1.は
じ め に
材料科学が目指すゴールとは何か.端的に説明すると, より良い機能をもつ物質を見つけること,物質をより簡 易に合成するルートをつくり出すことの二つである.つ まり,何をつくるか(What to make)とどうつくるか (How to make)の 2 種の問題に分けられる.これらの 問題解決のため,アカデミア・企業の両方において,膨 大な実験が繰り返されてきた.これらの膨大なデータと 情報科学の技術を融合させた“データ駆動型の材料科学” いわゆる“マテリアルズインフォマティクス”への展開 を目指した活動が活発化している.2011 年にアメリカ で始まったプロジェクト Materials Genome Initiative (MGI)を皮切りに,欧州では NOMAD,日本では情報 統合型物質・材料開発イニシアティブや NEDO 超先端 材料超高速開発基盤技術プロジェクトなど,さまざまな プロジェクトが立ち上がった.しかし,材料科学へのイ ンフォマティクス技術の適用には,いまだ大きな課題が 残っている.とにかくデータが足りないのだ.従来,良 い結果を示した実験のデータは蓄積・公開されてきたの に対し,良い結果を示さなかった実験のデータは,公開 はおろか,きちんと整理した形で蓄積されてこなかった からである.本稿では,材料科学におけるデータを蓄積 する取組みおよび,材料の記述法について概説し,最近 の適用事例を紹介する.2.データを
蓄積する取組み
2・1 既存のデータベース 材料の素材(化合物)のデータベースとして広く知ら れているのが,アメリカ衛生局の PubChem [Kim 19] とアメリカ化学会の Chemical Abstracts である.これ らのデータベースには,融点・沸点・溶解度といった化 合物の基礎的な特性をはじめ,バイオアッセイの情報な どが掲載されている.化合物の三次元構造の情報(化合 物の中の各原子の座標の情報:X 線結晶構造解析によっ て得られる)に特化したデータベースも多数存在して おり,新しい構造情報が論文発表されると,データベー スにも登録される仕組み(習慣)になっている.化合 物の種類によってデータベースが分かれており,分子 性結晶(有機化合物の結晶など)の場合は Cambridge Crystal Data Center(CCDC)[Groom 16] に,無機系 化合物の場合は Inorganic Crystal Database(ICSD) [Bergerhoff 87]や CRYSTMET [White 2002] に主に収 録される.また,材料としての特性に関わるデータを収 集したデータベースも増えてきている.例えば,米国国 立標準技術研究所(NIST)のデータベース [Bale 09] に は化合物の熱力学データやスペクトルデータが,国立研 究開発法人物質・材料研究機構(NIMS)の MatNavi には金属から高分子まで幅広い材料のデータが収録さ れ て い る [MatNavi]. 前 述 の Chemical Abstracts の SciFinderや Elsevier 社の Reaxys には化学反応のデー タが収録されており,化学の研究・開発の現場で広く利 用されている. 2・2 文献からのデータ抽出 ターゲットとする材料に関わるデータベースが存在 しない場合,次に考えられるのが文献からのデータ抽出 であろう.ここで必須となるのが,材料の名称の認識で ある.化合物には,複数の命名法があり,文献ごとに異 なる命名が用いられているというケースが多々ある.そ のため,文献からさまざまな命名法で名付けられた化 合物の情報を抽出し,同じ化合物か否かを判別するシ化学におけるマテリアルズインフォマティクス
の現状と課題
Achievements and Challenges of Materials Informatics in Chemistry
畑中 美穂
奈良先端科学技術大学院大学研究推進機構研究推進部門,Miho Hatanaka 先端科学技術研究科データ駆動型サイエンス創造センター Institute for Research Initiatives, Division for Research Strategy, /
Data Science Center, Graduate School of Science and Technology, Nara Institute of Science and Technology. [email protected], http://mswebs.naist.jp/LABs/hatanaka/index-j.html
