寄稿論文
多環状エーテル系天然物の合成戦略
理化学研究所 有機合成化学研究室 主任研究員中田 忠
1. はじめに 近年,興味ある顕著な生物活性を有し,複雑な化学構造を持つ天然有機化合物(天然物) が報告されている。これら天然物は,その特異な化学構造から合成的にも興味深く,また天 然から微量しか得られないことからその詳細な活性試験,活性発現機構の解明にとっても, 有機合成化学の果たす役割は重要なものとなっている。これら化合物を化学合成するには 斬新な合成戦略のもとに,かつその合成を実現する真に有用な化学反応の開発が必要であ り,その合成を達成することは天然物合成分野に限らず,有機合成化学の進展,更に周辺関 連分野の発展をうながすものである。 当研究室では,複雑な化学構造を有する多官能性生物活性天然物を標的化合物として合 成研究を展開してきた。今回,多環状エーテル系天然物の全合成研究における我々の最近の 成果について,特にその合成手法の開発に焦点を絞って紹介する。 メキシコ湾で多発する赤潮の原因種Gymnodinium breveの毒ブレベトキシンB(1)1 が1981 年に単離構造決定されて以来,ヘミブレベトキシンB(2 )2,イェッソトキシン(3 )3,マイ トトキシン(4)4など数種の海洋産多環状エーテル系天然物が単離されている(図1)。渦鞭 毛藻 Gambierdiscus toxicus の生産するマイトトキシン(4)は,生体高分子を除けば天然物 として最大の分子量(342 2)を持ち,非蛋白質として最強の毒性を有していることから大き な注目を集めている。これらの天然物は,特異な trans-縮環多環状エーテル構造を持ち,ま た強力な毒性と同時にイオンチャンネルに特異的に反応する顕著な生物活性を有しており 合成的にも,薬理活性の面でも極めて興味深い。 ブレベトキシンB(1)の単離構造決定の報告以来,本系天然物の合成を目指した研究が 世界の化学者により挑戦されている。Nicolaou らが本系天然物合成として初めてヘミブレ ベトキシンB(2)の全合成に成功し5 ,更に 12年の歳月をかけたブレベトキシンB(1)の 全合成が 1 99 5 年に報告され6,ついでブレベトキシンAの全合成が達成された7。その後, 山本ら8及び我々9がヘミブレベトキシンB(2)の全合成を達成し,森ら10及び Rainier ら11 がその形式全合成を報告した。また,ごく最近,平間らは 12 年の努力の末にシガトキシン CTX3Cの最初の全合成に成功している12。 我々は,多環状エーテル系天然物の強い生物活性に関する興味と同時に,合成化学的にも その立体選択性,効率性において解決しなければならない問題点を多く含んでいると考え その合成研究に着手した。マイトトキシン(4) ブレベトキシンB(1) ヘミブレベトキシンB(2) イェッソトキシン(3) CHO O O O O O O O O O O O O Me H Me H H Me Me H H H Me OH H H H Me H H H H H H Me J G E A B C D K I H F O O O O CHO OH Me H H HO H H H H Me A B C D NaO3SO O O O O O O O O O O O H H H H H Me Me Me H H Me H Me H H H H H H H H H H H NaO3SO OH MeOH B A C D E G J F H I K O O O O OH OH NaO3SO O O O O O O O Me OH OH Me OH OSO3Na Me OH Me H HO OH H Me H Me H H H H H H OH O O H HO OH H H H OH HO H H H HOHH H OH H H H H OH HOH OH OH O O HO H H OH H OH O O O O O O O O O O O O O O O O O Me Me H H H H H Me Me Me H H Me H Me H Me H Me H Me OH H H H H Me Me Me H H H H H H OH OH Me Me OH HO OH H Y A B C D F H I K M N O P Q S R T U V W X E G J L Z A' B' C' D' E' F' 図1 海洋産多環状エーテル系天然物 2. 転位−環拡大反応 海洋産多環状エーテル系天然物の合成において,まず解決しなければならない最も重要 な課題はその基本構成単位である 2,3-trans- 環状エーテル iii の構築である(図2)。その 直截な合成法の一つはエポキシアルコール i のエンド環化であると考えられる。またこの 反応は生合成合成ルートとも考えられている。しかし,Baldwin 則から予測されるように, この環化反応は通常エキソ環化反応が進行し,エーテル ii が生成する。そこで,我々はこ のエーテル ii の側鎖水酸基を脱離することによる転位−環拡大により目的とする iii を得 ようと考えた。
