Synthesis and Metalation of Doubly o-Phenylene-Bridged Cyclic Bis(dipyrrin)s with Highly Bent Skeleton of Dibenzoporphyrin(2.1.2.1)
10
0
0
全文
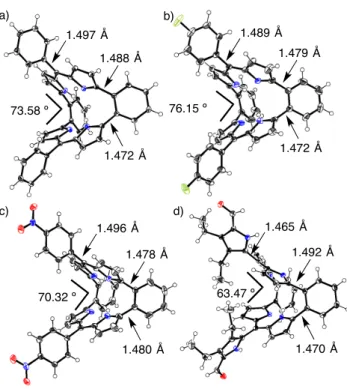
(2) FULL PAPER were isolated as stable compounds. These compounds exhibited highly planar structures with a 22p aromaticity. Furthermore, Andrioletti was able to synthesize the bis(quinoxaline)-fused porphyrins(2.1.2.1) by multi–step synthesis.[24] This compound showed a saddle-shaped bent conformation, providing non-macrocyclic-aromaticity. This is similar to the nature of cyclooctatetraene that has a boat-like structure and no anti–aromaticity in spite of contribution as an 8p–electron anti-aromatic circuit. For the reasons mentioned above, the genuine porphyrin(2.1.2.1) with only pyrrole units are important outstanding targets. The aromatic ring-fused porphyrin(2.1.2.1) are expected to have highly bent structures. Recently, others and we reported the synthesis of the o-dipyrrolylbenzenes, which are useful building blocks to synthesize the pyrrole-based aromatic compounds.[25-28] Herein, we report the facile and simple synthetic method of dibenzoporphyrin(2.1.2.1) derivatives from o-dipyrrolylbenzene and various aldehydes by acid catalysis condensations. The dibenzoporphyrin(2.1.2.1) consists of two dipyrrin units connected with o-phenylene bridges and forms highly bent structures confirmed by single crystal X-ray diffraction analysis. In addition, dibenzoporphyrin(2.1.2.1) can act as bianionic tetradentate ligands to give various metal complexes. The optical and electrochemical properties, and Xray diffraction analysis of these compounds are also discussed.. Ar NH NH. i) ArCHO BF 3•OEt 2 ii) DDQ CH2Cl2. The synthetic scheme of dibenzoporphyrin(2.1.2.1) derivatives is shown in Scheme 1. The starting material of o-dipyrrolylbenzene 1 was synthesized by previously reported methods.[28] Compound 1 reacted with various arylaldehydes in the presence of boron trifluoride ethyl ether complex (BF3•OEt2) as an acid catalysis in dichloromethane, followed by oxidation with 2,3dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give the dibenzoporphyrin(2.1.2.1) derivatives 2a-f as main products in moderate yields (18-48%). These condensation reactions proceeded with benzaldehyde derivatives with electron-donating or withdrawing substituents at para-position. In addition, 3trimethylsilylpropynal and five-membered heterocyclic aldehydes were employed for this condensation reaction to give 2g (20%) and 2h (12%), respectively. Surprisingly, 3,4-diethyl-2,5diformylpyrrole also provided 2i in 4.5% instead of the formations of triphyrin(2.1.1) or large number of macrocyclic compounds. The structures of these compounds were confirmed by 1H and 13C NMR, high-resolution mass spectroscopy and single crystal X-ray diffraction analysis. Single crystals of 2a, 2e, 2f and 2i suitable for the X-ray diffraction analysis were obtained by vapor diffusion methods (See Experimental for details). Crystal structures are shown in Figure 2. All crystal structures have remarkably nonplanar structures that form the saddle-shaped bent geometries. The bent angles between the each plane of dipyrrin units are 73.58º for 2a, 76.15º for 2e, 70.32º for 2f and 63.47º for 2i. The crystal structure of 2i shows smaller bent angle than other compounds because of formations of the intermolecular interactions via. N HN. N. Ar 2X. 1 f:. a: (40%). g:. (48%) c:. TMS (33%). OMe. h:. CH3. i:. (18%) H 3C d:. NO 2 (36%). CH3. b:. H 3C (20%). S (12%) Et Et N CHO H (5%). F. e:. Results and Discussion. NH. (48%) Scheme 1. Synthesis of dibenzoporphyrins(2.1.2.1).. multiple hydrogen bonds between nitrogen atoms in the macrocycles and 5-formylpyrrole units (Figure S7). These bent angles are larger than that of quinoxaline fusedporphyrin[2.1.2.1] (59º)[24] because of steric hindrances between the protons of phenylene bridges and pyrrolic b-positions. The inner nitrogen atoms positioned almost rectangular shapes with the nitrogen-nitrogen distances of 3.156, 3.001, 2.783 and 2.766 Å for 2a, 3.083 and 2.730 Å for 2e, 3.095 and 2.723 Å for 2f and 3.435, 3.291, 2.828 and 2.821 Å for 2i. These distances are slightly longer than those of tetraphenylporphyrin (2.908 and 2.930 Å).[29] The bond distances between benzene rings of phenylene bridge and dipyrrin unit are approximately 1.47 Å that show the typical single bond distance, indicating that the electronic structure of dibenzoporphyrins(2.1.2.1) has a very weak contribution of macrocyclic conjugation pathways through whole of molecules. The 1H NMR spectrum of 2a is shown in Figure 3b. The inner NH proton signal of 2a is observed as a broad singlet peak at 12.23 ppm which chemical shift is similar to that of 5-phenyl4,6-dipyrrin (12 ppm as a very broad signal).[30] The protons of bpositions of the pyrrole moieties of 2a are observed at 6.50 and 6.27 ppm. These results suggested that electronic property of dibenzoporphyrin(2.1.2.1) is close to the dipyrrin analogous without contributions of macrocyclic p-conjugation, although it has a macrocyclic porphyrinoid-like structure. This electronic.
