博士論文
卵子特異的インプリントにおける
DNA メチル基転移酵素の役割
農学研究科バイオサイエンス専攻
論文題目 マウス卵母細胞におけるメチル化インプリント確立の分子機構 博士論文 目次 第一章 緒論・・・・・・・・・・・・・・・・・・・・・・・・・・・・・3 付表および付図 第二章 卵子形成過程におけるDNA メチル基転移酵素の発現解析 第一節 緒言 ・・・・・・・・・・・・・・・・・・・・・・・・14 第二節 材料および方法 ・・・・・・・・・・・・・・・・・・17 第三節 結果 ・・・・・・・・・・・・・・・・・・・・・・・23 第四節 考察 ・・・・・・・・・・・・・・・・・・・・・・・25 付表および付図 第三章 2A peptide を用いた雌性生殖系列における遺伝子過剰発現系の構築 第一節 緒言 ・・・・・・・・・・・・・・・・・・・・・・・26 第二節 材料および方法 ・・・・・・・・・・・・・・・・・・30 第三節 結果 ・・・・・・・・・・・・・・・・・・・・・・・38 第四節 考察 ・・・・・・・・・・・・・・・・・・・・・・・42 付表および付図 第四章 Dnmt3a2 および Dnmt3L コンディショナルトランスジェニックマウス におけるDNA メチル化解析 第一節 緒言 ・・・・・・・・・・・・・・・・・・・・・・・43 第二節 材料および方法 ・・・・・・・・・・・・・・・・・・47 第三節 結果 ・・・・・・・・・・・・・・・・・・・・・・・56 第四節 考察 ・・・・・・・・・・・・・・・・・・・・・・・66 付表および付図 第五章 総合考察 ・・・・・・・・・・・・・・・・・・・・・・・74
第六章 総括 ・・・・・・・・・・・・・・・・・・・・・・・85
Summary ・・・・・・・・・・・・・・・・・・・・・・・95
謝辞 ・・・・・・・・・・・・・・・・・・・・・・・104
第一章 緒論 すべての生物は、種を次世代に残すために生殖を行う。生殖は無性生殖と有 性生殖の二種類に別れており、体細胞分裂を基本として親と全く同一の個体を 増殖させる無性生殖に対して、有性生殖では異なる細胞あるいは個体間のDNA を交雑することにより親とは異なる多様な次世代を作出し、環境変化への適応 あるいは進化を促す(1)。このような役割を持っている細胞が配偶子である。一 般に、哺乳類のような高等生物の場合、大きさの異なる異型配偶子を形成し、 大型で栄養を蓄えるものを卵子、小型で運動性に富むものを精子とよぶ。両親 が正常な次世代を生産するためには、当然、これらの精子および卵子が機能的 でなくてはならない。機能的な精子とは、卵子まで辿り着くことのできる運動 能を持ち、受精時に卵子と接着および融合が可能な精子である。それに対し機 能的な卵子とは、母性因子とよばれるmRNA あるいはタンパク質を細胞質に蓄 積させながらサイズの増大を伴って発育し、受精時においては同種の精子と特 異的に結合・融合して減数分裂を再開させることで、その後の発生過程を円滑 に行うことのできる卵子のことを指す。また、雌雄どちらにも必要な機能とし て、減数分裂時に正しく染色体が分離していることが挙げられる。これらのど れか一つが欠けるだけでも受精あるいはその後の発生が正常に行われることは なく、仮に出生したとしても、染色体異常を伴った出生後の成長異常を示すこ とから、機能的な配偶子の生産は生物の繁栄に必須の機構といえる。 哺乳類における異型配偶子の特異性はそのサイズや機能だけではなく、ゲノ
ムにおいても付与されている。哺乳類以外の生物は、単為発生によって単独の生殖 細胞で受精を経ることなく個体を生産できる(2-5)。しかし哺乳類において、片親由 来のゲノムのみからなる単為発生胚が正常な発生をすることはない。その理由 は発生工学的な手法により証明されてきており、前核期の受精卵の前核を交換 し、雄性前核のみで構成された二倍体胚である雄性発生胚および雌性前核のみ の雌性発生胚を作出すると、それぞれの胚は同一のゲノムをもつ二倍体である にもかかわらず、雄性発生胚は胎仔の成長不全および胎盤の肥大化を示す。そ れに対して、雌性発生胚は胎仔の成長に影響は少ないものの、胎盤が顕著に矮 小化するという、全く異なる表現型を示していずれも妊娠中期で致死となる (6-8)。このことから、雌性および雄性前核にはゲノム配列以外に明確な差異が 存在すること、正常な発生には雌雄のゲノムがどちらも必須であることが示さ れた。またヒトにおいても、片親由来の染色体を重複して持つ片親性ダイソミ ーであった場合、生後の発育不全、精神疾患といった異常を示すことが知られ ている。典型的な例としては、ヒト15 番染色体を父親から受け継いだ場合肥満 および知的障害を示すPrader-Willi 症候群(PWS)、母親から受け継いだ場合重 度の精神遅滞、てんかんおよび歩行障害を示すAngelman 症候群(AS)という、 全く異なった症状を示すことが知られる(9, 10)。それらもまた、両親から由来す るゲノムの不等価性を裏付けるひとつの証拠である。こうした親の性に応じて 染色体上に付与された情報が子に影響する現象は、「ゲノムへの刷込み(ゲノム インプリント)」と称される。また、PWS および AS の違いは、ヒト 15 番染色 体において発現異常あるいは発現抑制を示す遺伝子の違いであることが示され
ている。このことは、父親あるいは母親から受け継いだ染色体(父方および母 方アレル)からしか発現しない遺伝子が存在し、かつ染色体の一部の領域が父 母アレル間で異なる役割を与えられていることを意味している。
以上のようなゲノムインプリントの影響を受けて片親性の発現を示す遺伝子、 すなわちインプリント遺伝子とよばれるものとして、最初にインスリン様成長 因子II 型(Insulin-like growth factor type II; Igf2)遺伝子が発見された(11)。Igf2 をホモで欠損したマウスは、生後の成長不全を呈するが、Igf2 の欠損アレルを 父親から受け継いだ場合、その仔は同様の表現型を示すのに対して、欠損アレ ルを母親から受け継いだ場合、仔は何の表現型も示さない。このように、常染 色体上に存在するにもかかわらず、欠損したアレルの由来が父か母かにより次 世代の表現型が変化するインプリント遺伝子は、Igf2 を発端とし、現在までに 100 以上発見されている。Kono らは、完全にゲノムインプリントを欠如した胚 を核移植技術によって作出し、そのすべてが妊娠初期で致死となっていること を示すと同時に、インプリント遺伝子の発現プロファイルをより精子様の状態 に近づけた卵母細胞を用いることにより、雌性発生胚の発生能を延長させ、妊 娠満期まで到達させることに成功した(12-15)。このことから、父母アレルの決 定的な差はゲノムインプリントの違いによって説明できること、父母アレル特 異的なゲノムインプリントの確立は、受精後のインプリント遺伝子の片親性発 現および正常な胚発生に必須であることを示した。このことは、部分的にイン プリントを欠如した胚が妊娠満期まで到達するものの、その後の成長異常を伴 うことからも、ゲノムインプリントが正常な発生に不可欠な存在であることが
裏付けられている(16)。 インプリント遺伝子の片親性発現が仮に塩基配列の変異を伴ったものである ならば、それは次世代へ遺伝し、同様の異常を示すはずである。そうではない という事実は、こうした雌雄間の違いは、塩基配列ではないものによって付与 されていること、且つこの違いは生殖細胞において一旦リセットされ、自身の 性に応じて再編成されることを意味している。現在このような塩基配列以外の 修飾による遺伝子発現制御機構は、後成的発現制御(エピジェネティクス)と よばれる。 今日までに、DNA のシトシン–グアニンと続く配列(CpG 配列)におけるシ トシンのメチル化が、特に代表的なエピジェネティック制御の分子機構として 挙げられる。特にCpG 配列のメチル化とゲノムインプリントとの関係は、トラ ンスジェニックマウスの導入遺伝子が親のアレル特異的にメチル化を受け、片 親性の発現を示したことから着目されはじめた。その後、インプリント遺伝子 の近傍に存在する父母アレルの間でメチル化状態が異なる領域(Differentially Methylated Regions; DMRs)が発見され、内在性の遺伝子内にも同様な領域が 存在することが示された(17-20)。DMR を欠損したマウスは、その領域中に含ま れるインプリント遺伝子の発現制御が崩壊する(21-23)。また DNA メチル基転移 酵素Dnmt1 を欠損したマウスにおいて DMR のアレル特異的メチル化が消失し、 H19 および Igf2 を始めとした多くのインプリント遺伝子の片親性発現が崩壊す ることが示された(24)。現在までに、22 の DMR がインプリント遺伝子を制御
することが知られており、精子特異的なメチル化インプリントを受け継ぐ父方 メチル化DMR が 3 つ、卵子特異的なメチル化インプリントを受け継ぐ母方メチ ル化DMR が 19 存在する(25)。
DNA メチル化インプリントが、何によって付加されているのかについては、 DNA メチル基転移酵素(DNA methyltransferase; DNMT)遺伝子の研究から明 らかにされてきた。マウスにおけるDNMT ファミリーとして DNMT1、DNMT2 およびDNMT3 の 3 種に分岐し、現在までにアイソフォームを含めて 8 種類が 同定されている。維持型メチル化酵素DNMT1 は、1988 年に Bestor らにより同 定され、最も古くから研究されている DNMT である。DNMT1 は非常に強いヘ ミメチル化DNA へのメチル化活性を有し、体細胞分裂の際の DNA 複製によっ て希釈されるメチル化状態を修正するという役割を担っている。またDnmt1 を 欠損するとゲノム全体の低メチル化を引き起こし、着床後に致死となることが わかっている(24, 26)。DNMT2 は tRNA のアルギニンへのメチル化活性を持ち、 DNA へのメチル化活性は持たないこと、それと一致して欠損した ES 細胞では DNA メチル化に対する表現型がないことが知られている(27)。 DNMT3 は 1998 年に Okano らによって同定され、DNMT3A および DNMT3B の2 種類が存在している(28)。DNMT3A あるいは DNMT3B は、DNMT1 に比べ て酵素活性は弱いものの、非メチル化状態のCpG 配列にメチル化を付与するこ とのできる新規メチル化活性を持っている。Dnmt3a を全身で欠損すると、出生 後3 週間以内に生育不全や行動異常を伴い致死となる。また Dnmt3b の欠損は、
