この 事業 は 、競輪 の 補助金を受 けて 実 施したも のです 。 URL : http://ringring-keirin.jp/
システム開 発 21- F-4
臨床検査用分析装置における自動校正システム の開発に関するフィージビリティスタディ
報 告 書
- 要 旨 -
平成 22 年 3 月
財団法人 機械システム振興協会
委託先 特定非営利活動法人 日本臨床検査標準協議会
序
わが国経済の安定成長への推進にあたり、機械情報産業をめぐる経済的、社 会的諸条件は急速な変化を見せており、社会生活における環境、都市、防災、
住宅、福祉、教育等、直面する問題の解決を図るためには技術開発力の強化に 加えて、多様化、高度化する社会的ニーズに適応する機械情報システムの研究 開発が必要であります。
このような社会情勢の変化に対応するため、財団法人機械システム振興協会 では、財団法人JKAから機械工業振興資金の交付を受けて、システム技術 開発調査研究事業、システム開発事業、新機械システム普及促進事業を実施し ております。
このうち、システム技術開発調査研究事業及びシステム開発事業については、
当協会に総合システム調査開発委員会(委員長:東京大学名誉教授 藤正 巖氏)
を設置し、同委員会のご指導のもとに推進しております。
本「臨床検査用分析装置における自動校正システムの開発に関するフィージ ビリティスタディ」は、上記事業の一環として、当協会が特定非営利活動法人 日本臨床検査標準協議会に委託し、実施した成果をまとめたもので、関係諸分 野の皆様方のお役に立てれば幸いであります。
平成 22 年 3 月
財団法人 機械システム振興協会
はじめに
本報告書は、財団法人機械システム振興協会より、特定非営利活動法人日本 臨床検査標準協議会(JCCLS)が平成 21 年度事業として受託した「臨床検査用 分析装置における自動校正システムの開発に関するフィージビリティスタデ ィ」の成果をまとめたものである。
JCCLS は平成 16 年度、平成 17 年度、平成 18 年度には(財)機械システム振
興協会より「医療診断システム構築のための基盤整備に関するフィージビリテ ィスタディ」事業を受託し、臨床検査用標準物質の作製や、都道府県を構成単 位とする臨床検査の標準化活動により施設間の測定値差を解消する、いわゆる パッチワーク方式による臨床検査の標準化を提案し、大きな成果をあげてきた。
このパッチワーク方式は、平成 19 年度からは(財)日本臨床衛生検査技師会の 事業として拡大して展開され、施設間差の解消に寄与している。
JCCLS が平成 19 年度から受託した本事業は今年度が最終年度の 3 年目であ
る。初年度の平成 19 年度には平成 20 年度から厚生労働省で新たに施行予定の
「標準的な健診・保健指導プログラム」に必要な内臓脂肪症候群(メタボリッ クシンドローム)に必須な検査 8 項目を、平成 20 年度には日常検査として汎用 されている 22 項目について、臨床検査用分析装置を使用し標準化を推進するた めの自動校正システムの開発に関する F/S を実施した。
平成 21 年度はこの成果をさらに推し進めるべく、平成 19 年度及び平成 20 年 度の結果をもとに臨床検査用分析装置の性能表示規格(案)あり方について検 討を行った。その概要としては、臨床検査用分析装置基礎性能及び測定性能を 検討するための手順書の作成を行い、測定試料等を作製後、これに基づいて参 加企業が所定の作業を実施して統一的な分析装置の性能表示規格(案)のあり 方について検討を行った。この成果は本紙で報告をする。
本 F/S を実施するにあたり、経済産業省のご指導と財団法人機械システム振 興協会のご高配に深謝するとともに、本 F/S にご協力いただいた委員各位に心 より感謝申し上げる次第である。
平成 22 年 3 月
特定非営利活動法人 日本臨床検査標準協議会
目次 序
はじめに
1 F/S の目的
...11.1 事業目的 ...1
1.2 事業概要 ...2
2 F/S の実施体制
...43 F/S の内容
...81章 F/Sの背景 ...8
1.1 装置の性能に関する国内の取り組み状況 ...8
1.2 装置の性能に関する国外の取り組み状況 ...10
2章 検討用試料の調製 ...11
2.1 装置基礎性能の検討用試料の調製 ...11
2.1.1 吸光度の比例性の検討用試料 ...11
2.1.2 測光繰り返し精度の検討用試料 ...11
2.1.3 セル残水の検討用試料 ...11
2.1.4 サンプルプローブ外側及び内側のキャリーオーバの検討用試料 ...11
2.1.5 試薬プローブ内側のキャリーオーバの検討用試料 ...12
2.1.6 撹拌機構(スターラなど)プローブのキャリーオーバの検討用試料 ...12
2.1.7 分注混合能の検討用試料 ...12
2.1.8 試薬プローブの水希釈の検討用試料 ...12
2.1.9 セルのコンタミネーションの検討用試料 ...12
2.1.10 サンプルのクロスコンタミネーションの検討用試料 ...12
2.1.11 サンプル容量の検討用試料 ...12
2.1.12 試薬容量の検討用試料 ...13
2.1.13 サンプルプローブの水持ち込みの検討用試料 ...13
2.1.14 試薬プローブの水持ち込みの検討用試料 ...13
2.1.15撹拌機構(スターラなど)プローブの水持ち込みの検討用試料 ...13
2.2 装置測定性能の検討用試料の調製 ...14
2.2.1 物質濃度測定系(項目:ブドウ糖(GLU))の検討用試料 ...14
2.2.2 酵素活性測定系(項目:AST)の検討用試料 ...14
2.2.3 免疫成分測定系(項目:CRP)の検討用試料 ...15
2.2.4 イオン電極法(項目:Na、K、Cl)の検討用試料 ...16
3章 装置基礎性能の検討手順 ...17
3.1 吸光度の比例性の検討 ...17
3.2 測光繰り返し精度の検討 ...19
3.3 セル残水の検討 ...20
3.4 サンプルプローブのキャリーオーバの検討 ...21
3.4.1 サンプルプローブ外側のキャリーオーバ...21
3.4.2 サンプルプローブ内側のキャリーオーバ...22
3.4.3 サンプルプローブのキャリーオーバ(高感度法による)...24
3.5 試薬プローブ内側のキャリーオーバの検討 ...25
3.6 撹拌機構(スターラなど)プローブのキャリーオーバの検討 ...27
3.7 分注混合能の検討 ...28
3.8 試薬プローブの水希釈の検討 ...29
3.9 セルのコンタミネーションの検討 ...30
3.10 サンプルのクロスコンタミネーションの検討 ...32
3.11 サンプル容量の検討 ...32
3.11.1 平均的な組成のサンプル容量...32
3.11.2 特殊な組成のサンプル容量...34
3.12 試薬容量の検討 ...35
3.13 プローブの水持ち込みの検討 ...36
3.13.1 サンプルプローブの水持ち込み...36
3.13.2 試薬プローブの水持ち込み...37
3.13.3 撹拌機構(スターラなど)プローブの水持ち込み...38
3.14 測定温度の検討 ...39
3.15 血清情報の検討 ...40
3.16 総合性能―日立340キットの検討 ...41
3.17 写真掲載 ...42
4章 装置測定性能の検討手順 ...47
4.1 物質濃度測定系の検討 ...47
4.1.1 項目:ブドウ糖(GLU)...47
4.1.2 精密さの評価 ...47
4.1.2.1 併行精度 ...47
4.1.2.2 日間・日内精密度(室内精密度)...48
4.1.3 精確さの評価(濃度の異なる3種類以上の標準物質を用いた精確さの評価)...49
4.1.4 測定値の比例性 ...50
4.2 酵素活性測定系の検討 ...51
4.2.1 項目:AST ...51
4.2.2 精密さの評価 ...51
4.2.2.1 併行精度 ...51
4.2.2.2 日間・日内精密度(室内精密度)...51
4.2.3 精確さの評価 ...51