(3) FULL PAPER a). b) 1.489 Å. 1.497 Å. 1.479 Å. 1.488 Å. 76.15 º. 73.58 º. x 30. b). x6. 1.472 Å. 1.472 Å. c). a). c) 12.5. d). 12.0 8. 7. 6. 5. 4. 3. 2. chemical shift (ppm). 1.465 Å. 1.496 Å. 1.492 Å. 1.478 Å. 1. Figure 3. H NMR spectra of 2d (a), 2a (b) and Ni-2a (c) in CDCl3 at room temperature.. 63.47 º. 70.32 º. 1.480 Å. 1.470 Å. Figure 2. Crystal structures of 2a (a), 2e (b), 2f (c) and 2i (d). Thermal ellipsoids represent for 50% probability.. character is comparable to the meta-phenylene bridged bis(dipyrrin) (dibenziamethyrin).[13-15] To interpret the aromaticity, nucleus-independent chemical shifts (NICS) of 2a was calculated.[31] The NICS(0) value at the midpoint of four nitrogen atoms was 1.8 ppm, indicating very weak contributions of macrocyclic anti-aromaticity (Figure S1). In order to investigate the internal hydrogen transfer of 2d, 1H NMR spectra were measured at low temperatures in CD2Cl2. Dibenzoporphyrins(2.1.2.1) could be expected to form four tautomers with different positions of hydrogen atoms except for enantiomer. When decreasing the measurement temperature from 20 ºC to –90 ºC, the NH protons sharpened a bit associated with the maintenance to be a singlet (Figure S2). This result indicates that the NH protons are delocalized between the nitrogen atoms in the time scale of NMR conditions and barriers of tautomerizations are quite low even in low temperatures..On the other hand, three methyl protons of mesityl groups of 2d were independently observed as three singlet peaks at 1.96, 2.14 and 2.34 ppm associated with separated two part of doublet mesityl peaks at 6.86 and 6.92 ppm (Figure 3a). This indicates that the rotations of mesityl groups are prevented by steric hindrance and flip motion of bent structure is also fixed at NMR time scale.. Furthermore, we have examined variable temperature NMR measurements for the investigation of molecular dynamics of dibenzoporphyrin(2.1.2.1) (Figure S4). The NMR spectra of 2d at 70 and 120 ºC showed very similar signals to the spectrum at room temperature. It is concluded that dibenzoporphyrin(2.1.2.1) has rigid structure and conducts no flip-flop motions in the solution state even in high temperatures. The absorption spectra of dibenzoporphyrins(2.1.2.1) are shown in Figure 4. The meso-phenyl substituted dibenzoporphyrins(2.1.2.1) showed strong absorption band around 450 nm associated with broadened absorption band around 550 nm. The substitutions on para-positions slightly affect the maximum absorption peaks: 434 nm for 2a, 434 nm for 2b, 436 nm for 2c, 434 nm for 2e and 439 nm for 2f. On the other hand, maximum absorption peaks of 2g, 2h and 2i were observed at 455, 446 and 442 nm, respectively, which values were red-shifted compared to 2a because of the expansion of the p-conjugations with ethynyl and heterocyclic rings. We have measured the fluorescence of dibenzoporphyrins(2.1.2.1), however, all of them showed no emission..
(4) FULL PAPER magnetic measurement fields indicates that Ni-2a has diamagnetic property. As a result of metal insertion, the NH signal was disappeared. The signals of pyrrolic protons were observed at 6.12 and 6.62 ppm as the similar regions of 2a. It is suggested that metal complexes hold a similar electronic property with the free-base form. The 1H NMR of other metal complexes of Pd-2a, Pt-2a and Sn-2a showed similar tendencies.. -1. 4. 4. ε (10 M cm ). 6. -1. a). 2 0 300. 350. 400. 500. 550. 600. 650. Ph. wavelength (nm). -1. 6. -1. b). 4. 2a. metal salt. N. N M. N. N. 4. ε (10 M cm ). 450. 2 0 300. 400. 500. 600. 700. wavelength (nm) Figure 4. UV-Vis absorption spectra in dichloromethane of a) 2a (black), 2b (red), 2c (blue), 2e (green) and 2f (purple), b) 2a (black), 2g (red), 2h and 2i (green).. The results of 1H NMR spectroscopy and X-ray diffraction analysis revealed that dibenzoporphyrin(2.1.2.1) has a cavity inside of the macrocycle made by four inner nitrogen atoms which form dianionic tetradentate coordination structure. It is well known that porphyrin can be converted to varied metal complexes with most of the elements on the periodic table and the optical and electrochemical properties can be tuned. However the report of porphyrins(2.1.2.1) metal complexes have been limited. Andrioletti reported the metal complexes of quinoxaline fused-porphyrins(2.1.2.1),[24] however, they did not show any details of the properties of porphyrin(2.1.2.1) metal complexes except for absorption spectra. Therefore, we have developed the synthesis of metal complexes of compound 2a to investigate in term of the perturbation for structural geometry and optical and electrochemical properties by coordination with metal ions (Scheme 2). The nickel and copper complexes of 2a were synthesized by general protocols of metal insertion method of porphyrins. Compound 2a is reacted with methanol solutions of corresponding metal acetates Ni(OAc)2 and Cu(OAc)2 in chloroform to give metal complexes Ni-2a (92%) and Cu-2a (93%). The palladium Pd-2a and platinum Pt-2a complexes were prepared with PdCl2 and PtCl2 in chloroform and benzonitrile under reflux conditions in 57% and 74%, respectively. The tin complex Sn-2a was obtained by treatment of 2a with SnCl2 in pyridine under a reflux condition. The structures of the obtained metal complexes were determined by 1 H and 13C NMR, high-resolution mass spectroscopy and X-ray diffraction analyses. The 1H NMR spectrum of Ni-2a is shown in Figure 2c. The observation of NMR spectrum of Ni-2a in usual. Ph M = Ni (Ni-2a) [92%] M = Pt (Pt-2a) [74%] = Cu (Cu-2a) [93%] = SnCl2 (Sn-2a) [11%] = Pd (Pd-2a) [57%] Scheme 2. Synthesis of metal complexes of 2a.. Single crystals of Ni-2a, Pd-2a, Pt-2a and Sn-2a suitable for the X-ray diffraction analysis were obtained (See Experimental for detail). Crystal structures of Ni-2a, Pd-2a, Pt-2a and Sn-2a are shown in Figure 5. The coordinated metals of these complexes sited on the center of the cavity and formed square-planar coordination geometries the same as porphyrin metal complexes. Compound Sn-2a showed an octahedral geometry in which tin was coordinated with four nitrogen atoms and two chloride atoms (Figure 5d). All metal complexes also consisted of saddle shaped structures in the same manner of free-base one. The bent angled of metal complexes between each dipyrrin units are 83.03º for Ni-2a 88.38º for Pd-2a, 86.90º for Pt-2a and 105.80º for Sn-2a which values are larger than that of 2a (73.58º). In addition, the dihedral angles of two benzene rings between dipyrrin units are 57.09º for Ni-2a 59.26º for Pd-2a, 61.83º for Pt-2a and 70.85º for Sn-2a. The nitrogen-nitrogen distances of these metal complexes are 2.688, 2.669, 2.647 and 2.636 Å for Ni-2a, 2.915, 2.867, 2.795 and 2.762 Å for Pd-2a, 2.905, 2.875, 2.802 and 2.759 Å for Pt-2a and 3.111 and 2.912 Å for Sn-2a. The cavity shapes changed from rectangular (2a) to approximately square fashion due to square-planar coordination geometry of nickel (II), palladium (II) and platinum (II), and octahedral coordination geometry of tin (IV). These observations of structural changes of macrocycles by coordination to metal ions indicated that dibenzoporphyrin(2.1.2.1) is adaptable to metal radii and curvatures of dibenzoporphyrins(2.1.2.1) can be controlled depending on the metal geometry and metal ion sizes. On the other hand, there have been reported dipyrrin metal complexes in which they usually formed tetrahedral or highly bended square-planer coordination geometries because of composition of orthogonally positioned dipyrrin units.[30].