胎齢13.5–16.5 日(days post coitum; dpc)で胎生致死を引き起こすことがわか っている(29)。それと同時に、Dnmt3a および Dnmt3b の両欠損マウスは Dnmt1 欠損マウスと同様に妊娠中期で致死となること、Dnmt3a および Dnmt3b の両欠 損 ES 細胞は継代を重ねるにつれてゲノム全体のメチル化レベルが低下するこ とから、これらは DNMT1 を補うかたちで維持メチル化にも寄与していること が知られている(29, 30)。また、この両欠損マウスと Dnmt3a あるいは Dnmt3b 単独の欠損マウスとの 9.5 dpc におけるメチル化状態の違いを比較すると、 Dnmt3a 欠損マウス胚ではほとんど野生型と違いが認められないのに対し、 Dnmt3b 欠損マウス胚では内在性レトロウィルスやサテライト配列のメチル化 状態が顕著に低下することから、DNMT3A および DNMT3B はある程度の補償 的な関係にあること、DNMT3A と DNMT3B の間にはメチル化する DNA 領域の 嗜好性の違いが存在することがわかっている(29)。 前述の通り、Dnmt3a あるいは Dnmt3b を全身で欠損した場合は性成熟に達す る前に致死となることから、生殖細胞における解析を行うことが困難であった。 2004 年、Kaneda らは Cre/loxP システムを用いることで、Dnmt3a あるいは Dnmt3b を生殖細胞特異的に欠損するコンディショナルノックアウトマウスを 作出した。その結果、Dnmt3a を生殖細胞で欠損すると雄は Rasgrf1 以外の DMR の低メチル化および減数分裂の異常をもたらして無精子症となるのに対し、雌 においては一見表現型がなく、受精可能な卵子が形成されるものの、それらの 仔は卵子におけるメチル化インプリントを欠如しており、インプリント遺伝子 の発現異常を呈して胎生致死となることが示された(31, 32)。一方、Dnmt3b の
生殖細胞における欠損は野生型と比較しても違いが認められなかった。以上の ことから、生殖細胞におけるメチル化インプリントに必須なのはDNMT3A であ り、DNMT3B は着床後、内在性レトロウィルスやリピート配列のメチル化に寄 与することが明らかになっている(31, 32)。 DNMT3 ファミリーには、DNMT3A や DNMT3B と高い相同性を持つにもかか わらず、メチル化酵素活性を欠如したものが存在し、DNMT3L(DNMT3-like) とよばれている(33)。Dnmt3L を欠損したマウスは、Dnmt3a コンディショナル 欠損マウスと同様、雄ではIgf2-H19 領域の低メチル化および無精子症を示し、 卵子形成過程においても母方メチル化インプリントを欠如し、仔がすべて胎生 致死となる(34-38)。このことから、DNMT3L もまた DNMT3A とともにゲノム インプリントの確立に重要な役割を担っていることが示されている。同時に、 生化学的研究により、DNMT3L は DNMT3A あるいは DNMT3B と直接相互作用 し、その酵素活性を大きく上昇させることが明らかになっている(39)。 以上のことから、DNMT3A および DNMT3L は生殖細胞における DNA メチル 化インプリントの確立に必須であると考えることができる。しかし、現在まで に生殖細胞においてDNMT3A/DNMT3L が領域特異的に DNA メチル化インプリ ントを行う分子機構については明らかにされておらず、DNMT3A/DNMT3L の生 殖細胞における発現がDNA メチル化インプリント確立の十分条件であるか否か は明らかにされていない。 生体内におけるエピジェネティック修飾は世代ごとに一過的なものであるた
め、親の代のゲノムインプリントは、子の生殖細胞では一旦消去される必要が ある。マウスにおいては、生殖細胞の前駆体である始原生殖細胞(Primordial Germ Cell; PGC)は 7.5 dpc 付近に出現する。これらは 9.5 dpc までの移動期に かけて一度メチル化レベルが減少し、10.5 dpc 付近において完全に消去される (40, 41)。PGC を in vitro で培養して樹立された多能性幹細胞(Embryonic Germ Cell; EG 細胞)と体細胞との細胞融合によって、体細胞側のゲノムのメチル化 状態が低下したことから、PGC における脱メチル化は能動的なものであること が示されている(42)。しかし、植物における DEMETER 遺伝子のような、メチ ル化シトシンを除去してシトシンへ修復する脱メチル化酵素が哺乳類に保存さ れていないこと(43, 44)、メチル化シトシンを脱アミノ化し、チミンへの変化を 誘導する酵素Aid を欠損した PGC においても同様にメチル化インプリントが消 去されることから(45)、これがどのような機構で行われているのかについては、 長い間不明なままであった。近年、メチル化シトシンを酸化させる酵素TET フ ァミリーの発見(46, 47)、あるいは従来の解析方法ではメチル化シトシンと区別 できなかった新たな修飾であるヒドロキシメチル化シトシンが生体内で検出さ れたことにより、メチル化シトシンをヒドロキシル化させて最終的にDNA 修復 によってシトシンへ戻すという間接的な脱メチル化機構が提唱され(48, 49)、特 にPGC におけるゲノムインプリントの消去に、ヒドロキシル化酵素 Tet1 が重 要なはたらきを持っていることが明らかにされている(50)。 生殖隆起へ到達したPGC は 11.5 dpc 付近に起こる性決定の後に、自分の性 に特異的なゲノムインプリントを新たに確立する。精子形成過程においては、
14.5–16.5 dpc にかけての体細胞分裂休止期に入った前精原細胞において、 H19-Igf2、Dlk1-Dio3(IG-DMR)、Rasgrf1 といった 3 領域が高メチル化される (51)。 一方、卵子特異的なゲノムインプリントがいつ、どのようにして確立されて いくのかは、発生工学的手法を用いてKono らにより証明され、出生後すぐの非 成長期(non-growing; ng)卵母細胞においてはまったく確立されていないが、 成長期を経て徐々に確立されていき、フルサイズ(fully-grown; fg)卵母細胞ま でに完全に確立することを示した(12, 14, 15, 52)。その後、Obata ら(2002)、 Hiura ら(2006)は各日齢の卵母細胞をサイズごとに分類し、各 DMR について 詳細なメチル化解析を行った。その結果、卵子特異的インプリントの確立は日 齢ではなく卵母細胞のサイズに依存して起こっていることを証明した(53, 54)。 また確立されるタイミングは画一的なものではなく、インプリント領域ごとに 特異的なタイミングをもち、早いタイミングで確立される Igf2r、Zac1 および Lit1 DMR は卵母細胞の直径 50 µm を境に急激にメチル化レベルが増大する一方 で、Mest DMR のように卵母細胞の直径が 60 µm を超える成長後期のステージ でも低メチル化状態である領域も存在することを示した(16, 54)。しかしながら、 こうした領域特異的なタイミングの違い、あるいは雌雄間のメチル化される領 域の特異性の違いは未だ全く明らかにされてはいない。 近年、DNMT3A/DNMT3L 以外にもメチル化インプリント確立に必要な因子が 明らかにされてきている。Ciccone らは、ヒストン H3 Lys4 ジメチル(H3K4me2)
脱メチル化酵素であるKDM1B を欠損した卵子において、Grb10、Peg1、Zac1 および Impact 領域で卵子特異的なメチル化インプリントが確立されないこと、 一方、Igf2r、Lit1 および Snrpn 領域では卵子特異的な DNA メチル化インプリン トが確立されることを明らかにした(55)。また、in vitro における解析では、 DNMT3L が非メチル化 H3K4 に高い結合能を持つのに対し、メチル化 H3K4 に はほとんど結合しないことも示された(56)。また、Kobayashi らは fg 卵母細胞 における全ゲノムのDNA メチル化および全転写産物の解析を行い、転写が活性 化されている領域のgene body とよばれる遺伝子内部の CpG 配列は比較的高メ チル化されおり、このことから、転写によりクロマチン構造の変化を起こすこ とで、DNMT のアクセスが可能となり gene body の DNA がメチル化される可 能性を示唆した(57)。さらに、卵子形成過程において卵子特異的エキソンからの DMR をまたぐように起こる転写産物が、主要な母方インプリント領域で共通し て存在すること(58)、卵子形成過程で Snrpn や Gnas といったインプリント領域 において転写産物を短くし、DMR をまたがないようにすると Snrpn および Gnas 領域のメチル化インプリントが欠如することが明らかにされた(58, 59)。これら の結果から、DNMT3A/DNMT3L による DNA メチル化インプリントの確立には、 DMR が DNMT3A/DNMT3L のアクセスを許容し、メチル化を付加することの可 能なクロマチン状態へと変化することが重要であり、その移行には、KDM1B に よるインプリント領域のH3K4 の脱メチル化や DMR をまたぐ転写が起こること が必要であるという仮説が立てられる。しかし、インプリント獲得の詳細なメ カニズムは十分に理解されておらず、これらの役者でインプリント獲得機構を
全て説明できるか否かは不明である。 本研究では、DNMT3A および DNMT3L の存在は卵子特異的 DNA メチル化イ ンプリント確立に必要かつ十分であるか否か、また、DNA メチル化インプリン トを確立するために必要な分子機構は何であるのかを明らかにするために、1) Dnmt3a/Dnmt3L を卵母細胞において早期に過剰発現させるトランスジェニッ クマウスの作出および解析 2) KDM1B および DMR をまたぐ転写と DNA メチ ル化インプイリントとの関連性の解析 3) 過剰発現した成長期卵母細胞ゲノム におけるインプリント機能の評価 を行い、卵子特異的ゲノムインプリントに おけるDNA メチル基転移酵素の役割について考察することにより、卵子特異的 インプリントの分子機構の解明に迫った。
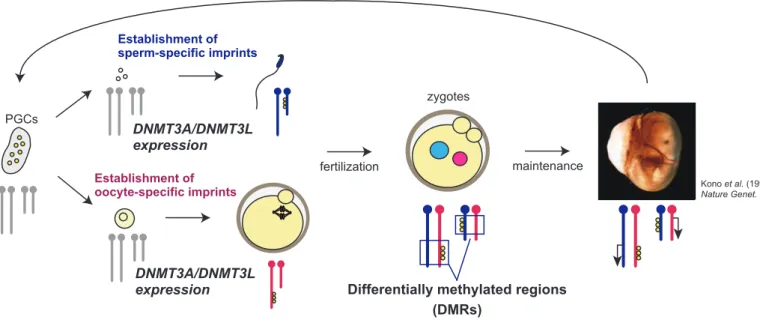
erasure of genomic imprints Establishment of sperm-specific imprints Establishment of oocyte-specific imprints fertilization PGCs zygotes maintenance
Differentially methylated regions (DMRs)
Epigenetic modification
(DNA methylation, histone modification)
DNMT3A/DNMT3L expression DNMT3A/DNMT3L expression
Fig. 1 Schematic representation of genomic imprinting in mice.