4.2.4 測定値の比例性 ...52
4.3 免疫成分測定系の検討 ...53
4.3.1 項目:CRP ...53
4.3.2 精密さの評価 ...53
4.3.2.1 併行精度 ...53
4.3.2.2 日間・日内精密度(室内精密度)...53
4.3.3 精確さの評価 ...54
4.3.4 検出限界 ...54
4.3.5 抗原過剰チェック ...55
4.4 イオン電極法の検討 ...55
4.4.1 項目:Na、K、Cl ...55
4.4.2精密さの評価 ...55
4.4.2.1 併行精度...55
4.4.2.2 日間・日内精密度(室内精密度)...55
4.4.3 精確さの評価(濃度の異なる3種類以上の標準物質を用いた精確さの評価)...55
4.4.4 測定値の比例性 ...56
4.4.5 Cl測定に対する妨害イオンの影響 ...57
5章 装置基礎性能の検討結果 ...58
5.1 吸光度の比例性の検討結果 ...58
5.2 測光繰り返し精度の検討結果 ...59
5.3 セル残水の検討結果 ...60
5.4 サンプルプローブのキャリーオーバの検討結果 ...61
5.4.1 サンプルプローブ外側のキャリーオーバ...61
5.4.2 サンプルプローブ内側のキャリーオーバ...62
5.4.3 サンプルプローブのキャリーオーバ(高感度法による)...63
5.5 試薬プローブ内側のキャリーオーバの検討結果 ...64
5.6 撹拌機構(スターラなど)プローブのキャリーオーバの検討結果...66
5.7 分注混合能の検討結果 ...67
5.8 試薬プローブの水希釈の検討結果 ...68
5.9 セルのコンタミネーションの検討結果 ...69
5.10 サンプルのクロスコンタミネーションの検討結果 ...71
5.11 サンプル容量の検討結果 ...72
5.11.1 平均的な組成のサンプル容量...72
5.11.2 特殊な組成のサンプル容量...73
5.12 試薬容量の検討結果 ...75
5.13 プローブの水持ち込みの検討結果 ...76
5.13.1 サンプルプローブの水持ち込み...76
5.13.2 試薬プローブの水持ち込み...76
5.13.3 撹拌機構(スターラなど)プローブの水持ち込み...77
5.14 測定温度の検討結果 ...78
5.15 血清情報の検討結果 ...80
5.16 総合性能―日立340キットの検討結果 ...82
6章 装置測定性能の検討結果 ...83
6.1 物質濃度測定系の検討結果 ...83
6.1.1 項目:ブドウ糖(GLU)...83
6.1.2 精密さの評価 ...83
6.1.2.1 併行精度 ...83
6.1.2.2 日間・日内精密度(室内精密度)...84
6.1.3 精確さの評価(濃度の異なる3種類以上の標準物質を用いた精確さの評価)...86
6.1.4 測定値の比例性 ...89
6.2 酵素活性測定系の検討結果 ...91
6.2.1 項目:AST ...91
6.2.2 精密さの評価 ...91
6.2.2.1 併行精度 ...91
6.2.2.2 日間・日内精密度(室内精密度)...91
6.2.3 精確さの評価 ...93
6.2.4 測定値の比例性 ...93
6.3 免疫成分測定系の検討結果 ...96
6.3.1 項目:CRP ...96
6.3.2 精密さの評価 ...96
6.3.2.1 併行精度 ...96
6.3.2.2 日間・日内精密度(室内精密度)...96
6.3.3 精確さの評価 ...98
6.3.4 検出限界 ...98
6.3.5 抗原過剰チェック ...100
6.4 イオン電極法の検討結果 ...101
6.4.1 項目:Na、K、Cl ...101
6.4.2 精密さの評価 ...101
6.4.2.1 併行精度 ...101
6.4.2.2 日間・日内精密度(室内精密度)...103
6.4.3 精確さの評価(濃度の異なる3種類以上の標準物質を用いた精確さの評価)...107
6.4.4 測定値の比例性 ...116
6.4.5 Cl測定における妨害イオンの影響 ...124
4 F/S の成果
...1264.1 成果の概要 ...126
4.2 目的に照らし合わせた達成状況 ...128
5 F/S の今後の課題及び展開
...1295.1 自動校正用試料 ...129
5.2 規格化のプロセス:JAIMAS、ISO, JIS ...130
5.3 JAIMA規格性能表示項目(案)...130
5.4 JAIMA規格性能表示 (案)の具体例 ...132
1 F/S の目的
1.1 事業目的
臨床 検査 で汎 用さ れて いる 多く の項 目は 現在 臨 床検 査用 分析 装置 を用 いて 病院 や健診 機関などで測定されているが、末端の施設間で検査データの互換性の確保が要望されてい る。この理由は、例えば、平成 20年4月より施行された40~74歳の国民を対象にする「標 準的な健診・保健指導」では全国で一律の判定値で保健指導が実施されているからである。
しかし平成20年度日本医師会精度管理調査結果でも、健診で実施されている生化学検査8 項目のばらつきは、項目によって異なるが 0.16 %~20.88 %(LDL-C)となっており、互 換性が確保されているとは言い難い。平成19年度本F/Sでは互換性を確保するための手段 として臨床検査用分析装置における自動校正システム構築とその条件を調査検討したとこ ろ、前述の 8 項目のばらつきは 0.9 %~7.8 %に縮小でき、将来、施設間の互換性確保の 目標値として掲げられると考えられる。平成 20年度は22項目について検討を行い、ばら つきは0.1 %~3.5 %であった。
本年度は、臨床検査用分析装置の検査データの信頼性向上に必要である装置の性能表示 規格のあり方に関して検討を行うことを目的とする。この理由としては、現在、性能表示 は各社に委ねられており、統一した規格は定められていない。性能表示に関する規格を設 定することで、性能についての内容が明確になるため、装置の使用者である第三者にも十 分に理解ができ、場合によっては確認や検証が可能となる。使用者間での情報の共有化が できるようになるので、装置企業や機種が異なっても、性能表示規格に関する情報提供が 可能となり、最終的には検査データの信頼性向上に寄与すると考えられる。また、性能表 示の規格は国内外でもシェアリーダーである日本の臨床検査用分析装置の競合力を今後も 維持発展することにもつながる。
本 F/S では(社)日本分析機器工業会の協力を得て、規格案を策定するために、装置企 業各社に直接反映されることになり、さらに使用者である第三者にも装置性能について理 解できるようになる。本 F/S では性能表示規格(案)をまず(社)日本分析機器工業会規 格(JAIMAS)とし、その後 ISO規格原案やJISなみの規格(案)として提案していきたい。
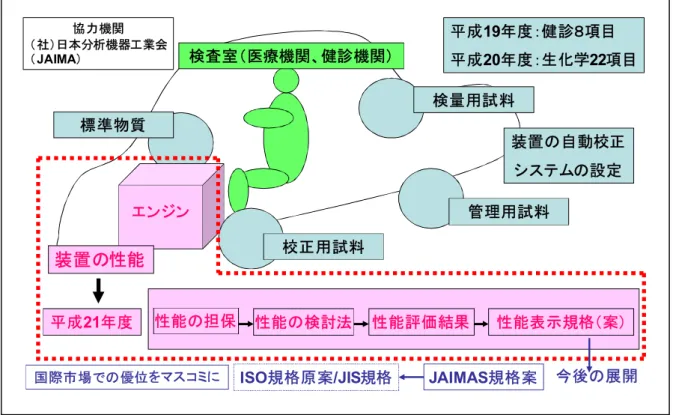
以上を要約すると前年の 2年間で我が国で汎用されている臨床検査用分析装置の自動校 正システムの構築と施設間の互換性確保の検討を実施した。これらの結果に立脚して本年 度は車のエンジン部分に相当する装置性能の表示規格(案)のあり方を策定することとし た。
この規格(案)に関しては将来、JAIMAS規格案さらにISO規格原案やJISなみの規格案 への提案を想定している(図 1.1)。
本 F/S の成果は、検査データの互換性を確保することにつながり、最終的には臨床検査 値のデータベース化が可能になると考えられる。これにより個人の検査データの蓄積によ り、わずかな変化を捉え早期に対策が打てる、また、医療機関での重複検査の削減にもつ ながり、ひいては増大する医療費削減に寄与すると考えられる。
図1.1 F/Sの事業目的とその概要
1.2 事業概要
臨床 検査 用分 析装 置の 検査 デー タの信 頼性 を 高める ため に必 要な 性能 表示 規格 のあり 方に関して検討するために、以下の内容を選定した。各々の実験は(社)日本分析機器工 業会 医療機器委員会に所属している7社が担当する。
