1 別紙3
厚生労働行政推進調査事業費補助金(厚生労働科学特別研究事業)
総括・分担研究報告書
新型コロナウイルス感染症治療薬等に係る開発情報の 収集・評価・提供手法の構築
研究代表者 斎藤嘉朗 国立医薬品食品衛生研究所 医薬安全科学部 部長 研究分担者 佐井君江 国立医薬品食品衛生研究所 医薬安全科学部 室長 研究分担者 青木良子 国立医薬品食品衛生研究所 医薬安全科学部 主任研究官 研究分担者 田中庸一 国立医薬品食品衛生研究所 医薬安全科学部 主任研究官
A.研究目的
新型コロナウイルス(SARS-CoV-2)による 感染症(COVID-19)は世界的流行を見せてお り、治療薬やワクチンの開発に向けた取組 が各国で加速している。このような新興・再 興感染症のアウトブレイクは、近年なかっ たものであり、行政関係者、製薬企業、アカ デミアにとっても未経験な点が多く、種々 の取り組みがなされている。治療薬開発に
関しては、ドラッグ・リポジショニング、回 復者血漿、さらに新規開発など、日米欧を中 心に臨床試験・臨床研究がなされており、ワ クチンについてもDNAワクチン等の新規モ ダリティの臨床開発が進められている。こ れら企業治験や医師主導治験等の治療薬開 発に関する取り組みや成果は、LANCETやNew England Journal of Medicine等の各種学術 誌に発表される他、臨床試験登録データベ 研究要旨:
行政施策への利用のため、新型コロナウイルス感染症治療薬・ワクチンの、主として臨床試験・
臨床研究を調査対象に、様々な情報源を検索すると共に、報道資料と文献資料に関してその信頼性 を評価する方法(検討項目や留意点)を構築すること、さらに入手した情報を関係者と共有する際 に用いる様式、日々追加・更新される情報の整理及び提供方法に関して検討を行い、標準様式を確 立することを目標とした。令和2年11月1日から、平日毎日、全国紙、業界紙、医療関係ネットニュ ース、学術論文、 規制当局のホームページ、開発製造販売企業のホームページ、臨床試験データベ ースを検索し、情報を収集した。またこれらの情報を効率良くまとめると共に、確認してわかりや すく提供しうる様式を、提供される側の規制当局者と協議して決定し、情報を平日毎日、厚生労働 省や国立感染症研究所の関連部署に提供した。またICMRA、WHO及び日米欧の行政機関から発出され た指針やステートメントを対象に、新型コロナウイルス感染症特有の評価項目である有効性エンド ポイントや安全性評価の時期に関する情報をまとめると共に、文献情報やメディア情報の信頼性を 評価する際の検討項目や留意点をリスト化した。
2 ース、企業からのプレスリリースやそれに 基づくメディア(一般紙、業界紙、医療関係 のインターネットニュース、等)の報道など、
様々な媒体で発表されているのが実情であ る。しかしながら、学術誌掲載論文に関して は、データソースの信頼性欠如に基づく撤 回が一流紙においても発生しており、また プレプリントの普及やこれに基づく報道な ど、拙速と思われる事例も認められている。
また新聞報道に関しても、根拠となる情報 ソースが記載されていないケースがあるな ど、その信頼性を確認すべき事例もある。従 って、情報の迅速な把握と共に、その信頼性 について検討することが必要となる。その 上で、これら新型コロナウイルス感染症治 療薬及びワクチン(以下、治療薬等という)
の世界的な開発状況を、時宜を得て適切に 把握・利用することは、本邦の新型コロナウ イルス感染症対策においても、早期に有望 な治療薬等候補の開発を後押し、加速する とともに、供給量を確保する等の観点から、
不可欠な作業である。
本研究では、新型コロナウイルス感染症 治療薬(再生医療等製品を含む)等の主とし て臨床試験・臨床研究を調査対象に、様々な 情報源を効率的、定期的、継続的に検索する と共に、報道資料と文献資料に関してその 信頼性を評価する方法(検討項目や留意点)
を構築する。さらに、行政関係者と意見交換 を行いながら、入手した情報を関係者と共 有する際に用いる様式、日々追加・更新され る情報の整理及び提供方法に関して種々の 検討を行い、行政施策への反映を目的とし た標準化をめざす。
B.研究方法
本研究は、新型コロナウイルス感染症治 療薬(再生医療等製品を含む)やワクチンの 臨床試験・臨床研究を主な対象として情報 を収集すると共に、報道の場合と文献の場 合を対象にそれぞれの信頼性評価に必要な 項目や留意事項を検討し、新型コロナウイ ルス感染症に関し適切な評価手法の構築を 行う。また関係者に共有する様式等、日々追 加・更新される情報の整理及び提供の方法 に関して種々の検討を行い、行政施策への 反映を目的とした標準化をめざすことを目 的としている。
1. 情報提供様式の構築:
以下の項目をプロトタイプとし、情報の 更新方法を含め、情報提供先である行政関 係者に情報を提供して、その内容、詳細さ、
行政課題への充足性等について意見交換を 行い、改訂の上、最終化する。
固定情報: 医薬品・ワクチン名(一般名・
販売名)、製造販売業者・開発企業名、開 発国/地域、治療薬では薬理作用・既存効 能(新型コロナウイルス感染症に対する 作用を含む)
各試験に依存する情報: 開発フェーズ、試 験の開始時期、終了(予定)日、試験デ ザイン・目標症例数(国別)、試験結果の 概要、海外の承認状況、ニュースソース
2. 情報検索方法:
下記の情報ソースに関し、調査を行い、対 象とする医薬品及びワクチン情報を収集し た。なお、調査の対象国は主として日米欧と した。
1) 検討対象とする情報ソース
<報道>
1. 全国紙: 日経、読売、朝日、毎日、産
3 経、東京
2. 業界紙: 日刊薬業、RISFAX
3. 医療関係ネットニュース: Medical tribune, m3, 日経メディカル, 日経 バイオテク, BioToday, CareNet
<文献>
4. 学術論文: PubMed でのキーワード検 索、Lancet, New England Journal of Medicine, JAMA, Nature Medicineの ホームページ等
<規制・開発関連>
5. WHO, 米国FDA, 欧州EMAのホームペー ジ
6. 開発製造販売企業のホームページ(プ レスリリース等)
7. 臨 床 試 験 デ ー タ ベ ー ス : UMIN、 IyakuSearch、医師会サイト(以上、日 本の治験登録サイト)、JRCT(臨床研究 法 に 基 づ く 臨 床 研 究 登 録 サ イ ト )、
ClinicalTrials.gov(米国の治験・臨床 研究登録サイト)
2)検索方法
新型コロナウイルス感染症+各治療薬・ワ クチン名(レムデシビル、デキサメサゾン、
ファビピラビル、トシリズマブ、サリルマ ブ、バリシチニブ、シクレソニド、ナファモ スタット、イベルメクチン、回復者血漿(血 漿分画製剤)、カモスタット、ロピナビル/リ トナビル、VIR-7831 等:各商品名も対象)
+臨床試験(または臨床研究)*とする。開 発状況に応じて、医薬品及びワクチン名は 追加する。
*Clinical Trial, Clinical Study, Randomized Controlled Trial, Cohort Study, Meta-Analysis, Retrospective
OR Observational Study
3) 情報収集頻度
平日に関し、原則1日1回
3. 信頼性評価手法の構築:
新型コロナウイルス感染症治療薬やワク チン開発という事例に特化した場合の信頼 性手法(評価項目や留意点)を構築する。既 に、ICMRA、米国FDA、PMDAから、ステード メントやガイダンス・指針が発出されてい るため、これらを参考に以下の検討を行っ た。
1) 一般紙、業界紙、医療関係ネットニュー ス:
それぞれ複数の情報に関し、利用する際に 必要となる情報項目の充足性、実際の根拠 データとの照合等を行い、その信頼性を評 価すると共に、情報収集にあたって留意す べき項目等についてリスト化を行う。
2) 科学論文:
複数の論文に関し、一般的に指摘されて いる試験デザイン項目(対照群の設定、無作 為性、盲検性、被験者数、統計解析手法)や 検証性(システマティックレビュー、メタ解 析)に加え、エンドポイントとしての評価項 目、治療薬では肺への分布性やin vitro活 性濃度との相関など新型コロナウイルス感 染症に特化した場合に適した留意すべき項 目等についてリスト化を行う。