図3 転位−環拡大反応 O Me OX O OH H OAc OR O Me OX OH H Me OAc OR O Me OX OR H Me OAc OAc Me Me O O Me Me Me 95% (R=Ac: 53%, R=H: 42%) 0% (SM 60%) 87% (R=Ac:19%, R=H: 68%) 90% (R=H) 19% (R=H) (SM 64%) 92% (R=H) 90% (R=Ac:10%, R=H: 80%) reflux, 8 h 80 °C, 6 h 80 °C, 3.5 h reflux, 6 h 50 °C, 24 h 50 °C, 4 h 5a: X=SO2Me 5b: X=SO2CH2Cl 75% (after acetylation) 13% (SM 67%) 85% (R=Ac: 8%, R=H: 77%) 9a: X=SO2Me 9b: X=SO2CH2Cl 80 °C, 8 h rt, 4 d rt, 26.5 h 50 °C, 2 h without Zn(OAc)2 8 10 6 7a: X=SO2Me 7b: X=SO2CH2Cl Zn(OAc)2 AcOH-H2O Zn(OAc)2 AcOH-H2O Zn(OAc)2 AcOH-H2O エポキシ化 環化 エキソ エンド 転位−環拡大 図2 2,3-trans- 環状エーテルの合成戦略 既にラサロシドA全合成13 において5員環エーテルの6員環エーテルへの転位−環拡大 反応を見いだしているが,本反応を更に詳細に検討することとした。エポキシ化及びエキソ 環化をへて得たメシレート 5a,7a を反応基質として環拡大反応を検討した(図3)。メシ レート 5a,7a をラサロシドA全合成と同様な反応条件(Ag2CO3,アセトン中還流)に付 すと,転位−環拡大反応が進行し求める 6,8 が得られたがその収率はそれぞれ 46%,17 %と低収率であった。種々反応条件を検討した結果,Zn(OAc)2存在下,酢酸−水溶液中還流 すると,環拡大した6及び7員環エーテル 6,8 がそれぞれ高収率で得られることを見いだ した14a。 更に我々は別の研究課題を進展中,モノクロロメタンスルホネート(モノクレート)が優 れ た脱 離能 を有 し, 2 級水 酸基 の反 転な ど 各種 反応 に極 めて 有 効で ある こと を見 い だした1 5。そこで上記転位−環拡大反応においてメシレートの代わりにモノクレート 5 b , 7b,9b を用いたところ,より温和な条件(室温,50-80 ℃)で反応が速やかに進行し,より 効率的な環拡大反応を開発することができた14b。本反応はヘミブレベトキシンB(1)のC D環構築に有効に利用された(後述)。また,この環拡大反応を繰り返すことによりマイト トキシン(4)のST環及びXY環の合成ができた16。 OH R2 OH R2 O O R2 OH R1 H O R2 OH R1 R1 R1 n n n n i ii iii
図4 4種の立体異性体の転位反応 3. スチリルエポキシドのエンド環化反応 ヘミブレベトキシンB(2)のB環構築のためにエーテル ii(R1 = H)の転位反応を試み たが(図2),R1 = H の場合,望む環拡大反応は収率良く進行しないことが判明した 18。そ こで,直接エンド環化反応によりB環を構築することとした。既に,Nicolaou らはエポキシ 基の隣にビニル基を配置した i(R1 = H, R2 = CH2)を酸処理すると,エンド環化が選択的 に進行し目的の 2,3-trans-iii が生成することを報告していた19。しかし,本手法をヘミブ レベトキシンB(2)のBC環構築のためにモデル化合物 11 (R= H)に応用したが(図5), 目的とする 6- エンド環化体 12 (R = H)を選択性良く得ることはできなかった (12:13 = 1:1)20。 本反応の反応機構を解明すべく,可能な4種の立体異性体を合成しその転位反応を検討 した(図4)17。その結果,この転位−環拡大反応は立体特異的であり,オキソニウムイオ ンを経由して反応が進行していると考えられる。オキソニウムイオンが形成しづらい基質 では,別のコンホメーションからの転位−開環反応が進行しケトン体が生成した。 図5 BC環構築のモデル反応 O Me Me Me OH H CHR O O Me Me Me O H CHR OH H H Me MeO Me O H OH H CHR PPTS CH2Cl2, rt + 86% 86% 50 : 50 100 : 0 Yield 11 12 13 12 : 13 B C R=H R=Ph O t-Bu Me H Me OMs OR Me H Me O t-Bu H O t-Bu Me H Me OMs OH t-Bu Me H O COMe Me OH t-Bu H Me 9% Zn(OAc)2 aq AcOH reflux, 10 min Zn(OAc)2 aq AcOH reflux, 1 hr O t-Bu Me H Me OMs COMe Me OH t-Bu H OR Me H Me O t-Bu H 77% 22% 58% (R=H, 49%; R=Ac, 9%) Zn(OAc)2 aq AcOH reflux, 5 min syn - equatorial anti - axial 89% (R=H, 