Kono et al. (1996)
第二章 卵子形成過程におけるDNA メチル基転移酵素の発現解析 第一節 緒言 哺乳類の生殖細胞形成過程において、雌雄特異的に付加されるゲノムインプ リントは、受精後の正常な発生に不可欠であることが知られている(2-4)。ゲノ ムインプリントの本質はエピジェネティックな修飾であり、その中でも最も良 く知られているのが DNA のシトシン–グアニンと続く配列のシトシンへのメチ ル化である。父母のアレルのどちらか一方からしか発現しないインプリント遺 伝 子 の 近 傍 に は 父 母 ア レ ル 間 で メ チ ル 化 状 態 の 異 な る 領 域 (Differentially methylated region; DMR)が存在し、DMR のメチル化状態はインプリント遺伝 子の片親性発現を制御することが知られている。こうしたDMR のメチル化は、 生殖細胞へ分化した際に一旦消去され、雌雄それぞれの生殖細胞形成過程にお いて自身の性に応じて新たにメチル化されることが知られている(55)。
DNA メ チ ル 化 を 行 う 分 子 と し て 、 DNA メ チ ル 基 転 移 酵 素 ( DNA methylatransferase; DNMT)の存在が知られている。マウスにおいては DNMT ファミリーとして、現在までに 5 種類が確認されている。最初に同定された DNMT は維持型メチル化酵素 DNMT1 であり、非常に強いヘミメチル化 DNA へ のメチル化活性を有している(24)。非メチル化状態の生殖細胞のゲノムに新規の インプリントを付与するとして、DNMT3A および DNMT3B の 2 種類が知られ ている(26)。コンディショナルノックアウトマウスを用いた Kaneda らの研究か ら、生殖細胞におけるゲノムインプリント確立に必須であるのはDNMT3A であ
り、DNMT3B は着床後の体細胞におけるメチル化に寄与することが明らかにな っている(22)。 DNMT3 ファミリーには、DNMT3A や DNMT3B と高い相同性を持つにもかか わらず、メチル化酵素活性を欠如したものが存在し、DNMT3L(DNMT3-like) とよばれている(30)。Dnmt3L を欠損したマウスは、Dnmt3a を生殖細胞におい て欠損したマウスとほとんど遜色ない表現型を示したことから(21)、DNMT3L もまた、DNMT3A とともにゲノムインプリントの確立に重要な役割を担ってい ることが示されている。その後の研究により、DNMT3L は DNMT3A あるいは DNMT3B と直接相互作用し、その酵素活性を大きく上昇させることが明らかに なった(35)。 2002 年、Chen らは未分化 ES 細胞において発現している Dnmt3a のアイソ フォームであるDnmt3a2 を同定した(56)。DNMT3A2 は DNMT3A の N 末端の 210 ア ミ ノ 酸 を 欠 如 し た 配 列 で あ り 、 ES 細 胞 に お い て は 未 分 化 状 態 で DNTM3A2 が発現し、分化するとともに DNMT3A1 の発現に切り替わることが 報告されている(56)。また、未分化状態の ES 細胞において DNMT3A2 が DNMT3L と相互作用し、核内に局在することがわかっている(57)。さらに Chen らは Dnmt3a および Dnmt3b の両欠損 ES 細胞へ DNMT3A あるいは DNMT3A2 のみ を発現させ、Igf2-H19 DMR が DNMT3A2 特異的にメチル化を回復したことを示 した(29)。このことから、DNMT3A および DNMT3A2 間には何らかの役割の違 いが示唆されているが、それが何であるかは明らかになっていない(Fig. 2-1)。 こうした DNMTs の発現レベルは、mRNA においてはノーザンブロットある いはRT-PCR を用いて、組織あるいは雌雄の生殖細胞において古くから解析が
進められている(58)。雄性生殖細胞においては、ゲノムインプリントの確立が始 まる14.5 dpc 付近から発現を開始し、インプリントの完了する 16.5 dpc 付近で その発現はピークとなる。この時Dnmt3a2 が特に強く発現し、Dnmt3a はほと んどまったく発現しないことが知られている(59)。卵子形成過程においては、成 長期卵母細胞において、mRNA レベルで Dnmt3a2 の発現が認められている。し かしながら、雄性生殖細胞とは異なり、その発現は Dnmt3a もまた同程度であ ることが報告されている(60)。一方で、タンパク質レベルの発現解析は、そのサ ンプルの微量さから免疫染色を用いた解析がほとんどであるが、インプリント の完了する16.5 dpc の精巣における免疫沈降を用いた解析から、雄性生殖細胞 におけるDNA メチル化インプリントの責任分子は DNMT3A2 および DNMT3L であることが確かめられている(57)。しかし卵子形成過程においてはいずれの DNMT3A が発現しているか定かではなく、DNMT3A および DNMT3A2 を区別し て、それぞれを定量的に解析することは行われてきていない。
そこで本章では、卵子形成過程においてメチル化インプリントを付与してい る責任分子を正確に把握することを目的として、Dnmt3a、Dnmt3a2 および Dnmt3L の卵子形成過程における発現を、定量 PCR およびウェスタンブロット によって解析した。
第二節 材料および方法 1. 供試動物 mRNA およびタンパク質の発現解析には、すべて野生型 C57BL/6N(B6)マ ウス(日本クレア)を用いた。 本研究で用いたすべてのマウスは、午前8 時点灯、午後 8 時消灯の日照条件 で、温度20-26℃および湿度 40–60%の SPF 環境下で飼育した。餌は滅菌済み 固形餌料を、水は滅菌済み水道水を不断給餌し、自由摂取させた。なお、これ 以降のすべての実験は、東京農業大学の動物実験指針に従い行った。 2. 卵母細胞の採取 2-1. ng 卵母細胞の採取 ng 卵母細胞は、生後 1–3 日の雌マウスから採取した。マウスを断首により安 楽死させた後、開腹し、卵巣を摘出した。卵巣はM2 培地中で余分な組織を取り 除いて1.5 ml エッペンチューブに移した後、0.1%コラゲナーゼ(Wako)添加 L15 培地を加えて 37℃で 15 分インキュベートした。その後 10000 rpm で 10 秒 程度遠心し、上清を除いて0.05% Trypsin-EDTA(Gibco)を加え、再び 37℃で 15 分インキュベートした。M2 培地を加えて反応を停止させた後、10000 rpm で10 sec 程度遠心して上清を除き、mRNA 解析の場合、5 µg/ml サイトカラシ ンB(Sigma)添加 M2 培地を加えてピペッティングにより懸濁した。この懸濁 液からマイクロマニピュレーター (NARISHIGE) を接続した倒立顕微鏡下で、 直径20 µm 程度のマイクロピペットを用いて ng 卵母細胞のみを回収した。
2-2. 成長期卵母細胞の採取 成長期卵母細胞は、生後10、15 および 20 日の雌マウスから採取した。マウ スを頚椎脱臼により安楽死させ、前項と同様に摘出および余分な組織を除去し た後、200 µl の 0.1%コラゲナーゼ添加 L15 培地ドロップに 2–4 卵巣/ドロップ となるよう移し、37℃で 1 時間インキュベートした。その後ピペッティングに より卵胞を分離させ、上清をできるだけ除き0.05% Trypsin-EDTA を 200 µl 加 えて37℃で 15–20 分インキュベートした。次いで M2 培地を加えて反応を停止 させ、激しくピペッティングすることにより卵胞から卵母細胞を裸化させた。 上清を除き、透明帯を除去するため0.1%プロナーゼ(Sigma)添加 L15 培地を 200 µl 加えて 37℃で 15-20 分インキュベートした。再び上清を除き 240 µM dibutyryl cyclic AMP(Sigma)添加 M2(dbcAMP+M2)培地を加える操作を数 回行って培地を置換し、卵母細胞を実体顕微鏡下でマウスピースキャピラリー により洗浄用のdbcAMP+M2 培地ドロップへ移して体細胞が完全に除去される まで洗浄した。 これらの成長期卵母細胞は、サイズによる違いを比較するため卵子直径40–49 µm、50–59 µm および 60–65 µm の 3 区分に分類した。マイクロマニピュレー ター付き倒立顕微鏡に対物ミクロメーターを用いて卵母細胞を各サイズに分類 した。 2-3. fg 卵母細胞の採取 fg 卵母細胞は、生後 8–12 週齢の性成熟した成体雌マウスから採取した。過排 卵処理のために5 IU の妊馬血清性性腺刺激ホルモン(PMSG)を腹腔内投与し、 その44-46 時間後に頚椎脱臼により安楽死させ、卵巣を摘出した。dbcAMP+M2