現在は装置企業が公開している性能表示が統一されていない、また、解釈が異なるため に使用者間で情報の交換が難しい場合がある。そこで公開する項目の統一や性能を評価す る方法を定め、装置企業や機種が異なっても使用者間で共通理解を図るための性能表示規 格(案)を策定することとした。
(1) 装置基礎性能の検討
装置基礎性能として、以下の項目などの検討を行う。
1) 吸光度の比例性の検討
溶液の希釈系列を作製し、340 nm、410 nm付近の吸光度の比例性がどの程度であるか の測定方法を検討確認する。
2) 測光繰り返し精度の検討
340 nm、410 nm付近の吸光度繰り返し精度の測定方法を検討確認する。
3) セル残水の検討
セルの自動洗浄が終了した後に、セルに残った残水量の測定方法を検討確認する。
4) キャリーオーバの検討
プローブ内外に付着したサンプル・試薬が洗浄後に次の試料へどの程度影響(キャ リーオーバ)するかなどの測定方法を検討確認する。
検査室(医療機関、健診機関)
装置の性能
検量用試料 標準物質
管理用試料
JAIMAS規格案 性能評価結果
性能の検討法 性能の担保
ISO規格原案/JIS規格 エンジン
平成21年度
平成19年度:健診8項目 平成20年度:生化学22項目
校正用試料
装置の自動校正 システムの設定
性能表示規格(案)
今後の展開 国際市場での優位をマスコミに
協力機関
(社)日本分析機器工業会
(JAIMA)
5) 分注混合能及び試薬プローブの水希釈の検討
色素液を用い、分注後混合及び試薬プローブによる水希釈の測定方法を検討確認する。
6) サンプルのクロスコンタミネーションの検討
高濃度と低濃度サンプルのクロスコンタミネーションの測定方法を検討確認する。
7) サンプル容量、試薬容量の検討
サンプル、試薬溶液の容量の正確さと精密さの測定方法を検討確認する。
8) プローブの水持ち込みの検討
サンプルプローブ、試薬プローブの水洗浄後にプローブに付着している洗浄水の量の 測定方法を検討確認する。
9) 測定温度の検討
反応槽及びセル内の温度の正確さ、精密さなどの測定方法を検討確認する。
(2) 装置測定性能の検討
装置測定性能として、以下の項目などの検討を行う。
1) 物質濃度測定系(イオン電極法を除く)の検討
物質濃度測定系の代表として、ブドウ糖(グルコース(GLU))の測定を実施して精 密さ、精確さ、測定値の比例性を検討確認する。
2) 酵素活性測定系の検討
酵素活性測定の代表として、ASTの測定を実施して、精密さ、精確さ、測定値の比例 性を検討確認する。
3) 免疫成分測定系の検討
免疫成分測定の代表として、CRPの測定を実施して、精密さ、精確さ、測定値の比例 性(検出限界)を検討確認する。
4) イオン電極法(Na、K、Cl)の検討
イオン電極法の代表として、Na、K、Clの測定を実施して精密さ、精確さ、妨害イオ ン(ハロゲンイオンなど)の影響などを検討確認する。
2 F/S の実施体制
実施体制として、財団法人機械システム振 興協 会内に総合シ ステム調 査開発委員 会を 、 特定非営利活動法人日本臨床検査標準協議会(JCCLS)内に学識経験者及び技術者からなる 委員会を設置し、(社)日本分析機器工業会(JAIMA)の協力を得て本委託事業を実行する委 員会を組織し、実施計画を策定した。
具体的にはJCCLSの中に設けられた委員会で、実験方法の立案、実験結果の解析や評価 などの検討を行い、各々のデータ取得作業については協力機関である(社)日本分析機器 工業会医療機器委員会に所属する企業が実施した。
社団法人
日本分析機器工業会 協力機関
委員会 特定非営利活動法人 日本臨床検査標準協議会
実験方法の立案、実験結果の 解析や評価などの検討
実験による各々のデータ 取得作業
委託
総合システム調査開発委員会 財団法人機械システム振興協会
表2-1 総 合 シ ス テ ム 調 査 開 発 委 員 会 委 員 名 簿
(順不同・敬称略)
委員長 東京大学 藤 正 巖
名誉教授
委 員 埼玉大学 太 田 公 廣 総合研究機構
教授
委 員 独立行政法人産業技術総合研究所 金 丸 正 剛
エレクトロニクス研究部門
研究部門長
委 員 独立行政法人産業技術総合研究所 志 村 洋 文
デジタルものづくり研究センター
招聘研究員
委 員 早稲田大学 中 島 一 郎 研究戦略センター
教授
委 員 東京工業大学大学院 廣 田 薫 総合理工学研究科
教授
委 員 東京大学大学院 藤 岡 健 彦 工学系研究科
准教授
表 2-2 委員 名簿
(順不同・敬称略)
委員長 独立行政法人 産業技術総合研究所 桑 克 彦 計測標準研究部門
副委員長 日本電子株式会社医用機器事業部 稲 次 稔 医用機器本部
委員 独立行政法人 産業技術総合研究所 千 葉 光 一 計測標準研究部門副部門
委員 香川県立保健医療大学 細 萱 茂 実 委員 九州大学大学院医学研究院保健学部門 大 澤 進 委員 天理よろづ相談所病院臨床病理部 山 本 慶 和 委員 一般社団法人 検査医学標準物質機構 谷 渉 委員 富士レビオ株式会社販売部門プロダクト推進部 望 月 克 彦
委員 積水メディカル株式会社検査薬営業統括部 鈴 木 英 明 カスタマーサポートセンター分析グループ
委員 和光純薬工業株式会社臨床検査薬研究所 花 田 寿 郎 委員 株式会社シノテスト品質管理部 梅 原 実 委員 ニットーボーメディカル株式会社学術部 笠 原 正
委員 株式会社日立ハイテクノロジーズ 矢 辺 良 平 ナノテクノロジー製品事業本部那珂事業所
委員 株式会社島津製作所分析計測機器事業部 玉 井 哲 男 委員 東芝メディカルシステムズ株式会社 山 岸 和 年
検体検査システム事業部
委員 東芝メディカルシステムズ株式会社 田 中 晶 子 検体検査システム事業部
委員 株式会社日立ハイテクノロジーズ 三 村 智 憲 ナノテクノロジー製品事業本部那珂事業所
委員 株式会社日立ハイテクノロジーズ 福 薗 真 一 ナノテクノロジー製品事業本部那珂事業所
委員 ベックマン・コールター・バイオメディカル株式 間 部 杉 夫 会社学術部
委員 ベックマン・コールター・バイオメディカル株式 今 井 直 也 会社商品開発部アプリケーショングループ
委員 日本電子株式会社医用機器事業部 柏 木 泰 敏 医用機器本部応用研究グループ
委員 古野電気株式会社システム機器事業部 山 内 和 夫 MEビジネスユニット開発課
委員 株式会社エイアンドティー 櫻 井 義 久 CA開発ユニット
委員 株式会社エイアンドティー 榊 徹 CA開発ユニット
委員 株式会社常光医療機器開発部 秋 山 英 時
委員 社団法人日本分析機器工業会 柾 谷 榮 吾 (2009年7月9日まで)
委員 社団法人日本分析機器工業会 林 健 太 郎
(2009年7月10日より) 委員 一般社団法人 検査医学標準物質機構 皮籠石 宏 親
(2009年12月4日まで)
3 F/S の内容 1章 F/S の背景
1.1 装置の性能に関する国内の取り組み状況
臨床検査用の自動分析装置は、日常臨床に用いる大部分の臨床検査項目の測定を迅速に かつ精密に行うことができる主要な分析装置である。中でも自動分析装置は、診療におけ るスクリーニング検査や健診などに用いられている主力装置になっている。したがって自 動分析装置によるこれら臨床検査の質は、自動分析装置の性能に大きく依存している。
これらの自動分析装置の性能に関する基礎検討は、自動分析装置が臨床検査の領域に適 用されて以降本格的に開始されている1)。これらの基礎検討の内容は学会レベルでまとめら れるようになり、なかでも日本臨床化学会の教育委員会及び機器専門委員会では、臨床検 査室における機器試験法マニュアル 2)としてまとめ、一般に用いられるきっかけとなった。
その後、自動分析装置の性能及びこれに適用する日常検査法の性能に関しては、日本臨床 検査自動化学会科学技術委員会で、遺伝子検査を除く臨床検査の自動化と情報に関する標 準化の作業を開始した。その結果、2001 年には装置メーカ及び試薬メーカの協力を得て汎 用自動分析装置の性能確認試験法マニュアル3)としてまとめられた。これは主としてユーザ レベルでの試験法マニュアルとして日常検査に必要となる作業あるいは教育マニュアルと して使用されている。