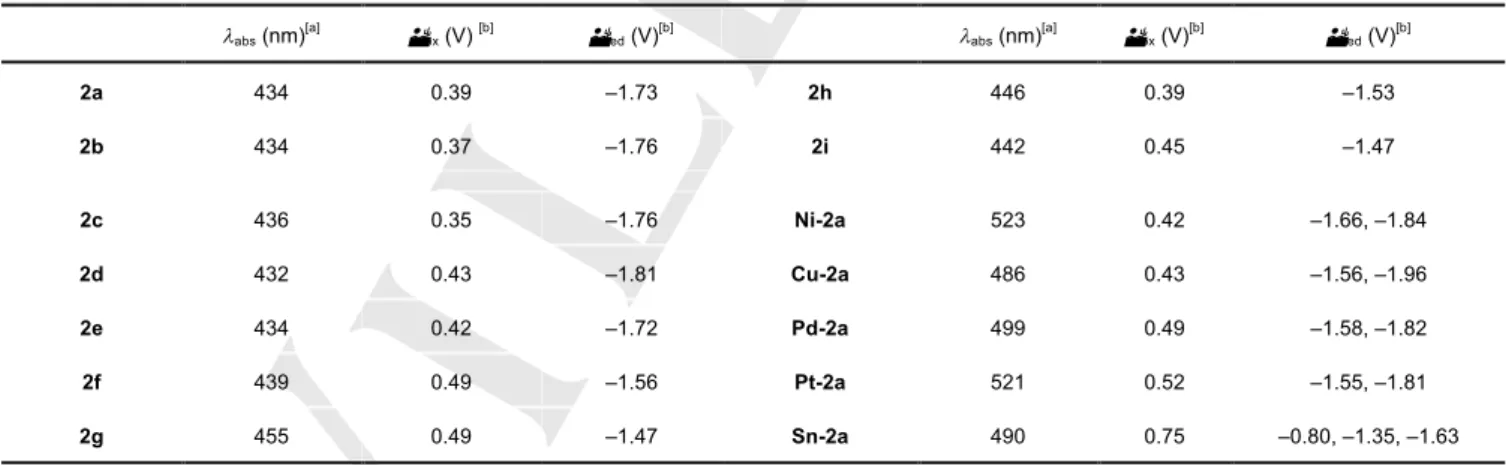
(5) FULL PAPER b) 1.494 Å. 8. 1.505 Å -1. ε (10 M cm ). a). 1.457 Å. 6. 4. -1. 1.468 Å. 83.03 º. 88.38 º. 1.470 Å. 1.467 Å. 4 2 0 300. 400. 500. 600. 700. wavelength (nm) c). d) 1.502 Å. 1.500 Å 1.457 Å. 1.472 Å. 86.90 º 105.80 º 1.464 Å. Figure 6. UV-Vis absorption spectra of 2a (black), Ni-2a (green), Cu-2a (purple), Pd-2a (blue), Pt-2a (red) and Sn-2a (purple) in dichloromethane.. 1.475 Å. Figure 5. Crystal structures of Ni-2a (a), Pd-2a (b), Pt-2a (c) and Sn-2a (d). Thermal ellipsoids represent for 50% probability. The solvent molecules of Pd–2a, Pt–2a and Sn–2a are omitted clarity.. The optical properties of metal complexes were evaluated by absorption spectra in dichloromethane. (Figure 6) All metal complexes showed red-shifted absorption compared to freebase 2a. The values of maximum absorption peak shifted depend on the coordinated metals, which peak tops were observed at 523 nm for Ni-2a, 486 nm for Cu-2a, 499 nm for Pd2a, 521 nm for Pt-2a and 490 nm for Sn-2a. The results of crystal structures suggested that the dibenzoporphyrin(2.1.2.1) ligand is adjusted to the coordination geometries of each metals promoting the different structures. These structural changes might generate varied absorption properties depending on the central metals. This phenomenon is commonly observed for the family of porphyrinoids.. The redox properties were measured by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as an electrolyte. The redox properties are summarized in Table 1 and Figures S5 and S6. All free-base dibenzoporphyrins(2.1.2.1) exhibited irreversible oxidation signals. The reduction signals were also irreversible expected for 2d and 2f. Presumably, monoradical anion of 2d and 2f were stabilized by bulky mesityl group and attached nitro groups worked as strong electron–withdrawing substituent. We have investigated the effects of p-substituents for first oxidation and reduction potentials with Hammett substituents constant (sp). The first oxidation and reduction potentials make linear slope with r = 0.063 and 0.100, respectively (Figure 7). These results concluded that these redox reactions occurred on macrocyclic dibenzoporphyrin(2.1.2.1) p-systems. On the other hand, the metal complexes employed one irreversible oxidation signal and multi reversible reduction signals. The reduction signals of Ni-2a, Cu-2a, Pd-2a and Pt-2a were observed at approximately same potentials (–1.66 and –1.84 V for Ni-2a, –1.56 and –1.96 V for Cu-2a, –1.58 and –1.82 V for Pd-2a and –1.55 and –1.81 V for Pt-2a). On the other hand, Sn-2a showed three reduction signals as reversible (–0.80, –1.35 and –1.63 V) which exhibited the cathode-shift compared with other metal complexes because of coordination with electron negative two chloride ions on the axial positions..