培地内で 25G 注射針にて卵胞を裂いて卵丘細胞-卵子複合体を得て、新しい dbcAMP+M2 培地内でピペッティングにより卵丘細胞を完全に除去した。
3. mRNA 発現解析
3-1. total RNA 抽出および cDNA 合成
各卵母細胞total RNA の抽出は、RNeasy Micro Kit(QIAGEN)を用いて行っ た。卵母細胞については、ng 卵母細胞を 300–400 個、成長期あるいは fg 卵母 細胞を100–200 個程度プールしたものを解析に供した。洗浄した卵母細胞は、 75 µl の 1% β-Mercaptoethanol 添加 RLT Buffer へ移し、使用時まで-80℃で保存 した。その後はキットの定法に従いDNase 処理済みの total RNA を得た。DNase によりゲノムDNA が完全に排除されているか否かは PCR によって判定した。 RNA サ ン プ ル 1 µl を Gapdh 遺 伝 子 特 異 的 プ ラ イ マ ー ( Forward ;5’-ACCACAGTCCATGCCATCAC-3’ 、 Reverse; 5’-TCCACCACCCTGTTGCTGTA-3’)を用いてアニーリング温度 55℃、40 サイ クルの条件でEx Taq(Takara Bio)により PCR 反応を行い、電気泳動後バンド が現れないことで確認した。その後cDNA は oligo (dT)12-18 primer(Invitrogen)
を用いて、SuperScript III Reverse Transcriptase(Invitrogen)の定法に従い 55℃ で1 時間反応を行うことで合成した。
3-2. 定量的 RT-PCR
Dnmt3a、Dnmt3a2 および Dnmt3L の mRNA 発現解析は、7500 Realtime PCR System(Applied Biosystems)および各遺伝子特異的に設計された TaqMan Probe(Applied Biosystems)を用いて行った。TaqMan Probe は、それぞれ
Dnmt3a ( Mm00432870_m1 )、 Dnmt3a2 ( Mm00463987_m1 )、 Dnmt3L (Mm00457635_m1)および Gapdh(Mm99999915_g1)を用いた。また検量 線を作成するため、増幅領域周辺をpGEM-T Easy Vector (Promega)にクロ ーニングしたプラスミドを作成し、そのコピー数をもとに1×107〜103コピー/µl の希釈系列を作成し、サンプルと同時にPCR 反応に供した。得られた各遺伝子 の数値はGapdh の数値で補正し、相対値として発現量を算出した。 4. ウェスタンブロッティング 4-1. SDS-PAGE 新生仔の卵巣組織はPBS(-)で洗浄し 10000 rpm で 10 秒程度遠心した後、上 清を除いて液体窒素に投入し、使用時まで-80℃で保存した。融解後 2×Sample Buffer ( 0.25M Tris-HCl (pH 6.8), 40% Glycerol, 0.8% SDS, 1% β-Mercaptoethanol)を加え、氷上で 5 分静置後ピペッティングにより組織を崩 し、95℃で 5 分インキュベート後すぐに氷上に静置して熱変性させた。その後 4℃, 15000 rpm で 5 分間遠心し、上清を新しいチューブに移してサンプルとし た。また卵母細胞は0.1% PVA 添加 PBS(-)ドロップで 3 回洗浄し、100 個ずつ を2×Sample Buffer 10 µl の入った 1.5 ml チューブへ移して溶解し、熱変性処 理せず使用時まで-80℃で保存した。融解後、熱変性処理を行い、そのままサン プルとして用いた。
SDS-PAGE には、5–20% e-PAGEL gradient gel(ATTO)を用いた。卵巣組 織は0.1–0.5 個/レーン、卵母細胞は 100 個/レーン(全量)をアプライし、プロ テインマーカーPrestained XL-Ladder(APRO science)と共に 150V で 90 分電
気泳動を行った。ランニングバッファーにはEzRun C+(ATTO)を用いた。泳 動したゲルは PVDF メンブレン(Roche)へ転写するため、タンク式ブロッテ ィング装置を用いて、氷上で冷却ながら100V で 60 分間泳動し、トランスファ ー反応を行った。
4-2. 抗体抗原反応
ブロッキング反応は、PVDF Blocking Reagent for Can Get Signal(TOYOBO) を用いて 4℃で一晩振盪しながら行った。その後の抗体抗原反応は、Can Get Signal Immunoreaction Enhancer Solution(TOYOBO)を用いて、定法に従っ て反応を行った。各遺伝子に対する一次抗体は、DNMT3A モノクローナル抗体 (1:2000, Imgenex)、DNMT3L ポリクローナル抗体(1:2000, 大阪大学 田島 正二先生より分与)を、二次抗体はAnti-Mouse IgG HRP Linked(1:10000, GE Healthcare)および Anti-Rabbit IgG HRP Linked(1:10000, GE Healthcare)を 用いた。その後の化学蛍光検出には ECL Select Western Blotting Detection Reagent(GE Healthcare)を用いて、LAS-1000 plus(Fuji Film)により検出し た。 ま た 同 じ メ ン ブ レ ン で 異 な る 抗 体 を 反 応 さ せ る た め 、Stripping Buffer (62.5mM Tris-HCl (pH 6.8), 2% SDS, 0.75% β-Mercaptoethanol)にメンブレン を浸し55℃で 40 分インキュベートして抗体を除去し、TBS-T(25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.01% Tween20)で 5–6 回洗浄した後に再度ブロッキ ング反応から同様の手順を繰り返した。 取得した画像は、Image J を用いて各バンドのシグナルの数値化を行い算出し た。その後fg 卵母細胞の値を 1 とした相対値を各サンプルについて求めた。
5. 統計処理
すべてのサンプルは、独立したプールからそれぞれ 3 検体用意し、解析に供 試した。得られた値から各サンプル間の平均値および標準偏差を求めグラフを 作成後、t 検定による有意差検定を行った。なお有意水準は P < 0.05 とした。
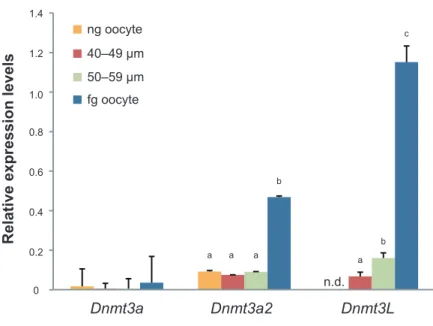
第三節 結果 1. 卵子形成過程における Dnmt3a および Dnmt3L の mRNA 発現解析 最初に、DNA メチル化インプリントに必須である Dnmt3a および Dnmt3L の、 卵子形成過程における発現ダイナミクスの把握を試みた。Dnmt3a には 2 種類の バリアント(Dnmt3a および Dnmt3a2)が存在していることから、そのどちら が主に発現しているかを明らかにするため、定量PCR はそれぞれのバリアント に特異的なTaqMan probe を用いた。その結果、mRNA レベルの解析において、 Dnmt3a 特異的なプローブを用いた場合は ng、40–49 µm、50–59 µm および fg 卵 母 細 胞 ま で 一 貫 し て 発 現 レ ベ ル は わ ず か な も の で あ っ た の に 対 し て 、 Dnmt3a2 特異的なプローブではそれよりも顕著に高レベルな発現が検出された。 このことから、卵母細胞におけるDnmt3a の発現は、Dnmt3a2 が主要に発現し ていることが確かめられた。また、それぞれの成長段階における発現は、ng 卵 母細胞においてもわずかに存在しており、そのレベルは 50–59 µm まで一定で あるものの、fg 卵母細胞において約 3 倍程度有意に増加した。同様に Dnmt3L は、ng 卵母細胞の時点ではまったく検出されなかったのに対して、成長段階を 経るに従ってその発現は徐々に有意差を持って増大し、fg 卵母細胞にかけてそ れまでの約6 倍程度にまで増加した(Fig. 2-2)。 2. 卵子形成過程における DNMT3A および DNMT3L タンパク質の定量発現解析 次いで、DNMT3A および DNMT3L のタンパク質レベルでの発現を、ウェスタ ンブロットにより解析した。卵母細胞は含まれるタンパク量がサイズごとに大
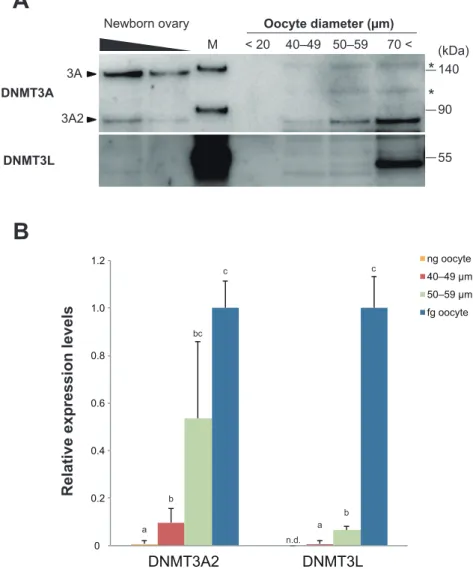