汎用自動分析装置の性能確認試験法マニュアルは、生化学成分検査や免疫成分検査に用 いられている自動分析装置の性能を確認するためのものである。いわゆる生化学自動分析 装置は、多様なニーズへ対応できる機能が搭載されたランダムアクセス型が主流である。
このような装置の測定原理は、大部分が吸光光度法である。この測定原理による自動分析 装置の性能因子は大別すると正確さと精密さになり、それぞれについて測光系と測定系に なる。測光系には吸光度の比例性、測光繰り返し精度、見かけのモル吸光係数が、また、
測定系にはキャリーオーバ、クロスコンタミネーション、分注・混合能、プローブの水持 ち込み、サンプル容量、試薬容量、測定温度などがある。
分析装置の性能には、個別特性と総合特性がある。前者はメーカにより部品ごとに把握 されている。後者はメーカにより仕様として提示されるものである。これに対してユーザ が日常検査で用いる場合は、後者の総合特性としての性能をメーカの仕様を試験して確認 することになる。
その後、引き続いて日本臨床検査自動化学会科学技術委員会では、2002 年に試験法マニ ュアルの第2冊目として日常検査法の性能試験法マニュアル4)をまとめた。この日常検査法 の性能試験法マニュアルは、生体試料を分析する日常検査法の測定性能を把握するための ものである。このマニュアルで取り上げた対象測定領域は、生体試料分析の物質濃度測定 系、酵素活性測定系、免疫成分測定系である。また、これらの測定系の試薬は自動分析装
置を用いた日常検査法に用いる市販キットである。
このマニュアルでの試験内容は、これまで報告された試験法を実際に測定できるように 組み立て、現状で一般化した方法である。試験項目の構成は、基本性能試験として物質濃 度測定系で、精密さの評価(併行精度、日間・日内精密度、実試料測定値の精密さ)、精確 さの評価(標準を測定する方法、濃度の異なる 3 種以上の標準物質を用いる方法、比較対 照法による分析法間比較)、測定値の比例性(純物質の濃度系列、実試料の混合比系列)、
反応タイムコース、干渉試験(干渉試料による方法、干渉薬物の影響試験)、回収試験及び 相関性試験とした。次いで他の測定系に追加される項目として、酵素活性測定系では酵素 反応性、免疫成分測定系では測定精密さの評価(RER法、PP法)、検出限界(SD法、定量 限界、実効感度)、干渉試験(共存物質の影響試験)、抗原過剰チェック(反応速度比法)
及び採血管の影響とした。
装置基礎性能の各検討法は、現状で一般的に適用可能の範囲のものであり、装置の特性 を必ずしも完全に表現できるものではないが、実用上の性能として把握することは可能で ある。また、試験法の実施に当たっては、分析装置の運転を日常の運転方式と変更して行 う必要があるので必ずメーカの技術者との連携が重要である。
これら臨床検査用の自動分析装置は、現在では高性能化し、国内外で日本製のものが大 活躍している。また、医療の多様化により自動分析装置の担当者も必ずしも専従ではない ことから、装置それ自体に性能の維持・監視機構などが求められ、いつでも安心して操作 でき、精密で安定な測定結果を迅速に得ることの保証が強く求められるようになった。こ のような背景から、これらの自動分析装置には、メーカによる性能仕様表示の標準化の必 要性が求められている。
文献
1)桑 克彦責任編集:実践臨床検査機器マニュアル.サイエンスフォーラム、東京、1985.
2)日本臨床化学会教育委員会・機器専門委員会:臨床検査室における機器試験法マニュ アル、ありい出版、大阪、1993.
3)日本臨床検査自動化学会科学技術委員会. 汎用自動分析装置の性能確認試験法マニュア ル.日本臨床検査自動化学会会誌26(Suppl.1)、2001.
4)日本臨床検査自動化学会科学技術委員会. 日常検査法の性能試験法マニュアル.日本臨 床検査自動化学会会誌27(Suppl.1)、2002.
1.2 装置の性能に関する国外の取り組み状況
装置の性能に関する国際的な規格としては、ISO 15197:2003 In vitro diagnostic systems – Requirements for blood-glucose monitoring systems for self-testing in managing diabetes mellitus
(体外診断検査システム - 糖尿病管理における自己測定のための血糖モニターシステムに
対する要求事項)、及びISO 17593:2007 Clinical laboratory testing and in vitro diagnostic test systems – Requirements for in vitro monitoring systems for self-testing of oral anticoagulant therapy
(臨床検査と体外診断検査システム - 抗凝固薬治療の自己測定のための体外モニターシス
テムに関する要求事項)がISO/TC212 Clinical laboratory testing and in vitro diagnostic systems から発行されているが、本F/Sの対象である汎用の生化学自動分析装置に対する国際規格は 制定されていない。日本の臨床検査用分析装置は世界的に高い製品競争力を有しているた め、日本が中心となって性能規格をまとめることにより、グローバルなデジュールスタン ダードとなることが期待できる。
2 章 検討用試料の調製
今回の検討で使用した試料は全て(社)検査医学標準物質機構(ReCCS)にて調製した。
2.1 装置基礎性能の検討用試料の調製
2.1.1 吸光度の比例性の検討用試料
試料液は、340 nm付近はβ-NADHの0.1 Mトリス塩酸緩衝液(pH7.8)、410 nm付近は 4-NPのアルカリ性(0.2 M NaOH)溶液とした。試料液は分光光度計を用いて、スペクトル バンド幅(SBW)2 nm以下、光路長1 cmのキュベットを使用して自動分析装置の測定波長 でABS 3.0~3.1の範囲に入るように濃度を調整した。ABS 3.0(410 nm)試料液は 4-NP 23.7 mg /L、ABS 3.0(340 nm)試料液はβ-NADH 375 mg /Lとした。
溶媒として用いた0.2 M NaOHは8.0 g のNaOHを水に溶かして 1 Lにした。また、0.1 M トリス塩酸緩衝液はトリス(ヒドロキシメチル)アミノメタン 12.1 gを約0.9 Lの水に溶
かし、6 M HClを加えてpH7.8 にして水で1 Lに合わせて作製した。それぞれに界面活性
剤(0.01 %相当のトリトン X-100)を添加した。
次にβ-NADHのABS 3.0の試料液に0.1 Mトリス塩酸緩衝液を希釈液として加えて、0/10
から10/10 の11段階の容量比希釈系列を作製した。4-NP についても0.2 M NaOHを希釈 液として同様に作製した。(表2.1.1)
表2.1.1 吸光度の比例性の検討用試料作製のための容量比希釈系列
No. 1 2 3 4 5 6 7 8 9 10 11
希釈液 10 9 8 7 6 5 4 3 2 1 0
試料液(ABS 3.0) 0 1 2 3 4 5 6 7 8 9 10
2.1.2 測光繰り返し精度の検討用試料
吸光度の比例性で調製したABS 3.0のβ-NADHの試料液を希釈液でそれぞれ3倍、1.5 倍に希釈して、測定主波長で ABS 1.0及び 2.0の試料液を調製した。4-NPの試料液につい ても同様に希釈してABS 1.0 及び2.0の試料液を調製した。
2.1.3 セル残水の検討用試料
ABS 2のオレンジG(OG)水溶液を調製した。478 nmの場合、130 mg/L OG水溶液のABS は5になるので、これに希釈水(脱イオン水に界面活性剤として0.01 %相当のトリトン
X-100 を添加)を加えて2.5倍に希釈した。
2.1.4 サンプルプローブ外側及び内側のキャリーオーバの検討用試料
