1.8.2 添付文書(案)設定根拠 ボリコナゾールはカンジダ属,アスペルギルス属,クリプトコックス属に対して極めて高い抗 真菌活性を有し,また,まれな真菌であるフサリウム属やスケドスポリウム属に対しても抗真菌 活性を有する広域スペクトルのトリアゾール系抗真菌剤である。ボリコナゾールの剤型は経口剤 と注射剤があり,経口剤は安定して生物学的利用率が高いため(96%),医師の判断で注射剤か ら経口剤へスイッチすることにより不要な医学的侵襲を減らすことができ,また医療費の削減が 可能である。さらに,ボリコナゾールは髄液を含む全身の組織移行性に優れる。 ボリコナゾールは,2002 年より米国・欧州を含む諸外国で承認され,重症又は難治性の深在 性真菌症治療に使用されている。特に致死的な疾患であるアスペルギルス症に対して,40 年以 上にわたる標準薬とされてきたアムホテリシン B を上回る有効性・安全性成績が確認されたため, 第 1 選択薬として承認されている。また,本邦・外国とも極めてまれな日和見感染症ではあるも のの,既存薬では難治で,かつ,死亡率が高いため新たな薬剤が強く必要とされてきたフサリウ ム症とスケドスポリウム症に対して効能を有する世界で唯一の薬剤である。 なお,本剤はスイッチ投与できることが有用性のひとつである。したがって,経口剤(ブイフ ェンド錠)と注射剤(ブイフェンド静注用)の添付文書(案)の内容は剤型特異的な点及び食道 カンジダ症の使用を除き,できる限り同じとした。 以下では,(1)効能・効果(案),(2)用法・用量(案),(3)使用上の注意(案)について設 定根拠を述べる。 (1) 効能・効果(案)及びその設定根拠 以下ではまず,本剤の in vitro 感受性試験成績及び組織移行性について論じ,その後国内及び 外国臨床試験成績に基づいて,効能・効果(案)における対象疾患の設定の根拠を述べる。 1) 効能・効果(案)対象疾患における原因真菌のボリコナゾール感受性 in vitro 試験成績 ボリコナゾールはフルコナゾール低感受性あるいは非感受性の C. glabrata 及び C. krusei を含 むカンジダ属,アスペルギルス属,クリプトコックス属に対して極めて高い抗真菌活性を有する。 また,まれな真菌であるフサリウム属やスケドスポリウム属に対しても活性を有する。表 1 及 び表 2 は,それぞれ国内臨床分離保存株(1995∼1997 年度分離)及び国内第 3 相試験における 分離株(1999 年∼2002 年分離)に対するボリコナゾールの MIC を示す。なお,ボリコナゾール は MIC の約 2 倍の濃度で,アスペルギルス属に対して殺真菌的作用を示す。
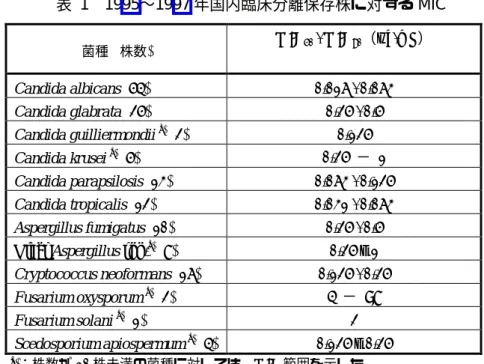
表 1 1995∼1997 年国内臨床分離保存株に対する MIC 菌種 (株数) MIC50 / MIC90(µg/mL) Candida albicans (54) 0.016 / 0.063 Candida glabrata (25) 0.25 / 0.5 Candida guilliermondii a) (2) 0.125 Candida krusei a) (5) 0.25 − 1 Candida parapsilosis (13) 0.063 / 0.125 Candida tropicalis (12) 0.031 / 0.063 Aspergillus fumigatus (10) 0.25 / 0.5 Other Aspergillus spp.a) (8) 0.25 – 1 Cryptococcus neoformans (16) 0.125 / 0.25 Fusarium oxysporum a) (2) 4 − >8 Fusarium solani a) (1) 2 Scedosporium apiospermum a) (4) 0.125 – 0.25 a):株数が 10 株未満の菌種に対しては,MIC 範囲を示した。 99M-109,496-MIC-1 最終報告書 Table1-2 より引用 表 2 国内第 3 相試験における分離株に対する MIC 臨床分離菌 分離 株数 MIC(µg/mL) 株数 0.25 7 0.5 3 A. fumigatus 14 1 4 アスペルギルス属 A. niger 1 0.5 1 ≦0.0039 1 0.0078 3 0.016 2 0.031 3 0.063 1 C. albicans 11 0.125 1 0.063 1 0.125 4 C. glabrata 6 0.25 1 C. guilliermondii 1 0.063 1 C. krusei 1 0.25 1 0.031 1 カンジダ属 C. parapsilosis 2 0.063 1 0.063 1 0.125 2 クリプトコックス属 C. neoformans 4 0.25 1 フサリウム属 Fusarium sp. 1 4 1 A1501001 総括報告書 Table 5.11.1 より引用 国内第 3 相試験で測定された各患者のボリコナゾール平均血中濃度は,平均値 3.4 µg/mL (SD=1.8 µg/mL),中央値 2.9 µg/mL(範囲 0.6 µg/mL∼8.4 µg/mL)であった。本血中濃度と上述の MIC を比較すると,ボリコナゾールはカンジダ属,アスペルギルス属,クリプトコックス属,フ サリウム属,スケドスポリウム属による深在性真菌症に対して臨床効果が期待できると考えられ