Keywords:
database, description of materials, chemoinformatics, quantum chemical calculation, first principle calculation.ステムが不可欠である.これを可能にするものとして, CHEMDNER [Krallinger 15]があげられる.また,文 献の中で重要なデータは,文章・表・図の中に散らばっ ている.これらのデータを抽出・分類・タグ付けするシ ステムの構築も,この分野において不可欠な要素の一つ だ.これを行うシステムの一つが ChemDataExtractor [Swain 16]であり,実際に,ChemDataExtractor を用 いて文献から材料の合成条件を抽出し,そのデータを もとに機械学習による解析をした例が報告されている [Krallinger 17]. しかし,文献からのデータ抽出には決定的な弱点があ る.前述のとおり,多くの文献において,材料としての 特性が良い場合のデータしか記載されないことである. 例えば,ある化合物の合成方法に着目すると,文献に記 載されるのは,合成に成功した際の情報のみである.合 成に失敗した際の情報は公開されないことがほとんどで ある.どのような条件で実験すると合成に失敗するのか の情報がなければ,合成の可否を予測する分類器をつく ることが不可能であることはいうまでもない.この問題 を解決するために,失敗した実験のデータを蓄積する試 みもなされている.Dark Reaction Project がその筆頭で あろう [Raccuglia 16].しかし,失敗データの蓄積・収 集の機運が高まっているとは言いがたいのが現状である. 2・3 計算データのデータベース化 実験データの代わりになり得るデータの一つが,量子 化学計算で得られる計算データである.量子化学計算と は,化合物の中の電子の状態を記述する方程式を解く計 算である(分野によって呼び名が異なり,分子を取り扱 う分野では量子化学計算,固体などを取り扱う分野では 第一原理計算・バンド計算ともいう場合が多い).こち らのデータは比較的蓄積が進んでおり,前述の MGI に よる Materials Project [Jain 13] や AFLOW [Curtarolo 12],PubChemQC [Nakata 17] など,さまざまな化合 物の計算データを蓄積した汎用的データベースが構築さ れてきた.また,材料の特定の性質の計算値に特化した データベースも構築が進んできている.有機太陽電池や 有機エレクトロニクスの素材に特化したデータを集め た Harvard Energy Clean Project がその代表格である [Hachmann 11].実験データの蓄積に比べ,計算データ の蓄積は自動化が容易であるうえに,統一した計算手法 を用いることで均質なデータが集められるという利点が ある.また,材料の特性が発現するメカニズムの理解の 一助にもなり得る.しかし,この「計算」はけっして万 能ではない.第一に,方程式そのものの厳密解が得られ ないため,すべて近似解であるという弱点がある.第二 に,計算を行うためのモデル化が必要であるため,モデ ルに含まれない効果の議論はできない.第三に,材料の すべての特性が「計算できるとは限らない」という問題 がある.例えば,太陽電池の中の素材の電気伝導率を見 積もるためには,フロンティア軌道のエネルギー準位を 計算すればよいことが既知であるし,その計算に適切な 近似方法が何であるかも既知である.しかし,計算に非 常に長い時間がかかる場合も,計算すべき量が未知の場 合もある.いくつか例をあげると,触媒や高分子の特性, 溶剤への溶けやすさなどがある.そのため,このような 計算が難しい系に対しては,実験のデータの蓄積が必須 となる. 2・4 実験データのデータベース化 現在,多くの材料開発の現場において,一つ一つの実 験が手動で行われているため,実験データのデータベー ス化は容易ではない.実験データを効率良く集めるため, 実験ノートの電子化や,実験の自動化・ロボット化が議 論されているが,まだまだ始まったばかりという状態だ. 製造業の現場ではロボットによるライン生産が一般的に なってきているにもかかわらず,なぜ実験のライン化が 遅れているのか不思議に思う読者もいるかもしれない. この理由の一つが,実験の行程が日々変わり得ることに ある.