(倫理面への配慮)
本研究は、公開資料のみを対象とした研 究であり、特に倫理申請等は不要と考えら れた。
4 C.研究結果
1. 情報提供様式の構築
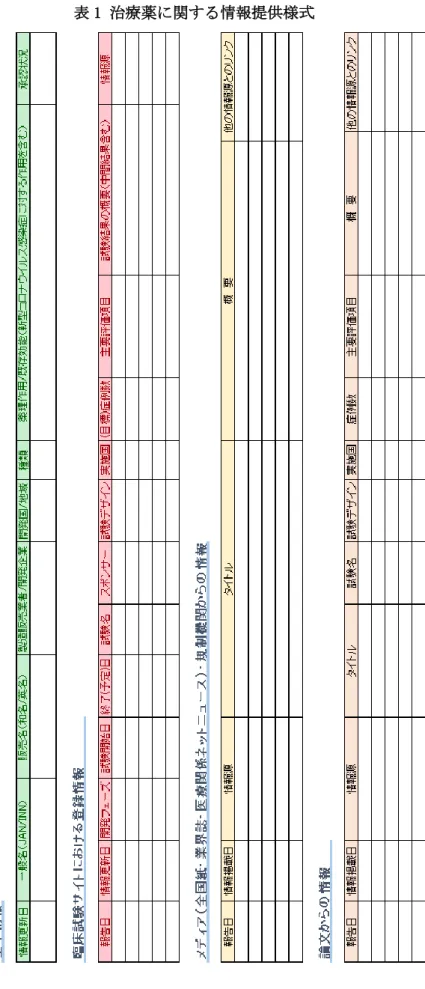
1-1. メール配信用の情報提供様式の作成 厚生労働省医政局研究開発振興課との協 議の結果、添付の様式(表1及び表2)を最終 版として、情報を提供した。即ち、研究方法 に記載した項目を表形式にし(プロトタイ プの表を作成)、試行期間中に情報収集を行 いながら表に改良を加えた。表は、「治療薬 開発情報」報告用と「ワクチン開発情報」報 告用の2種類のエクセルファイルを用意し た。また、医薬品またはワクチンの製品1つ に対し1シートを作成し、新たな開発候補製 品の情報を得た場合には、シートを追加す ることとした。収集情報は、固定情報(①治 療薬・ワクチンの基本情報)と随時更新され る情報(②臨床試験情報、③メディアからの 情報、④論文情報)に分け、同一シート上に 4つの表を配置した。試行期間中の検討の結 果、上記プロトタイプの項目に、以下の情報 を追加した。
①基本情報:医薬品・ワクチン名には一般 名・販売名としてそれぞれの英名と和名 を記載し、また必要に応じて開発コード も記載した。治療薬・ワクチンの種類(薬 効分類、作用機序による分類、薬剤クラス など)、各国での承認状況〔承認日、承認 の種類(EUA、特例承認、条件付き承認な ど)〕、情報更新日を表に追加した。ワク チンでは備考欄を設け、ワクチン製剤の 特徴、開発や製造に関する企業間の提携 に関する情報などを記載した。
②臨床試験サイトにおける登録情報:臨床 試験名、スポンサー、試験実施国、試験の
主要評価項目、情報源(試験登録番号、情 報元へのリンク)、臨床試験登録サイトの 情報更新日を表に追加した。
③メディア(全国紙・業界誌・医療関係ネッ トニュース)・規制機関からの情報:情報 のタイトル、概要、情報元へのリンク、他 の関連情報(論文、開発企業や大学等のプ レスリリース)へのリンク、情報公開日を 記載した。
④論文からの情報:情報源(ジャーナル書誌 事項、情報元へのリンク)、論文タイトル、
試験名、試験デザイン、実施国、症例数、
主要評価項目、概要、他の情報源とのリン ク、情報掲載日(発行日、ePub掲載日)を 記載した。
1-2. 情報収集用作業ファイルの作成 情報収集は複数人で手分けして行ったた め(基本的には2名体制)、情報ソースごと に作業ファイルを2つずつ分けて作成した。
具体的には、①臨床試験情報は、治療薬とワ クチン、②メディアからの情報は全国紙と、
ネットニュース・業界誌、③論文情報は、
PubMed検索情報と医学四大誌とにそれぞれ 分け、同時に作業してもファイルへの入力 を支障なく行えるようにした。また、作業フ ァイルの様式は、収集した情報をそのまま コピーして報告用シートにペーストできる ようにするため、報告用の表と同一形式と した。
1-3. 配信メールの本文に、当日収集した最 新情報をまとめた表を作成
本研究では、新型コロナウイルス感染症 の治療薬・ワクチンの開発情報を必要とす る厚生労働省及び国立感染症研究所の関連
5 部署に毎日収集した情報をメールで提供し た。メールには、これまで収集した情報をす べて累積したエクセルファイルを添付する が、その日に収集した最新情報は、表に簡易 にまとめてメール本文に貼り付け、一目で 確認できるようにした。メール本文への記 載内容は、治療薬・ワクチンの一般名(記号)、
情報源(URLをリンク)、タイトル、概要と し、各情報には対応するエクセルファイル のシート名を付与して参照可能にした。
2. 情報検索方法
令和2年11月1日の研究開始より、平日の 毎日、新型コロナウイルス感染症治療薬及 びワクチンの開発情報を調査し、厚生労働 省大臣官房、医薬・生活衛生局、健康局、国 立感染症研究所等の関連部署の担当者に、
情報を提供した。令和3年3月31日時点での 収集情報を別添として添付した。
例えば、ファイザー/ビオンテック製ワク チン(トジナメラン)の開発・承認状況に関 しては、以下の内容が調査期間中に明らか となった。
令和2年11月9日: ファイザー社は治験 中間データとして、9割以上の予防効果の確 認を発表
11 月18日;ファイザー社は第3 相試験の 最終有効性解析にて、主要評価項目全てを 達成したことを発表
11 月 20 日:ファイザー社は米食品医薬品 局(FDA)へ緊急使用許可を申請
11月30日:ファイザー/ビオンテック社は 欧州医薬品庁(EMA)に条件付き販売許可 を申請
12 月 2 日:英国医薬品・医療製品規制庁
(MHRA)による緊急承認
12月8日:英国でワクチン接種開始 12月8日:米FDAは、1回のワクチン投与 でも有効性を確認したとの評価書を公表 12月9日:英MHRA によりワクチン投与 後のアレルギー反応を報告、注意呼びかけ 12月9日:カナダ保健省による緊急使用の 承認
12月11日:米FDAによる緊急使用許可の 承認
12月14日:米国でワクチン接種開始 12 月 16 日:米国でのワクチン接種後のア レルギー反応の報告
12 月 18 日:ファイザー社は日本の厚生労 働省へ承認申請
12月19日:スイス保健省による承認(通常 審査)
12月21日:EU EMAによる承認勧告、欧 州委員会(EC)による条件付き販売の承認 12月27日:EUにおけるワクチン接種開始 12月31日:世界保健機関(WHO)による 緊急使用の承認
令和 3 年 1 月 6 日:米疾病対策センター
(CDC)は、アナフィラキシーのリスクは 非常に低いことを報告
1月19日:ワクチン接種者の血清が英国型 変異株(B.1.1.7)にも中和活性を示す研究論 文公開(bioRxiv Jan. 19, 2021 doi.org 10.1101 2021.01.18.426984)
1月27日:ワクチン接種者の血清の中和活 性に関し、英国型及び南アフリカ型の変異 の影響はわずかであることを示す研究論文 公開 (bioRxiv Jan. 27, 2021 doi.org 10.1101 2021.01.27.427998
⇒ 2 月 8 日: Nature Medicine volume 27, 620–621, 2021に掲載)
1月21日:初回接種後のアナフィラキシー
6 発現頻度は100万回投与当たり11.1例であ る研究論文公開(JAMA insights Jan. 21, 2021) 1月29日:英国B.1.1.7変異株に対する中和 活 性 を 示 す 研 究 論 文 公 開(Science Jan. 29 eabg6105)
1月29日:EMAは、既知のワクチンの安全 性プロファイルと一致し、新しい副作用は 確認されなかったと結論
2 月 1 日:コロナウイルス感染歴の有る人 ではワクチン1 回接種による抗体反応は感 染歴の無い人の2回接種を上回ることを示 す 研 究 論 文 公 開(medRexiv Feb. 1, 2021 doi.org/10.1101/2021.01.29.21250653)
⇒3月10日:NEJM correspondence Mar. 10, 2021 DOI10.1056NEJMc2101667に追加デー タとともに掲載)
2 月 14日:厚生労働省により、『コミナテ ィ筋注』の製造販売が特例承認
2月17日:日本国内でのワクチン接種開始 2月17日:ワクチン接種者の血清は、英国 型(B.1.1.7)、南アフリカ型(B.1.351)及び ブラジル型(P.1)の変異株に対しても中和 活性を有するが、南アフリカ型(B.1.351)
への効果は低めであることを示す研究論文
公 開 (NEJM Feb. 17, 2021
DOI10.1056NEJMc2102017 ⇒ 3月8日:
N Engl J Med. 2021 Mar 8. doi:
10.1056/NEJMc2102017として、追加データ とともに掲載)
2月18日:接種1回で85%の有効性を示す イスラエルにおける研究論文公開(Lancet.