る。 2) ボリコナゾールの組織移行性 ボリコナゾールは髄液も含めた組織移行性に優れた薬剤である。 ポピュレーション PK 解析から,ボリコナゾールの定常状態における分布容積は 4.6 L/kg と推 定され,極めて高い組織移行性が示唆される。 通常薬剤移行性が最も問題になる部位として,髄液がある。外国人患者において,ボリコナゾ ール投与後 1∼10 時間のボリコナゾールの血漿中濃度と髄液中濃度の関係を検討した。血漿中ボ リコナゾール濃度に対する髄液中ボリコナゾール濃度の比の範囲は 0.22∼1.0(比の中央値 0.46)であり優れた髄液移行性が確認された。 また,外国において治験責任医師より依頼があった場合に,生検又は剖検からの組織及び髄液 サンプルにおけるボリコナゾールの組織内及び髄液中濃度が測定されている。外国人患者での肝 臓,腎臓,胃,肺,心臓,脳,脾臓,リンパ節,眼球硝子体,髄液,膵臓,結腸,小腸,食道及 び甲状腺の組織サンプルの解析において,いずれの組織においてもボリコナゾール濃度は 1000 ng/mL(又は 1000 ng/g)以上であり,主要真菌に対する最小発育阻止濃度を上回っていた。 以上から,ボリコナゾールは髄膜炎及び脳感染症を含む広範囲の感染部位における深在性真菌 症において臨床効果が期待できると考えられる。 3) 臨床試験有効性成績 効能・効果(案)の根拠となる国内及び外国臨床試験有効性成績を表 3 に示す。なお,部位 がその他(播種性,椎間板,リンパ節,膝関節,肝,口腔咽頭,骨,気管支,縦隔,角膜,脾, 耳,前立腺炎)に分類された症例は除いて集計した。 表 3 国内・外国臨床試験有効性成績 開発相 (治験№) 有効例/症例 菌種 疾患名 国内第3相 (1501001) 外国第3相 (150-307/602) 外国第3相 (150-309/604) 外国第3相 (150-608) カンジダ属 カンジダ血症 食道カンジダ症 カンジダ腹膜炎 気管支・肺カンジダ症 カンジダ副鼻腔炎 1/2 5/5 4/4 1/1 - - - - - - 11/21 23/38 - 1/2 1/1 162/248 - - - - 小 計 11/12 (91.7%) - 36/62 (58.1%) 162/248 (65.3%) アスペルギ ルス属 侵襲性肺アスペルギルス症 慢性壊死性肺アスペルギル ス症 肺アスペルギローマ アスペルギルス膿胸 アスペルギルス脳感染症 アスペルギルス副鼻腔炎 アスペルギルス皮下感染症 10/16 3/5 14/18 1/1 - - - 67/119 - - - 1/2 2/7 1/1 40/86 - - - 4/12 5/9 4/7 - - - - - - - 小 計 28/40 (70.0%) 71/129 (55.0%) 53/114 (46.5%) -
開発相 (治験№) 有効例/症例 菌種 疾患名 国内第3相 (1501001) 外国第3相 (150-307/602) 外国第3相 (150-309/604) 外国第3相 (150-608) クリプトコ ックス属 クリプトコックス髄膜炎 肺クリプトコックス症 クリプトコックス胸膜炎 クリプトコックス脳感染症 クリプトコックス血症とクリ プトコックス髄膜炎の併発 クリプトコックス血症とク リプトコックス脳感染症の 併発 1/1 7/7 - - - - - - - - - - 1/4 - 0/1 3/9 1/2 1/1 - - - - - - 小 計 8/8 (100%) - 6/17 (35.3%) - フサリウム 属 フサリウム血症 フサリウム副鼻腔炎 フサリウム眼内炎 肺フサリウム症とフサリウ ム皮下感染症の併発 - - - - - - - - 1/2 1/1 2/4 0/1 - - - - 小 計 - - 4/8 (50.0%) - スケドスポ リウム属 スケドスポリウム脳感染症 肺スケドスポリウム症 スケドスポリウム皮下感染症 スケドスポリウム脳感染症 とスケドスポリウム皮下感 染の併発 スケドスポリウム血症 - - - - - - - - - - 0/3 1/2 2/2 0/1 0/1 - - - - - 小 計 - - 3/9 (33.3%) - 合 計 47/60 (78.3%) 71/129 (55.0%) 102/210 (48.6%) 162/248 (65.3%) (a) 効能・効果(案)対象疾患について 国内臨床試験における有効率はカンジダ血症 1/2,食道カンジダ症 5/5,カンジダ腹膜炎 4/4, 気管支・肺カンジダ症 1/1,カンジダ属全体で 11/12(91.7%),侵襲性肺アスペルギルス症 10/16, 慢性壊死性肺アスペルギルス症 3/5,肺アスペルギローマ 14/18,アスペルギルス膿胸 1/1,ア スペルギルス属全体で 28/40(70.0%),クリプトコックス髄膜炎 1/1,肺クリプトコックス症 7/7, クリプトコックス属全体で 8/8(100%)であり高い有効性成績が確認された。外国試験でもこれら の感染症に対して優れた有効性成績が確認された。 国内臨床試験ではスケドスポリウム属による感染症の症例は組み入れられなかった。スケドス ポリウム症は国内及び外国とも非常にまれな日和見感染症である。一方で既存薬では難治であり, 非常に重篤な疾患のため新たな薬剤に対する医療上の必要性は高い。スケドスポリウムに対して ボリコナゾールは非常に高い in vitro 活性を有する(国内臨床分離株 MIC=0.125∼0.25 µg/mL)。 また,外国臨床試験でスケドスポリウム症に対して 3/9(33.3%)の有効性成績が確認されている。 フサリウム属による感染症については,国内で 1 例のみ全身性フサリウムソラニ感染症が組み 入れられた(対象感染部位ではないため表 3 には含めていない)。本症例の総合効果は「無効」 であった。また,本症例の分離株に対するボリコナゾールの MIC は 4 µg/mL であり,真菌学的
効果は「消失」であった。フサリウム属に対するボリコナゾールの MIC は他の菌種と比較して 高いものの,外国臨床試験ではフサリウム症に対して 4/8(50.0%)の有効率が得られている。フサ リウム症は国内及び外国とも非常にまれな日和見感染症である。一方で既存薬では難治であり, 非常に重篤な疾患のため新たな薬剤に対する医療上の必要性は高い。 対象真菌に対するボリコナゾールの in vitro 活性及び臨床試験成績からカンジダ血症,食道カ ンジダ症,カンジダ腹膜炎,気管支・肺カンジダ症,侵襲性アスペルギルス症,肺アスペルギロ ーマ,慢性壊死性肺アスペルギルス症,クリプトコックス髄膜炎,肺クリプトコックス症,フサ リウム症,スケドスポリウム症を添付文書(案)における対象疾患とした。なお,食道カンジダ 症については,注射剤の使用経験がないため,注射剤の対象疾患から除外した。 4) スケドスポリウム属における効能・効果(案)設定根拠 (a) In vitro感受性試験成績 ボリコナゾールはスケドスポリウム属に対して高い抗真菌活性を示す。外国における感受性試 験(試験番号 DI/022/0)では,Scedosporium apiospermum に対するボリコナゾール,イトラコナ ゾール,フルコナゾール及びアムホテリシン B の MIC(幾何平均)はそれぞれ,0.39,1.56,25 及び 71 µg/mL であり,ボリコナゾールの高い抗真菌活性が確認されている注1)。また,1995∼