製造業の場合,同じ材料をつくり続けるため,一 度ラインを構築すれば,その後,数年(数十年 ?)にわたっ て使い続けることができるのに対し,実験の現場では, さまざまな材料をつくり試す必要があるため,同じ工程 を繰り返せばよいとは限らないのだ.非常に似た材料を つくろうとしても,出来上がったものが,粉の場合もあ れば,結晶の場合も,何だかよくわからない“ねばねば” の場合すらある.できたものの形状によって,次にやる べき操作が異なるため,同じラインを繰り返し使うこと ができないというわけだ.そこで,注目を集めているの が,人の動きを再現するヒューマノイド型ロボットであ る.生命科学の分野では,ヒューマノイド型実験ロボッ ト「まほろ」が開発されている [現代化学編集グループ 18].材料科学分野でも,このような試みがいくつか報 告されており,例えば,Cronin らは,化学反応の進行 具合をリアルタイムで評価する装置(核磁気共鳴測定や 赤外分光測定,質量分析)と化学反応の実験を行うロボッ トをつなぎ合わせ,実験データを集めながら,反応が進 行するか否かを予測する分類器をつくり,それをロボッ トの作業に反映させるという戦略を取ることで,新しい 反応の発見に至ったことを報告している [Granda 18]. 先ほど,出来上がったものが何だかよくわからない“ね ばねば”の場合があると述べたが,これは化学系材料分 野では,比較的よくある現象の一つで,これが自動化を 難しくする一因にもなっている.“ねばねば”は,多く の場合,高分子化した何かなのだが,一度できてしまう と,容器にへばりついて,取り除くことができない(溶 剤を入れようと,強い酸を入れようと,超音波にかけよ うと,取れないときは本当に取れない).つまり容器を 捨てるしかない状態になってしまうのだ.そのため,自 動実験で用いる容器は,使い捨てても惜しくない程度の
価格で設計する必要がある.また,ねばねばができるよ うな実験に用いる化合物は往々にして水に溶けず,有機 溶剤の使用が必須だったりするため,容器はガラスやほ うろうなど,溶解や腐食に耐え得るものでなければなら ない.容器の素材を安くするという方向性には,限界が ありそうだ.このような諸々の問題を鑑みると,材料の 自動合成装置に一番向いているのは,容器のサイズを限 りなく小さくした“マイクロチップ”ではないかと個人 的には考えている. また,自動実験とは少し趣向が異なるが,実験に 3D プリンタを使うという試みも注目を集めている.最近の 面白い例をあげると,3D プリンタを使ってさまざまな 形の多孔性の触媒をつくるという試みや,3D プリンタ によって反応容器そのものをつくるという試みが報告さ れている(注:反応容器の形状によって反応を制御する ことができる)[Parra-Cabrera 18, Rossi 17]. このようにロボットによる自動実験の分野は,まだま だ萌芽期にある.材料科学の研究者自らがロボットをつ くるのは困難な場合が多いので,ロボットづくりの腕に 覚えのある研究者・技術者は,ぜひ,材料科学者の話を 聴きに行ってほしい.これらの分野の融合によって,自 動実験システムの構築,および実験結果のビッグデータ が構築されることを切に期待している.
3.
化合物をどう記述するか
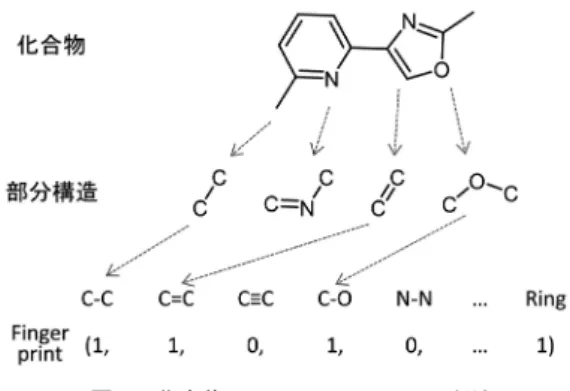
さて,材料の特性に関するデータが蓄積されてきたら, 次は,その特性を予測する機械学習・深層学習モデルを 構築したい.材料の特性は,どのような材料をつくるか (What to make)に依存するのはもちろんのこと,どの ようにつくるか(How to make)にも依存する.そのた め,材料の特性を出力する機械学習モデルの入力には, 「材料そのもの」や「材料をつくる際の条件」が使われ ることとなる.これらの情報をどのような形で入力すれ ばよいのだろうか? 材料を数値化・文字列化する試み は,主にケモインフォマティクスと呼ばれる分野で長年 研究が積み重ねられてきた [Chen 06](数値化・文字列 化されたものを「記述子」と呼ぶ).本章では,材料科 学におけるさまざまな記述子を紹介する. 3・1 実験条件を記述子に 材料をどうつくるか(How to make)の問題におけ る記述子の設定は比較的簡単だ.材料をつくる際の条件 ─実験条件,つまり,反応時間・温度・圧力・添加剤の 有無─そのものが,数値で表現されるためである.