7.58 (d, J = 6.9 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 14.0, 44.8, 107.2, 120.5, 125.9, 126.3, 127.8, 128.6, 128.6, 128.9, 136.4, 136.8, 138.5, 160.4, 165.2. IR (neat) 3060, 3030, 1784, 1654, 1449, 1308, 1148, 967, 747, 695. HRMS (ESI): m/z: calcd for C19H18NO2+ [M+H]+ 292.1338, foundx292.1329. [α]D26- 6.5 (c 1.9, CHCl3) (89% ee). Enantiomer ratio was determined by HPLC using a Daicel CHIRALCEL OJ-H (hexane/2-propanol = 25/1, flow: 1.0 mL/min, 254 nm, 35 ˚C, tR 26.3 min (minor); tR 34.4 min (major)).