2021 Feb 18:S0140-6736(21)00448-7〉
2月25日:厚生労働省の予防接種・ワクチ ン分科会副反応検討部会にて、先行接種 2 万2000人における副反応の疑いは3件であ り、継続に「重大な懸念がない」と評価
3月1日:日本アレルギー学会は、ワクチン 接種後の重い副作用「アナフィラキシー」に 対する指針を公表
3月1日:ワクチン接種後のヒトT細胞が、
英国型や南アフリカ型変異株も認識するこ とを示す研究論文公開(BioRxiv Mar. 1, 2021 doi.org 10.1101 2021.02.27.433180)
3 月 2 日:高齢者におけるワクチンの有効 性を示す研究論文公開(medRxiv. 2021 Mar 2.
doi: 10.1101/2021.03.01.21252652)
3月8日: 妊娠及び授乳中の女性への接種 により免疫反応が誘導され、新生児への免 疫移行 を示した研究論文公開 (medRxiv.
2021 Mar 8;2021.03.07.21253094)
3 月 9 日:ワクチン接種者のリンパ節に免 疫を持続させる胚中心の確立を示す研究論 文公開(Nature Portfolio Mar. 9, 2021 DOI 10.21203 rs.3.rs-310773 v1)
3月12日:厚生労働省の予防接種・ワクチ ン分科会副反応検討部会にて、アナフィラ キシーが疑われる事例が 3/11までに 36 件 報告されたが、「安全性に重大な懸念は認め られない」と評価
3月15日:固形臓器移植者への単回接種後 の免疫原性は低いことを示す研究論文公開 (JAMA. 2021 Mar 15. doi:
10.1001/jama.2021.4385)
3月25日:ワクチンの1回接種で、強い免 疫応答を誘導することを示す研究論文公開
(Preprints with THE LANCET. 2021 Mar 25.
ID: 3812375)
3月25日:高齢者への接種により、フレイ ルや障害の有無に関わらず、抗体が生成さ れることを示す研究論文公開(J Am Geriatr Soc. 2021 Mar 25. doi10.1111jgs.17153)
3月26日:厚生労働省の予防接種・ワクチ
7 ン分科会副反応検討部会にて、健康状況調 査の中間報告として、発熱や痛みなどの副 反応が2回目の接種後に多くみられること が報告された。
3月29日:米疾病対策センター(CDC)は ワクチン(モデルナ製を含め)の感染を防ぐ 高い有効性を確認したことを発表(1回目で 80%低下、2回目で90%)
2-1.調査対象の情報の集計結果
検索対象とした各コロナ感染症治療薬 35品目及びワクチン15品目のリスト(一般 名・商品名)を表3に示す(2月末時点)。メ ディア(新聞、業界紙、医療関係ネットニュ ース)ならびに文献情報検索式は、収集情報 の内容をもとに、定期的に見直しを行い、新 聞に関しては、記載が簡略なケースが多い、
等の理由から2紙の優先順位を下げ、2021年 1月より報告は6社から4社に絞ることとし た。また、文献検索のキーワードに関しては、
データの重複性や、世界的な開発動向とし て、ワクチン開発情報にシフトしているこ とも考慮し、“Meta-analysis”、 治療薬に 関 し て は”Retrospective, Observational study”を除くこととした(2月中旬より)。
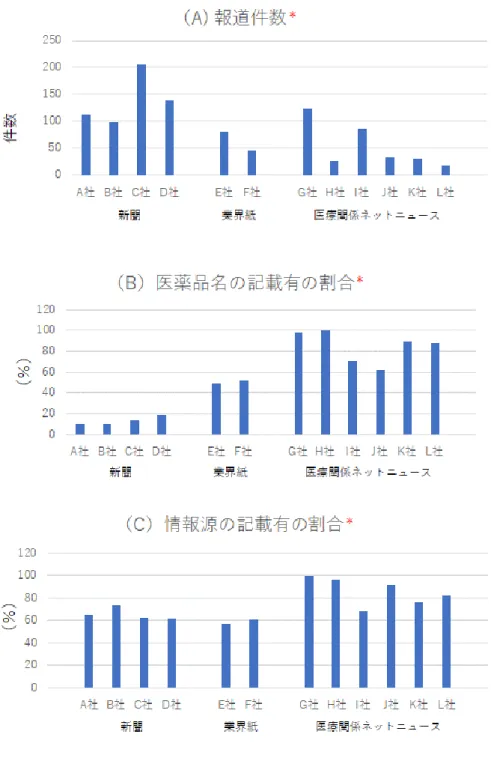
上記の提供様式にて、関係部署へ報告した 件数(2020年11月~2021年2月)は、新聞(4 社)は555件、業界紙(2社)は126件、医療 関係ネットニュース(6社)は315件であり、
各メディア別の報告件数は図1Aに示す通り である。また、論文(抄録翻訳)は105件、
規制当局からの情報は115件、開発・製造販 売企業プレスリリースは164件、臨床試験登 録情報は146件を収集・報告した。
2.2. メディア別の情報の特性
報道(新聞、業界紙、医療関係ネットニュ ース)の情報の質の指標として、開発医薬品 名(一般名、開発コードまたは商品名)の明 記、及び情報のソース(臨床試験名、論文書 誌情報、情報発信元の組織、またはそれらを 特定できる情報等)の記載の有無について も確認し、情報記載の有る割合をメディア 別に集計した。医薬品名称の記載有の割合 は、新聞(4 社)では 10%~20%であり、
業界紙(2社)では約50%、医療関係ネッ トニュース(6社)では60%以上であり、
大半は90%~100%であった(図1B)。掲載
情報内に情報ソースの記載の有る割合は、
新聞ならびに業界紙は約60%、医療関係ネ ットニュースでは 80%~100%であった
(図1C)。
2-3.主なトピックおよび報道の迅速性比較 調査期間におけるトピックとして、ワク チン及び治療薬に関する各国の承認・申請 に関する報道があった。主なものとして、フ ァイザー/ビオンテック製ワクチンの承認・
申請(米国:2020年12月緊急使用許可(EUA)、 英国:2020年12月承認、日本:2020年12 月承認申請)、モデルナ製ワクチンの承認
(米国:2020年12月EUA取得、英国:2021 年1月承認)、アストラゼネカ製ワクチンの 承認・申請(英国:2020 年 12 月承認、日 本:2021年2月承認申請)、レムデシビルの バリシチニブとの併用の承認(米国:2020 年11 月)、バムラニビマブの承認(米国:
2020年11月)などが挙げられる。ファイザ ー/ビオンテック製及びモデルナ製ワクチ ンの承認関連情報は、医療関係ネットニュ ースおよび日本の新聞各社においても、海 外情報を含めて広く報道されており、12月
8 に報道された件数で比較すると、医療関係 ネットニュースでは26件、新聞では24件 あった。なお、新聞では同じ情報について、
複数回報道するものも含む。治療薬の米国 における承認情報は、医療関係ネットニュ ースでは 8件あったが、今回対象の新聞で は報道はなかった。
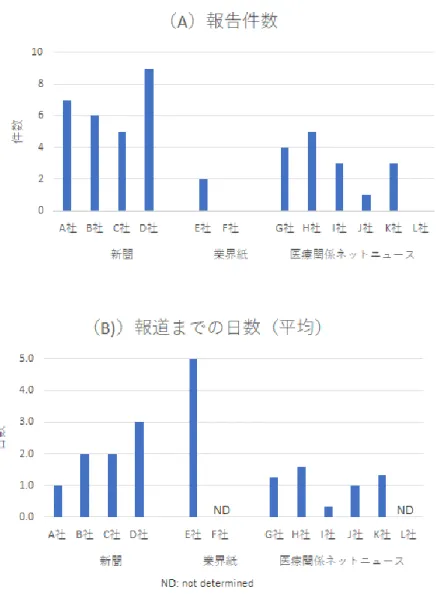
メディア別の報道の迅速性についても比 較するため、ファイザー/ビオンテック製ワ クチンの各国の承認・申請状況(12/2~
12/20付けの掲載情報)を例として、メディ