例え ば,Dark Reaction Project の Norquist らは,さまざま な合成条件におけるバナジウムセレナイトの結晶生成実 験を行い,合成時の反応時間や添加する材料の基礎物性 値,加える添加剤の有無(0/1 表現)を記述子とした生 成物の結晶構造の学習器を構築することで,未検討の 合成条件に対する生成物の構造の予測に成功している [Raccuglia 16].Moliner らも,ゼオライトの合成条件 144に対する生成した構造の実験データを蓄積し,合成 条件に対するゼオライトの結晶化のしやすさや結晶構造 の学習器をつくることで,予測モデルの構築に成功して いる [Moliner 05].化学工学の分野においても,製造プ ラントの異常検出・ソフトセンサなど,幅広い分野に応 用されている [船津 14]. 3・2 構造式を元にした記述子 次に化合物そのものの記述子─つまり What to make の問題に利用する記述子─について議論する.化合物の 記述法として最も想像しやすいのは,合金であろう.合 金に含め得る全元素種に対する含有率を用いれば,さま ざまな合金を一次元のベクトルとして表現することが可 能である.実際に,合金を構成する元素種とその割合を 最適化することで,高活性な合金触媒を設計したという 報告が多数ある.実際に,この手法は多くの研究グルー プで用いられており,遺伝的アルゴリズムを用いて,元 素の含有率を最適化することで,安価なプロパンから他 の化合物へ変換する触媒 [Le 16] の高効率な設計を達成 している. 着目する化合物が分子の場合は,少し複雑になる. 図 2 に示す化合物(ベンゼン C6H6とシクロヘキサン C6H12)は,いずれも六つの炭素原子からなる環状の化 合物であり,構造式は似ているが,その立体構造・化学 的性質は大きく異なる.このような(ただの画像とは異 なる)化合物特有の記述の方法論は,古くから提案され 図 1 合金の一次元ベクトルによる記述 図 2 ベンゼン・シクロヘキサンの構造式と化学的性質ており,さまざまな流儀がある. この化学的性質の違いは,主に,原子どうしの結合に 関わる「電子の状態」の違いに由来する.そのため,化 学的性質の情報を簡便に記述子に落とし込む方法の一 つとして,原子や結合,部分構造の有無を(1, 0)で表 現する fingerprint が広く用いられている(図 3).部 分構造の定義の仕方は一意ではなく,部分構造の定義 の異なる fingerprint が多数開発されている.また,原 子間の結合をグラフとして取り扱う方法 [Takigawa 13, Yamashita 14]や,各原子の記述に周囲の原子の影響の 重みを含めて取り扱う方法,原子配置の三次元構造情報 を取り入れる方法 [Axen 17] など,さまざまな方法が考 案されている. 全く別の観点による分子の記述法に,文字列による表 現がある.特に,広く用いられるのが SMILES と呼ばれ る記述子である.これは,原子間の結合の種類および結合 のつながり方を表現したものであり,比較的単純なルー ルで決定できる.いくつか代表的なルールをあげよう. (1)元素記号を利用する.ただし水素原子は省略する. (2)構造式上で隣り合う原子は隣に書く. (3)原子間の結合の種類を記号で表す. (二重結合,三重結合はそれぞれ =,# で表すなど) (4)分岐は( )で表す. (5)環を形成する原子どうしに数字のラベルを付ける. 実際に,いくつかの類似分子の構造式と SMILES 表 現を図 4 に示す.ケモインフォマティクス以外の化学の 専門家が初めて SMILES 表現を目にすると,面食らう ことが多いのだが,しばらく眺めていると,構造式と文 字列が 1 対 1 に対応しているという感覚がつかめてく る.多くの化学の専門家は,分子の構造式を見て,さま ざまな化合物や化学反応のパターン認識を無意識のうち に行うことができるので(しかも,異なるバックグラウ ンドをもつ化学者どうしであっても,同じようにパター ンを認識している),構造式と 1 対 1 に対応している SMILES表現を用いても,同様のパターン認識が可能な はずだ.実際に,SMILES を入力表現とし,分子の毒性 などを予測する機械学習モデルが構築できたという例も 報告されている [Karwath 06].また,SMILES は機械 学習モデルの入力のためだけでなく,類似構造をもつ化 合物の検索や,新規化合物を自動生成するという目的で も広く利用されている [Maldonado 06]. このほかにも,さまざまな記述子が設計されており, その応用例も多数報告されている.