(6) FULL PAPER Figure 7. Plot of first oxidation (a) and reduction (b) potentials of porphyrins(2.1.2.1) against 2sp values of the substituents (E = spr).. a) 0.50. 2f (NO 2). Conclusions. E1/2 (V). 0.45. 2e (F) 0.40. 0.35. 2b (Me). ρ = 0.063. We have succeeded in the preparation of dibenzoporphyrin(2.1.2.1) derivatives by simple condensation reactions from o-dipyrrolylbenzene in the presence of acid. This method makes it possible to synthesize the varied dibenzoporphyrins(2.1.2.1) derivatives with various aldehydes including the electron–donating and –withdrawing units substitutes phenyl, five members heterocycles and ethynyl groups. In addition, dibenzoporphyrin(2.1.2.1) is able to work as porphyrin–like dianionic tetradentate ligands to give diverse metal complexes with nickel (II), copper (II), palladium (II), platinum (II) and tin (IV) ions. Dibenzoporphyrin(2.1.2.1) derivatives and their metal complexes consisted of unusually highly bent conformations by confirming the X-ray diffraction analysis. We have demonstrated that the electronic property of dibenzoporphyrins(2.1.2.1) can be controlled by the psubstituents on attached phenyl groups and insertion of the metals. Extensions of this synthetic method to other functional groups and heterocycle-included porphyrins(2.1.2.1) are actively in progress in our laboratory.. 2a (H). 2c (OMe) -0.5. 0.0. 0.5. 1.0. 1.5. 2σp. b). 2f (NO 2). -1.60. E1/2 (V). -1.65. ρ = 0.100. -1.70. 2e (F) 2a (H). 2b (Me) -1.75 -1.80. 2c (OMe) -0.5. 0.0. 0.5. 1.0. 1.5. 2σp. Table 1. UV-Vis absorption peaks (labs) and oxidation and reduction potentials (Eox and Ered) of dibenzoporphyrins(2.1.2.1) and metal complexes.. labs (nm). [a]. Eox (V). [b]. Ered (V). [b]. labs (nm). [a]. Eox (V). [b]. Ered (V). [b]. 2a. 434. 0.39. –1.73. 2h. 446. 0.39. –1.53. 2b. 434. 0.37. –1.76. 2i. 442. 0.45. –1.47. 2c. 436. 0.35. –1.76. Ni-2a. 523. 0.42. –1.66, –1.84. 2d. 432. 0.43. –1.81. Cu-2a. 486. 0.43. –1.56, –1.96. 2e. 434. 0.42. –1.72. Pd-2a. 499. 0.49. –1.58, –1.82. 2f. 439. 0.49. –1.56. Pt-2a. 521. 0.52. –1.55, –1.81. 2g. 455. 0.49. –1.47. Sn-2a. 490. 0.75. –0.80, –1.35, –1.63. [a] in dichloromethane. [b] These oxidation and reduction potentials were determined by DPV.. Experimental Section. Instrumentation and Materials: 1H NMR and 13C NMR spectra were recorded on a JEOL JNM-ECX 400 spectrometer at ambient temperature using tetramethylsilane as an internal standard. High-resolution electrospray ionization (HR-ESI) mass spectra and matrix assisted laser desorption/ionization (MALDI) were measured on a JEOL JMS-700.
(7) FULL PAPER spectrometer and JEOL spiralTOF JMS-S3000 spectrometer, respectively. Single-crystal X-ray diffraction analysis data collection was carried out at 90 K on a Bruker APEX II X-Ray diffractometer equipped with a large area CCD detector by using graphite monochromated Mo-Ka radiation (l = 0.71073 Å). UV-Vis absorption spectra were measured on a JASCO UV/VIS/NIR Spectro-photometer V-570. CV measurements were conducted in a solution of 0.1 M TBAPF6 in dry-dichloromethane with a scan rate of 0.1 V s–1 in an argon-filled cell. A glassy carbon electrode and a platinum wire were used as a working and a counter electrode, respectively. An Ag/AgNO3 electrode was used as reference electrodes, which were normalized with the half-wave potential of ferrocene/ferrocenium+ (Fc/Fc+) redox couple. Thin-layer chromatography (TLC) and gravity column chromatography were performed on Art. 5554 (Merck KGaA), and Silica Gel 60N (Kanto Chemical Co.), respectively. All solvents and chemicals were reagent grade quality, obtained commercially and used without further purification. For spectral measurements, spectral grade were purchased from Nacalai tesque Inc. Synthesis General procedures of synthesis of dibenzoporphyrin(2.1.2.1): A solution of 1 (100 mg, 0.480 mmol) in dichloromethane (50 mL) was degassed by an argon bubbling for 15 min and then aldehyde (1 eq.) was added to the reaction mixture. BF3・OEt2 (0.3 mol%) was slowly added. The reaction mixture was stirred at room temperature and then DDQ (1 eq) was added. After stirring for another 2 h, the solution was concentrated under a reduced pressure. The residue was purified by alumina column chromatography (dichloromethane) and silica gel column chromatography (dichloromethane to dichloromethane: methanol = 20:1). Recrystallization from dichloromethane/methanol gave the desired products 2a-2i. 5,16-Diphenyl-dibenzo[j,u]porphyrin(2.1.2.1) (2a): Reaction time: 5 h; Yield: 40%; The single crystal suitable for X-ray diffraction analysis was obtained from a solution of ethyl acetate and hexane. 