ヒトプール血清(PS)にオレンジG(OG)を加え124 g/Lとしたものを 0.2 μm メンブ ランろ過して ABS 5,000(478 nm)のOG/PS液を調製した。
2.1.5 試薬プローブ内側のキャリーオーバの検討用試料
脱イオン水(トリトンX-100 を0.01 %相当含む)にオレンジG(OG)を加え130 g/Lに したものを0.2 μm メンブランろ過してABS 5,000(478 nm)のOG 水溶液を調製した。
2.1.6 撹拌機構(スターラなど)プローブのキャリーオーバの検討用試料
試薬プローブ内側のキャリーオーバの検討用試料であるABS 5,000 のOG 水溶液をその まま用いる。
2.1.7 分注混合能の検討用試料
試薬プローブ内側のキャリーオーバの検討用試料であるABS 5,000 のOG 水溶液を脱イ オン水(トリトンX-100を0.01 %相当含む)で 1,000倍に希釈して ABS 5オレンジG(OG) 水溶液を調製した。
2.1.8 試薬プローブの水希釈の検討用試料
分注混合能の検討用試料で用意したABS 5 OG 水溶液を脱イオン水(トリトンX-100を
0.01 %相当含む)で 5倍に希釈して ABS 1オレンジG(OG)水溶液を調製した。
2.1.9 セルのコンタミネーションの検討用試料
リン酸ナトリウムを脱イオン水(トリトン X-100を0.01 %相当含む)で希釈して以下の 4濃度のリン酸塩水溶液を調製した。
① 0.1 Mリン酸塩(NaH2PO4・2H2O): NaH2PO4 (MW156.01)1.56 g/dL水溶液
② 1.0 Mリン酸塩(NaH2PO4・2H2O): NaH2PO4 (MW156.01)15.6 g/dL水溶液
③ 200 mMリン酸塩: 1.0 Mリン酸塩を脱イオン水で5倍希釈
④ 20 mMリン酸塩: 0.1 Mリン酸塩を脱イオン水で5倍希釈
2.1.10 サンプルのクロスコンタミネーションの検討用試料
LD添加PS原液: LD標品(ブタ心臓由来、5,000 U/mL)15 mLをプール血清(PS)25 mL に添加して、0.2 μm メンブランろ過して調製した。このLD添加 PS原液のLD活性値は 140万U/Lであった。LD添加PS原液の活性値は生理食塩水で1,000 倍に希釈した試料を 測定して求めた。
2.1.11 サンプル容量の検討用試料
(1) 平均的な組成のサンプル容量の検討用試料
オレンジG 血清液(色素原液):オレンジ G を血清に30 g/L相当加え、ゆっくり撹拌して
溶解させ0.2 μm メンブランろ過して ABS 1,200(478 nm)の色素原液を調製した。比重瓶
(ピクノメータ-)を用いて測定した色素原液の比重は1.037(25 ℃)であった。
(2) 特殊な組成のサンプル容量の検討用試料
オレンジG サンプル原液(色素原液):溶媒としてプール血清、マトリックス補正溶液
(PEG200 添加、粘度1.6)、脂質成分調整血清の 3種を以下のとおり用意した。
① プール血清:健常ヒトプール血清の凍結品(-60 ℃以下保管)。
② マトリックス補正溶液: 約75 mLの水に 16.2 gのPEG200、0.238 gのHEPESを加え て溶解させ1 M NaOH でpH7.4に調整後、水を加えて 100 mLに合わせて作製した16.2 (w/v)% PEG 200+10 mmol/L HEPES(pH7.4)溶液(冷蔵保管)。
③ 脂質成分調整血清:前記のプール血清をシリカ吸着法で脱脂処理して血清中の脂質成分 を完全に除去した凍結品(-60 ℃以下保管)。
これら3種の材料にオレンジGを30 g/L相当加え、ゆっくり撹拌して溶解させ0.2 μm メ ンブランろ過してABS 1,200(478 nm)の OG血清液(平均的組成血清)、マトリックス調 整OG溶液、OG 脂質成分調整血清液(校正用試料血清)をそれぞれ調製した。比重瓶(ピ クノメータ-)を用いて測定した各色素原液の比重及びウベローデ粘度計を用いて測定し た粘性率は以下のとおりである。(表2.1.11)
表 2.1.11 サンプル容量の検討用試料の物理的性状
色素原液 比 重(25 ℃) 粘性率(mPa s、20 ℃)
OG血清液(平均的組成血清) 1.0370 1.75 マトリックス調整OG溶液 1.0368 1.77 OG脂質成分調整血清液
(校正用試料血清) 1.0362 1.74
2.1.12 試薬容量の検討用試料
オレンジG 水溶液(OG 色素原液):オレンジ Gを水(0.01 %相当のトリトンX-100を含
む)に 30 g/L相当加え、ゆっくり撹拌して溶解させ 0.2 μm メンブランろ過してABS 1,200
(478 nm)のOG色素原液を調製した。比重瓶(ピクノメータ-)を用いて測定した色素
原液の比重は 1.032(25 ℃)であった。
2.1.13 サンプルプローブの水持ち込みの検討用試料
ABS 1.0のOG/生食液:生理食塩水(0.01 %相当のトリトンX-100を含む0.9 % 塩化ナ トリウム水溶液)にオレンジG(OG)を加え26 mg/Lとしたものを0.2 μm メンブランろ
過してABS 1.0(478 nm)の OG/生食液を調製した。
2.1.14 試薬プローブの水持ち込みの検討用試料
ABS 1.0のOG生食液:サンプルプローブの水持ち込みの検討用試料と同様の材料を用
いた。
2.1.15 撹拌機構(スターラなど)プローブの水持ち込みの検討用試料
ABS 2.5のOG 生食液:生理食塩水(0.01 %相当のトリトン X-100を含む0.9 % 塩化ナ トリウム水溶液)にオレンジG(OG)を加え65 mg/Lとしたものを0.2 μm メンブランろ 過してABS 2.5(478 nm)の OG/生食液を調製した。
2.2 装置測定性能の検討用試料の調製
2.2.1 物質濃度測定系(項目:ブドウ糖(GLU))の検討用試料
(1) 精密さの検討用試料
測定試料は限外ろ過とグルコースを添加する方法により、ヒトプール血清のグルコース 濃度を基準範囲の下限値付近(L)、基準範囲の上限値付近(M)、及び臨床的な異常濃度域
(H)の3種類の濃度に調整したものをメンブランろ過(0.2 μm)して作製し、検討実験 期間中に目的物質の値が変化しないように-60 ℃以下で凍結保管した。測定試料のグルコ ース濃度は以下を準備した。
L:50~80 mg/dL程度 M:110 mg/dL程度 H:350 mg/dL程度
(2) 精確さの検討用標準物質
本試験に用いる標準物質は、含窒素・グルコース常用標準物質(JCCRM 521-9:ReCCS) の3濃度を用いた。グルコース濃度の認証値は低、中、高濃度で各々93.4±0.9、152.1±1.4、
248.0±2.2 mg/dLである。
(3) 測定値の比例性検討用試料
限外ろ過法とグルコースを添加する方法により、ヒトプール血清のグルコース濃度を 20、
1,000 mg/dL付近に調整したものをメンブランろ過(0.2 μm)して、それぞれ低値試料(L)、
高値試料(H)とした。これらのLとH の試料を用い、以下の11段階の混合比系列(容量 比)を作製して凍結保管(-60 ℃以下)した。(表2.2.1)
表2.2.1 測定値(GLU)の比例性検討用試料作製のための混合比系列
No. 1 2 3 4 5 6 7 8 9 10 11
低値試料(L) 10 9 8 7 6 5 4 3 2 1 0 高値試料(H) 0 1 2 3 4 5 6 7 8 9 10
2.2.2 酵素活性測定系(項目:AST)の検討用試料
(1) 精密さの検討用試料
ヒトプール血清にAST(ヒト組換え体、胚型遺伝子)を添加する方法により、ASTの活
性単位を15、35、500 U/L付近に調整したものをメンブランろ過(0.2 μm)して、それぞ
れL、M、H の測定試料とした。これらの試料はドライアイス中で急速凍結して、フリー ザー(-80 ℃以下)で保管した。
(2) 精確さの検討用標準物質
本試験に用いる標準物質は、常用酵素標準物質(JCCLS CRM-001b)の1濃度を用いた。