きく異なることから、今回はすべてのサイズの卵母細胞を 100 個/レーンとし、 卵母細胞1 個あたりの絶対量を求めた。その結果、約 130 kDa 付近に存在する DNMT3A は、新生仔卵巣では検出されたものの卵子形成過程を通して卵母細胞 ではシグナルが検出されなかったのに対し、約85 kDa 付近の DNMT3A2 は卵子 形成過程を通して徐々に発現を増大させた。このことから、卵巣において認め られたDNMT3A は、顆粒層細胞を始めとした体細胞に由来するものであると考 えられた。一方、約50 kDa 付近に存在する DNMT3L は、新生仔卵巣、ng 卵母 細胞および40–49 µm の卵母細胞まで全く検出されず、50–59 µm の卵母細胞で わずかに認められた。その後急速にタンパクの蓄積が開始され、fg 卵母細胞で のみ強いシグナルが検出された(Fig. 2-3A)。 またDNMT3A2 および DNMT3L についてシグナルを数値化し、fg 卵母細胞の シグナルに対する相対値を算出したところ、DNMT3A2 の発現レベルは、40–49 µm と 50–59µm 間、50–59 µm と fg 卵母細胞間に有意差は認められないものの 発現上昇する傾向が認められ、40–49 µm と fg 卵母細胞間に有意な発現上昇が 認められた。DNMT3L については、その発現が各サイズ間において有意に上昇 した(Fig. 2-3B)。
第四節 考察 DNMT3A と DNMT3A2 の役割の違いについては、様々な考察はあるものの具 体的な証明には至っていない。Suetake らは、DNMT3A の N 末端側 211 アミノ 酸がDNA と結合する活性をもつことを示し、それが核内への局在性を強めてい ることを示唆した(61)。また Kotini らは ES 細胞における DNMT3A の機能を解 析するため、DNMT3A の N 末端側を過剰発現させた ES 細胞を作出し、具体的 に結合する遺伝子領域や相互作用するタンパク質が DNMT3A2 と異なることを 示した(62)。同様の解析はヒト培養細胞においても行われ、ヒト DNMT3A およ び DNMT3A2 においてもメチル化する領域がわずかに異なることが報告されて いる(63)。しかしながらいずれの報告においても、ゲノムインプリントに関与す ることが予想されるようなタンパク質や DMR のメチル化との関係性はないこ とから、未だに役割の違いについての議論に決着はついていない。 2012 年、O’Doherty らはウシの卵母細胞を用いて DNMT3 ファミリーのタン パク質発現解析を行い、インプリントが確立される時期の卵母細胞においては DNMT3A2 だけでなく、DNMT3A もまた同等に発現していることを明らかにし た(64)。しかしウシとは異なり、マウスにおいては DNMT3A ではなく DNMT3A2 が精子形成過程と同様に卵子形成過程においても強く発現していた。このこと から、マウス生殖細胞系列におけるゲノムインプリントは、雌雄共通の責任分 子 DNMT3A2 および DNMT3L(以下 DNMTs)によって行われていることが考 えられた。 卵子形成過程における卵母細胞のサイズごとの DNA メチル化状態は、Hiura
らの詳細な解析により、40–49 µm のステージにおいて、Igf2r、Lit1 および Zac1 といった領域が、既に 20–40%程度メチル化を受けていることがわかっている (48)。しかし意外なことに、DNMT3L タンパク質は、このステージの卵母細胞 においては検出限界以下であったことから、40–49 µm の段階では DNMT3A2 の存在比がDNMT3L に対し非常に高いと考えることができる(Fig. 2-2A および B)。Kobayashi らは Dnmt3L 欠損マウス fg 卵母細胞を用いた全ゲノムメチル化 解析の結果、Dnmt3L を欠損した fg 卵母細胞においてメチル化されていた DMR は Snrpn 領域だけであり、その他の領域は低メチル化状態であったことから、 ほとんどのDMR における卵子特異的メチル化インプリント確立が DNMT3L 依 存的であることを示した(52)。よって、これらの領域のメチル化には検出限界以 下のわずかなDNMT3L であっても DNMT3A2 の酵素活性を上昇させる可能性が 示唆された。それと同時に、DNMT3L の発現は fg 卵母細胞以降急激に低下し、 そのレベルは受精後も胚盤胞期まで増加しないことが報告されたことから(65)、 DNMT3L の強く発現する時期は、卵子形成過程において非常に限定されている ことが示唆された。 一方、ng 卵母細胞において、DNMT3A2 の mRNA は存在するもののタンパク 質は検出限界以下、DNMT3L は mRNA およびタンパク質のいずれのレベルでも まったく検出されなかった。よって、このステージにおけるng 卵母細胞におけ る非メチル化状態は、DNMT3A2 および DNMT3L が存在していないことに起因 する可能性が示唆された。 2014 年、ng 卵母細胞における非メチル化状態について、大変興味深い結果が Obata らにより報告された。彼らは裸化した fg 卵母細胞に卵胞細胞を再び接着
させて培養する方法を開発し、裸化した状態では 48 時間以内に死滅する fg 卵 母細胞を 5–6 日間にまで延長させることを可能にした。この技術を用いて、核 移植によって ng 卵母細胞のゲノムを fg 卵母細胞の細胞質に長期間曝露させた 際、DNMTs の発現により ng 卵母細胞ゲノムのインプリント領域に部分的なメ チル化が誘導されたことを示した(66)。この事実から、ng 卵母細胞ゲノムは DNMTs の存在によりインプリントを確立する可能性が示唆された。 通常、培養細胞に DNMTs を過剰発現させた場合、エピソーム由来の外来性 DMR をメチル化することはできても、生体内の DMR の父母アレル特異的メチ ル化状態を崩し、両アレルとも高メチル化状態にすることができない(67)。これ は、体細胞におけるDMR 内の非メチル化 CpG が何らかの機構により、DNMTs のDNA への結合あるいはメチル化活性を阻害することで非メチル化状態を保護 していると考えられる。一方、生殖細胞に分化した細胞は、10.5 dpc 付近で起 こるゲノムワイドな脱メチル化と同じタイミングでDNMTs の発現を減少させ、 以降、DNMTs の発現および DNA メチル化状態を ng 卵母細胞まで低レベルに維 持している(68)。Obata らの研究は、そうした ng 卵母細胞のゲノムが、少なく ともfg 卵母細胞の細胞質内で保護されていないことを示しているが、核移植と いう人工的な手段を用いて、ng 卵母細胞ゲノムを異所的な環境に曝露している のも事実である。よって、仮にng 卵母細胞においての DNA メチル化からの保 護が存在しないならば、ng 卵母細胞において DNMTs を過剰発現させることで メチル化インプリントを早期に行うことが可能になると考えられた。 以上のことから、DNMT3A2 および DNTM3L が卵子形成過程における DNA メチル化インプリントの責任分子であることが改めて確かめられた。しかしな
がら、卵母細胞におけるDNA メチル化インプリント確立に、DNMT3A2/DNMT3L の存在は必要かつ十分な因子であるのか否かについては議論の余地が残された。
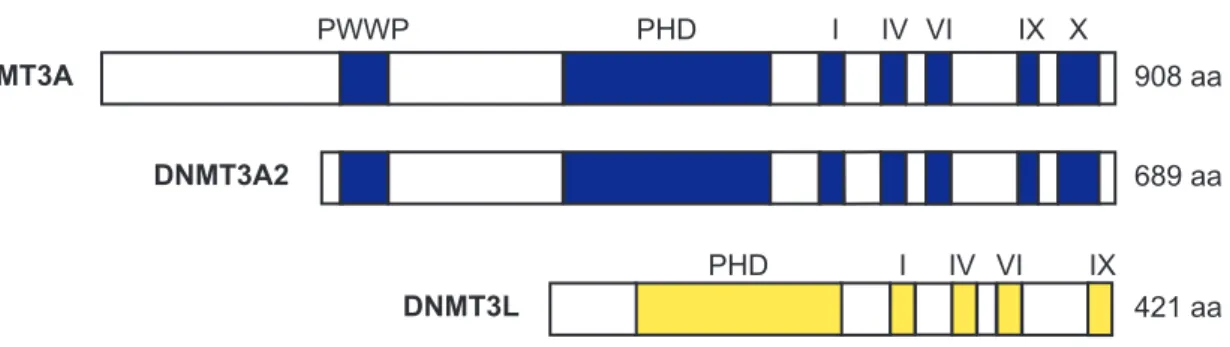
DNMT3A DNMT3A2 DNMT3L PWWP PHD I IV IX X PHD I IV IX VI VI
FIg. 2-1 Schematic represents of DNMT3A, DNMT3A2 and DNMT3L protein.
Functional domains of DNMT3A and DNMT3L are indicated by blue (DNMT3A) and yellow (DNMT3L) box.
908 aa 689 aa
ng oocyte fg oocyte 40–49 µm 50–59 µm 0 0.2 0.4 0.6 0.8 1.2 1.0 1.4 Dnmt3a Dnmt3a2 Dnmt3L
Relative expression levels
n.d. a b a a a b c
Fig. 2-2 mRNA expression analysis of Dnmt3a, Dnmt3a2, and Dnmt3L in mouse oocytes.
qRT-PCR analysis of Dnmt3a, Dnmt3a2, and Dnmt3L in differently sized WT oocytes. Error bars represent standard deviation. A significant difference was observed between a, b, and c (P < 0.05).