1H NMR (400 MHz, CDCl3) δ = 12.23 (brs, 2H), 7.51 (m, 4H), 7.47-7.39 (m, 14H), 6.50 (d, J = 4.1 Hz, 4H), 6.27 (d, J = 4.1 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ = 155.63, 141.94, 140.87, 137.79, 134.49, 131.40, 129.40, 129.14, 128.85, 127.98, 127.41, 118.77; HRMS (ESI): m/z = 588.2296. calcd. for C42H28N4: 588.2314 [M]+. Crystal data: orthorhombic; space group Pbca (No. 61); a = 12.5737(13) Å, b = 20.898(2) Å, c = 23.367(2) Å; V = 6140.0(11) Å3; Z = 8; rcalcd = 1.274 g cm–3; T = 90 K; R1 = 0.0432 [I > 2s(I)]; Rw = 0.0607 (all data); GOF = 1.019.[32] 5,16-Di(4-methylphenyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2b): Reaction time: 2.5 h; Yield: 48%; 1H NMR (400 MHz, CDCl3) δ = 12.15 (brs, 2H), 7.45-7.38 (m, 12H), 7.21 (d, J = 8.2 Hz, 4H), 6.52 (d, J = 4.1 Hz, 4H), 6.26 (d, J = 4.1 Hz, 4H), 2.43 (s, 6H); 13C NMR (100 MHz, CDCl3) δ = 155.50, 142.26, 140.92, 138.97, 134.93, 134.50, 131.54, 129.38, 129.10, 128.15, 127.92, 118.60, 21.36; HRMS (ESI): m/z = 616.2612. calcd. for C44H32N4: 616.2627 [M]+. 5,16-Di(4-methoxyphenyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2c): Reaction time: 3.5 h; Yield: 18%; 1H NMR (400 MHz, CDCl3) δ =12.06 (brs, 2H), 7.46-7.39 (m, 12H), 6.93 (d, J = 9.2 Hz, 4H), 6.53 (d, J = 4.1 Hz, 4H), 6.28 (d, J = 4.1 Hz, 4H), 3.87 (s, 6H); 13C NMR (100 MHz, CDCl3) δ = 160.49, 155.43, 142.10, 140.90, 134.46, 133.24, 130.24, 129.36, 129.02, 127.92, 118.53, 112.92, 55.37; HRMS (ESI): m/z = 648.2525. calcd. for C16H16N2Na: 648.2517[M+H]+. 5,16-Di(2,4,6-trimethylphenyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2d): Reaction time: 72 h; Yield: 20%; 1H NMR (400 MHz, CDCl3) δ = 12.16. (brs, 2H), 7.46-7.26 (m, 8H), 6.92 (s, 2H), 6.86 (s, 2H), 6.28 (d, J = 4.1 Hz, 4H), 6.17 (d, J = 4.1 Hz, 4H), 2.34 (s, 6H), 2.14 (s, 6H), 1.96 (s, 6H); 13 C NMR (100 MHz, CDCl3) δ = 155.35, 140.65, 140.38, 137.27, 136.85, 136.71, 134.63, 134.00, 129.36, 127.87, 127.65, 127.54, 127.45, 118.75, 21.13, 19.98, 19.90 ; HRMS (ESI): m/z = HRMS (ESI): m/z = 673.3275. calcd. for C48H40N4: 672.3253 [M+H]+. 5,16-Di(4-fluorophenyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2e): Reaction time: 4.5 h; Yield: 48%; The single crystal suitable for X-ray diffraction analysis was obtained by a slow vapour diffusion of hexane into dichloromethane solution. 1H NMR (400 MHz, CDCl3) δ = 12.28 (brs, 2H), 8.30 (d, J = 8.7 Hz, 4H), 7.70 (d, J = 8.7 Hz, 4H), 7.47-7.42 (m, 8H), 6.41 (d, J = 4.1 Hz, 4H), 6.31 (d, J = 4.1 Hz, 4H); 13C NMR (100 MHz, CDCl3) [typical signails] δ =164.49, 162.01, 155.82, 140.82, 140.69, 134.32, 133.73, 133.70, 133.14, 129.39, 128.97, 128.07, 118.95, 114.68, 114.63; HRMS (ESI): m/z = 624.2117. calcd. for C42H26F2N2: 624.2126 [M]+. Crystal data: monoclinic; space group C2/c (No. 15); a = 21.360(5) Å, b = 7.3279(15) Å, c = 22.217(5) Å; b = 113.877(4)°; V = 3180.0(12) Å3; Z = 4; rcalcd = 1.305 g cm–3; T = 90 K; R1 = 0.0470 [I > 2s(I)]; Rw = 0.0752 (all data); GOF = 1.025.[32] 5,16-Di(4-nitrophenyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2f): Reaction time: 1.5 h; Yield: 36%; The single crystal suitable for X-ray diffraction analysis was obtained by a slow vapour diffusion of hexane into dichloromethane solution.1H NMR (400 MHz, CDCl3) δ = 12.17 (brs, 2H), 7.51-7.40 (m, 12H), 7.11 (t, J = 8.7 Hz, 4H), 6.48 (d, J = 4.1 Hz, 4H), 6.28 (d, J = 4.1 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ =156.47, 148.11, 144.32, 140.24, 138.58, 134.34, 132.04, 129.49, 128.77, 128.36, 122.77, 119.71; HRMS (ESI): m/z = 678.2025. calcd. for C42H26N6O4: 678.2016 [M]+. Crystal data: monoclinic; space group C2/c (No. 15); a = 21.397(3) Å, b = 7.9114(13) Å, c = 21.859(4) Å; b = 113.244(3)°; V = 3400.0(10) Å3; Z = 4; rcalcd = 1.326 g cm–3; T = 90 K; R1 = 0.0507 [I > 2s(I)]; Rw = 0.0790 (all data); GOF = 1.011.[32] 5,16-Di(trimethylsilylethynyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2g): Reaction time: 2 h; Yield: 33%; 1H NMR (400 MHz, CDCl3) δ = 11.88 (brs, 2H), 7.42-7.37 (m, 8H), 7.14 (d, J = 4.1 Hz, 4H), 6.26 (d, J = 4.1 Hz, 4H), 0.27 (s, 18H); 13C NMR (100 MHz, CDCl3) δ = 156.47, 142.41, 134.58, 129.65, 128.42, 128.15, 121.25, 119.44, 104.95, 101.84, 0.30; HRMS (ESI): m/z = 629.2517. calcd. for C40H36N4Si2: 628.2479 [M+H]+. 