84%; R=Ac, 5%) syn - axial O t-Bu Me H Me OMs COMe Me OH t-Bu H OH Me H Me O t-Bu H anti - equatorial 50% Zn(OAc)2 aq AcOH reflux, 1 hr 28% O t-Bu H OMs H O t-Bu Me H H Me antiperiplaner oxonium ion O t-Bu Me H H nOe nOe O t-Bu Me H H Me nOe O t-Bu Me H H OMs nOe O t-Bu H Me Me H O t-Bu Me H Me H O t-Bu H Me H Me H2O Zn2+ OMs Me Me Me OMe Me
そこで,本反応を改良すべくエポキシ基の隣りにスチリル基を配置させたスチリルエポ
キシド 1 4 の環化反応を検討した(図6)。 スチリル基の位置及び立体選択性への効果は予
想以上に高く,14 を酸処理(CSA またはPPTS)あるいは塩基(NaH, DMSO)処理すると求め
るエンド環化反応のみが進行し,2,3-trans-15 が立体選択的に得られることが判明した21。 なお,エポキシド 16 では 5- エキソ環化体のみが生成し,ビニルエポキシド 17 も 5- エキソ 及び 6- エンド環化体の混合物を与えたことから,スチリル基の regio-controller としての能 力は極めて高いものであった。本反応を用いるとモデル化合物 11 (R = Ph)は完全な 6- エ ンド環化で進行し,求めるBC環 12(R = Ph)のみを立体選択的に与えた(図5)20。 14 15 6-endo 5-exo CSA (PPTS), CH2Cl2 or NaH, DMSO OH O Me H CHPh Me O HO CHPh 17 5-exo 6-endo HO Me O 16 5-exo 6-endo cf. Me O HO OH 図6 スチリルエポキシドのエンド環化反応 4. ヘミブレベトキシンB (2) の全合成 Gymnodinium breve の生産する毒素ヘミブレベトキシンB(2)は ,10 個の不斉中心を 有する特異な4環性 6 , 6 , 7 , 7環状エーテル骨格と,側鎖に Z - ジエン,α- ビニル アルデヒド構造を有している。 これらの開発した手法を駆使してヘミブレベトキシンB(2)の全合成を計画した。CD
環は相対的に同じ立体配置を有していることから,ii の iii への環拡大反応 (R1 = Me) に
より構築できると考えられる。我々は当初からこの全合成における鍵反応として,CD環の 構築を段階的ではなく一段階で効率的に構築したいと考えていた。即ち,6 , 6員環エー テルのダブル環拡大反応により一挙にCD環に相当する7 , 7員環エーテルを構築しよう とするものである。 ゲラニオールより Sharpless の不斉エポキシ化をへて6 , 6員環エーテル 18 を合成した。 当初開発したメシレートを用いる環拡大反応は,反応は進行するものの低収率でそれ以上 全合成を進めるのは困難であり1年ほどの研究停滞を余儀なくされた。しかし,後に改良し たモノクレートを用いる方法によりこの困難を克服することができた。即ち,ジオール 1 8 をビスモノクレートとし,Zn(OAc)2 を作用させるとダブル転位−環拡大反応が高収率で進 行し求める7 , 7員環エーテル 19 を立体選択的に得ることができた(図7)。B環の構築 には,開発したスチリルエポキシドの 6- エンド環化反応を適用した。Sharpless 不斉エポ キシ化をへて導いたスチリル α- エポキシド 20 に CSA を作用させるとB環に相当する6員 環エーテル 2 1 を立体選択的に得ることができた。A環をラクトールとして構築し,側鎖 (Z) - ジエン部を導入後, 22 に TMSOTf 存在下,CH2=C(CH2OAc)CH2TMS を作用させる と一挙に側鎖4炭素ユニットが立体選択的(β-axial)に導入された。アセテートを加水分解 しアリルアルコールを得,最後に MnO2で酸化しヘミブレベトキシンB(2)を合成するこ とができた。ここに他のグループとは全く異なる極めてユニークな合成ルートによる全合 成が達成された。
図7 ヘミブレベトキシンB(2)の全合成 さて,ヘミブレベトキシンB(2 )の全合成に成功したが,より大きな分子であるブレベト キシンB(1)等の全合成を達成するためには更に効率的合成法の開発が必要であった。その ために,これまでと全く異なる発想でのより画期的合成戦略の開発を行うこととした。 5. SmI2 による還元的環化反応 5.1 繰返し型合成 これまで開発した合成法はエポキシドの開環による C - O 結合の形成をへていることか ら,新たに C - C 結合の形成で多環状エーテル i v を構築できないかと考えた。逆合成的に C-C結合を点線部位で切断すると重要前駆体として vi が浮かびあがってくる。vi の C-C 結 合は分子内にアルデヒドとβ- アルコキシアクリレートを有する vii の環化反応で構築でき