ア各社の報道件数、及び承認日(または申請 日)から初回報道までの日数(タイムラグ)
の平均を算出した(図2)。なお、新聞各社 では、同一の情報を複数回報道しているた め、それぞれ初回の報道までの日数を用い た。医療関係ネットニュースでは、0.3~1.6 日、新聞各社では1~3日であり、業界紙で は5日であった。
3. 信頼性評価手法の構築
まず医薬品及びワクチンの2種に関し、
ICMRA、WHO、PMDA、FDA、EMAから発出された、
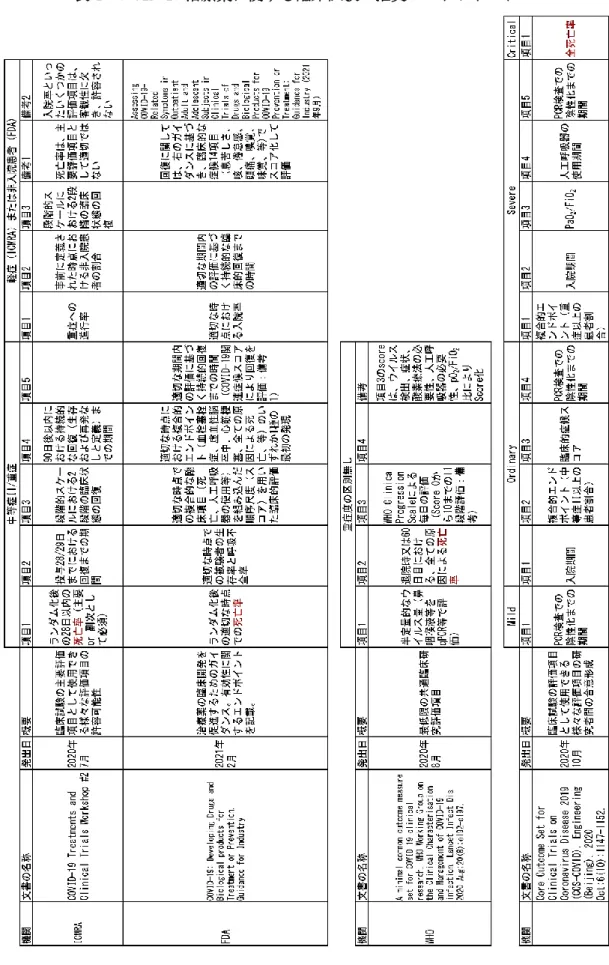
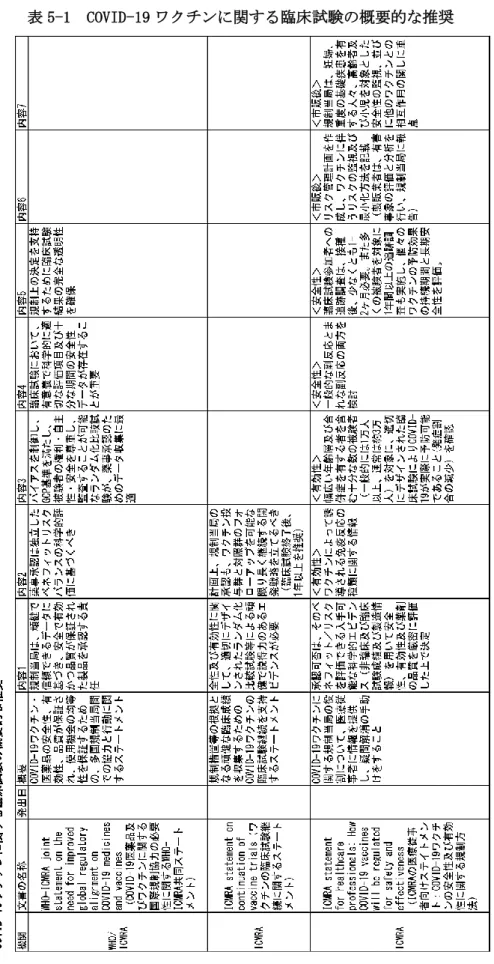
承認審査に関する指針やステートメントの 調査(一部は、学術論文調査)を行い、臨床 試験等に関する要求事項、特に評価すべき エンドポイントに関してまとめた。医薬品 に関する結果を表4に、ワクチンに関する結 果を表5に示す。なお、ワクチンに関しては、
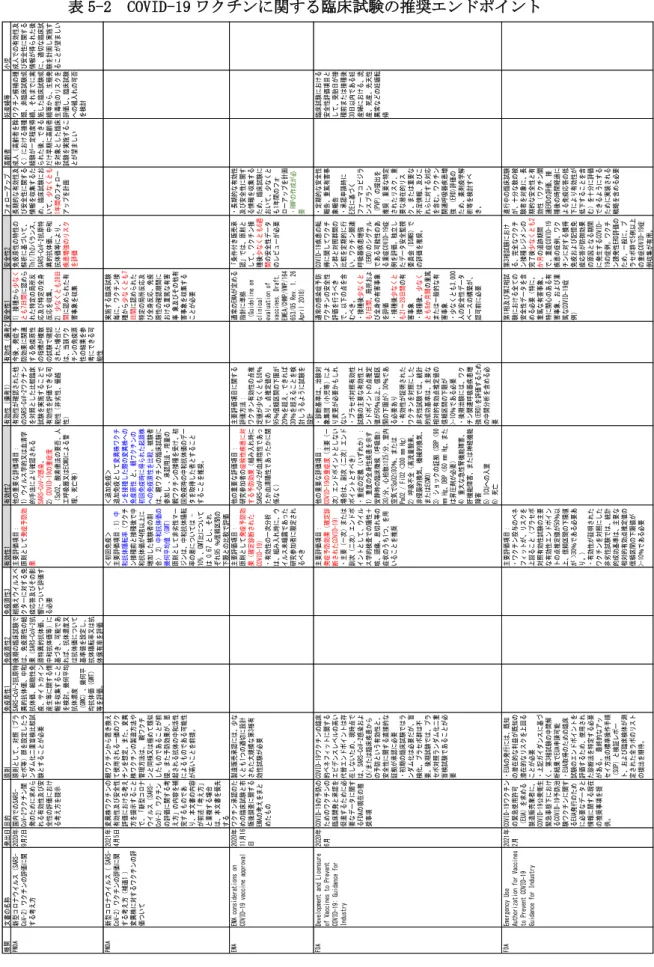
概要的な内容を表5-1に、エンドポイントに 関する内容を表5-2に、リアルワールドデー タを利用した評価に関し表5-3に分割して 記載した。
医薬品・ワクチンに共通した点として、製 造販売承認を得るためには、ランダム化(無 作為化)した二重盲検による対照を用いた
試験が必要であり、特に今回のCOVID-19の ように対照となる既存薬・ワクチンがない、
又は(確保できないことを含め)不足してい る状況では、プラセボを対照とした試験デ ザインを用いる重要性が挙げられた。また 検出力に基づき、十分な被験者数を確保す ることも必要である。
医薬品に関しては、「死亡率」が共通して、
最重要なエンドポイントとしてあげられて いた。また「複合的な臨床項目を組み込んだ スコア」、「持続的な回復までの期間」も用 いられていた。従って、これらの評価を行っ た臨床試験・研究の結果は、承認のために用 いられる可能性が高いと考えられ、論文に 本解析項目の結果が含まれる場合、情報と して有用なものとなると考えられた。一方、
入院率は国・地域や貧困率等により異なる 可能性があるため、多施設共同の試験では バイアスに注意が必要と考えられた。また ドラッグ・リポジショニングによる医薬品 開発では、in vitroでの薬理作用評価に加 え、現行の適応症に対して認められている 投与量により到達する血中濃度と薬効濃度 との関連や、感染臓器(特に肺)への分布に 関し、留意することが重要と考えられた。な お、米国FDAのガイダンスでは、臨床試験中 の安全性モニタリングを補足するため、リ アルワールドデータの利用検討が記載され ていた。
ワクチンに関しては、ICMRAより承認に必 要とされる臨床試験等に関する概要的な内 容を記した3種の文書が発表されていた。頑 健で信頼性が保証されたエビデンスを得る ことの重要性が謳われており、また承認後 も1年以上の長期的なフォローアップを推 奨していた。日米欧から公表されているガ
9 イドライン等における、臨床試験の有効性 のエンドポイントに関しては、いずれも「発 症 予 防 効 果 」 を 推 奨 し て お り 、 さ ら に
「COVID-19重症度」、「症候性疾患の予防効 果」、「SARS-CoV-2感染」等が用いられてい た。従って、論文に本解析項目の結果が含ま れる場合、情報として有用なものとなると 考えられた。また安全性に関しては、接種か ら少なくとも「7日間に認められた特定の局 所反応及び特定の全身反応」、「少なくとも 28日間に認められた有害事象」をそれぞれ 収集すること、が推奨されていた。さらに、
「ワクチン関連呼吸器疾患増強の評価」を 求めていた。日本同様、EMAでは、リスク管 理計画の提出が明記されていた。
前項の検討結果を含め、下記が、その信頼 性評価において、特に留意すべき点と考え られた。
1) 一般紙、業界紙、医療関係ネットニュー ス:
a) 出典の記載、及びその出典との照合 b) 医薬品・ワクチン名の記載、開発企業名・
研究者所属機関等の記載
c) 発表の信頼性に関する考察等の記載
2) 科学論文:
a) 臨床試験の場合:
a-1) 共通項目として、試験デザイン(対 照群の設定、無作為性、盲検性、被験者 数、統計解析手法)や別群を用いた検証 結果の有無。
a-2)治療薬の場合は、エンドポイントと して「死亡率」、「複合的な臨床項目を 組み込んだスコア」、「持続的な回復ま での期間」の評価項目。加えて、治療薬 では肺への分布性、血中濃度とin vitro
活性濃度との相関が信頼性評価に重要 と考えられる。
a-3) ワクチンの場合は、エンドポイント として、「発症予防効果」,「COVID-19重 症度」、「症候性疾患の予防効果」、「SARS- CoV-2感染」の評価項目。安全性に関して は、「接種から少なくとも7日間に認めら れた特定の局所反応及び特定の全身反 応」、「少なくとも28日間に認められた 有害事象」の評価項目。
b)システマティックレビューのメタ解析の 場合:文献の選択方法、出版バイアスの 可能性検討、被験者数、原著の掲載誌の レベル、等
D.考察
1. 情報提供様式の構築:
収集する情報量が膨大なため、如何に効率 的かつ迅速に、間違いなく情報を整理し、見 やすい形で提供できるかを重要視した。そ のため、エクセルのフィルター機能を活用 し、作業用ファイルから報告用ファイルへ 情報を転記する際、必要な情報を的確に振 り分けられるよう工夫した。報告用ファイ ルは作成後、配信前に記載漏れや分類間違 いがないかを点検するが、この作業にもフ ィルター機能を用い、人為的ミスを防いだ ことが正確な情報提供に役立ったと思われ る。
2. 情報検索方法:
本調査では、コロナ感染症に関する幅広 いメディア情報から、コロナウイルス感染 症治療薬及びワクチンの開発・承認情報に 特化した情報を収集するため、予め情報サ
10 イト、検索キーワード、検索式を検討し、実 際の収集情報については、個別にそれらの 医薬品情報や情報源(論文、臨床登録情報、
情報発信元のサイト、等)の掲載情報と照合 し、内容の確認とともに、情報源の詳細情報 も収集した。また、後述のように各メディア の情報の質(開発製品の名称、情報源の記載)
についても精査し、情報ソースの見直しも 行った。