1997 年の国内臨床分離保存 Scedosporium apiospermum 4 株に対するボリコナゾールの MIC は 0.125∼0.25 µg/mL であり,外国と同様に高い抗真菌活性を示した。
(b) In vivo試験成績
外国における,in vivo 試験(試験番号 DI/09/98,試験番号 DI/002/98)では注2),免疫正常及び
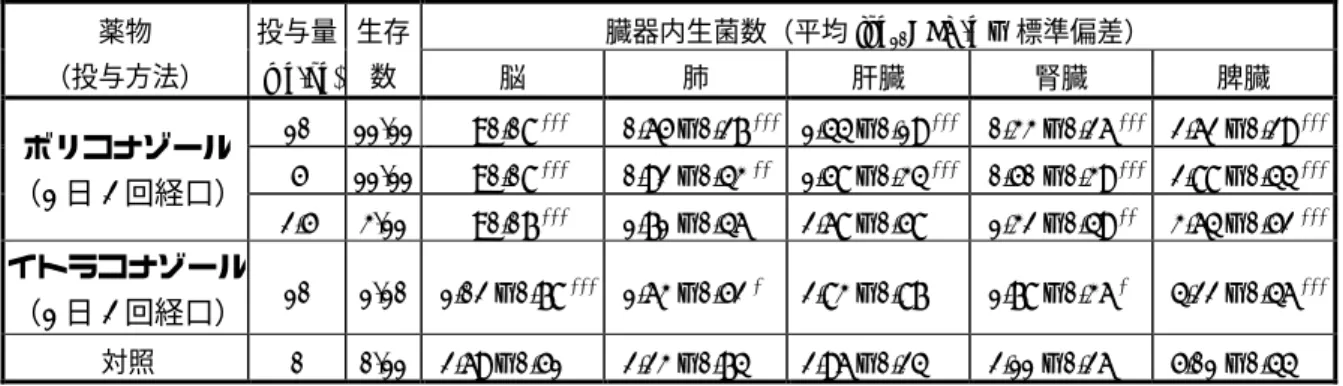
免疫抑制モルモットにおいて,Scedosporium apiospermum を静脈内投与して,播種性真菌症のモ デルを作成し,感染防御効果を検討した。以下にその成績を示す。 a) 免疫正常モルモットにおける播種性スケドスポリウム症(試験番号 DI/09/98) 免疫正常モルモットに S. apiospermum H76.03 を静脈内接種して全身感染(播種性スケドスポリ ウム症)モデルを作製し,薬物の感染防御効果を感染動物の生存数及び各種臓器内生菌数に基づ いて評価した。 ボリコナゾール投与群(2.5∼10 mg/kg 1 日 2 回 10 日間経口投与)では,脳,肺,肝臓,腎臓 及び脾臓内生菌数の減少並びに生存数の増加が認められた(表 4)。一方,イトラコナゾール投 与群(10 mg/kg 1 日 2 回 10 日間経口投与)では,脳,肺,腎臓及び脾臓内生菌数の減少が認め られたものの,肝臓内生菌数の変化は認められなかった。 注1)CTD 2.6.2.2 (1) 1) 感受性試験(試験番号 DI/022/0)の項参照。 注2)CTD 2.6.2.2 (2) 4) Scedosporium 感染モデルの項 p32∼33 参照。
表 4 免疫正常モルモットにおける全身感染(S. apiospermum H76.03) 臓器内生菌数(平均 log10CFU/g ± 標準偏差) 薬物 (投与方法) 投与量 (mg/kg) 生存 数 脳 肺 肝臓 腎臓 脾臓 10 11/11 <0.08 *** 0.65 ± 0.27 *** 1.44 ± 0.19 *** 0.33 ± 0.26 *** 2.62 ± 0.29 *** 5 11/11 <0.08 *** 0.92 ± 0.43 ** 1.58 ± 0.34 *** 0.50 ± 0.39 *** 2.88 ± 0.44 *** ボリコナゾール (1 日 2 回経口) 2.5 3/11 <0.07 *** 1.71 ± 0.46 2.68 ± 0.58 1.32 ± 0.49 ** 3.64 ± 0.52 *** イトラコナゾール (1 日 2 回経口) 10 1/10 1.02 ± 0.78 *** 1.63 ± 0.52 * 2.83 ± 0.87 1.78 ± 0.36 * 4.22 ± 0.46 *** 対照 0 0/11 2.69 ± 0.51 2.23 ± 0.74 2.96 ± 0.24 2.11 ± 0.26 5.01 ± 0.44 対照群との比較:* = P<0.05, ** = P<0.01, *** = P<0.001(対応のない t 検定) (出典;DI/09/98,Table 2 から抜粋) b) 免疫抑制モルモットにおける播種性スケドスポリウム症(試験番号 DI/002/98) シクロホスファミド(腹腔内投与)及びデキサメタゾン(経口投与)により免疫抑制を施した モルモットに,S. apiospermum H76.03 を静脈内接種して全身感染(播種性スケドスポリウム症) モデルを作製し,薬物の感染防御効果を各種臓器内生菌数に基づいて評価した。 ボリコナゾール投与群(1∼10 mg/kg 1 日 2 回 5 日間経口投与)では,用量依存的な脳,肺, 肝臓,腎臓及び脾臓内生菌数の減少が認められた(表 5)。これに対し,イトラコナゾール投与 群(1∼10 mg/kg 1 日 2 回 5 日間経口投与)及びアムホテリシン B 投与群(2.5 mg/kg 1 日 1 回 5 日間腹腔内投与)では,臓器内生菌数の減少がほとんど認められなかった。 表 5 免疫抑制モルモットにおける全身感染(S. apiospermum H76.03) 臓器内生菌数(平均 log10CFU/g ± 標準偏差) 薬物 (投与方法) 投与量 (mg/kg) 生存数 脳 肺 肝臓 腎臓 脾臓 10 11/11 <0.08 *** <0.63 * 2.20 ± 0.81 ** <0.84 ** 3.16 ± 0.70 ** 5 9/11 0.61 ± 0.39 ** 0.76 ± 0.56 * 2.42 ± 0.68 * 0.95 ± 0.55 ** 3.36 ± 0.69 ** ボリコナゾール (1 日 2 回経口) 1 7/11 2.85 ± 1.36 1.47 ± 0.88 2.85 ± 0.72 2.06 ± 0.84 4.09 ± 0.95 10 4/11 <2.66 <1.30 2.62 ± 0.72 2.05 ± 0.91 3.66 ± 1.14 5 4/11 3.33 ± 1.05 1.26 ± 0.95 2.93 ± 0.77 2.15 ± 0.85 4.37 ± 0.59 イトラコナゾール (1 日 2 回経口) 1 1/6 3.53 ± 0.77 0.63 ± 0.30 * 2.43 ± 0.26 2.24 ± 0.51 3.80 ± 0.38 アムホテリシン B (1 日 1 回腹腔内) 2.5 3/10 <2.09 1.53 ± 1.30 2.84 ± 1.01 1.67 ± 0.74 * 4.62 ± 0.90 対照 0 5/11 3.60 ± 1.24 1.50 ± 0.89 3.07 ± 0.51 2.48 ± 0.73 4.34 ± 0.56 対照群との比較:* = P<0.05, ** = P<0.01, *** = P<0.001(対応のない t 検定) (出典;DI/002/98,Table 3 から抜粋) 以上の非臨床試験成績より,ヒトにおける,スケドスポリウム属による真菌血症(播種性真菌 症)及び脳真菌症に対して,臨床効果が期待できると考えられる。