ると考えた。vii は,エステル viii から構築可能であろう。この vii の環化反応が求める vi を 立体選択的に与えるなら,生成物は出発基質 v i i i と同じ構造を有しているので同様な一連 の反応の繰返しにより次々とエーテル環が構築でき多環状エーテル iv が合成できると考え
られる。AIBN 存在下,Bu3SnHによる vii のラジカル環化が報告されていたが,立体選択性
は高くなく,かつ主生成物は我々の望む trans- 体 vi ではなく相当する cis- 体が主生成物 であった(cis:trans = 65:35)22。 我々は,この環化反応に SmI2を用いるラジカル還元的環 化反応が有効に働くだろうと予想した。 O OAc OH Me H H O Me HO O OH OH O H Me H H H Me HO C Double ring-expansion D 61% (3 steps) 1) ClCH2SO2Cl 2) Zn(OAc)2 aq. AcOH, 80 °C 3) K2CO3, MeOH 18 19 endo-Cyclization of styrylepoxide CSA O OH O H Me H H H Me RO O O Me Me CHPh O O O O H Me H H H Me RO O O Me Me OAc H HCHPh B 20 21 C D D C O O O H Me H H H Me RO O H H OH OMe 1) TMSOTf 2) K2CO3 3) MnO2 O O O O CHO OH Me H H HO H H H H Me OAc TMS A Direct introduction of C-4 unit 22 C D B C B D A ヘミブレベトキシンB(2)
図8 trans-縮環多環状エーテルの繰返し型合成戦略
まず,テトラヒドロピラン 23 とエチルプロピオレートとのヘテロ Michael 反応,続く脱チ
オアセタール化により反応基質となるアルデヒド 2 4 を合成した(図9)。 ついで,2 4 に
THF中,MeOH 存在下2等量の SmI2を作用させると,望む 2,6-syn-2,3-trans- 配置を有す
る6員環エーテルが構築され,2環性エーテル 25 が単一生成物として得られた23。本反応 は7員環エーテルの構築にも有効で,アルデヒド 2 6 に室温下 S mI2を作用させると求める 環化反応が進行すると同時にラクトン化が生じ,2,7-syn-2,3-trans- オキセパン 27 を立体 選択的に得ることができた。 本環化反応は,まず SmI2による一電子還元で生成したケチルラジカルが Sm(III) とエステ ルとのキレーション遷移状態 (A-1)をへて,立体選択的に C-C 結合が形成し A-2 が生成
する。ついで2等量目の SmI2により A-2 のラジカルがアニオンに還元され,MeOH による
プロトネーション化により 25が生成したと考えられる。オキセパン 27もキレーション遷移 状態 B をへて立体選択的に生成したと考えられる。 H H O O H H O H H O H H H H H H O O H H O H H O H H H H iv v O H H O CO2R H H O H H O O CO2R OH O H H CO2R OH vi vii viii O OH H H S S CO2Et O CHO O H H 1) N-Me morpholine
2) MeI, aq. MeCN
CO2Et 87% 23 24 SmI2 THF, MeOH 0 °C, 10 min O H H 92% O OH CO2Et H H 25 SmI2, MeOH THF rt, 10 min 84% O O CO2Et H H 26 CHO O H H 27 O O O H H 図9 SmI2による立体選択的環化反応 O O O Sm I2 O OEt H H H H O O O Sm I2 O OEt H H H H A-1 A-2 O O O H H H H SmI2 O OEt B
本反応は,鎖状化合物からでも立体選択的に 2,3-trans- 環状エーテルが得られることか らも,その完全な立体選択性発現には Sm(III) とエステル基とのキレーションが重要な役割
を果たしていることが明らかである24。
一方,SmI2を用いる各種の反応は HMPA の添加により反応が促進されることがよく知ら
れている。上記環化反応に HMPA を加えると生成物は全く異なる化合物を与えた(図 10)。
即ち,24 の THF-HMPA (3:1) 溶液中,SmI2を作用させると 2,6-syn-2,3-cis- 及び
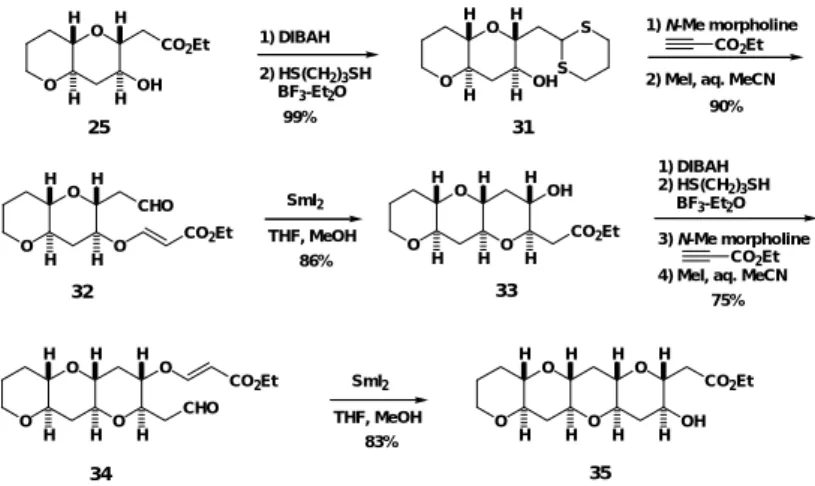