0 0.2 0.4 0.6 0.8 1.0 1.2 DNMT3A2 DNMT3L ng oocyte 40–49 µm 50–59 µm fg oocyte DNMT3A DNMT3L Newborn ovary 40–49 50–59 70 < < 20 Oocyte diameter (µm) 3A 3A2 140 90 55 * * (kDa) M
A
n.d. a b bc c a b cRelative expression levels
B
Fig. 2-3 DNMT3A and DNMT3L expression in mouse oocytes.
(A) Western blotting analysis of DNMT3A and DNMT3L expression during oocyte growth. Ovarian samples derived from newborn mice were diluted to 50% (left) and 25% (right). One hundred oocytes were pooled according to their diameter: <20 µm, 40–49 µm, 50–59 µm, and >70 µm. DNMT3A, 130 kDa; DNMT3A2, 85 kDa; DNMT3L, 50 kDa; M, molecular weight marker; and asterisk, non-targeted bands predicted by the molecular weight marker.
(B) Relative expression levels of DNMT3A2 and DNMT3L. Each expression level is relative to the level in fg oocytes. Error bars indicate standard deviation. A significant difference was observed between a, b, and c (P < 0.05).
第三章 2A peptide を用いた雌性生殖系列における遺伝子過剰発現系の構築 第一節 緒言 第二章において、DNMT3A2 および DNMT3L(DNMTs)がメチル化インプリ ントを付加する責任分子であることが強く示唆されると共に、それらはng 卵母 細胞ではほとんど発現していないことを示した。しかし、ng 卵母細胞における 非メチル化状態のインプリント領域は、DNMTs を発現させることによりメチル 化されうるのかは不明である。 この問いに対する答えは、ng 卵母細胞において Dnmt3a2/Dnmt3L を過剰発現 させることで明らかになると考えられた。しかし外来性の遺伝子を導入するた めのエレクトロポレーションやリポフェクションといった一般的な方法は体細 胞をターゲットとしている。これまでに我々の研究室では、あらゆる遺伝子導 入法により新生仔卵巣あるいはng 卵母細胞への遺伝子導入を試みてきた。しか しその導入効率はいずれの方法で行っても 1%未満と極めて低いことが示され ている(2004 年度 山田宗弘修士論文)。よって、トランスジェニック(Tg) マウスを用いた生体内での過剰発現系が必要と考えられた。 哺乳類の発生生物学研究において、受精卵へのマイクロインジェクションに より外来性の遺伝子を導入したトランスジェニック(Tg)マウスを作出し、そ の表現型から標的遺伝子の機能を推測する機能獲得型(gain of function)の遺伝 子操作技術は、二十年以上前から用いられてきた重要な方法である(69, 70)。機 能獲得型は、Cytomegalovirus(CMV)や Chicken β actin(CAG)に由来する
恒常的プロモーターから標的遺伝子を発現させるベクターを導入することで、 全身で標的遺伝子を強く発現する過剰発現モデルを作出することが可能である が、遺伝子によっては過剰発現による胎生致死によって出生後の解析を行うこ とが困難な場合がある。従って、致死性を回避し、且つ標的遺伝子の過剰発現 を時空間的に制御するため、組織や時期特異的に遺伝子を発現させるコンディ ショナルTg マウスを用いた解析は、現在では機能獲得型研究において非常に重 要な手法となっている。 バクテリオファージP1 で発見された Cre/loxP システムは、同じ向きの loxP 配列で挟まれたDNA 配列を、ゲノム組み換え酵素である Cre リコンビナーゼに よりDNA 配列上から除去する機構である。一般に、組織特異的に標的遺伝子を 発現させる際に、組織特異的プロモーターを用いたTg マウスを作出する方法を 用いるが、組織特異的プロモーターは恒常的プロモーターに比べて遺伝子の発 現レベルが低い場合がある。そこで、Cre/loxP システムと恒常的プロモーター を組み合わせたコンディショナルTg マウスをデザインすることにより、組織特 異的に標的遺伝子を十分量発現させることが期待できる。まず、恒常的プロモ ーターと標的遺伝子の間にloxP で挟んだ別の遺伝子のカセット(floxed-polyA) を挿入したベクターを導入した Tg マウス(flox マウス)を作出する。次いで、 組織特異的にCre を発現する Cre マウスを flox マウスと交配させることにより、 Cre の発現した組織だけで floxed-polyA が脱落し下流の標的遺伝子が発現する。 これによって、時空間的に制御された遺伝子の高レベルな過剰発現の誘導が可 能になる。
現させる場合、組織非特異的アルカリフォスターゼ(Tissue non-specific alkaline phosphatase; TNAP)遺伝子上に Cre を挿入した TNAP-Cre ノックインマウス が用いられてきている(71)。近年、Vasa(Ddx4 あるいは Mvh としても知られ る)遺伝子プロモーター下にCre を接続した、Vasa-Cre Tg マウスが作出され た(72)。Vasa-Cre は雌雄の生殖細胞において強い特異性を示すことから、出生 前後の未熟な卵母細胞における研究を行う上で有効なツールになると考えられ る。 本研究では、標的遺伝子をコンディショナルに発現させる実験系に加えて、 Dnmt3a2 と Dnmt3L を同時発現するシステムが要求される。また、標的遺伝子 を発現している細胞を生きたまま可視化することができるように蛍光レポータ ー遺伝子を導入することも、その後の解析に有効となる。蛍光レポーター遺伝 子を生体内に導入する場合、標的遺伝子との融合タンパクにするのが一般的で あるが、この方法では不適切なフォールディングによりタンパク質が機能しな くなる可能性がある。また原核細胞とは異なり、動物細胞はほとんどの場合遺 伝子ごとにそれぞれ単独のmRNA が転写される。このため、複数の遺伝子を発 現させるには複数のプロモーターを持ったベクターを構築する必要があるが、 それぞれのプロモーターが等しく機能する保障はない。現在までに、1 つのプロ モーターから複数の標的遺伝子を同時に発現させるシステムには、internal ribosome entry sites(IRES)がよく用いられてきた。IRES は mRNA の 5’非翻 訳領域に存在するタンパク質発現制御配列であり、これにより2つの遺伝子を 連結することで並列に翻訳を行うことが可能である(73)。しかし IRES で連結し た場合、それぞれの発現量が低下することが知られている。
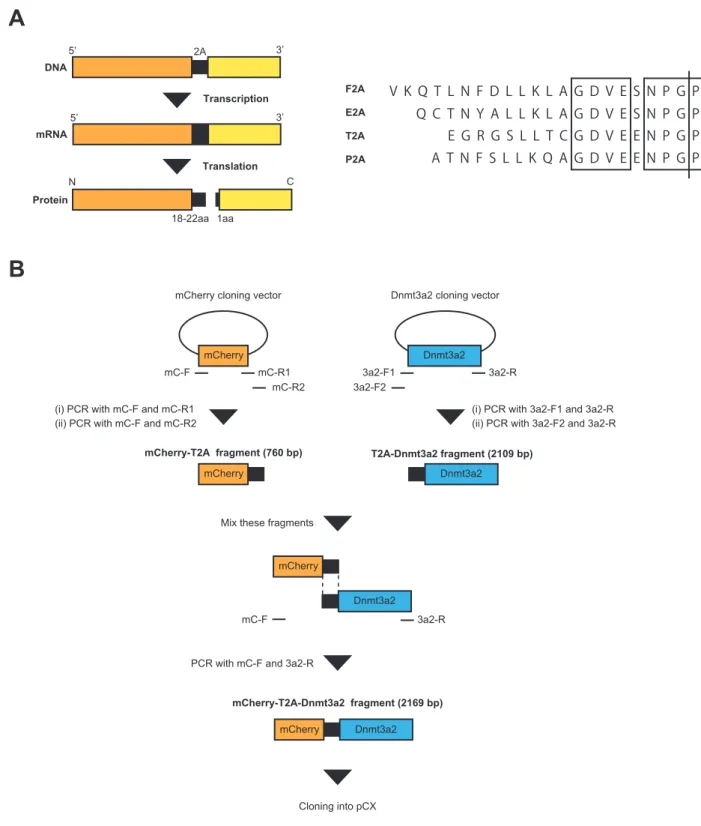
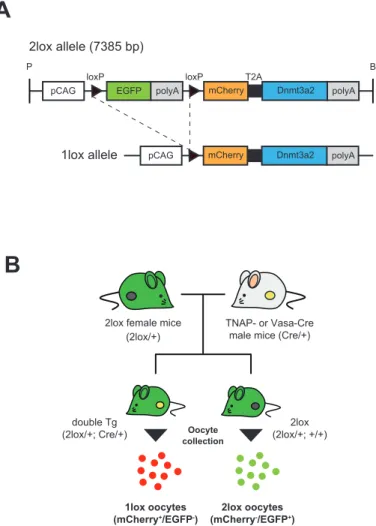
近年注目されている2A peptide はウィルスに由来するアミノ酸配列であり、 その由来に応じて4 種類の配列(F2A, T2A, P2A および E2A)が報告されてい る 。2A peptide で 連 結 さ れ た 配 列 は 、 2A 配 列 に 共 通 し て 存 在 す る Asp-Val/Ile-Glu-X-Asn-Pro-Gly-Pro のモチーフによって翻訳時にリボソームが スキップすることでアミノ酸が分断され、結果として同一のプロモーターから 複数の遺伝子を高発現させることが可能である(Fig. 3-1A)(74, 75)。これによ り、IRES とは異なり、複数の遺伝子を偏りなく、且つ IRES よりも強く発現さ せることが期待できる。 マウスにおいては、現在までに2A peptide を用いて複数の Tg マウスやノッ クインマウスの作出が報告されている(76-78)。しかし、これらはすべて体細胞 系列における2A の機能を確認したものであり、雌性生殖細胞系列において 2A 配列が機能したという報告は現在までにない。そこで本章では卵母細胞におけ るDnmt3a2/Dnmt3L 過剰発現マウス作出の端緒として、Cre/loxP システムによ り雌性生殖系列において2A 配列で連結された標的遺伝子 Dnmt3a2 とレポータ ー遺伝子を共に発現するようにデザインされたコンディショナル Tg マウスを 作出し、2A 配列の機能および挿入遺伝子の発現解析を行った。加えて、 TNAP-Cre マウスと Vasa-Cre マウスのいずれが卵子形成過程におけるコンディ ショナルな発現に有効かを検証した。