(c) 臨床における知見 臨床試験評価資料において,スケドスポリウム症の症例は外国における治験№ 150-309/604 よ り得られた 10 例のみである。これら 10 例の詳細を表 6 に示す。 表 6 治験№ 150-309/604 におけるスケドスポリウム症の一覧(MITT) 被験者 ID 性別 年齢 基礎疾患 感染部位 菌名 MIC(µg/mL) 総合効果 30901441181 男 58 腎移植 皮下 S. apiospermum 0.25 治癒 30901441182 男 62 肺移植 脳 S. apiospermum 0.125 悪化 30902031130 男 27 ALL 播種性 S. prolificans 4 悪化 30902031131 男 29 ALL 血液 S. prolificans 4 悪化 30902031132 男 70 AML 皮下 Scedosporium sp. N/A 改善 30902201701 女 27 肺移植 脳 S. apiospermum 0.25 悪化 30902251481 男 33 外傷 皮下・脳 S. apiospermum 0.25 −a) 60410056037 男 51 リンパ腫 脳 S. apiospermum N/A 悪化 60410406087 男 38 AML 肺 S. apiospermum 0.125 悪化 60410186026 女 46 CML 肺 Pseudallescheria boydiib) 0.125 改善 CTD 2.5.4 p69 表 35 より引用 a) 治験責任医師による判定がなく,無効として取り扱った。 b) S. apiospermum の有性生殖型 10 例中 3 例に臨床効果が確認されているが,これらの中には,効能・効果(案)に含まれる, 脳真菌症及び真菌血症(播種性真菌症)の有効例は含まれていない。 ボリコナゾールは脳・髄液移行に優れた薬剤であり,脳アスペルギルス症に対して治験№ 150-307/602,150-309/604 でそれぞれ 1/2,4/12 の有効性成績が得られている注1)ことから,脳ス ケドスポリウム症に対しても臨床効果が期待できる。 また,ボリコナゾールは十分な血漿中濃度が静注及び腸管吸収により得られ,また,カンジダ 血症に対して治験№ A1501001,150-309/604 及び 150-608(中間成績)でそれぞれ 1/2,11/21 及 び 23/31 の有効性成績が得られている注2)ことから,スケドスポリウム血症(播種性スケドスポ リウム症)に対しても臨床効果が期待できる。 以下では,ボリコナゾールにより治療が成功した脳スケドスポリウム症,スケドスポリウム 血症(播種性スケドスポリウム症)の症例を提示した。 a) 脳スケドスポリウム症 脳スケドスポリウム症をボリコナゾールで治療し,臨床効果が確認された症例は 2 文献で報告 されている。以下では,その要約を示す。 注1)CTD 1.8.2 (1) 3) 臨床試験有効性成績の項参照。 注2)CTD 1.8.2 (1) 3) 臨床試験有効性成績の項参照。
Thomas J. Walsh et al, Pediatric Infectious Disease Journal 2002; 21: 240-248
本文献は,小児のアスペルギルス症及びスケドスポリウム症をボリコナゾールで治療した成績 の報告である。脳スケドスポリウム症(文献上 CNS と記載されており,脳∼髄膜の感染と考え られる)5 例中,治癒 1 例,改善 2 例であり,全体として 60%(3/5)の有効率であった。
Mark A. Nesky et al, Clinical Infectious Disease 2000; 31: 673-677
本文献は,ボリコナゾールにより治療成功した,脳スケドスポリウム症(Pseudallescheria boydii(Scedosporium apiospermum の有性生殖型)による脳膿瘍)の 1 例報告である。
症例は腎移植後の免疫抑制剤投与中の 59 歳 男性であった。頭部 CT 所見及び膿瘍ドレナージ の培養結果から,確定診断に至っている。開頭ドレナージ後,アムホテリシン B リポソーム製剤 で治療開始されたが,神経所見等の臨床症状は悪化した。その後,ミコナゾール注射剤による治 療に変更し,臨床症状の悪化は見られなくなった。しかしながら 2 日間の治療で薬剤の在庫が尽 きたため,イトラコナゾール経口剤による治療に変更した。その結果,臨床症状は再度悪化し始 め,また,画像所見上も悪化が認められた。次に,ボリコナゾール注射剤による治療に移行した。 その結果,神経所見の改善が認められ,また,ボリコナゾール開始後 14 日目の画像所見では, 膿瘍の悪化は認められなかった。その後 2 回目の開頭ドレナージが施行され,さらに 14 日間ボ リコナゾール注射剤による治療を実施した。その後,長期にわたる経口ボリコナゾール治療に移 行している。ボリコナゾール治療を開始して 12 ヵ月後の時点で,臨床所見および MRI 所見とも, 脳膿瘍は認められないため,治療は終了した。
なお,本 Pseudallescheria boydii に対するボリコナゾールとミコナゾールの MIC は不明であり, アムホテリシン B,イトラコナゾールの MIC はそれぞれ,>16,0.25 µg/mL であった。
b) 播種性スケドスポリウム症
播種性スケドスポリウム症をボリコナゾールで治療し,臨床効果が確認された症例は 4 文献で 報告されている。以下では,その要約を示す。