2,6-anti-2,3-trans- テトラヒドロピラン 28, 29 が生成した。また,26 も 2,7-syn-2,3-cis- オキセパ ン 30 のみを生成した。これは,HMPA が Sm 原子に配位し,キレーション遷移状態をとれ ず,それぞれ遷移状態 C, D 及び E をへて反応が進行したと説明できる。 図 10 HMPA 存在下の SmI2による環化反応 SmI2, MeOH THF-HMPA (3:1) -78 °C, 30 min O O CO2Et H H OH H H 28 (56%) O O CO2Et H H OH H H 29 (17%) O CHO O H H CO2Et 24 + SmI2, MeOH THF-HMPA (2:1) rt, 10 min 84% O H H O H H OH CO2Et O O CO2Et H H 26 CHO 30 O O H O OEt H H H OSmI2 (HMPA)n O O H H H OSmI2 (HMPA)n H CO2Et H O O H H H H OSmI2 O OEt (HMPA)n C D E SmI2による還元的環化反応が完全な立体選択性で求める trans- 体を与えたことから,更 に本反応を基盤とする多環状エーテルの繰返し型合成法を確立することとした(図 11)。エ ステル 25 を DIBAH 還元,チオアセタール化により出発物質 23 に相当する 31 に変換できた。 更に同様な一連の反応を繰返すことにより3環性エーテル 3 3 および4環性エーテル 3 5 を 立体選択的,高収率で得ることができた。
図 11 trans- 縮環 4 環性エーテルの繰返し型合成 本繰返し型合成法により6 , 7 , 6員環エーテル 37 及び6 , 7 , 7 , 6員環エーテル 39も立体選択的かつ効率的に合成できた(図 12)。 O H H O O O H H 1) DIBAH 2) HS(CH2)3SH 3) 4) MeI O H H O O CHO CO2Et H H 73% CO2Et 27 36 86% O H H O O CO2Et H H OH H H 37 SmI2 O H H O H H O H H CHO O CO2Et O H H O H H O H H 88% O OH CO2Et H H 38 39 SmI2 THF, MeOH O H H O OH CO2Et H H 1) DIBAH 2) HS(CH2)3SH BF3-Et2O O H H O OH H H S S CO2Et 1) N-Me morpholine
2) MeI, aq. MeCN
99% 90% 25 31 O H H O O CHO H H CO2Et SmI2 THF, MeOH O H H O O H H CO2Et OH H H 1) DIBAH2) HS(CH 2)3SH BF3-Et2O 3) N-Me morpholine 4) MeI, aq. MeCN
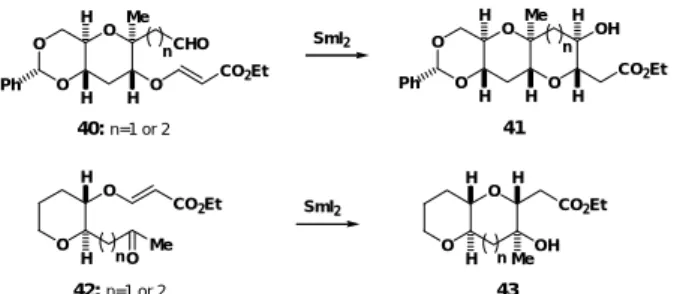
CO2Et 86% 75% 32 33 O H H O O H H CHO O H H CO2Et SmI2 THF, MeOH O H H O O H H O H H CO2Et OH H H 83% 34 35 図 12 trans- 縮環多環状エーテルの合成 SmI2を用いる環化反応は,核間メチル基を有するエーテル環構築(40 → 41, 42 → 43)に も有効である(図 13)。また,1,3-diaxial メチル基を有するエーテル環の合成にも有効で, 44に SmI2を作用させるとマイトトキシンのC ' D ' 環に相当するエーテル 45 を収率 99%で 与えた(図 14)25。即ち,1,3-diaxial メチル基の立体障害を克服して,キレーション遷移状 態 F をへて環化反応が進行したことになる。これは 1,3-diaxial メチル基を有するエーテル 環の直截な合成の初めての例である。
図 14 1,3-diaxial メチル基を有する環状エーテルの合成 以上,S m I2 を用いる環化反応は種々のタイプのエーテル環を容易に合成できることか ら,多環状エーテル化合物の一般的合成法として極めて有効であると期待できる。 5.2 2 方向型合成 SmI2による環化反応を基盤とする繰返し型合成は,各工程とも極めて簡便な反応である ことから,本手法は2方向型合成法にも効果的に使用できると期待できた。事実,C2-対称 ジオール 46 へのダブル Michael 反応は収率よく進行し,ジチオアセタールの脱保護で得た ビスアルデヒド 4 7 に S mI2を作用させると,ダブル環化反応が立体選択的に進行し3環性 エーテル 48 を一挙に得ることができた(図1 5 )。 更に,同様な手法の繰返しによりビスア ルデヒド 4 9 に導き,再び S mI2を作用させるとダブル環化が同様に進行し5環性エーテル 50を短工程で合成することができた2 6。ここに,極めて効率的な2方向型合成法の開発に 成功した。本手法は現在,ブレベトキシンB(1)及びイェッソトキシン(3)の合成に効果的に 用いられている。 