ここでは紹介しきれ ないが,ケモインフォマティクスの分野で開発されて きた化合物の記述子については,この分野を牽引してき た Gastiger らの書籍で詳しく紹介されている [Gastiger 18].なお,化合物の記述子への変換を行うソフトウェ アは有償・無償を含め,すでに数多く公開されている. 3・3 量子化学計算の計算結果を記述子に 材料のデータには,他のデータにはない特徴がある. それは,データの裏に「物理法則がある」ことである. 例えば,化学反応の進みやすさは,反応の前・途中・後 の安定性によって決まるが,この安定性を決めるのが三 つの因子のバランス(化合物の中の電子の偏り,フロン ティア軌道のエネルギー差,立体反発)であることが知 られている.そのため,これらの三つの因子を反映する ような量を記述子に使えば,比較的理解しやすいモデル を構築できる可能性がある.このような量の算出を可能 にするのが,量子化学計算である.これらの物理量の計 算結果のデータベースは前述のとおり,最近急速に増え ていて,分子性化合物であれば PubChemQC に,固体 系材料であれば Materials Project や AFLOW にまとめ られている. また,特殊な化合物や特殊な記述子をターゲットとす る場合は,量子化学計算のソフトウェアを使って自前で データベースを構築することも可能だ.量子化学計算の 実行には,原子のXYZ座標の入力が不可欠である.この座 標の取得の自動化ソフトもある程度整備されている.例 えば,化合物データベース PubChem を用い,特定の 部分構造を含む化合物を検索し,SMILES 表記でデー タをダウンロードする.ここから,ケモインフォマティ クスの無償ソフトウェア(例えば OpenBabel [O’Boyle 11])を用いることで,SMILES から XYZ 座標に変換で きる.この方法を用いれば,ターゲットとする化合物群 のデータを自動的にためていくことが可能である. 図 3 化合物の fingerprint による記述 図 4 さまざまな分子の構造式と SMILES 表現
事例紹介 化合物の量子化学計算の結果を記述子に用い,実験 結果の予測器を構築した例に,Doyle らの研究がある [Ahneman 18].Doyle らは,特定の触媒反応に着目し, 反応物や触媒,添加剤の種類を変えることで,反応物の うち,何割が生成物に変換されるか(収率)を予測する システムの構築を目指した.反応系に含まれる化合物の さまざまな物理量(分子量や電荷,NMR シフト,分子 振動の情報など)を量子化学計算によって求め,これら を説明変数,反応の収率を目的変数とした学習を行った ところ,ランダムフォレスト回帰を用いたときに,特に 高い精度で収率を予測できることを見いだした.この回 帰において重要度の高い説明変数は,添加剤の特定の部 分の NMR シフト値や電荷,軌道エネルギーなどであっ た(この論文には問題もあり,後に反論 [Chuang 18, Estrada 18]も出版されたのだが,化学反応のデータを 自ら集め,機械学習による予測が可能なのかを検証した 最初の論文であり,化学におけるインフォマティクスの 現状・課題を示す良い例であるため,ここで紹介するこ ととした). ここからは化学者の腕の見せどころである.重要な記 述子を総合的に考えると,求電子的な性質をもつ添加剤 を用いるときに,特に収率に影響を及ぼすという傾向が あると捉えることができる.Doyle らはこの情報から, 添加剤が触媒と反応してしまうことで,触媒反応を妨げ ているのではないかと仮説を立てた.実際に,サンプル の中で最も求電子性の高いものを添加剤として用いる と,触媒と添加剤による副生成物が得られることが確認 された.このように,メカニズムがわからない状態であっ ても,大量のデータを集めれば,メカニズムの予想に役 立つパラメータを抽出できる可能性がある. さて,この例からわかるとおり,材料に関わるデータ には,何かしらの物理法則やメカニズムに関わる情報が 内在している.メカニズムに関わる情報がなくても,適 切な学習方法と研究者の科学的センスを生かすことで, メカニズムに関わる情報を発見できる可能性があること を表している.しかし,あらかじめ反応のメカニズムに 関わる情報が少しでもあれば,もっと少ないデータ量か らでも生成物の収率の予測器を構築することが可能だっ たかもしれない. 