Thomas J. Walsh et al, Pediatric Infectious Disease Journal 2002; 21: 240-248
本文献は,1)脳スケドスポリウム症で述べた小児での報告と同じものである。播種性スケド スポリウム症は 1 例で,本症例の臨床効果は治癒であった。
Corrado Girmenia et al, Journal of Clinical Microbiology 1998; 36: 1436-1438
本文献は,ボリコナゾール治療により効果が認められた播種性スケドスポリウム症の 1 例報告 である。 症例は化学療法不応の急性骨髄性白血病を基礎疾患に有する 25 歳の男性であった。抗菌剤不 応の発熱及び複数の顔面皮膚病変が初発所見の播種性スケドスポリウム症である。鼻粘膜ぬぐい 液及び皮膚病変の培養から,原因真菌は Scedosporium apiospermum であることが判明した。さら に,CT 所見から,Scedosporium apiospermum は,副鼻腔,肺,肝臓,脾臓,膵臓にも播種してい ることが判明した。アムホテリシン B リポソーム製剤で 7 日間治療したが,解熱せず,また,皮
膚病変の悪化が認められたため,ボリコナゾール注射剤による治療に移行した。ボリコナゾール による治療開始から数日で解熱し,また,皮膚病変の改善も認められ,治療は著効した。しかし ながら,ボリコナゾールによる治療開始から約1ヵ月後,消化管穿孔が原因と推測される消化管 出血により死亡した。
なお,本 Scedosporium apiospermum に対するボリコナゾール,アムホテリシン B の MIC はそ れぞれ,0.25,4 µg/mL であった。
Patricia Munoz et al, Clinical infectious Disease 2000; 31: 1499-1501
本文献は,ボリコナゾール治療により効果が認められた播種性スケドスポリウム症の 1 例報告 である。 症例は重度の喘息のため低用量のステロイド治療管理下にある 75 歳の女性であった。熱発, 胸痛,喀血,白血球数増加,胸部 CT 異常所見が認められ,病変組織の培養から,Scedosporium apiospermum による感染症であることが判明した。その後さらに真菌は播種し,多数の皮膚病変 も出現している。イトラコナゾールによる 7 日間の治療では解熱せず,さらに胸部 CT 所見も悪 化した。したがって,ボリコナゾールによる治療(注射剤 40 日間治療の後,経口剤 63 日間治療 へスイッチした)に変更された。治療開始から数日で臨床症状,皮膚病変,胸部画像所見は著し く改善しはじめた。治療開始から 8 ヵ月の時点で,スケドスポリウム症は再発していない。
なお,本 Scedosporium apiospermum に対するボリコナゾール,イトラコナゾールの MIC はそ れぞれ,0.12,2 µg/mL であった。
B.P Howden et al, European Journal of Clinical Microbiology and Infectious Disease 2003; 22: 111-113 本文献は,ボリコナゾールとテルビナフィンの併用治療により効果が認められた播種性スケド スポリウム症の 1 例報告である。 症例は多発性骨髄腫に対して化学療法の後,ミニ移植(nonmyeloablative BMT)を受けた 53 歳 の女性であった。右眼球の突出,MRI 上認められる副鼻腔病変,熱発が初発症状である。外科的 に病変を 3 回減圧し,病変組織の培養から Scedosporium prolificans による感染症であることが判 明した。本症は,イトラコナゾールによる治療と外科的減圧にて,一旦は軽快して治療を終了し ている。その後,真菌は播種し,Scedosporium prolificans による腰椎∼仙椎の椎間板・骨髄炎, 肝動瘤を形成している。これらに対して,イトラコナゾールとテルビナフィン併用による治療で は十分な効果は得られなかった。その後,経口ボリコナゾールとテルビナフィンによる併用治療 に移行した。15 ヵ月の経口ボリコナゾールとテルビナフィン併用治療で,腰痛は認められるも のの神経所見は消失し,MRI 所見上特に変化はなく,また,炎症所見も正常範囲内であった。 なお,本 Scedosporium prolificans に対するボリコナゾール,テルビナフィン,イトラコナゾー ルの MIC はそれぞれ,2,4,>16 µg/mL であった。また,テルビナフィンとボリコナゾールの 併用で相乗効果が認められ,併用時の MIC はボリコナゾール:0.008 µg/mL,テルビナフィン: 0.5 µg/mL であった。
(d) 結論 スケドスポリウム症は国内外ともに極めて稀な疾患である。しかしながら既存薬では十分な臨 床効果が得られず,新たな薬剤が必要とされている。非臨床における知見及び少数例ながら,以 上の脳スケドスポリウム症及び播種性スケドスポリウム症の臨床文献報告から,ボリコナゾール 効能・効果(案)における「スケドスポリウム属による脳真菌症及び真菌血症」は妥当と判断し ている。 5) 効能・効果(案) 効能・効果(案)を以下に示す。本剤の有効性・安全性プロファイルを考慮すると,深在性真 菌症のうちでも特に,重症又は難治性真菌感染症治療に本剤を投与した場合,得られるベネフィ ットはリスクを上回り,臨床的有用性は極めて高いと考えられる。 したがって効能・効果(案)には本剤の対象が重症又は難治性真菌症である旨を明示した。 また,カンジダ感染の治療については,米国及び EU の添付文書の記載内容を参考にし,他の 抗真菌剤が無効あるいは忍容性に問題があると考えられる場合に使用する旨,〈効能・効果に関 連する使用上の注意〉(案)に追記した。 食道カンジダ症に関しては,注射剤の使用経験がないため,注射剤の効能・効果(案)から除 外した。