なお、論文の種類として、ドラッグ・リポ ジショニングをテーマとした臨床試験のメ タ解析、ならびにレトロスペクティブの観 察研究の論文もこの期間に増える傾向にあ った。特に、ドラッグ・リポジショニングに 関するメタ解析の論文も複数報告されてい たが、大規模な臨床試験の数は限られてい たため、メタ解析等の統合した結果は大規 模な試験データに依存し、同じ結果が報告 されるものが多く見受けられた。また、ラン ダム化臨床試験以外の種々のデータソース によるレトロスペクティブ研究の論文も増 える傾向にあったが、エビデンスレベルは 一般に低いとされており、また世界的な開 発動向や薬事行政においてもワクチン開発 に焦点がシフトしていることを考慮し、2月 中旬以降は、論文の検索条件から、”Meta-
analysis” 、 治 療 薬 に つ い て
は”Retrospective, Observational study”を 除いた。
なお、論文情報の収集に当たっては、報告 の重複が生じないように留意した。例えば、
オンライン版での掲載後に冊子版として書 誌情報が変更される場合も、日々の更新情 報として検索されるため、冊子版について は既報告の有無も確認した。また、本調査で は、プレプリント版も対象としたが、情報の
報告後に雑誌掲載版として発刊される場合 もある。タイトル、抄録、掲載データに追 加・変更を認める場合は、これも新たな報告 対象とし、情報提供ファイルにはプレプリ ントに関する履歴も追記した。また、内容変 更がない場合には、掲載版の書誌情報につ いて、情報提供ファイルに追記することと した。
本調査では、各メディアの特性や質の違 いを把握するため、医薬品名称や情報源の 記載の有無、及びワクチン承認・申請に関す る報道を例として、報道までのタイムラグ についても比較した。医薬品名称及び情報 源の記載は、医療関係ネットニュースでは 大半が記載されており、報道までのタイム ラグも比較的短かった。新聞では名称記載 の割合は低く(<2割)、業界紙では5割で あったが、新聞及び業界紙では情報源の記 載も、ネットニュースと比較して低い傾向 にあった。新聞および医療関係ネットニュ ースにおける報道までの日数は、平均で 1
~2 日以内であったが、業界紙は情報量と しては多いが、報道までの時期は遅れる傾 向にあった。全般的に、海外における情報 は、海外の主要メディア情報を網羅する医 療関係ネットニュースが比較的迅速であっ た。新聞は、治療薬に関しては、日本の開 発・承認情報の報道が多く、海外情報は少な い傾向にあったが、ワクチンに関しては、海 外情報も多く報道されていた。
以上のように、メディアにより、報道の内 容や質、報道回数などにも違いが見られた ことから、これらの特性をもとに、調査対象 とするメディアについても再度見直し行い 反映することとした。
11 3. 信頼性評価手法の構築
治療薬やワクチン開発に関しては、まず 各国規制機関から求める情報を把握するこ とが重要であるが、規制機関としてはデー タを評価する場合、治療薬・ワクチン開発に おいて一般的に求める臨床試験の信頼性確 保の要件以外に、新型コロナウイルス感染 症を対象にした評価に特化した内容も重要 である。後者に関しては、有効性のエンドポ イントや安全性評価の時期が考えられる。
そ こ で ま ず 日 米 欧 の 規 制 機 関 、WHOや ICMRAを対象に、指針やステートメントを 調査した。今回は、日米欧を始めとする各国 規制当局が、ICMRAでの概論的な議論に参 加しており、概ね主なエンドポイントは調 和されていると考えられた。
今後は、各国におけるワクチン接種率が 上昇するため、未感染・未ワクチン接種者は 少なくなり、これまでのような「発症予防効 果」による評価が困難になると予想される。
中和抗体価の上昇と維持期間など、別の有 効性エンドポイントの設定が必要と考えら れ、中和抗体価と従来の臨床指標との関連 解析結果の蓄積が特に必要と考えられる。
なお、変異株に対するワクチンの有効性エ ンドポイントとしては、欧州EMAからは令和 2年2月23日に、日本では令和3年度に入った 4月5日に指針が発出されており、初回免疫 の場合の主要な有効性評価項目として、1) 中和抗体陽転率(ワクチン接種前と接種後 で中和抗体価が4倍以上に増加した被験者
の割合) 2) 中和抗体価の幾何平均値(GMT)
が設定されている。
E.結論
行政施策への利用のため、新型コロナウ
イルス感染症治療薬・ワクチンの、主として 臨床試験・臨床研究を調査対象に、様々な情 報源を検索すると共に、報道資料と文献資 料に関してその信頼性を評価する方法(検 討項目や留意点)を構築すること、さらに入 手した情報を関係者と共有する際に用いる 様式、日々追加・更新される情報の整理及び 提供方法に関して検討を行い、標準様式を 確立することを目標とした。令和2年11月1 日から、平日毎日、全国紙、業界紙、医療関 係ネットニュース、学術論文、規制当局のホ ームページ、開発製造販売企業のホームペ ージ、臨床試験データベースを検索し、情報 を収集した。またこれらの情報を効率良く まとめると共に、確認してわかりやすく提 供しうる様式を、提供される側の規制当局 者と協議して決定し、情報を平日毎日、厚生 労働省や国立感染症研究所の関連部署に提 供した。またICMRA、WHO及び日米欧の行政機 関から発出された指針やステートメントを 対象に、新型コロナウイルス感染症特有の 評価項目である有効性エンドポイントや安 全性評価の時期に関する情報をまとめると 共に、文献情報やメディア情報の信頼性を 評価する際の検討項目や留意点をリスト化 した。
F.健康危険情報 該当なし
G.研究発表 1. 論文発表
該当なし 2. 学会発表
12 該当なし
H.知的財産権の出願・登録状況 (予定を含む。)
1. 特許取得 該当なし
2. 実用新案登録 該当なし
13
表1 治療薬に関する情報提供様式
14
表2 ワクチンに関する情報提供様式
15
表3 調査対象の治療薬、ワクチン(2021年2月時点)
一般名または開発コード(商品名)
治 療 薬 ワクチン
レムデシビル (ベクルリー) 回復期血漿 トジナメラン(コミナティ)
デキサメタゾン(デカドロン) コルヒチン (コルヒチン) AZD1222
ファビピラビル (アビガン) エクリズマブ (ソリリス) mRNA-1273/TAK-919