5,16-Di(2-thienyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2h): Reaction time: 72 h; Yield: 12%; 1H NMR (400 MHz, CDCl3) δ = 11.94 (brs, 2H), 7.54 (m, 2H), 7.48-7.39 (m+m, 2H+8H), 7.14 (dd, J = 5.0, 3.7 Hz, 2H), 6.87 (d, J = 4.1 Hz, 4H), 6.33 (d, J = 4.1 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ =155.95, 140.74, 139.05, 134.47, 134.17, 132.34, 132.34, 129.32, 129.05, 128.03, 126.96, 118.81; HRMS (ESI): m/z = 601.1476. calcd. for C38H24N4Si2: 600.1442 [M+H]+. 5,16-Di(3,4-diethyl-5-formyl-2-pyrrolyl)-dibenzo[j,u]porphyrin(2.1.2.1) (2i): Reaction time: overnight ; Yield: 4.5%; The single crystal suitable for X-ray diffraction analysis was obtained by a slow vapour diffusion of hexane into dichloromethane solution. 1H NMR (400 MHz, C2D2Cl4, at 120 °C) δ = 9.74 (s, 2H), 8.90 (brs, 2H), 7.52-7.50 (m, 4H), 7.46-7.43 (m, 4H), 6.55 (d, J = 4.2 Hz, 4H), 6.32 (d, J = 4.2 Hz, 4H), 2.83 (q, J = 7.6 Hz, 4H), 2.38 (m, 4H), 1.33 (t, 6H), 0.94-0.89 (m, 6H); HRMS (ESI): m/z = 735.3445. calcd. for C48H42N6O2: 735.3442 [M+H]+. Crystal data: monoclinic; space group P21/n (No. 11); a = 12.955(2) Å, b = 16.168(2) Å, c = 19.515(3) Å; b = 107.522(3)°; V = 3897.9(10) Å3; Z = 4; rcalcd = 1.252 g cm–3; T = 90 K; R1 = 0.0533 [I > 2s(I)]; Rw = 0.1368 (all data); GOF = 1.012.[32].
(8) FULL PAPER Synthesis of 5,16-Diphenyl-dibenzo[j,u]porphyrinate(2.1.2.1) nickel (Ni-2a): A solution of Ni(OAc)2·4H2O (63.6 mg, 0.256 mmol) in methanol (8 mL) was added to a solution of compound 2a (29.0 mg, 0.0493 mmol) in chloroform (23 mL). After being stirred for 2 h at room temperature, the solvent was concentrated. The crude material was purified by silica gel column chromatography (dichloromethane). Recrystallized from chloroform/methanol gave Ni-2a as dark green solids in 92% (29.2 mg, 0.0452 mmol). The single crystal suitable for X-ray diffraction analysis was obtained by a slow vapour diffusion of methanol into chloroform solution. 1H NMR (400 MHz, CDCl3) δ = 7.67 (m, 4H), 7.49-7.44 (m, 10H), 7.38-7.36 (m, 4H), 6.62 (d, J = 3.7 Hz, 4H), 6.12 (d, J = 3.7 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ = 162.11, 146.16, 138.92, 136.97, 133.30, 131.85, 130.52, 129.51, 128.63, 128.35, 127.49, 11.60; HRMS (ESI): m/z = 644.1515. calcd. for C42H26N4Ni: 644.1511 [M]+. Crystal data: orthorhombic; space group Pca21 (No. 29); a = 21.356(4) Å, b = 6.8556(12) Å, c = 20.954(4) Å; V = 3067.8(9) Å3; Z = 4; rcalcd = 1.397 g cm–3; T = 90 K; R1 = 0.0499 [I > 2s(I)]; Rw = 0.0834 (all data); GOF = 1.000.[32] Synthesis of 5,16-Diphenyl-dibenzo[j,u]porphyrinate(2.1.2.1) copper (Cu-2a): A solution of Cu(OAc)2·H2O (49.2 mg, 0.246 mmol) in methanol (6 mL) was added to a solution of compound 2a (30.7 mg, 0.0503 mmol) in chloroform (10 mL). After being stirred for 1.5 h at room temperature, the solvent was concentrated. The crude material was purified by silica gel column chromatography (chloroform). Recrystallized from chloroform/methanol gave Cu-2a as blue solids in 93% (32 mg, 0.0113 mmol). HRMS (ESI): m/z = 650.1490. calcd. for C42H27N4Cu: 650.1526 [M+H]+. Synthesis of 5,16-Diphenyl-dibenzo[j,u]porphyrinate(2.1.2.1) palladium (Pd-2a): A solution of compound 2a (30.4 mg, 0.0516 mmol) and Pd(OAc)2 (18.4 mg, 0.0820 mmol) in chloroform (4 mL) was refluxed for 1.5 h, and then the solvent was concentrated. The crude material was purified by silica gel column chromatography (chloroform). Recrystallized from chloroform/methanol gave Pd-2a as dark green solids in 57% (20.5 mg, 0.0296 mmol). The single crystal suitable for Xray diffraction analysis was obtained by a slow vapour diffusion of methanol into 1,1,2,2-tetrachloroethylene solution. 1H NMR (400 MHz, CDCl3) δ = 7.58 (m, 4H), 7.47-7.46 (m, 6H), 7.42-7.40 (m, 4H), 7.37-7.36 (m, 4H), 6.65 (d, J = 4.4 Hz, 4H), 6.39 (d, J = 4.4 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ =160.99, 147.24, 138.37, 137.60, 132.92, 131.41, 130.79, 130.50, 128.52, 128.48, 127.36, 118.95; HRMS (ESI): m/z = 692.1196. calcd. for C42H26N4Pd: 692.1192 [M]+. Crystal data: monoclinic; space group P21/n (No. 11); a = 16.265(3) Å, b = 11.660(2) Å, c = 18.620(4) Å; b = 114.285(3)°; V = 3218.7(11) Å3; Z = 4; rcalcd = 1.532 g cm–3; T = 90 K; R1 = 0.0488 [I > 2s(I)]; Rw = 0.0705 (all data); GOF = 1.023.