図 13 核間メチル基を有する環状エーテルの合成 O O Ph O O CHO H Me H H CO2Et O O Ph O O H Me H H CO2Et OH H H 40: n=1 or 2 41 n SmI2 n O O H H CO2Et Me O O H H O OH H Me CO2Et 42: n=1 or 2 43 n n SmI2 HO O H H OH H H S S S S CO2Et 1) 2) MeI, aq MeCN 90% 46 OHC O O CHO H H O H H CO2Et EtO2C 47 SmI2 THF, MeOH 84% O O H H O H H CO2Et EtO2C OH HO H H H H 48 O O H H O H H CHO OHC H H H H CO2Et EtO2C 1) DIBAH 2) HS(CH2)3SH 3) 4) MeI CO2Et 49 42% SmI2 THF, MeOH 77% O O H H O H H O O H H H H CO2Et EtO2C HO OH H H H H 50 図 15 2方向型合成 C' O O O Ph H H Me OH Me O EtO2C 99% SmI2 THF, MeOH 44 C' D' O O O Ph H H Me OH HO EtO2C Me H 45 F O O O O H H H H Me Me O Sm Ph O OEt I2
6. 収束型合成 多環状エーテル系天然物の効率的合成には収束型合成法が不可欠である。我々は,最も効 率的な収束型合成法として,二つのエーテル環 ix 及び x をアセチリド経由で結合後,ジケ トン xi に導き,その分子内ケタール化により6 , 6員環ジケタール xii が得られるなら,そ の還元により一挙に trans- 縮環6 , 6 , 6 , 6員環エーテル xiii が得られると考えた(図 16)。問題は,このケタール化の段階で xii の他に5 , 5員環ジケタール xiv 及び5 , 6員環
スピロケタール xv が得られる可能性があることである。ab initio 計算の結果は目的の xii が 最も生成熱エネルギーが低くその生成の可能性を示した。この結果に力を得て,実際の合成 研究を開始した。 O O OP X PO H H H H + ix x O OP PO O H H H O H O xi O O O O H OMe H H MeO H xii: 0.0 kJ/mol O O O O H H H H OMe MeO xv: 28.7 kJ/mol O O O O H H OMe MeO H H O O O O H H H H H H xiv: 57.4 kJ/mol xiii カップリング ダブルケタール化 ラジカル還元 図 16 trans- 縮環多環状エーテルの収束的合成戦略 アセチリド 51 とトリフレート 52 を結合した後,得られた 53 のルテニウム酸化によりジ ケトン 54 を収率良く得ることができた(図 17)。 ジケトン 54 に MeOH-CH2Cl2中,CSA, CH(OMe)3を作用させダブルケタール化を試みると,反応途上いくつかのスポットが現れる が最終的に一つの生成物に収束し,望む6 , 6員環ジケタール 55 が定量的に生成した。つ
いで,55 に TMSOTf 存在下,Et3SiHを反応させるとジケタールが立体選択的に還元され,目
的とする trans- 縮環4環性エーテル 56 を得ることができた。ここに,アセチリド 51 より4
図 17 trans- 縮環エーテルの収束的合成 7. おわりに ブレベトキシンB(1)に代表される多環状エーテル系天然物の合成を目指して,効率的 な種々の環状エーテル合成法の開発に成功した。特に,SmI2を用いるラジカル環化反応は エーテル環合成の一般的手法として利用できることが高く期待できる。実際,本手法は,他 グループによってもエーテル環構築に有効に利用されている28。 我々は現在,これらの開発した反応を駆使した海洋産多環状エーテル系天然物の全合成 研究を展開している。既に環拡大反応,スチリルエポキシドのエンド環化反応,SmI2環化 反応を基盤としてブレベトキシンB(1)のABC環,EFG環,及びIJK環の立体選択 的合成を報告した29。現在更に,SmI2環化反応を用いる繰返し型及び2方向型合成戦略を 駆使して,より効率的なABCDEF環及びIJK環の合成を既に達成し,それらの結合に よる 1 の全合成を目指している3 0。また,S m I2環化反応及び収束型合成法を基盤とする イェッソトキシン(3)のABCDEF環の合成,SmI2環化反応を駆使してマイトトキシン (4)のC ' D ' E ' F ' 環の合成25 及びWXYZA ' 環の合成30 を達成している。これら開発 した合成手法を基盤として,海洋産多環状エーテル系天然物の全合成研究,各種誘導体合成 による生物活性発現機構の解明が飛躍的に進展することを期待している。 O O O O H OMe H H MeO H O OTBS H H TfO TBSO O H H O OTBS H H O H H TBSO O O H H O O H H O O TBS TBS O O O O H H H H H H Li THF-HMPA CSA, CH(OMe)3 MeOH-CH2Cl2 reflux, 26 h 100% Et3SiH, TMSOTf CH2Cl2, rt, 1 h 98% RuO2, NaIO4 CCl4-MeCN-H2O (2:2:3) rt, 21 h 94% 51 52 + 53 54 55 56 84%