トシリズマブ (アクテムラ) フルボキサミン (デプロメール・ルボック
ス) NVX-CoV2373
サリルマブ (ケブザラ) SNG001 Gam-COVID-Vac (スプートニクV)
バリシチニブ (オルミエント) AV-001 CVnCoV シクレソニド (オルベスコ) AZD7442 bacTRL-Spike
ナファモスタット (フサン) ラナデルマブ (タクジーロ) JNJ-78436735/Ad26.COV2.S イベルメクチン (ストロメクトー
ル) ジルコプラン MT-2766
カモスタット (フオイパン) 高度免疫グロブリン製剤(CoVIg-19) AG0301-COVID19 ロピナビル・リトナビル (カレト
ラ) アドレノメデュリン S-268019
VIR-7831, VIR-7832 MK-7110 INO-4800
バムラニビマブ ルキソリチニブ (ジャカビ) VLA2001 エリトラン アナキンラ (キネレット) SCB-2019 アプレミラスト(オテズラ) ADG20 SP0253
BI764198 AT-527
アスピリン レグダンビマブ (レッキロナ) カシリビマブ+イムデビマブ
16
図1 報道(新聞、業界し、医療関係ネットニュース)の特性の比較 2020年11月~2021年2月における調査対象情報のメディア別の集計
*H社, J社, K社, L社は2月14日までの集計データ
17
図2 報道(新聞、業界紙、医療関係ネットニュース)の迅速性の比較 2020年12月のファイザーワクチン承認・申請に関する報道のメディア別の集計
対象情報;英国:一時使用許可(12月2日)、米国:緊急衣装許可(12月11日)、 日本:承認申請(12月18日)
18
表4 COVID-19治療薬に関する臨床試験の推奨エンドポイント
19
表5-1 COVID-19ワクチンに関する臨床試験の概要的な推奨
20
表5-2 COVID-19ワクチンに関する臨床試験の推奨エンドポイント
COVID-19ワクチンに関する臨床試験の推奨エンドポイント 機関文書の名称発出日目的原則免疫原性1免疫原性2免疫原性3有効性1有効性2有効性(備考1)有効性(備考2)安全性1安全性2フォローアップ高齢者妊産婦等小児 PMDA新型コロナウイルス(SARS- CoV-2)ワクチンの評価に関 する考え方 2020年 9月2日国内でのSARS- CoV-2ワクチン開 発のために求めら れる有効性及び安 全性の評価におけ る考え方を提示 原則として、対照(プラ セボ等)群を設定したラ ンダム化二重盲検比較試 験とすることが必要
SARS-CoV-2抗原特 異的抗体価、中和 抗体価、細胞性免 疫、サイトカイン 産生等に関する情 報を収集すること を検討。幾何平均 抗体濃度 (GMC)、幾何平 均抗体価(GMT) 等を評価。
後期の臨床試験で は、免疫原性の結 果(SARS-CoV-2抗 原特異的抗体価、 中和抗体価等)に 基づき、可能であ れば、抗体濃度又 は抗体価について 基準値を設定し、 抗体陽転率又は抗 体保有率を評価 組換えウイルスベ クターに対する免 疫応答及びその影 響について評価す る必要 主要評価項目: 原則として発症予防効 果 他の重要な評価項目: 1) ウイルス学的又は血清学 的手法により確認される SARS-CoV-2感染、 2) COVID-19の重症度 (SpO2、酸素療法の要否、人 工呼吸器又はECMOによる管 理、死亡等)
有効性が確認された他 のSARS-CoV-2ワクチン を対照とした比較臨床 試験を実施することで 有効性を評価できる可 能性(非劣性、優越 性)
今後、発症予 防効果に関連 する免疫原性 の指標が複数 の試験で確認 された場合に は、当該ワク チンの免疫原 性の結果を参 考にできる可 能性 1) 接種から少なく とも7日間に認めら れた特定の局所反 応及び特定の全身 反応を収集、 2) 少なくとも28日 間に認められた有 害事象を収集 免疫原性の特性の 解析に基づいて、 Th1/Th2バランス、 SARS-CoV-2抗原特 異的抗体価、中和 抗体価等により、 疾患増強のリスク を評価 長期的な有効性及 び安全性に関する 情報を収集するた め、臨床試験にお いて、少なくとも 1年間のフォロー アップを計画 成人(高齢者を除 く)における接種 経験が一定程度得 られた後、できる だけ早期に高齢者 を対象とした臨床 試験を実施するこ とが望ましい ワクチン候補の種 類、非臨床試験成 績、それまでに実 施した臨床試験成 績等から、生殖発 生毒性のリスクを 評価し、臨床試験 への組入れの可否 を検討
成人での有効性及 び安全性に関する 情報が得られた後 に、適切な臨床試 験を計画し実施す ることが望ましい PMDA新型コロナウイルス( SARS- CoV-2)ワクチンの評価に関 する考え方(補遺1 ) 変異株に対するワクチンの評 価ついて
2021年 4月5日変異株ワクチンの 有効性及び安全性 評価における考え 方を提示すること で、「新型コロナ ウイルス(SARS- CoV-2)ワクチン の評価に関する考 え方」の内容を補 完するものであ り、本文書の内容 が前述「考え方」 と重複する場合 は、本文書を優先 する 親ワクチンから置き換え て使用される一価のワク チンを想定。また、変異 株ワクチンの製造方法や 管理方法は、 親ワクチ ンと同様又は極めて類似 したものであることが前 提。また予防効果が、惹 起される抗体の中和活性 によるものである可能性 が高いことを前提。
<初回免疫> 主要評価項目:1) 中 和抗体陽転率(ワクチ ン接種前と接種後で中 和抗体価が4倍以上に 増加した被験者の割 合) 2) 中和抗体価の 幾何平均値(GMT) 原則として非劣性マー ジンは、中和抗体陽転 率の差については - 10% 、GMT比について は 0.67 とし、それ ぞれ95 %信頼区間の 下限との比較で評価
<追加免疫> 追加免疫として変異株ワクチ ンを接種した際の変異株への 免疫原性 と、親ワクチンの 初回免疫時に得られた起源株 への免疫原性を比較。被験者 は、親ワクチンの臨床試験に 参加し 、承認用法・用量の 親ワクチンの接種を受け、初 回免疫時の中和抗体価のデー タを取得した者とすること することを推奨。