[32] Synthesis of 5,16-Diphenyl-dibenzo[j,u]porphyrinate(2.1.2.1) platinum (Pt-2a): A solution of compound 3 (30.4 mg, 0.0516 mmol) and PtCl2 (16.0 mg, 0.0602 mmol) in benzonitrile (4 mL) was refluxed for 2 h, and then the solvent was concentrated. The crude material was purified by silica gel column chromatography (chloroform). Recrystallized from chloroform/methanol gave Pt-2a as brown solids in 74% (29.9 mg, 0.0382 mmol). The single crystal suitable for X-ray diffraction analysis was obtained by a slow vapour diffusion of methanol into chloroform solution. 1H NMR (400 MHz, CDCl3) δ = 7.59 (s, 4H), 7.47-7.35 (m, 14H), 6.63 (d, J = 4.4 Hz, 4H), 6.33 (d, J = 4.4 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ = 160.85, 147.05, 138.44, 137.34, 132.40, 130.86, 130.47, 128.50, 127.39, 118.72; HRMS (ESI): m/z = 782.1881. calcd. for C42H26N4Pt: 782.1878 [M+H]+. Crystal data: monoclinic; space group P21/n (No. 11); a = 16.201(11) Å, b = 11.720(8) Å, c = 18.269(13) Å; b = 112.641(12)°; V = 3202(4) Å3; Z = 4; rcalcd = 1.688 g cm–3; T = 90 K; R1 = 0.0588 [I > 2s(I)]; Rw = 0.1044 (all data); GOF = 1.000.[32]. Synthesis of 5,16-Diphenyl-dibenzo[j,u]porphyrinate(2.1.2.1) tin(IV) chloride (Sn-2a): A solution of 2a (51.2 mg, 0.0870 mmol) and SnCl2·2H2O (391.9 mg, 1.74 mmol) in pyridine (6 mL) was refluxed for 12 h, and then the solvent was concentrated. The crude material was extracted with chloroform. The combined organic layer was washed with water, 3.0 M HCl aq. and brine, and dried over Na2SO4. The crude compound was recrystallized from dichloromethane/acetone to give Sn2a as dark green solids in 11% (7.9 mg, 9.79 µmol). The single crystal suitable for X-ray diffraction analysis was obtained by a slow vapour diffusion of acetone into dichloromethane solution.1H NMR (400 MHz, CDCl3) δ = 7.71 (brs, 2H), 7.45-7.48 (m, 12H), 7.35-7.38 (m, 4H), 6.72 (d, J = 4 Hz), 6.41 (d, J = Hz, 6H); 13C NMR (100 MHz, CDCl3) δ = 147.44, 143.14, 137.19, 135.51, 135.11, 133.10, 132.15, 130.20, 129.66, 128.90, 127.47, 119.57; HRMS (MALDI): m/z = 776.0541 calcd. for C42H26Cl2N4Sn: 776.0556 [M]+. Monoclinic; space group I2/a (No. 15); a = 14.9615(13) Å, b = 17.8497(16) Å, c = 15.6826(10) Å; b = 113.275(6)°; V = 3847.6(6) Å3; Z = 4; rcalcd = 1.483 g cm–3; T = 90 K; R1 = 0.0342 [I > 2s(I)]; Rw = 0.0411 (all data); GOF = 1.081.[32]. Acknowledgements This work was partly supported by CREST JST and Grants-in-Aid for Scientific Research (Nos. 25288092, 2628803, 26105004 and 26288038), the Green Photonics Project in NAIST, and the program for promoting the enhancement of research universities in NAIST supported by MEXT. The authors thank Ms. Y. Nishikawa, Mr. S. Katao, and Mr. F. Asanoma in NAIST for the measurement of mass spectra, single-crystal structure analysis, and NMR measurement, respectively.. Keywords: porphyrin(2.1.2.1) • bent structure • dipyrrin • metal complex • porphyrinoid [1] [2]. [3] [4] [5] [6] [7] [8] [9] [10]. [11] [12] [13] [14]. T. Kojima, T. Nakanishi, R. Harada, K. Ohkubo, S. Yamauchi, S. Fukuzumi, Chem. Eur. J. 2007, 13, 8714–8725. T. Kojima, T. Honda, K. Ohkubo, M. Shiro, T. Kusukawa, T. Fukuda, N. Kobayashi, S. Fukuzumi, Angew. Chem. 2008, 120, 6814–6818; T. Kojima, T. Honda, K. Ohkubo, M. Shiro, T. Kusukawa, T. Fukuda, N. Kobayashi, S. Fukuzumi, Angew. Chem. Int. Ed. 2008, 47, 6712–6716. T. Honda, T. Kojima, S. Fukuzumi, Chem. Commun. 2009, 4994–4996. T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima, S. Fukuzumi, J. Am. Chem. Soc. 2010, 132, 10155–10163. S. Shimizu, S. Nakano, T. Hosoya, N. Kobayashi, Chem. Commun. 2011, 47, 316–318. K. Berlin, E. Breitmaier, Angew. Chem. 1994, 106, 1356–1357; K. Berlin, E. Breitmaier, Angew. Chem. Int. Ed. 1994, 33, 1246–1247. M. Stępień, L. Latos-Grażyński, J. Am. Chem. Soc. 2002, 124, 3838– 3839. M. Stępień, L. Latos-Grażyński, L. Szterenberg, J. Panek, Z. Latajka, J. Am. Chem. Soc. 2004, 126, 4566–4580. M. Stępień, L. Latos-Grażyński, Acc. Chem. Res 2005, 38, 88–98. M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz, L. Szterenberg, Angew. Chem. 2007, 46, 8015–8019; M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz, L. Szterenberg, Angew. Chem. Int. Ed. 2007, 46, 7869–7873. M. Stępień, B. Szyszko, L. Latos-Grażyński, J. Am. Chem. Soc. 2010, 132, 3140–3152. B. Szyszko, N. Sprutta, P. Chwalisz, M. Stępień, L. Latos-Grażyński, Chem. Eur. J. 2014, 20, 1985–1997. R. J. P. Corriu, G. Bolin, J. J. E. Moreau, C. Vernhet, J. Chem. Soc. Chem. Commun. 1991, 211–213. F. H. Carré, R. J. P. Corriu, G. Bolin, J. J. E. Moreau, Organometallics 1993, 12, 2478–2486..