3・4 化合物は動く・変化する メカニズムに直接的に関わる情報をもっと積極的に記 述子に含めるためにはどうすればよいだろうか? 着目 する材料の特性ごとに考慮すべき点は異なるが,化学反 応に限っていえば,ここまで紹介してきた記述子には決 定的に欠けている視点がある.それは,反応の前後で化 合物の構造や結合パターンが変化するという点である. 前節の触媒反応の例では,触媒と添加剤が副生成物に変 化するという情報が事前になかったわけだが,もしも, 副生成物への変化のしやすさに関わるパラメータが明示 的に含まれていれば,より解釈しやすいモデルになるか もしれない. 変化のしやすさ─つまり反応のしやすさ─を表すパラ メータとは何か? それは,化学反応の進行に伴う化合 物の構造(各原子の XYZ 座標)やエネルギー(安定性) の変化の情報だ.このような反応前後,または反応途中 の情報を効率良く取得できれば,メカニズムの情報を内 在する記述子を上手に選ぶことができるはずだ. しかし,化合物がどのように構造変化し得るか・ど のような結合の組換えが起こるかの情報を集めるのは難 しい.化合物の中の原子の座標やその安定性を調べるシ ミュレーションは存在するが,ほとんどの場合,化学反 応を観ることができない.なぜなら,化学反応は非常に まれな現象で,とても遅い時間スケールでしか進まない ことが多いため,現実的に実行可能なシミュレーション の時間の中ではけっして起こらないからである. 従来,どのように化学反応の様子を見ようとしてきた かというと,研究者が化学の直感を生かし,どのような 構造変化が起こり得るかを予測したうえで,それを元に 化合物内の原子の XYZ 座標を(半手動で)動かしなが らその安定性の変化を追うことで,その反応が起こり得 るかどうかを調べてきた(何という職人芸!). これを完全自動化する試みの中で最も成功したもの の一つが,反応経路自動探索(Global Reaction Route Mapping:GRRM)である [Maeda 13].詳細は割愛す るが,この方法は,反応前の情報を入力するだけで,自 動的に反応途中の構造変化や,安定性の変化を調べるこ とができるものである.特筆すべきことは,反応後の情 報を入力する必要がないことである(多くの自動探索を 謳う方法は,反応前と反応後の情報を入力して,その間 を自動探索している).そのため,一番多く得られる主 生成物に至る反応だけでなく,ほんの少ししか得られな い副生成物に至る反応の情報も余すことなく調べ上げる ことができるのだ.これを使えば,前節で述べた Doyle らの例でも,副生成物の得られやすさをあらかじめ入力 パラメータとして用意することも可能であろう. この方法は,比較的新しい方法であるため,これを駆 使した化学反応のメカニズムの解析は行われているが, この情報を記述子に用いた機械学習モデルの構築例は, まだほとんどない(我々のグループで現在,推進してい る最中である).このような反応の進みやすさに直結す る記述子─つまりメカニズムの根幹となるパラメータ─ を入力変数に入れることで,比較的解釈しやすい学習モ デルが構築できるのではないかと期待している.
4.お
わ り に
本稿では,化学分野におけるマテリアルズインフォ マティクスの現状について概説するため,材料に関するデータベースや材料を記述する方法について事例ととも に紹介した.後半部分では,解釈可能かどうかに重点を 置いていたが,これはマテリアルズインフォマティクス において,解釈可能かどうかが,最重要項目であると言 おうとしているわけではない.現時点で存在する材料の データに,サンプル数が少なく,偏りが大きいという問 題があるために,その弱点を補うべく,物理・化学の知 見を最大限に活用しようとしているのである.今後,材 料の合成や測定といった実験のさまざまな面が自動化さ れ,データが蓄積していけば,物理的・化学的解釈を伴 わないモデルでも,材料の特性を予測することが可能に なっていくと考えている.絨毯爆撃的に行われている材 料開発現場におけるさまざまな実験が,よりスマートに 進められるようになり,新しい材料の開発が加速される なら(例えそのメカニズムがわからなかったとしても), 材料科学にとって大きな進展であると思う.ただ,著者 は理論化学者なので,大量のデータの蓄積および材料 の特性の予測器の構築の先に,新しい物理法則・化学法 則を見いだすことを虎視眈々と狙いたいとも思っている.