実施する臨床試験 毎に、ワクチン接 種から少なくとも7 日間に認められた 特定の局所反応及 び全身反応、免疫 原性の確認期間に おける重篤な有害 事 象及びその他有 害事象を収集する ことが必要 EMAEMA considerations on COVID-19 vaccine approval2020年 11月16 日
ワクチン承認のた めの臨床試験と市 販後調査に関する EMAの考えをまと めたもの 製造販売承認には、少な くとも1つの適切に設計 された大規模な第3相有 効性試験が必要 主要評価項目: 原則として発症予防効 果(確定診断された COVID-19) ・有効性の一次分析 は、組み入れ時に、ウ イルス未曝露であった 研究参加者に限定され るべき 他の重要な評価項目: 研究参加者の症候性疾患に対 する予防効果(組み入れ時に SARS-CoV-2が血清陰性であっ たか血清陽性であったかに関 係なく)
主要評価項目に関する 評価方法: ワクチン有効性の点推 定値が少なくとも50% であり、点推定値の 95%信頼区間の下限が 20%を超え、できれば 30%を超えることを検 討しうるように試験を 設計 通常のEMAが定める 指針に準拠 (Guideline on clinical evaluation of vaccines, Draft, EMEA/CHMP/VWP/164 653/05 Rev. 1, 26 April 2018)
「条件付き販売承 認」では、原則と して、ワクチン接 種後少なくとも6週 間の安全性データ のレビューが必要
・長期的な有効性 及び安全性に関す る情報を収集する ため、臨床試験に おいて、少なくと も1年間のフォ ローアップを計画 ・RMPの作成が必 要 FDADevelopment and Licensure of Vaccines to Prevent COVID-19: Guidance for Industry
2020年 6月COVID-19の予防の ためのワクチンの 臨床開発と承認を 促進するために必 要なデータに関す るFDAの現在の推 奨事項
COVID-19ワクチンの臨床 的ベネフィットに関する エビデンスレベルの高い 代替エンドポイントは存 在しないため、現時点で は、SARS-CoV-2感染およ び/または臨床疾患から の予防という有効性と、 安全性に関する直接的な 証拠が承認に必要 ・初期の臨床試験ではラ ンダム化は必要だが、盲 検化、プラセボ群は不 要。後期試験では、プラ セボ対照ランダム化二重 盲検試験であることが必 要 主要評価項目: 発症予防効果(確定診 断されたCOVID-19) ・主要(一次)または 副次(二次)エンドポ イントとして、ウイル ス学的検査での陽性+ 咳、頭痛、息切れ等の 症状のうち1つ、を用 いることを推奨
他の重要な評価項目: COVID-19の重症度(主要(一 次)エンドポイントとしない 場合は、副次(二次)エンド ポイントとすべき) ・重症の定義(いずれか): 1) 重度の全身性疾患を示す 安静時の臨床徴候(呼吸数≥ 30/分、心拍数≥125/分、室内 空気でSpO2≤93%、または PaO2 / FiO2 <300 mm Hg) 2) 呼吸不全(高流量酸素、 非侵襲的換気、機械的換気、 またはECMO) 3) ショックの証拠(SBP <90 mm Hg、DBP <60 mm Hg、また は昇圧剤が必要) 4) 重大な急性腎機能障害、 肝機能障害、または神経機能 障害 5) ICUへの入室 6) 死亡
・診断基準は、治験対 象集団(小児等)によ り変更が必要かもしれ ない ・プラセボ対照有効性 試験の主要な有効性エ ンドポイントの点推定 値が50%以上、信頼区 間の下限が> 30%であ る必要あり ・有効性が証明された ワクチンを対照にした 非劣性試験では、統計 的成功基準は、主要な 相対的有効点推定値の 信頼区間の下限が >-10%である必要 ・後期治験には、ワク チン関連呼吸器疾患増 強(ERD)を評価するため の中間分析を含める必 要 通常の感染症予防 ワクチンの安全性 評価を行うべき で、以下の点を含 むべき。 ・接種後少なくと も7日間、局所およ び全身の有害事象 を評価 ・接種後少なくと も21〜28日間の有 害事象 ・接種後、少なく とも6か月間の重篤 または一般的な有 害事象 ・少なくとも3,000 人の安全性データ ベースの構築が、 認可前に必要 COVID-19疾患の転 帰に関してワクチ ン群と対照群間の 比較を定期的に行 い、ワクチン関連 呼吸器疾患増強 (ERD)のシグナル である可能性のあ る重症COVID-19症 例を評価。独立し たデータ安全監視 委員会(DSMB)で の評価を推奨。
・定期的な安全性 報告、重篤有害事 象報告 ・承認申請時に E2Eに基づく ファーマコビジラ ンスプラン (PVP)の提出を 推奨:重要な特定 されたリスク、重 要な潜在的リス ク、または重要な 不足情報、及びこ れらに対する対応 を含む。ワクチン 関連呼吸器疾患増 強 (ERD)評価の ため、薬剤疫学解 析等を検討すべ き。
臨床試験における 安全性評価項目と して、受胎日が接 種前または接種後 30日以内である妊 産婦における、流 産、死産、先天性 異常などの妊娠転 帰 FDAEmergency Use Authorization for Vaccines to Prevent COVID-19 Guidance for Industry
2021年 2月COVID-19ワクチン の緊急使用許可 (EUA)を求める 製造販売業者に、 COVID-19公衆衛生 緊急事態下におけ るCOVID-19予防治 験ワクチンに関す るEUA発行のため に必要なデータと 情報に関する現在 の推奨事項を提 供。
・EUAの発行には、既知 の潜在的な利益が既知の 潜在的なリスクを上回る ことが必要 ・上記ガイダンスに基づ く、第3相試験の中間解 析結果でEUA申請可能 ・EUA取得のための臨床 試験のエンドポイントを 評価するため、使用され た診断法を特定する必要 がある。 最終的なアッ セイ方法の標準操作手順 (SOP)と検証レポー ト、および臨床検体が測 定された全ラボのリスト の提出を期待。