(9) FULL PAPER [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26]. J.-i. Setsune, M. Toda, T. Yoshida, K. Imamura, K. Watanabe, Chem. Eur. J. 2015, 21, 12715–12727. K. S. Anju, S. Ramakrishnan, A. Srinivasan, Org. Lett. 2011, 13, 2498– 2501. Z. Hu, J. L. Atwood, M. P. Cava, J. Org. Chem 1994, 59, 8071–8075. H. Zhongying, C. P. Michael, Tetrahedron Lett. 1994, 35, 3493–3496. T. Zhao, Z. Wei, Y. Song, W. Xu, W. Hu, D. Zhu, J. Mater. Chem. 2007, 17, 4377–4381. K. Singh, A. Sharma, J. Zhang, W. Xu, D. Zhu, Chem. Commun. 2011, 47, 905–907. K. Singh, T. S. Virk, J. Zhang, W. Xu, D. Zhu, Chem. Commun 2012, 48, 12174–12176. H. Liu, Y. Qin, D. Huang, W. Xu, D. Zhu, Tetrahedron 2014, 70, 1872– 1879. K. Singh, T. S. Virk, J. Zhang, W. Xu, D. Zhu, Chem. Commun. 2012, 48, 121–123. F. Szydlo, B. Andrioletti, E. Rose, Org. Lett. 2006, 8, 2345–2348. B. Chandra, S. P. Mahanta, N. N. Pati, S. Baskaran, R. K. Kanaparthi, C. Sivasankar, P. K. Panda, Org. Lett. 2013, 15, 306–309. M. Pawlicki, M. Garbicz, L. Szterenberg, L. Latos-Grażyński, Angew. Chem. 2015, 127, 1926–1929; M. Pawlicki, M. Garbicz, L. Szterenberg, L. Latos-Grażyński, Angew. Chem. Int. Ed. 2015, 54, 1906–1909.. [27]. F. Chen, Y. S. Hong, S. Shimizu, D. Kim, T. Tanaka, A. Osuka, Angew. Chem. Int. Ed. 2015, 127, 10785–10788; F. Chen, Y. S. Hong, S. Shimizu, D. Kim, T. Tanaka, A. Osuka, Angew. Chem. Int. Ed. 2015, 54, 10639–10642. [28] D. Kuzuhara, S. Miyake, H. Moriyama, Y. Tamura, N. Aratani, H. Yamada, Tetrahedron Lett. 2015, 56, 5564–5567. [29] K. Kano, K. Fukuda, H. Wakami, R. Nishiyabu, R. F. Pasternack, J. Am. Chem. Soc. 2000, 122, 7494–7502. [30] C. Brückner, V. Karunaratne, S. J. Rettig, D. Dolphin, Can. J. Chem. 1996, 74, 2182–2193. [31] P. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. E. Hommes, J. Am. Chem. Soc. 1996, 118, 6317–6318. [32] CCDC-1446076 (2a), CCDC-1446075 (2e), CCDC-1446079 (2f), CCDC-1446078 (Ni-2a), CCDC-1446080 (Pd-2a), CCDC-1446081 (Pt-2a) and CCDC-1446082 (Sn-2a) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif..
(10) FULL PAPER Entry for the Table of Contents (Please choose one layout) Layout 1:. FULL PAPER Highly bent porphyrins(2.1.2.1) have been synthesized by simple condensation reactions from odipyrrolylbenzene with various aldehydes. We found by X-ray diffraction analysis that porphyrins(2.1.2.1) have macrocyclic o-phenylene bridged bis(dipyrrin) structures which formed saddle shaped structures. Porphyrin(2.1.2.1) performed porphyrin-like dianionic ligand which led to the varied metal complexes.. Daiki Kuzuhara*, Wataru Furukawa, Aya Kitashiro, Naoki Aratani, Hiroko Yamada* Page No. – Page No. Synthesis and Metalation of Doubly o-Phenylene–Bridged Cyclic Bis(dipyrrin)s with Highly Bent Skeleton of Dibenzoporphyrin(2.1.2.1).
(11)
図
![Figure 1. Structures of benzene ring incorporated porphyrins and porphyrin(2.1.2.1) and core modified [22]porphyrin(2.1.2.1)](https://thumb-ap.123doks.com/thumbv2/123deta/8639530.1342128/1.892.455.822.609.886/figure-structures-benzene-incorporated-porphyrins-porphyrin-modified-porphyrin.webp)


+2
関連したドキュメント
The surrounding structure of Fe 2+ was examined using light, X-ray absorption spectroscopy, and molecular dynamics simulation.. The results suggest that Fe 2+ ions in Na 2
Apply Shafen Star as a post-emergence broadcast application in Regions 1, 2, 3, 4, and 5 for control or partial control of weeds listed in “APPLICATION RATES FOR WEED GROWTH
1 ミャンマー(ビルマ) 570 2 スリランカ 233 3 トルコ(クルド) 94 4 パキスタン 91 . 5
1号機 2号機 3号機 4号機 5号機
12月 1月 2月 3月 4月 5月 6月 2Q 3Q 4Q 1Q 2Q 3Q 4Q 新設ピッ.
画像 ノッチ ノッチ間隔 推定値 1 1〜2 約15cm. 1〜2 約15cm 2〜3 約15cm
1月 2月 3月 4月 5月 6月 7月 8月 9月 10月 11月 12月.
The IOUT pin sources a current in proportion to the total output current summed up through the current summing amplifier. The voltage on the IOUT pin is monitored by the internal