◇ 参 考 文 献 ◇
[Ahneman 18] Ahneman, D. T., Estrada, J. G., Lin, S., Dreher, S. D. and Doyle, A. G.: Predicting reaction performance in C-N cross-coupling using machine learning, Science, Vol. 360, pp. 186-190(2018)
[Axen 17] Axen, S. D., Huang, S. P., Caceres, E. L., Gendelev, L., Roth, N. L. and Keiser, M. J.: A simple representation of three-dimensional molecular structure., J. Med. Chem., Vol 60, pp. 7393-7409(2017)
[Bale 09] Bale, C., Bélisle, E., Chartrand, P., Decterov, S., Eriksson, G., Hack, K., Jung, I-H., Kang, Y-B., Melançon, J., Pelton, A., Robelin, C. and Petersen, S.: FactSage thermochemical software and databases ̶ Recent developments, Calphad, Vol. 33, pp. 295-311(2009)
[Belsky 2002] Belsky, A., Hellenbrandt, M., Karen, V. L. and Luksch, P.: Inorganic Crystal Structure Database(ICSD): Accessibility in support of materials research and design, Acta
Crystallogr. B, Vol. 58, pp. 364-369(2002)
[Chen 2006] Chen, W. L.: Chemoinformatics: Past, present, and future, J. Chem. Inf. Model., Vol. 46, p. 2230-2255(2006) [Chuang 18] Chuang, K. V. and Keiser, M. J.: Comment on
“Predicting reaction performance in C-N cross-coupling using machine learning”, Science, Vol. 362, p. eaat8603(2018) [Curtarolo 12] Curtarolo, S., Setyawan, W., Hart, G. L. W.,
Jahnatek, M., Chepulskii, R. V., Taylor, R. H., Wang, S., Xue, J., Yang, K., Levy, O., Mehl, M. J., Stokes, H. T., Demchenko, D. O. and Morgan, D.: AFLOW: An automatic framework for high-throughput materials discovery, Comp. Mat. Sci., Vol. 58, pp. 218-226(2012)
[Estrada 18] Estrada, J. G., Ahneman, D. K., Sheridan, R. P., Dreher, S. D. and Doyle, A. G.: Response to comment on ‘Predicting reaction performance in C-N cross-coupling using
machine learning’ , Science, Vol. 362, pp. eaat8763(2018) [船津 14] 船津公人,金子弘昌:ソフトセンサー入門─基礎から実
用的研究例まで─,コロナ社(2014)
[現代化学編集グループ 18] 現代化学編集グループ:夏目徹博士 に聞く 実験の自動化が科学を変える,現代化学,Vol. 568, pp. 18-21(2018)
[Gómez-Bombarelli 18] Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling,
B., Sheberla, D., Aguilera-Iparraguirre, J. Hirzel, T. D., Adams, R. P. and Aspuru-Guzik, A.: Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent.
Sci., Vol. 4, pp. 268-276(2018)
[Granda 18] Granda, J. M., Donina, L., Dragone, V., Long, D.-L. and Cronin, L.: Controlling an organic synthesis robot with machine learning to search for new reactivity, Nature, Vol. 559, pp. 377-381(2018)
[Groom 16] Groom, C. R., Bruno, I. J., Lightfoot, M. P. and Ward, S. C.: The cambridge structural database, Acta. Cryst. B, Vol. 72, pp. 171-179(2016)
[Hachmann 11] Hachmann, J., Olivares-Amaya, R., Atahan-Evrenk, S., Amador-Bedolla, C., Sanchez-Carrera, R. S., Gold-Parker, A., Vogt, L., Brockway, A. M. and Aspuru-Guzik, A.: The Harvard Clean Energy Project: Large-scale computational screening and design of organic photovoltaics on the world community grid, J. Phys. Chem. Lett., Vol. 2, pp. 2241-2251 (2011)
[Jian 13] Jain, A., Ong, S. P., Hautier, G., Chen, S., Richards, W. D., Dacek, S., Cholia, S., Gunter, D., Skinner, D., Ceder, G. and Persson, K. A.: Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., Vol. 1, p. 011002(2013)
[Karwath 2006] Karwath, A. and Raedt, L. D.: SMIREP: Predicting chemical activity from SMILES, J. Chem. Inf.