主要評価項目: ・ワクチン投与のベネ フィットが、リスクを 上回ること(プラセボ 対照有効性試験の主要 な有効性エンドポイン トの点推定値が50%以 上、信頼区間の下限値 が >30%である必要あ り。) ・有効性が証明された ワクチンを対照にした 非劣性試験では、統計 的成功基準は、主要な 相対的有効点推定値の 信頼区間の下限値が >-10%である必要 第1相及び第2相試 験における全ての 安全性データを含 める必要(特に、 重篤な有害事象、 特に関心のある有 害事象、および重 篤なCOVID-19症 例)
第3相試験におけ る、完全なワクチ ン接種レジメンの 完了後少なくとも2 か月の追跡期間 で、重症COVID-19 疾患の症例、ワク チンに対する獲得 免疫および記憶免 疫応答が防御効果 の原因となる期間 に発生するCOVID- 19の症例。ワクチ ン誘発性ERD評価の ため、一般に、プ ラセボ群で5例以上 の重症COVID-19症 例収集が有用。
進行中の臨床試験 が、十分な数の被 験者を対象に、長 期的な安全性と有 効性(ワクチン関 連ERDの評価、接 種後の時間経過に よる免疫応答の低 下により有効性が 低下することを含 む)を十分に評価 できるようにする ために実装される 戦略を含める必要
21
表5-3 リアルワールドデータを用いたCOVID-19ワクチンに関する観察研究用指針
リアルワールドデータを用いたCOVID-19ワクチンに関する観察研究用指針 機関文書の名称発出日目的概要有効性評価の原則有効性評価項目1有効性評価項目2有効性評価項目3有効性評価項目4有効性評価項目5有効性評価項目6 WHOEvaluation of COVID-19 vaccine effectivene ss (Interim guidance)
2021年 3月特に中・低所得 国で、観察研究 デザインを使用 してCOVID-19ワ クチンの有効性 (VE)を評価す る方法に関する 暫定的なベスト プラクティスガ イダンス(リア ルワールドデー タでは、データ の完全性と品質 が高い設定でも 偏った結果が生 じる可能性があ るため、COVID- 19 VE評価の設 計、分析、およ び解釈における 重要な考慮事項 について説明)
VE評価の目的は、リアルワールドでのワク チン性能評価を行い、臨床試験に基づくエ ビデンスで不足する情報への対応(サブグ ループでの有効性、変異株に対する有効 性、ワクチン接種による防護期間を含 む)、影響モデルへの入力提供、さらに条 件付きで承認された製品の有効性の確認を 提供すること。 以下は留意点 ・VE評価における評価項目として、症候性 疾患と重度の疾患を推奨する。 ・参加者の臨床検査は重要で、現時点で は、rRT-PCR結果を評価項目として利用する ことを推奨。検体は発症から10日以内に採 取する必要。 ・想起バイアスと製品の詳細の欠如のため に、ワクチン接種の自己申告は推奨しな い。 ・全ての観察研究デザインはバイアスの影 響を受けることに注意:バイアスとして は、ヘルスケアの追求または曝露リスクレ ベルによる交絡、診断エラーによる結果の 誤分類、以前のSARS-CoV-2感染等。分析に おける交絡バイアスを制御するための主要 な共変量の収集は重要。
・一次分析では、ワク チンの推奨用量を投与 された人とワクチン接 種を受けていない人を 比較する必要。 ・二次分析では、部分 的にワクチン接種され た人(2回接種のとこ ろ、1回接種のみ 等)、2つの異なるワ クチンの投与を受けた 人、標的とされたサブ グループ、ウイルス変 異株、および病歴等と の関連の解析を行う。
<CIVID-19による 死亡> 重要だが、評価困 難(理由:1) 死亡 者の感染・非感染 が区別困難、2) 評 価可能な死亡数症 例の確保困難、3) 死亡者がワクチン 接種していたか、 把握困難)。但し データベース等が あれば、可能。
<COVID-19による重症疾患> 多くの状況で評価できる可能 性がある。重症度評価として 「入院」は他のバイアスが大 きく利用は不適切。 1. WHOの重症・重篤疾患の症 例管理の定義: 肺炎の臨床的兆候(発熱、 咳、呼吸困難、呼吸困難)に 加え、次のいずれか1)呼吸数 >30呼吸/分、重度の呼吸困 難、または室内空気で SpO2<90%、2)急性呼吸窮迫症 候群(ARDS)、敗血症、敗血 症性ショック、3)死亡。 2.重症急性呼吸器疾患感染症 (SARI)のWHOサーベイランス 症例の定義: 38°C以上の発熱または発熱の 病歴があり、過去10日以内に 発症した咳を伴う急性呼吸器 感染症の入院患者。 可能な場合はカテゴリではな く、測定値として収集するこ とを推奨。
<COVID-19による症候性疾 患> ほとんどのワクチン臨床試 験の主要な評価項目である が、リアルワールドデータ ではバイアスを低減するた めに、以下の定義を選択す ることを推奨。 ・修正されたWHOサーベイ ランス症例の定義:過去10 日以内に以下の症状を示し た人:発熱及び咳の急性発 症。または次の兆候・症状 を3つ以上有する急性発 症:発熱、咳、倦怠感、頭 痛、筋痛、喉の痛み、鼻感 冒、呼吸困難、食欲不振/ 吐き気/嘔吐、下痢、精神 状態の変化。 ・インフルエンザ様症状の 定義: 38°C以上の発熱を伴う急 性呼吸器感染症、咳を過去 10日以内に発症。
<CIVID-19への 感染> 重要だが、評価 は困難。 長期 にわたりワクチ ン接種者と非接 種者の定期的な 核酸検査が必要 であり、バイア スが入る可能性 ある。感染のVE 評価は、疾患の 評価よりも数倍 費用がかかり、 ロジスティック 的に複雑。
<臨床検査で確認 された結果(核酸 検査によるSARS- CoV-2への感染)> 臨床徴候や症状に 基づく症候群の結 果よりも、COVID- 19疾患にはるかに 特異的であり、変 異株と従来株との 区別も可能である ため、また臨床試 験の結果との比較 も可能で有用性が 大きい。COVID-19 VE評価では、理想 的にはrRT-PCRを使 用した検査結果を 使用することを推 奨。
<予防可能期間> 臨床試験結果を フォローアップす るために重要な項 目。臨床試験で評 価する接種直後の 感染等に加え、経 時的な防御的抗体 濃度の低下を血清 学的に調査するこ とが考えられる が、細胞性免疫の 関与も示唆されて おり、関連評価指 標は確立されてい ない。ワクチン接 種群で経時的に増 加する疾患の症例 数又は割合を評価 する。比較群間の 背景情報と接種後 期間の一致が、バ イアス低減に重 要。