経口剤 効能・効果(案) 下記の重症又は難治性真菌感染症 ・侵襲性アスペルギルス症,肺アスペルギローマ,慢性壊死性肺アスペルギルス症 ・カンジダ血症,食道カンジダ症,カンジダ腹膜炎,気管支・肺カンジダ症 ・クリプトコックス髄膜炎,肺クリプトコックス症 ・フサリウム症 ・スケドスポリウム症 〈効能・効果に関連する使用上の注意〉(案) 1. カンジダ感染の治療については,他の抗真菌剤が無効あるいは忍容性に問題があると考えら れる場合に本剤の使用を考慮すること。 注射剤 効能・効果(案) 下記の重症又は難治性真菌感染症 ・侵襲性アスペルギルス症,肺アスペルギローマ,慢性壊死性肺アスペルギルス症 ・カンジダ血症,カンジダ腹膜炎,気管支・肺カンジダ症 ・クリプトコックス髄膜炎,肺クリプトコックス症 ・フサリウム症 ・スケドスポリウム症 〈効能・効果に関連する使用上の注意〉(案) 1. カンジダ感染の治療については,他の抗真菌剤が無効あるいは忍容性に問題があると考えら れる場合に本剤の使用を考慮すること。
(2) 用法・用量(案)及びその設定根拠 用法・用量(案) 経口剤の用法・用量(案) 通常,成人(体重 40 kg 以上)にはボリコナゾールとして初日に 1 回 300 mg を 1 日 2 回,2 日目 以降は 1 回 150 mg 又は 1 回 200 mg を 1 日 2 回食間投与する。なお,症状に応じて又は効果不十 分の場合には,増量できるが,初日投与量の上限は 1 回 400 mg 1 日 2 回,2 日目以降投与量の上 限は 1 回 300 mg 1 日 2 回までとする。 また,体重 40 kg 未満の患者には,ボリコナゾールとして初日は 1 回 150 mg を 1 日 2 回,2 日目 以降は 1 回 100 mg を 1 日 2 回食間投与する。なお,症状に応じて又は効果不十分の場合には 2 日目以降投与量を 1 回 150 mg 1 日 2 回まで増量できる。 注射剤の用法・用量(案) 通常,成人にはボリコナゾールとして初日は 1 回 6 mg/kg を 1 日 2 回,2 日目以降は 1 回 3 mg/kg 又は 1 回 4 mg/kg を 1 日 2 回点滴静注する。 経口剤の用法・用量に関連する使用上の注意(案)に関する記述 経口投与が可能な患者,注射剤から投与を開始し経口投与が可能となった患者及び腎機能障害の ある患者で注射剤の投与ができない患者に対しては,錠剤を使用すること。 用法・用量に関連する使用上の注意(案)でのスイッチ投与に関する記述 注射剤からボリコナゾールの投与を開始した患者において,経口投与可能であると医師が判断し た場合は,錠剤に切り替えることができる。 用法・用量に関連する使用上の注意(案)での維持投与量の調整に関する記述 投与期間中は血中濃度をモニタリングすることが望ましい。(「その他の注意」の項参照) その他の注意での血中濃度モニタリングと維持投与量調整に関する記述 外国人患者において,ボリコナゾールの血中濃度と肝機能検査値異常発現率の間に統計的に有意 な関連性が認められた。日本人健康成人においては,肝機能障害が発生した症例で,かつ,血中 濃度が測定されていた症例の血中濃度トラフ値はいずれも 4.5μg/mL 以上であった。また,国内 臨床試験では有効性及び安全性に応じた投与量の調整に加え,目安としてトラフ血中濃度が 4.5 μg/mL 以上の場合,必要に応じて投与量を減量する血中濃度モニタリングを実施した。国内外 の臨床試験データからは肝機能検査値異常の出現を予測する血中濃度の閾値は認められていな い。 上記の用法用量(案)は,国内及び外国臨床試験成績に基づいて設定した。 以下では,1)2 日目以降の維持投与量設定根拠,2)初日負荷投与量設定根拠,3)注射剤か ら経口剤へのスイッチ投与の根拠,4)体重 40 kg 未満での経口剤投与量の調整の根拠,5)初期 維持投与量開始後の投与量の調整法の根拠,6)投与期間,7)食間投与の順で論じる。また,参 考として最後に 8)外国における承認用法・用量を提示した。
1) 2日目以降の維持投与量について (a) 国内第 3 相試験(治験№ A1501001)で用いられた用法・用量と有効性・安全性の解析 a) 治験実施計画書に規定された用法・用量 国内第 3 相試験では以下に示す用法・用量を治験実施計画書で規定した。 血 漿 中 濃 度 測 定 用 検 体 採 取 全症例 投与3日目朝投与前 経口投与量 静注投与量 静注療法b) 6 mg/kg bid 4 mg/kg bid 3 mg/kg bid 4 mg/kg bid 3 mg/kg bid 2.5 µg/mL未 満 3 mg/kg bid 4 mg/kg bid 3 mg/kg bid 2.5 µg/mL以上 重篤 な 真菌 症 その 他 投与調整なし
経口療法a) 300 mg bid 200 mg bid 200 mg bid
200 mg bid 150 mg bid 2.5 µg/mL未 満 2.5 µg/mL以上 1日目 2日目 3日目 4日目 5∼7日目(投与量調整) 負荷投与 維持投与 c) bid:1 日 2 回 a) 体重が 40 kg 未満の場合は,1 日目の負荷投与を 150 mg 1 日 2 回,2 日目以降の維持投与を 100 mg 1 日 2 回と した。 b) 静注療法を 3 日間行った後,治験責任医師により経口療法が可能と判断された被験者においては,静注療法か ら経口療法への切り替え(スイッチ療法)を可能とした。その場合の経口投与は,静注療法時の 3 日目の血漿 中ボリコナゾール濃度に応じて,2.5 µg/mL 未満の場合は 200 mg 1 日 2 回,2.5 µg/mL 以上の場合は 150 mg 1 日 2 回とした。 c) 2 日目以降に経口剤 200 mg 1 日 2 回及び,注射剤 4 mg/kg 1 日 2 回を選択された被験者については第 3 日目の 血中濃度に応じて投与量を調整して「初期維持投与量」を決定した。原則として全ての被験者について,経口 200 mg 1 日 2 回,経口 150 mg 1 日 2 回,経口 100 mg 1 日 2 回(体重 40 kg 未満),注射剤 4 mg/kg 1 日 2 回, 注射剤 3 mg/kg 1 日 2 回のいずれかの「初期維持投与量」が 5∼7 日目までには決定された。その後,有効性・ 安全性に加えて血中濃度を目安(トラフ値で 4.5 µg/mL を目安)にして 84 日目まで,治験責任医師の判断で維 持投与量は適宜増減できるものとした。ただし,経口剤の上限は 300 mg 1 日 2 回,注射剤の上限は 4 mg/kg 1 日 2 回とした。下限は設定しなかった。 治験実施計画書作成時,有効性上の必要最低血中濃度の目安を,主要真菌に対する MIC を上 回る 1 µg/mL とした。安全性上の最高血中濃度の目安を 6 µg/mL(トラフ値で約 4.5 µg/mL に相 当)とした。これは,治験実施計画書作成時に外国における第 1 相試験(治験№ 150-230)から 最高血中濃度が 6 µg/mL を超えると肝機能検査値異常発現頻度が上昇する可能性が示唆されたた めである。 健康被験者でのポピュレーション PK 解析データに基づくシミュレーションでは,上記の用