AST活性の認証値は169±4 U/Lである。
(3) 測定値の比例性検討用試料
ヒトプール血清にAST(ヒト組換え体、胚型遺伝子)を添加する方法により、ASTの活
性単位15、1,000 U/L付近に調整したものをメンブランろ過(0.2 μm)して、それぞれ低値
試料(L)と高値試料(H)とした。これらLとH の試料を用いて、以下の11段階の混合 比系列(容量比)を作製してドライアイス中で急速凍結後、フリーザー(-80 ℃以下)で 保管した。(表2.2.2)
表2.2.2 測定値(AST)の比例性検討用試料作製のための混合比系列
No. 1 2 3 4 5 6 7 8 9 10 11
低値試料(L) 10 9 8 7 6 5 4 3 2 1 0 高値試料(H) 0 1 2 3 4 5 6 7 8 9 10
2.2.3 免疫成分測定系(項目:CRP)の検討用試料
(1) 精密さの検討用試料
ヒトプール血清(CRP 濃度:0.03 mg/dL)にCRP 高純度認証標準物質(NMIJ CRM 6201-a :認証値39.5±1.9 μmol/kg、日常濃度単位では91.5±7.4 mg/dLに相当)を添加する 方法により、CRP濃度0.1、1、3 mg/dL付近に調整したものに0.04 %相当のアジ化ナトリ ウムを添加した。その後メンブランろ過(0.2 μm)して、それぞれ L、M、Hの測定試料と した。これらの試料は冷蔵で保管した。
(2) 精確さの検討用標準物質
本試験に用いる標準物質は、CRP国際標準品DA472/IFCCの1濃度を用いた。CRP 濃度 の認証値は4.18±0.25 mg/dLである。
(3) 検出限界検討用試料
盲検試料:盲検試料としてCRPフリー血清(オリエンタル酵母工業製、CRP Free Serum G1: Lot 55,304,901)を用いた。本品はアフィニティ法により高度に精製処理されており、
現在市販のCRP測定試薬ではいずれも不検出レベルであった。
低値試料:盲検試料としたCRPフリー血清にCRP高純度認証標準物質(NMIJ CRM
6201-a)を添加する方法により、CRP濃度0.1 mg/dLに調製した試料を盲検試料で11段階
の混合比系列(容量比)に希釈した低値試料を以下のとおり作製し、冷蔵で保管した。(表 2.2.3)
表2.2.3 測定値(CRP)の検出限界検討用試料作製のための混合比系列
No. 1 2 3 4 5 6 7 8 9 10 11
0.1 mg/dL 試料 0 0.25 0.5 1 2 3 4 5 6 8 10 盲検試料 10 9.75 9.5 9 8 7 6 5 4 2 0
2.2.4 イオン電極法(項目:Na、K、Cl)の検討用試料 (1) 精密さの検討用試料
測定試料はイオン交換法と電解質を添加する方法により、ヒトプール血清の電解質濃度 を基準範囲の下限値付近(L)、基準範囲の上限値付近(M)、及び臨床的な異常濃度域(H) の3種類の濃度に調整したものをメンブランろ過(0.2 μm)して作製し、検討実験期間中 に目的物質の値が変化しないように-60 ℃以下で凍結保管した。測定試料の電解質濃度は 以下を準備した。
L :Na 135、 K 3.5、 Cl 95 mmol/L程度 M :Na 145、 K 4.8、 Cl 108 mmol/L程度 H :Na 160、 K 6.0、 Cl 120 mmol/L程度
(2) 精確さの検討用標準物質
本試験に用いる標準物質はイオン電極用一次実試料標準物質(JCCRM 111-6:ReCCS)
の3濃度を用いた。電解質濃度の認証値は低、中、高濃度でNaが各々123.9±0.3、141.3±0.4、 157.2±0.4 mmol/L、Kが各々3.246±0.010、4.473±0.017、5.662±0.018 mmol/L、Clが各々89.8±0.2、
106.4±0.3、120.8±0.4 mmol/Lである。
(3) 測定値の比例性検討用試料
イオン交換法と電解質を添加する方法により、ヒトプール血 清の電解 質濃度をNa 110、 K 2.0、 Cl 70 mmol/L 程度とNa 160、K 8.0、Cl 120 mmol/L程度に調整したものをメンブ ランろ過(0.2 μm)して、それぞれ低値試料(L)、高値試料(H)とした。
これらのLとHの試料を用いて、以下の11段階の混合比系列(容量比)を作製して、
凍結保管(-60 ℃以下)した。(表 2.2.4)
表2.2.4 測定値(Na、K、Cl)の比例性検討用試料作製のための混合比系列
No. 1 2 3 4 5 6 7 8 9 10 11
低値試料(L) 10 9 8 7 6 5 4 3 2 1 0 高値試料(H) 0 1 2 3 4 5 6 7 8 9 10
3 章 装置基礎性能の検討手順
日本臨床検査自動化学会が2001年に発行した「汎用自動分析装置の性能確認試験法マニ ュアル」を基に装置の各機能の基礎性能を表示する際のデータ取得に係わる検討手順を以 下に設定した。なお、各検討で必要な試料は本F/Sにおいては2章に記載の検査医学標準物
質機構(ReCCS)にて調製したものを使用したが、通常はそれぞれの検討項目に記載の調製
法にしたがい用意する。また、本章では説明の一部に写真を参照しているが、それらはま とめて3.17 写真掲載に掲載している。そこで使用した装置の外観と分析部全景は3.17 写 真掲載 写真1、2に示す。
3.1 吸光度の比例性の検討 (1) 目的
試料液の分光吸収スペクトルにおける極大吸収近傍の波長で試料液の濃度系列に対す る測定吸光度の比例性を確認する。
(2) 方法及び材料 1) 試料液の調製
試料液は、340 nm付近はβ-NADHのアルカリ性(0.2 M NaOH)溶液、410 nm付近は4-NP のアルカリ性(0.2 M NaOH)溶液を用いる。試料液は分光光度計を用いて、スペクトルバ ンド幅(SBW)2 nm以下、光路長1 cmのキュベットを使用して自動分析装置の測定波長で
ABS 3.0~3.1の範囲に入るように濃度を調整し、各々200 mL準備する。
340 nmで吸光度(ABS)3.0の試料液は73 mg β-NADH/0.2 M NaOH 200 mL(*)、410 nmで ABS 3.0の試料液は4.73 mg 4-NP/0.2 M NaOH 200 mL(*)を参考に調製する。なお、溶媒及 び希釈液の0.2 M NaOHは8.0 g NaOH/L(*)で作製する。
注1:β-NADHの溶媒及び希釈液に0.1 Mトリス塩酸緩衝液を使用すると調 製後 、 より安定である。0.1 Mトリス塩酸緩衝液はトリス(ヒドロキシメチル)アミ ノメタン12.1 gを約0.9 Lの水に溶かし、6 M HClを加えてpH7.8にして水で1 L(*)として調製する。
注2: (*)印の溶液には界面活性剤を添加(例えばトリトンX-100を0.01 %など)し たものを用いるとセルに対する親水性がよくなりセル内面に気泡が付着し にくくなる。
2) 希釈系列の作製(3.17 写真掲載 写真3参照)
ホールピペットを用いて0/10から10/10の11段階の希釈系列を作製する。(表3.1-1) 注3: 希釈系列の作製を重量法で行う場合は、試料液の比重を測定して容量換算
をする。
表3.1-1 希釈系列の作製例(容量単位:mL)
No. 1 2 3 4 5 6 7 8 9 10 11
β-NADH 0 1 2 3 4 5 6 7 8 9 10
希釈液 10 9 8 7 6 5 4 3 2 1 0
3) 測定
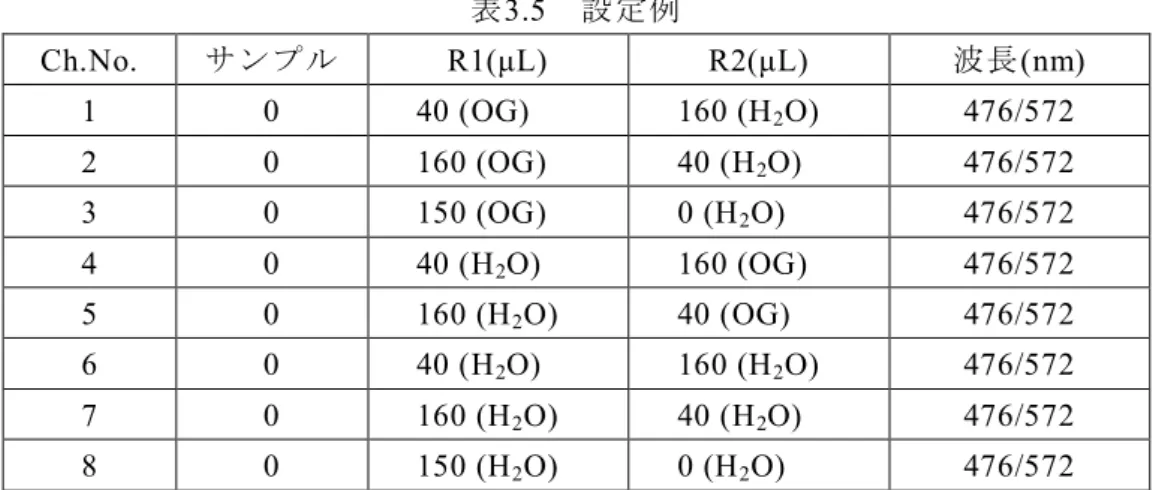
測定波長をセットして調製した希釈試料を直接(反応/測定)セルに用手分注(例えば200 μL)して吸光度を測定する。各試料液について5重測定する。(表3.1-2)
表3.1-2 設定例
試料 測定波長:二波長系(主/副波長)
β-NADH/0.2 M NaOH 340/412 nm
4-NP/0.2 M NaOH 404/500 nm
なお、測定の副波長は、試料の吸収スペクトルで吸収がない波長域を用いる。
注4: セルヘ分注した試料液は恒温状態で測定する。