参考文献
1. Y.-Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James, and K. Nakanishi, J. Am. Chem. Soc., 1981, 103, 6773.
2. A. V. K. Prasad and Y. Shimizu, J. Am. Chem. Soc., 1989, 111, 6476.
3. (a) M. Satake, K. Terasawa, Y. Kadowaki, and T. Yasumoto, Tetrahedron Lett., 1996, 37, 5955. (b) H. Takahashi, T. Kusumi, Y. Kan, M. Satake, and T. Yasumoto, Tetrahedron Lett., 1996, 37, 7087.
4. (a) M. Murata, T. Iwashita, A. Yokoyama, M. Sasaki, and T. Yasumoto, J. Am. Chem. Soc., 1992, 114, 6594. (b) M. Murata, H. Naoki, T. Iwashita, S. Matsunaga, M. Sasaki, A. Yokoyama, and T. Yasumoto, J. Am. Chem. Soc., 1993, 115, 2060. (c) M. Murata, H. Naoki, S. Matsunaga, M. Satake, and T. Yasumoto, J. Am. Chem. Soc., 1994, 116, 7089. (d) M. Satake, S. Ishida, T. Yasumoto, M. Murata, H. Utsumi, and T. Hinomoto, J. Am. Chem. Soc., 1995, 117, 7019. (e) M. Sasaki, N. Matsumori, T. Maruyama, T. Nonomura, M. Murata, K. Tachibana, and T. Yasumoto, Angew. Chem. Int. Ed. Engl., 1996, 35, 1672. (f) T. Nonomura, M. Sasaki, N. Matsumori, M. Murata, K. Tachibana, and T. Yasumoto, Angew. Chem. Int. Ed. Engl., 1996, 35, 1675. (g) W. Zheng, J. A. DeMattei, J. -P. Wu, J. J. -W. Duan, L. R. Cook, H. Oinuma, Y. Kishi, J. Am. Chem. Soc., 1996, 118, 7946.
5. (a) Nicolaou, K. C.; Reddy, K. R.; Skokotas, G.; Sato, F.; Xiao, X.-Y. J. Am. Chem. Soc. 1992, 114, 7935. (b) Nicolaou, K. C.; Reddy, K. R.; Skokotas, G.; Sato, F.; Xiao, X.-Y.; Hwang, C.-K. J. Am. Chem. Soc. 1993, 115, 3558.
6. (a) K. C. Nicolaou, E. A. Theodorakis, F. P. J. T. Rutjes, M. Sato, J. Tiebes, X. -Y. Xiao, C. -K. Hwang, M. E. Duggan, Z. Yang, E. A. Couladouros, F. Sato, J. Shin, H. -M. He, and T. Bleckman, J. Am. Chem. Soc., 1995, 117, 10239. (b) K. C. Nicolaou, F. P. J. T. Rutjes, E. A. Theodorakis, J. Tiebes, M. Sato, and E. Untersteller, J. Am. Chem. Soc., 1995, 117, 10252.
7. K. C. Nicolaou, Z. Yang, G.-Q. Shi, J. L. Gunzner, K. A. Agrios, and P. Gartner, Nature, 1998, 392, 264.
8. (a) I. Kadota, P. Jung-Youl, N. Koumura, G. Pollaud, Y. Matsukawa, and Y. Yamamoto, Tetrahedron Lett., 1995, 36, 5777. (b) I. Kadota and Y. Yamamoto, J. Org. Chem., 1998, 63, 6597.
9. (a) M. Morimoto, H. Matsukura, and T. Nakata, Tetrahedron Lett., 1996, 37, 6365. (b) T. Nakata, S. Nomura, H. Matsukura, and M. Morimoto, Tetrahedron Lett., 1996, 37, 217 (c) T. Nakata, J. Synth. Org. Chem., Jpn., 1998, 56, 940.