法・用量で本剤を投与すると,本剤の主要代謝酵素である CYP2C19 の遺伝子型にかかわらず, 全ての被験者で最低血中濃度は 1 µg/mL を上回り,最高血中濃度は 6 µg/mL を下回ると推定され た。 また,深在性真菌症は重篤な疾患であるため,負荷投与を行うことにより早期に血中濃度を定 常状態にすることが重要と考えられた。したがって,日本人における体重や第 1 相試験での忍容 性を考慮した上で,第 1 日目の負荷投与には経口剤 1 回 300 mg 1 日 2 回(体重 40 kg 未満の場合 は 150 mg 1 日 2 回),注射剤 1 回 6 mg/kg 1 日 2 回を用いた。 以上の用法・用量を用いると,初日負荷投与により早期に血中濃度は定常状態に達し,また, 3 日目の血中濃度に基づき 5∼7 日目までに決定される「初期維持投与量」のその後の反復投与 により,血中濃度は治験期間を通して CYP2C19 の遺伝子型にかかわらず 1 µg/mL から 6 µg/mL の間に維持できると推定された。 5∼7 日目に「初期維持投与量」が決定されてからは,有効性・安全性に加えて血中濃度を目 安にして,治験責任医師の判断で投与量を適宜増減できるものとした。血中濃度の目安として, トラフ血中濃度が 4.5 µg/mL 以上の場合は,必要に応じて投与量を減量することとした。 深在性真菌症の治療は通常長期に及ぶため,最長投与期間は 12 週間までとした。 b) 実際に用いられた用法・用量の解析 本試験では固定した用量が用いられたのではなく,被験者ごとに有効性・安全性に加え,血中 濃度に応じて投与量が適宜調整された。国内第 3 相試験で実際に用いられた用法・用量を以下に 示す。経口剤は体重 40 kg 以上及び 40 kg 未満で異なる用量を用いたため,解析結果は体重別に 分けて提示する。また,CYP2C19 遺伝子多型は,血中濃度を規定する重要な因子のひとつであ るため,可能な限り層別して解析結果を提示する。 100 例の症例ごとの投与量の推移(投与量調整時はその理由や血中濃度に関する情報を含む), CYP2C19 遺伝子型,体重,年齢,性別,DRC(データレビュー委員会)による最終診断名, DRC による総合効果を含む詳細な一覧表は 2.7.3 項付録に添付した。 体重 40 kg 以上の患者 体重 40 kg 以上の患者 86 例について,2 日目から 5∼7 日目までに用いられた 1 回用量,及び 3 日目の血中濃度に応じて 5∼7 日目に決定された初期維持投与量(1 回用量)を表 7 に示す。 また,初期維持投与量決定以降の平均 1 回投与量を表 8 に示す。 5∼7 日目に決定された初期維持投与量としては,3 日目の血中濃度に基づく調整の結果,150 mg 1 日 2 回及び 3 mg/kg 1 日 2 回が選択された患者が多かった。また,初期維持投与量決定以降 の投与量の調整の結果,EM 及び HEM では同量を継続,あるいは増量する傾向,PM では減量す る傾向が認められた。
表 7 体重 40 kg 以上の患者において選択された初期維持投与量(N=86) 1 回投与量 2 日目から 5∼7 日目(例数) 5∼7 日目の初期維持投与量決定時a)
(例数) CYP2C19 遺伝子型 EM HEM PM 不明 計 EM HEM PM 不明 計
経口 300 mg 0 0 0 0 0 1 0 0 0 1 経口 250 mg 0 0 0 0 0 0 0 0 1 1 経口 200 mg 16 23 10 5 54 3 13 1 1 18 経口 150 mg 0 0 0 0 0 12 6 8 4 30 経口 100 mg 1 0 0 0 1 1 2 1 0 4 注射 4 mg/kg 5 5 1 5 16 2 1 0 0 3 注射 3.5 mg/kg 0 0 0 0 0 0 1 0 0 1 注射 3 mg/kg 5 4 4 2 15 6 6 3 5 20 注射 2 mg/kg 0 0 0 0 0 0 1 1 1 3 合計 27 32 15 12 86 25 30 14 12 81 2.7.3.付録(1)Table 5.1 より引用 a)86 例中,5 例は 5∼7 日までに投与を終了・中止した。 表 8 体重 40 kg 以上の患者における初期維持投与量決定以降の平均 1 回投与量(N=81) 投与量 投与量 投与量(中央値) 例数 最小 最大 EM HEM PM 不明 全体 注射 28a) 1.6 mg/kg 4.0 mg/kg 3.0 mg/kg 3.0 mg/kg 2.6 mg/kg 3.0 mg/kg 3.0 mg/kg 経口 66a) 57.7 mg 300.0 mg 150.0 mg 200.0 mg 108.9 mg 150.0 mg 150.0 mg 2.7.3.付録(1)Table 5.2 より引用 a)初期維持投与量決定時には注射剤であった 27 例中 12 例は,その後経口剤へスイッチした。また,初期維持投 与量決定時には経口剤であった 54 例中 1 例は,その後注射剤へスイッチした。 体重 40 kg 未満の患者 体重 40 kg 未満の患者 14 例について,2 日目から 5∼7 日目までに用いられた 1 回用量,及び 3 日目の血中濃度に応じて 5∼7 日目に決定された初期維持投与量(1 回用量)を表 9 に示す。 また,初期維持投与量決定以降の平均 1 回投与量を表 10 に示す。 初期維持投与量として 100 mg 1 日 2 回及び 3 mg/kg 1 日 2 回が多くの場合選択された。 表 9 体重 40 kg 未満の患者において選択された初期維持投与量(N=14) 1回投与量 2 日目から 5∼7 日目(例数) 5∼7 日目の初期維持投与量決定時a)(例数) CYP2C19 遺伝子型 EM HEM PM 不明 計 EM HEM PM 不明 計