第二節 材料および方法
1. 供試動物
2lox アレルの全長を持つ Tg マウス(2lox マウス)は、すべて C57BL/6N マ ウスを遺伝的背景として作出され、維持した。作出の際に用いた偽妊娠マウス および精管結紮雄マウスは、性成熟した ICR マウス(日本クレア)を用いた。 また、TNAP 遺伝子に IRES-Cre が挿入されているマウス(TNAP-Cre マウス、 九州大学 佐々木裕之教授より分与)はC57BL/6N へ 10 世代以上戻し交配した ものを、Vasa プロモーター下で Cre Recombinase を発現するマウス(Vasa-Cre マウス、Jackson Lab.より導入、stock no. 006954)はすべて FVB/N マウスを 遺伝的背景として維持した。飼育条件は第二章の方法に準じた。
2. Dnmt3a2 コンディショナル発現ベクターの構築
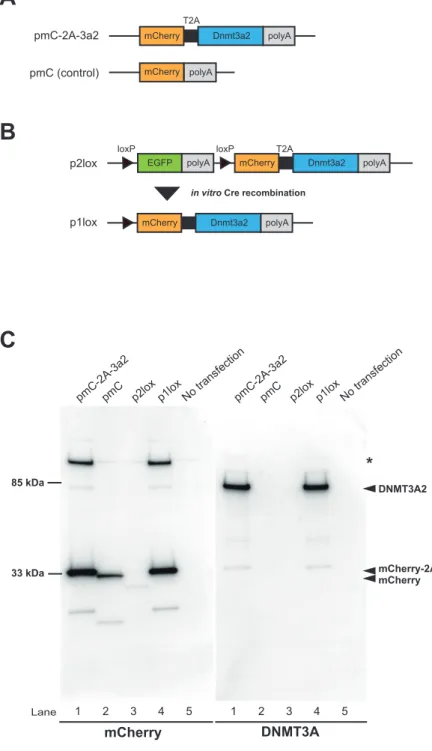
コンディショナル発現ベクターは、Cre/loxP システムによる組織特異的発現 を起こすためにChicken β-Actin(CAG)プロモーターを持つ pCX-EGFP を基 本骨格としてデザインされた。これによりEGFP 配列を持つ場合(2lox)は EGFP が発現し、Cre Recombinase により EGFP が脱落する(1lox)ことで下流の DNMT3A2 が mCherry と共に発現するようにした(Fig. 3-2B)。また EGFP 配 列を持ちながら下流の遺伝子が発現することを防ぐため、EGFP 配列の直後に pA カセットが挿入されている他、EGFP 配列の終止コドン手前の配列を点変異 (サイレント変異)させることでスプライシングを防いだ。
卵子形成過程において主要に発現するDNMT3A2 を用い、また 2A 配列は Thosea asigna virus に由来する配列(T2A)およびブタテッショウウィルス(Porcine Teschovirus-1)に由来する配列(P2A)を使用した。T2A 配列は N 末端側 20 アミノ酸、C 末端側 1 アミノ酸に分断されるため、20 アミノ酸が mCherry の C 末端側に付加され、1 アミノ酸が DNMT3A2 の N 末端側に付加されると予測さ れた。なお、T2A 配列は本来 54 bp の配列であるが、2A peptide の分断活性を 上昇させるため、T2A の 5’末端に Gly-Ser-Gly の 3 アミノ酸(5’-GGC AGT GGA-3’)を付加し、63 bp の配列としている。
2-1. loxP で挟まれた EGFP-pA(floxed-EGFP-pA)の挿入および点変異導入(平 成22 年 小肩実央氏との共同実験)
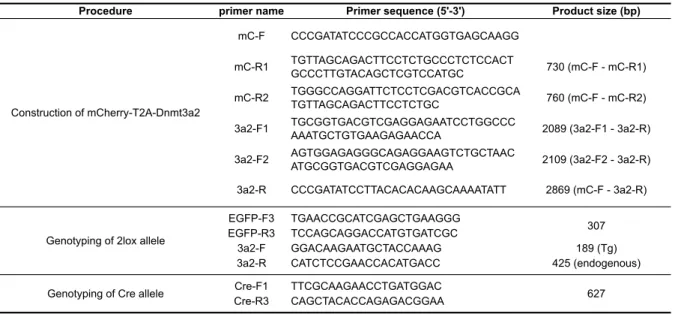
CAG プロモーター下流に floxed-EGFP-pA を挿入するため、オリジナルの pCX-EGFP から EGFP 配列を EcoRI により除去した(pCX)。また、EGFP お よび pA 配列をマルチクローニングサイトおよびクロラムフェニコール耐性遺 伝子を除去したpDNr-1r の EcoRI および挿入して floxed-EGFP-pA を作製後、 pCX の NotI サイトへ挿入した(pCX-floxed-EGFP)。 また、floxed-EGFP-pA より下流がリードスルーするのを防ぐため、スプライ シングのドナーサイトになり得る EGFP の終止コドン直前のグアニンをアデニ ンに置換した。 2-2. 2A 配列を含む DNA 断片の作製および挿入(平成 21 年 高野喬氏との共同 実験) mCherry-T2A-Dnmt3a2 あるいは mChery-P2A-Dnmt3a2 配列は、リコンビナ ントPCR により作製した。最初に、pmCherry-N1 あるいは Dnmt3a2 の cDNA
クローンからmCherry および Dnmt3a2 の DNA 断片を増幅した。この際、T2A 配列(63 bp)を 30 bp 程度付加したプライマー(Table 3-2)を用いて 2 段階の PCR を行うことにより 2 つの産物が T2A 配列の部分で 50 bp 程度オーバーラッ プするようにした。それぞれの産物を精製後、それらを1:1 で混合したものをテ ンプレートとし、両端のプライマーを用いてPCR を行い、2 つの産物をタンデ ムに連結した(Fig. 3-1B)。
精製した産物は EcoRV で処理し、pCX および pCX-floxed-EGFP の EcoRV サイトへ導入した。シークエンス解析により挿入されている向きを確認して、 正しいものをそれぞれ pmC-2A-3a2 および p2lox とした。また、それぞれのコ ントロールとして、mCherry のみを pCX の EcoRV サイトに挿入したベクター (pmC)を作製した。
また、p2lox 200 ng を Cre Recombinase (Clontech) と in vitro で反応させ、 floxed-EGFP-pA を脱落させた。この反応液の一部をコンピテントセル DH5α (TOYOBO)へ形質転換させ、EGFP が脱落したベクター(p1lox)を単離した。 すべてのベクターは、それぞれ大腸菌に形質転換後アンピシリン添加LB 液体培 地にて大量培養し、EndoFree Plasmid Maxi Prep Kit (QIAGEN) の定法に従って 精製した。
2-3. 培養細胞における一過的発現
コンディショナルベクターが正しく機能することを確認するため、培養細胞 において一過的に発現させた。pmC-2A-3a2、p2lox、p1lox および pmC を、培 養細胞NIH3T3 へ一過的に導入した。NIH3T3 は、10% FBS 添加 DMEM(Gibco) 培地にて、37℃、5% CO2および95% Air の条件で培養した。導入は Lipofectamine
LTX(Invitrogen)を用いたリポフェクション法により行い、1×105 cells / 35 mm dish の細胞に 2.5 µg のプラスミド DNA を加えた。導入後 48 時間で倒立顕微鏡 にて蛍光観察を行い、その後導入細胞をトリプシン処理により回収し、その後 の実験に供試した。
2-4. 2lox マウスおよびダブル Tg マウスの作出
2lox アレルをヘテロで持つ Tg マウス(2lox マウス)は、精子を DNA キャリ アーとした顕微授精により作出を行った。p2lox を制限酵素 BamHI および Psp1406I で処理することによりリニアライズし、7385 bp の DNA 断片を切り 出して精製した。断片は200–800 コピー/pl に調整した C57BL/N 雄マウスの凍 結-融解精子と HTF 培地内で共培養し、この精子頭部を C57BL/6N 雌マウスの第 二減数分裂中期(Metaphase II; MII)卵子に注入した。受精卵は KSOM 培地ド ロップにて洗浄および培養し、2-cell において偽妊娠雌マウスに卵管移植した。 次いで、性成熟した2lox 雌マウスを TNAP-Cre あるいは Vasa-Cre ヘテロ雄 マウスと自然交配させ、2lox アレルと Cre アレルを同時にヘテロに持ったマウ ス(dTg)を作出した。 2-5. PCR による Tg マウスの遺伝子型判別 マウスの遺伝子型はPCR を用いて決定した。ゲノム DNA の抽出はアルカリ -SDS 法により行った。Tg マウスの指あるいは尾の先端を外科鋏で採取し、組 織サンプルに50 mM NaOH を加えて 95℃で 10 分熱処理後、1M Tris-HCl (pH 8.0) を加えて中和させ、12000 rpm で 10 分遠心してその上清 1 µl を PCR 反応 の鋳型とした。PCR 反応は KOD-FX(TOYOBO)を用いて EGFP(307 bp)、 Dnmt3a2(189(Tg)+ 425(内在性)bp)および Cre(627 bp)のプライマー
でPCR を行い、すべてのバンドが現れた個体を dTg マウス、Cre 以外のバンド が現れた個体を2lox マウスと判定した。PCR に用いたプライマーおよび得られ るバンドの分子量はTable 3-2 に示した。