注5: セルヘの試料液の用手分注は、装置を通常運転して、所定の位置でセルに 入れる。使用するセル数の設定は、測定数より多めに入力しておくとよい。
また、分注器具は、例えばエッペンドルフ型ピペットを用いる。さらにあ らかじめ予備トレーニングしておく。(3.17 写真掲載 写真4参照)
注6: 装置により、例えば日本電子(JEOL)のBMシリーズなどでは、セルにあら かじめ試料液を分注しておき、装置を運転して測定することもできる。
注7: サンプルプローブ、試薬プローブ、撹拌機構(スターラなど)などの影響 をなくすために、これらははずすかあるいは作動しないように止めておく。
4) 分光光度計を用いた吸光度の測定
調製した希釈系列は、分光光度計を用いて試料に応じて340 nmまたは410 nmで吸光度 を測定しておく。これは調製した希釈系列の比例性を検証するのに用いる。
5) 計算
得られた測定結果とそれに基づき算出した相対吸光度(%)を集計用紙(様式は本編
6.5.1 装置基礎性能の検討結果データ集 1 吸光度の比例性、測定データ参照)に記入して
おく。
相対吸光度(%)
=(希釈系列ごとの平均吸光度:En)/{n×(希釈系列1/10の平均吸光度:E1)}×100 n: 1~10 は希釈系列(n/10)に対応
なお、所定の吸光度におけるずれの程度は相対吸光度偏差(%)で示し、これは作図を して求める。また、表示の方法は、相対値の幅で表す。(図3.1)
表示例: 100±3 %:ABS 0.000~2.000 100±5 %:ABS 0.000~3.000
100% 相対吸光度偏差(%)
希釈系列の吸光度 所定吸光度
図3.1 作図例 相対吸光度(%)
3.2 測光繰り返し精度の検討 (1) 目的
二波長での吸光度の繰り返し測定精度を確認する。
(2) 方法及び材料 1) 試料液の調製
測定の主波長でABS 1.0及び2.0の試料液を各50 mL調製する。用いる試料液の組成は 3.1 吸光度の比例性の検討の吸光度の比例性で使用のものと同じ。
2) 測定
測定波長をセットして調製した各試料液を用手法にて20個分のセルに分注(例えば200 μL)しその吸光度を測定する。(表3.2-1)
表3.2-1 二波長測定での設定例
試料 β-NADH β-NADH 4-NP 4-NP
吸光度(ABS) 1.0 2.0 1.0 2.0
波長:主/副 nm 340/412 340/412 404/500 404/500
注1: サンプルプローブ、試薬プローブ、撹拌機構(スターラなど)などの影響 をなくすために、これらははずすかあるいは作動しないように止めておく。
3) 計算
得られた測定結果とそれに基づき平均値(mean)、最大値(max)、最小値(min)、範囲(range)、 標準偏差(SD)、変動係数(CV(%))を計算してこれらを集計用紙(様式は本編6.5.1 装置基礎 性能の検討結果データ集 2 測光繰り返し精度、測定データ参照)に記入する。
注2: 測定値にはセルの光路長のばらつきも含まれる。
3.3 セル残水の検討 (1) 目的
装置による(反応/測定)セルの自動洗浄が終了した後に、セルに残った洗浄水の量を確認 する。
(2) 方法及び材料
注1: セル残水の試験法には、オレンジG水溶液を用い、対照のセルとして長時 間乾燥させたものを使用する方法もあるが、ここではセルの乾燥操作を伴 わない試験法を示す。
1) 対照セルの残水除去器具
注2: セル内の残水を除去するために用いる器具は、例えば残水吸収用の綿棒な どを準備する。この場合は綿棒(軸が紙製のもの)の綿球部を切り落とした 軸の先端に、キムタオル5枚分を三角形に切り取ったものを、先端が刷毛 のような状態に巻き付け、その上端を本綿糸で軸に縛りつける。これらは 1回の試験に予備試験を含めて少なくとも20本は必要になる。(3.17 写真 掲載 写真5参照)
なお、これらを用いる場合は、吸収の程度をみるために回収試験を行い、
分注水の回収率が97 %以上であることを確認しておく。
注3: 回収試験は洗浄後の一連のセルを取り出し、セル内を完全に乾燥させるか、
あるいは予備の乾燥させたセルを用いる。セルに分注した水の量から、吸 収用綿棒などで吸収して残水量を重量法で求める。用いる天秤の感量は0.1 mg以下のもの、マイクロピペットを用いて2 μL及び3 μLのH2O(脱イオン水 など)をそれぞれ5個のセルに分注して回収率を求める。
なお、吸収用綿棒によるセルヘの挿入は、セルの底面まですばやく挿入し、
装置での設定セル駆動時間に合わせた一定時間保持する。また、綿棒など の取り扱いは、吸収した水の蒸発を防げるように、スクリューキャップ付 きのチューブなどを用いる。
2) 試料の調製
ABS 2のオレンジG(OG)水溶液を作製する。478 nmの場合、15 mg/dL OG水溶液のABS は5になるので、これをH2O(脱イオン水など)で希釈する。
注4: 希釈に用いるH2Oには、界面活性剤を添加(例えばトリトンX-100を0.01 % など)したものを使用するとよい。
3) 測定
OG水溶液のABS測定ができるように装置の測定条件を設定する。
注5: 用手分注した試料は、十分混和後測定するようにする。
注6: サンプリングプローブ、試薬プローブのセルヘの吐出位置は、対照セルの 設定操作(対照セルの残水吸引操作など)がやりやすい別の位置に移動し てもよい。対照セルの設定操作を行い、次いでこのセルに試料を200 μL 用手分注して、ABSを測定する。これを対照ABSとする。10重測定を行う。
(3.17 写真掲載 写真6参照)
なお、対照ABSを測定した後は、被検セルとしても使用する。
また、この対照ABSは、残水除去処理をした所定セルに、OG水溶液を分 注して測定したABSである。
注7: 対照セルの設定操作は、サイクルタイムなどを延長しておくとやりやすい。
注8: 200 μLの分注は、再現性のよいエッペンドルフ型のピペットなどを用いる
とよい。なお、このとき重量法で容量を試験しておく(=セル用手分注量)。 次に装置を通常どおり運転してセルを自動洗浄させ、そのセルに試料を200 μL用手分注して、ABSを測定する。これを試料ABSとする。10重測定を行 う。
4) 計算
得られた測定結果から残水量を以下の計算式を用いて算出する。
残水量(μL)=(セル用手分注量)×{(対照ABS)/試料ABS)}-(セル用手分注量) 10個のセルについて求め、平均値(mean)、標準偏差(SD)、変動係数(CV(%))をそれぞれ 計算する。
算出結果を集計用紙(様式は本編6.5.1 装置基礎性能の検討結果データ集 3 セル 残水 、 測定データ参照)に記入する。
3.4 サンプルプローブのキャリーオーバの検討
3.4.1 サンプルプローブ外側のキャリーオーバ
(1) 目的
サンプルプローブの外側に付着したサンプルが、洗浄後に次のサンプルヘどの程度持ち 込み(キャリーオーバ)するかを確認する。
(2) 方法及び材料 1) 試料の調製
① ABS 5,000オレンジG(OG)/ヒトプール血清(PS)液:OG/PS)を作製する。(3.17 写真 掲載 写真7参照)
注1: OGの溶解は撹拌機構(スターラなど)で約 10分間攪拌し、十分に溶けた
ことを確認する。478 nm*1の場合はOG7.5 g/50 mL PSとなる。
*1:測定波長は機種によって適切な波長を選ぶ。
②ABS 5,000 OG/PS液を生食で5,000倍に希釈する。
注2: 希釈液の生食には界面活性剤を添加(例えばトリトン X-100を0.01 %な ど)したものを用いると良い。
2) サンプルを吸わないように操作する。
注3: サンプル吸引の停止の方法は、例えばサンプルシリンジのプランジャを外 すなどの操作を行う。(3.17 写真掲載 写真8参照)
3) サンプル容器の準備
注4: OG/PSはサンプルカップにABS 5,000のOG/PS液を適当量(例えば200 μLな ど)とる。
また生食はサンプルカップに生食200 μLをとる。生食の分注は、分注量を 重量法で求めておく(a)。
4) 測定