10. (a) Y. Mori, K. Yaegashi, and H. Furukawa, J. Am. Chem. Soc., 1997, 119, 4557. (b) Y. Mori, K. Yaegashi, and H. Furukawa, J. Org. Chem., 1998, 63, 6200.
11. J. D. Rainier, S. P. Allwein, and J. M. Cox, J. Org. Chem., 2001, 66, 1380.
12. M. Hirama, T. Oishi, H. Uehara, M. Inoue, M. Maruyama, H. Oguri, and M. Satake, Science, 2001, 294, 1904.
13. T. Nakata, G. Schmid, B. Vranesic, M. Okigawa, T. Smith-Palmer, and Y. Kishi, J. Am. Chem. Soc., 1978, 100, 2933.
14. (a) T. Nakata, S. Nomura, and H. Matsukura, Tetrahedron Lett., 1996, 37, 213. (b) N. Hori, K. Nagasawa, T. Shimizu, and T. Nakata, Tetrahedron Lett., 1999, 40, 2145.
15. (a) T. Shimizu, S. Hiranuma, and T. Nakata, Tetrahedron Lett., 1996, 37, 6145. (b) T. Shimizu, T. Ohzeki, K. Hiramoto, N. Hori, and T. Nakata, Synthesis, 1999, 1373.
16. (a) K. Nagasawa, N. Hori, R. Shiba, and T. Nakata, Heterocycles, 1997, 44, 105. (b) T. Nakata, S. Nomura, and H. Matsukura, Chem. Pharm. Bull., 1996, 44, 627.
17. Y. Sakamoto, H. Koshino, and T. Nakata, Heterocycles, 2002, 56, 177.
18. K. Nagasawa, N. Hori, H. Koshino, and T. Nakata, Heterocycles, 1999, 50, 919.
19. K. C. Nicolaou, C. V. C. Prasad, P. K. Somers, and C.-K. Hwang, J. Am. Chem. Soc., 1989, 111, 5330.
執筆者紹介 中田 忠(なかた ただし) 理化学研究所 有機合成化学研究室 主任研究員。 薬学博士。 [ご経歴] 1967年北海道大学薬学部薬学科卒。1969年北海道大学大学院薬学研究科修士課程 修了。理化学研究所研究員,副主任研究員を経て,1992 年より現職。1992年より埼玉大学大学 院客員教授を兼任。1985年日本薬学会奨励賞,2001年日本薬学会賞を受賞。 [ご専門] 有機合成化学,天然物合成化学。
21. H. Matsukura, M. Morimoto, H. Koshino, and T. Nakata, Tetrahedron Lett., 1997, 38, 5545. 22. E. Lee, J. S. Tae, Y. H. Chong, Y. C. Park, M. Yun, and S. Kim, Tetrahedron Lett., 1994, 35, 129. 23. (a) N. Hori, H. Matsukura, G. Matsuo, and T. Nakata, Tetrahedron Lett., 1999, 40, 2811. (b) N. Hori, H. Matsukura, and T. Nakata, Org. Lett., 1999, 1, 1099. (c) G. Matsuo, N. Hori, and T. Nakata, Tetrahedron Lett., 1999, 40, 8859.
24. G. Matsuo, H. Kadohama, and T. Nakata, Chem. Lett., 2002, in press.
25. Y. Sakamoto, G. Matsuo, H. Matsukura, and T. Nakata, Org. Lett., 2001, 3, 2749. 26. H. Matsukura and T. Nakata, in preparation.
27. G. Matsuo, H. Hinou, H. Koshino, T. Suenaga, and T. Nakata, Tetrahedron Lett., 2000, 41, 903. なお,同様な合成戦略が他グループからも報告された。(a) K. Fujiwara, H. Morishita, K. Saka, and A. Murai, Tetrahedron Lett., 2000, 41, 507. (b) Y. Mori, S. Mitsuoka, and H. Furukawa, Tetrahedron Lett., 2000, 41, 4161.
28. (a) I. Kadota, C. Kadowaki, H. Takamura, and Y. Yamamoto. Tetrahedron Lett., 2001, 42, 6199. (b) Y. Nagumo, H. Oguri, Y. Shindo, S. Sasaki, T. Oishi, M. Hirama, Y. Tomioka, M. Mizugaki, and T. Tsumuraya, Bioorg. Med. Chem. Lett., 2001, 11, 2037. (c) H. Takakura, K. Noguchi, M. Sasaki, and K. Tachibana, Angew. Chem. Int. Ed., 2001, 40, 1090.
29. (a) G. Matsuo, H. Matsukura, N. Hori, and T. Nakata, Tetrahedron Lett., 2000, 41, 7673. (b) G. Matsuo, N. Hori, H. Matsukura, and T. Nakata, Tetrahedron Lett., 2000, 41, 7677. (c) H. Matsukura, N. Hori, G. Matsuo, and T. Nakata, Tetrahedron Lett., 2000, 41, 7681.