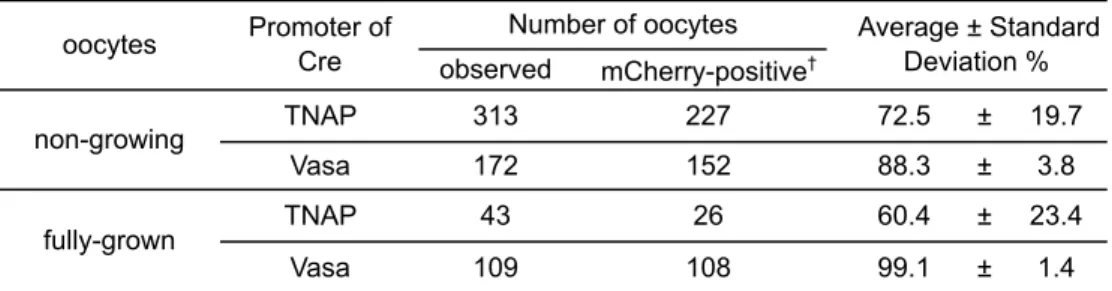
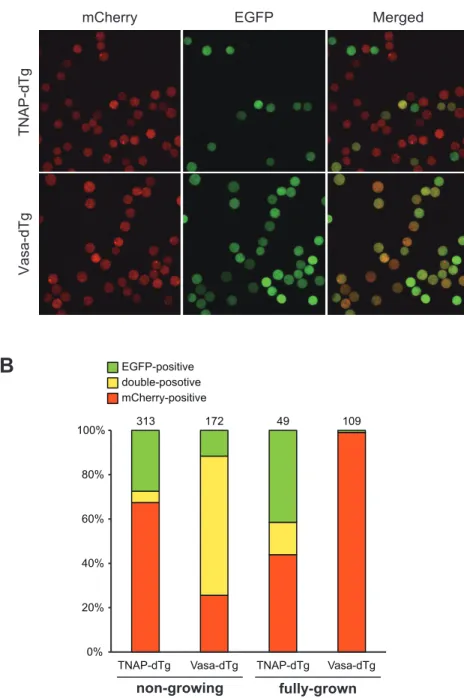
2-6. dTg マウス由来卵母細胞におけるレポーターアッセイ
dTg マウスにおける TNAP-あるいは Vasa-Cre による組換えの効果を評価す るため、TNAP-Cre を持つ dTg(TNAP-dTg)および Vasa-Cre を持つ dTg (Vasa-dTg)マウスより、第二章の方法に準じてそれぞれ ng および fg 卵母細 胞を回収した。これらの卵母細胞を、共焦点レーザー顕微鏡LSM710(Carl Zeiss) を用いてEGFP(励起波長 488 nm)および mCherry(励起波長 543 nm)の蛍 光を観察し、EGFP 陽性、mCherry 陽性および EGFP/mCherry 両陽性細胞の数 をカウントした。全体に占める mCherry 陽性および両陽性細胞、すなわち mCherry 陽性細胞の合計の割合を Cre 効率と定義し、TNAP-あるいは Vasa-dTg マウス卵母細胞におけるCre 効率および各卵母細胞の蛍光の分布を求めた。 3. ウェスタンブロットを用いた 2A 配列の機能解析 3-1. 培養細胞における解析 プラスミドを発現させた培養細胞は各レーン1×104細胞となるようにアプラ イし、第二章の方法に準じてSDS-PAGE を行った。PVDF メンブレンにトラン スファー後、5%スキムミルクで室温にて 1 時間振盪しブロッキングを行い、一 次抗体反応を 4℃で一晩行った。TBS-T で洗浄後、室温で 1 時間二次抗体反応 を行い、ECL Prime Western blotting detection system(GE healthcare)にて検 出した。一次抗体は、Living Color Anti-DsRed ポリクローナル(Clontech, 1:1000)、
Anti-DNMT3A モノクローナル(Imgenex, 1:2000)、Anti-GAPDH ポリクローナ ル(Trevigen, 1:2000)および Anti-αTubulin モノクローナル(Sigma, 1:2000) を用いた。 3-2. 新生仔卵巣および成長期卵母細胞における解析 新生仔卵巣はそれぞれ1 個分を、成長期卵母細胞は 150 個をプールしてアプ ライし、同様の条件でSDS-PAGE を行った。トランスファー後のブロッキング および抗体反応は第二章の方法に準じて行い、3-1.と同様の抗体を用いた。 4. dTg マウス由来 ng 卵母細胞におけるインプリント遺伝子メチル化解析 4-1. ng 卵母細胞の回収 第二章の方法に準じて、400–500 個の ng 卵母細胞を回収した。回収した卵母 細胞は1.5 ml チューブに移し、使用時まで-80℃にて保存した。 4-2. DNA 抽出およびバイサルファイト処理
融解したサンプルに18 µl の DNA Lysis Buffer (1% SDS, 1 mg/ml Proteinase K, 2 µg E. Coli tRNA) を加え、37℃で 1 時間インキュベートした。その後、反 応液の全量を用いてEpiTect Bisulfite Kit (QIAGEN) の定法に従い、ゲノム DNA 中のシトシンをチミンに置換した。
4-3. Nested PCR およびクローニング
バイサルファイト処理を行ったDNA を用いて、H19 および Lit1 領域に対して Nested PCR を行った。PCR 反応には Advantage cDNA Polymerase(Clontech)
を用いた。1st PCR には ng 卵母細胞 50 個分を鋳型 DNA とし、反応液 10%を 鋳型として2nd PCR を行った。各遺伝子のプライマーは Hiura らによって使用 されたものを用いた(48)(H19; 368 bp, 20 CpG sites, accession no. AF049091. Lit1; 337 bp, 17 CpG sites, accession no. AP001295)。
2nd PCR の反応液全量を 2%アガロースゲルにて電気泳動後、エチジウムブ ロマイドによりPCR 産物を検出した。その後標的の分子量のバンドを切り出し、 Wizard SV Gel and PCR Clean Up System(Promega)により DNA を精製し た。精製したDNA は pGEM-T Easy Vector(Promega)へライゲーションし、 TA クローニングを行った。その後ライゲーション反応液を用いてコンピテント セ ル DH5α ( TOYOBO ) へ 形 質 転 換 さ せ 、 100 mM IPTG ( isopropyl-β-D-thiogalactopyranoside; Wako ) お よ び 40 µg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside; Wako)を塗布した 50 µg/ml アン ピシリン(Wako)添加 LB 寒天培地へ播種し、37℃で 16 時間程度培養し、ブ ルーホワイトセレクションを行った。
4-4. シークエンス解析およびデータ処理
大腸菌コロニーからシークエンスに必要な量のプラスミドDNA を増幅させる ため、ホワイトコロニーを爪楊枝でピックアップし、Illustra TempliPhi DNA Amplification Kit(GE Healthcare)を通常の 1/5 の反応量で用いて反応させた。 その後、反応液に40 µl の DDW を加え、そのうちの 2 µl をシークエンス解析の 鋳型とした。シークエンスは、pGEM-T Easy Vector 中の配列を 6 塩基追加した Sp6 プライマー(5’-ACACTATAGAATACTCAAGCTATGC-3’)および BigDye
Terminator v3.1 Cycle Sequencing Kit(Applied Biosystems)を用いてシークエ ンスPCR 反応を行い、ABI PRISM 3100 Genetic Analyzer(Applied Biosystems) を用いて配列を取得した。 シークエンス解析で得られたデータをオンライン上のメチル化分析ツールで あるQUMA (http://quma.cdb.riken.jp/top/quma_main_j.html) へアップロードし、 メチル化率の算出を行った。この際、相同性 90%以上、バイサルファイト変換 効率95%以上の DNA ストランドのみを解析に用い、さらに重複したクローンは 完全に排除した。
第三節 結果
1. Dnmt3a2 コンディショナル発現ベクターの構築
最初に、2A 配列が培養細胞において正しく機能するかを確かめるため、CAG promoter の下流で mCherry と DNMT3A2 とを T2A 配列で連結したベクター (pmC-2A-3a2)および CMV promoter から mCherry を単独で発現するベクタ ー(pmC)を作製し、NIH3T3 細胞に導入して 48 時間後にウェスタンブロット を行った(Fig. 3-2A)。mCherry は DsRed に由来する変異体であることから、 DsRed 抗体を用いることで検出が可能である。その結果、33 kDa 付近に mCherry、 約85 kDa に DNMT3A2 に特異的なバンドが検出された。また、mCherry のみ 発現するベクター(pmC)導入細胞と比較してわずかに分子量が増加したが、 これはT2A 配列に由来する 20 アミノ酸の分子量によるものであると考えられ た(Fig 3-2C レーン 1 および 2)。なお、P2A 配列を用いた場合、mCherry お よびDNMT3A2 の融合タンパクが検出されたことから、2A 配列が機能していな いと考えられた。これらのことから、T2A 配列が培養細胞において正常に機能 すること、またDsRed および DNMT3A2 のどちらの抗体でも検出されるタンパ ク質、つまり切断されなかったタンパク質は存在しなかったことから、NIH3T3 細胞においてmCherry と DNMT3A2 の切断効率が非常に高いことが考えられた。 次 に 、Cre/loxP シ ス テ ム を 用 い た 過 剰 発 現 の 誘 導 を 行 う た め 、 mCherry-2A-Dnmt3a2 配列の上流に loxP で挟んだ EGFP と polyA 配列(floxed EGFP)を挿入したベクター(p2lox)および in vitro において Cre リコンビナー ゼによりEGFP を削除したベクター(p1lox)をそれぞれ構築して NIH3T3 に導