①作製したABS 5,000のOG/PS液の実際の吸光度(C)を5,000倍に希釈した液の測定吸光度 (A)から算出する。C=A×5,000
②サンプル吸引動作を1セットについて5回連続して行う。これを5セットについて実施 したのち、生食のサンプルカップ内の液を よく 混和後、これ を試料液と して採取 し、
セルに分注して吸光度を測定する(b)。
注5:「5回連続して行う」とは、OG/PS液から生食への持ち込みを5回行うこと である。
③生食液の吸光度を測定する(B)。
5) 計算
カップ内液量200 μLに対する汚染の影響の割合をOG/PS液の吸光度に対する汚染生食 吸光度の比として以下の計算式にしたがい汚染率K値を試料ごとに求め、n=5の平均値 を出す。
K= {(b-B)×(a/200)}/{5×(C-B)} K(%)=K×100
Kが10-6以下の場合、K×106として%ではなくppm表示とすることもできる。
集計用紙(様式は本編6.5.1 装置基礎性能の検討結果データ集 4-1 サンプルプローブ外 側のキャリーオーバ、測定データ参照)を用いて算出し、最終結果は集計用紙の該当欄に 記載しておく。
3.4.2 サンプルプローブ内側のキャリーオーバ
(1) 目的
サンプルプローブの内側に付着したサンプルが、洗浄後に次のサンプルヘどの程度持ち 込み(キャリーオーバ)するかを確認する。
(2) 方法及び材料 1) 試料の調製
①ABS 5,000オレンジG(OG)/ヒトプール血清(PS)液:(OG/PS)を作製する。(3.17 写真掲載 写真7参照)
注1: OGの溶解は撹拌機構(スターラなど)で約10分間撹拌する。478 nmの場合、
OG 7.5 g/50 mL PSとなる。
②ABS 5,000 OG/PS液を生食で5,000倍に希釈する。
注2: 希釈液の生食には界面活性剤を添加(例えばトリトンX-100を0.01 %など) したものを用いるとよい。
2) サンプル容器の準備
生食3本-OG/PS3本-生食3本を1セットとして、これを計5セット(3×3×5=45本)準備 する。すなわち次のごとくとなる。
(bi-1、bi-2、bi-3-ai-1、ai-2、ai-3-bi-4、bi-5、bi-6) i=1、2、3、4、5 (3.17 写真掲載 写真9参照)
注3: 生食液は注2に同じ。
3) ダイアグラム
サンプル量は可変(例えば2、3、5、10、15、20 μL)にする。(表3.4.2)
第一試薬(R1)は生食液の一定量(例えば200 μL)、第二試薬(R2)は0 μL、測定波長は2波長 (例 476/572 nm)にする。
注4: 測定用撹拌機構(スターラなど)は、撹拌の必要性を考慮して使用の可否 を決める。
注5: サンプルごとに実施チャンネルを設定する。
4) 測定
5,000倍希釈液のABSを5重測定し高濃度サンプルの吸光度を求める。
試料セット(45本)についてチャンネルごとにABS測定をする。
5) 計算
以下の計算式にしたがって汚染率K値(%)をサンプル量ごとに求め、n=5の平均値を出 す。
表3.4.2 設定例 Ch.No Sample Vol.
1 2 μL
2 3 μL
3 5 μL
4 10 μL
5 5 μL
6 20 μL
計算式
K(%)=[{(bi-4)-(bi-3)}/{A-(bi-3)}}×100 ここでi=1、2、3、4、5
A=高濃度サンプル吸光度×[(Sample Vol.)/{(Reagent Vol.)+(Sample Vol.)}]
高濃度サンプル吸光度=5,000倍希釈液の測定吸光度×5,000
集計用紙(様式は本編6.5.1 装置基礎性能の検討結果データ集 4-2 サンプルプローブ 内側のキャリーオーバ、測定データ参照)を用いて算出し、最終結果は集計用紙の該当 欄に記載しておく。
3.4.3 サンプルプローブのキャリーオーバ(高感度法による)
(1) 目的
サンプルプローブに付着したサンプルが、洗浄後に次のサンプルヘどの程度キャリーオ ーバするかを確認する。ここでは、一つの検体容器を共通にサンプリングして高感度な項 目も測定するケースを想定し、特に極微少量のキャリーオーバを高感度に検出することを 目的とする。
(2) 方法及び材料
1)試薬と別装置の準備
キャリーオーバ試験に供する装置(被試験装置)とは別にHBsAg測定用試薬キットと 測定装置を用意する。被試験装置にて高濃度 HBsAg試料と希釈液試料(ゼロ濃度試料)
を継続してサンプリングさせる。別装置では HBsAgの検量、コントロール測定を行った のち、希釈液を測定して測定値が装置の測定限界以下であることを確認する。
注1: 希釈液には専用希釈液もしくは陰性検体を用いるとよい。
2)試料の調製(表 3.4.3)
①最小試験濃度(ここでは0.01 ppm)で測定感度を確保できる程度のHBsAg高濃度試料
(リコンビナント検体もしくは陽性検体)を用意する。
具体例としてここではHBsAg濃度 480,000 IU/mLのリコンビナントとする。
②HBsAg高濃度試料と希釈液を使って、0.01 ppm、0.1 ppm、1 ppmの試 料を作製す る。
表 3.4.3希釈系列の例
HbsAg濃度(希釈比率) サンプル量 希釈液量
4,800 IU/mL (10000 ppm) HBsAg高濃度試料を10 μL 990 μL
48 IU/mL (100 ppm) 10,000 ppm試料を 10 μL 990 μL
0.48 IU/mL (1 ppm) 100 ppm試料を10 μL 990 μL
0.048 IU/mL (0.1 ppm) 1 ppm試料を100 μL 900 μL
0.0048 IU/mL (0.01 ppm) 0.1 ppm試料を100 μL 900 μL 注2: 濃度、希釈系列は最小試験濃度の目標値に応じて調整する。
3)HBsAg 0.01 ppm、0.1 ppm、1 ppmの試料を各3重測定する。
注3: ここで得られた結果は、低濃度域の直線性の確認とキャリーオーバの算 出に使用する。
4)サンプル容器の準備
HBsAg高濃度試料1検体に対して希釈液試料3検体を用意し1セットとする、これを5セ ット分、即ち希釈液15検体を準備する。(3×5=15本)
注4: サンプルカップに入れる量は、HBsAg高濃度試料は十分量(システムによ って異なる)、希釈液はサンプル吸引動作を行ったのちに、HBsAgが測定 できる最低量(例えば300 μL)が残る量とする。希釈液量により測定値が
変わるため、本手順では高感度測定を行う際に装置が必要とする必要試料 量に対しての汚染率を把握することとする。
5)測定
一つのHBsAg高濃度試料検体のサンプリング後に継続して希釈液3検体をサンプリン グするシーケンスを1セットとし、同様なシーケンスで5セットについてサンプル吸引動 作を実施する。(一つの高濃度試料と15本の希釈液試料)終了後、希釈液検体をよく混和 し、これを試料液として別のHBsAg測定装置でHBsAgを測定する。
注5: 被試験装置でのサンプル吸引量は設定可能な最大量とする。
6)計算
3)で測定したHBsAg 0.01 ppm、0.1 ppm、1 ppmの試料の結果 から直線回 帰式を求 め、
この式を使ってキャリーオーバ(ppm)を算出する。
注6: 計算にはHBsAgの濃度値では値が小さいため相対発光量:RLU (Relative
Light Unit)値を用いるとよい。
3.5 試薬プローブ内側のキャリーオーバの検討 (1) 目的
試薬プローブ固有のキャリーオーバの程度を確認する。
(2) 方法及び材料 1) 試料の調製
①ABS 5,000オレンジG(OG)水溶液を作製する。
注1: OGの溶解は撹拌機構(スターラなど)で約10分間撹拌する。478 nmの場合 はOG 30 g/200 mLとなる。
②ABS 5,000 OG水溶液は、H2O(脱イオン水など)で5,000倍に希釈する。
注2: 用いるH2O(脱イオン水など)には、界面活性剤を添加(例えばトリトン
X-100を0.01 %など)したものを用いるとよい。
2) ダイアグラム
次の例のごとく第一試薬(R1)と第二試薬(R2)の容量をセットする。(表3.5)