Model., Vol 46, pp. 2432-2444(2006)
[Kim 19] Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., Li, Q., Shoemaker, B. A., Thiessen, P. A., Yu, B., Zaslavsky, L., Zhang, J. and Bolton, E. E.: PubChem 2019 update: Improved access to chemical data, Nucleic Acids Res., Vol. 47, p. D1102-D1109(2019)
[Krallinger 15] Krallinger, M., Leitner, F., Rabal, O., Oyrzabal, J. and Valencia, A.: CHEMDNER: The drugs and chemical names extraction challenge, J. Cheminf., Vol. 7, pp. S1(2015) [Krallinger 17] Krallinger, M., Rabal, O., Lourenco, A., Oyrzabal,
J. and Valencia, A.: Information retrieval and text mining technologies for chemistry, Chem. Rev., Vol. 117, pp. 7673-7761 (2017)
[Le 16] Le, T. C. and Winkler, D. A.: Discovery and optimization of materials using evolutionary approaches, Chem. Rev., Vol. 116, pp. 6107-6132(2016)
[MatNavi] MatNavi: https://mits.nims.go.jp/index.html [Maeda 13] Maeda, S., Ohno, K. and Morokuma, K.: Systematic
exploration of the mechanism of chemical reactions: the global reaction route mapping(GRRM) strategy using the ADDF and AFIR methods, Phys. Chem. Chem. Phys., Vol. 15, pp. 3683-3701(2013)
[Maldonado 06] Maldonado, A. G., Doucet, J. P., Petitjean, M. and Fan, B. T.: Molecular similarity and diversity in chemoinformatics: From theory to applications, Mol. Divers., Vol. 10 pp. 39-79(2006)
[Moliner 15] Moliner, M., Serra, J. M., Corma, A., Argente, E., Valero, S. and Botti, V.: Application of artificial neural networks to high-throughput synthesis of zeolites, Micropor.
Mesopor. Mat., Vol. 78, pp. 73-81(2005)
[Nakata 17] Nakata, M. and Shimazaki, T.: PubChemQC Project: A large-scale first-principles electronic structure database for data-driven chemistry, J. Chem. Inf. Model., Vol. 57, pp. 1300-1308(2017)
[O’Boyle 11] O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T. and Hutchison, G. R.: Open Babel: An open chemical toolbox, J. Cheminfo., Vol. 3, p. 33(2011)
[Parra-Cabrera 18] Parra-Cabrera, C., Achille, C., Kuhn, S. and Ameloot, R.: 3D printing in chemical engineering and catalytic technology: structured catalysts, mixers and reactors, Chem.
Soc. Rev., Vol. 47, pp. 209-230(2018)
[Rossi 17] Rossi, S., Puglisi, A. and Benaglia, M.: Additive manufacturing technologies: 3D printing in organic synthesis,
ChemCatChem., Vol. 10, pp. 1512-1525(2018)
[Raccuglia 16] Raccuglia, P., Elbert, K. C., Adler, P. D. F., Falk, C., Wenny, M. B., Mollo, A., Zeller, M., Friedler, S. A., Schrier,
J. and Norquist, A. J.: Machine-learning-assisted materials discovery using failed experiments, Nature, Vol. 533, pp. 73-76 (2016)
[Swain 16] Swain, M. C. and Cole, J. M.: ChemDataExtractor: A toolkit for automated extraction of chemical information from the scientific literature, J. Chem. Inf. Model., Vol. 56, pp. 1894-1904(2016)
[Takigawa 13] Takigawa, I. and Mamitsuka, H.: Graph mining: Procedure, application to drug discovery and recent advances,
Drug Discovery Today, Vol. 18, pp. 50-57(2013)
[White 02] White, P. S., Rodgers, J. R. and Page, Y. L.: CRYSTMET: A database of the structures and powder patterns of metals and intermetallics, Acta Crystallogr., Sect.
B: Struct. Sci., Vol. 58, pp. 343-348(2002)
[Yamashita 14] Yamashita, H., Higuchi, T. and Yoshida, R.: Atom environment kernels on molecules, J. Chem. Inf. Model., Vol. 54 pp. 1289-1300(2014) 2019年 3 月 25 日 受理