経口 200 mg 0 1 0 0 1 0 0 0 0 0 経口 150 mg 0 0 0 0 0 0 0 0 1 1 経口 100 mg 0 4 1 2 7 1 3 0 1 5 経口 50 mg 0 0 0 0 0 0 1 0 0 1 注射 4 mg/kg 1 1 1 0 3 0 1 0 0 1 注射 3 mg/kg 0 2 1 0 3 0 2 1 0 3 合計 1 8 3 2 14 1 7 1 2 11 2.7.3.付録(1)Table 5.1 より引用 a) 14 例中,3 例は 5∼7 日までに投与を終了・中止した。
表 10 体重 40 kg 未満の患者における初期維持投与量決定以降の平均 1 回投与量(N=11) 投与量 投与量 投与量(中央値) 例数 最小 最大 EM HEM PM 不明 全体 注射 4a) 2.1 mg/kg 4.0 mg/kg - 3.0 mg/kg 3.0 mg/kg - 3.0 mg/kg 経口 11a) 50 mg 163.5 mg 142.5 mg 100.0 mg 100.0 mg 156.8 mg 100.0 mg 2.7.3.付録(1)Table 5.2 より引用 a) 初期維持投与量決定時には注射剤であった 4 例は全例その後経口剤へスイッチした。 要約と考察 国内第 3 相試験において,第 3 日目の血中濃度に応じて調整された,初期維持投与量は多くの 場合,経口剤は 1 回 150 mg 1 日 2 回(体重 40 kg 未満の場合は 100 mg 1 日 2 回),注射剤は 1 回 3 mg/kg 1 日 2 回であった。 第 3 日目の血中濃度に応じた投与量調整に基づき設定された初期維持投与量として経口剤 200 mg 1 日 2 回及び,注射剤 4 mg/kg 1 日 2 回が選択された被験者はそれぞれ 18 例,4 例であった。 これらの症例の CYP2C19 遺伝子型はそれぞれ,経口剤 EM3 例,HEM13 例,PM1 例,遺伝子型 不明 1 例,注射剤 EM2 例,HEM2 例であった。 5∼7 日目に初期維持投与量が決定された後,さらに有効性・安全性に加え,血中濃度も目安 にして治験責任医師による投与量の調整がなされた結果,EM 及び HEM では同量が用いられる か増量される傾向にあり,また,PM では減量される傾向が認められた。 第 1 日目から第 84 日目までを通し,用法・用量(案)における最高用量である経口剤の 300 mg 1 日 2 回,及び注射剤の 4 mg/kg 1 日 2 回投与が 7 日間以上行われた症例はそれぞれ 5 例 (EM2 例,HEM2 例,遺伝子型不明 1 例),7 例(EM2 例,HEM3 例,PM1 例,遺伝子型不明 1 例)であった。また,体重 40 kg 未満の被験者で維持投与量を増量し,150 mg 1 日 2 回投与が 7 日以上行われた症例は 3 例(EM1 例,HEM1 例,遺伝子型不明 1 例),200 mg 1 日 2 回投与が 7 日以上行われた症例は 1 例(遺伝子型不明)であった。 初期維持投与量として 5∼7 日目に経口 150 mg 1 日 2 回あるいは注射 3 mg/kg 1 日 2 回が選択 された患者が多かった理由は,CYP2C19 遺伝子型を問わず,第 3 日目の血中濃度が 2.5 µg/mL を超えた被験者が多かったためである(図 1)。治験実施計画書作成時の健康被験者でのシミュ レーションでは,PM の場合には第 3 日目の血中濃度は 2.5 µg/mL を超え,他の遺伝子型の被験 者の第 3 日目の血中濃度は 2.5 µg/mL を下回ると考えられた。PM に経口剤1回 200 mg 1 日 2 回 及び注射剤 1 回 4 mg/kg 1 日 2 回投与を続けると血中濃度が著しく上昇し,有害事象の発現頻度 が高くなる可能性が予想された。そのような状態を避けるため,第 3 日目の血中濃度を CYP2C19 遺伝子型の指標とし,PM と考えられる患者では 200 mg 1 日 2 回は 150 mg 1 日 2 回に 減量し,4 mg/kg 1 日 2 回は 3 mg/kg 1 日 2 回に減量して初期維持投与量とすることとした。 しかしながら深在性真菌症患者では EM や HEM でも血中濃度が 2.5 µg/mL を上回る被験者が 多く認められ,それらの被験者では 200 mg 1 日 2 回は 150 mg1 日 2 回に減量され,4 mg/kg 1 日 2 回は 3 mg/kg 1 日 2 回に減量されて初期維持投与量とされた。これは健康被験者と異なり深在 性真菌症患者では CYP2C19 以外の血中濃度に影響を与える複雑な背景因子があるためと考えら れ,その結果多くの患者で血中濃度は 2.5 µg/mL を上回り,経口 150 mg 1 日 2 回又は,注射 3
mg/kg 1 日 2 回が選択されたものと考えられる。第 3 日目の血中濃度が 2.5 µg/mL を上回るか否 かは CYP2C19 遺伝子型を判定する指標にはならなかった。 図 1 CYP2C19 遺伝子多型別第 3 日目トラフ血中濃度 箱ひげ図について:箱の下端,中央,上端の水平線は,それぞれ 25%点,50%点(中央値),75%点を 表す。垂直線(ひげ)は箱の両端から四分偏差(25%点から 75%点までの距離)の 1.5 倍以内で最も離 れた点まで引いている。 国内第 3 相試験では,第 3 日目及び第 8 日目にはトラフ血中濃度が測定されており,また,そ の後もランダムに血中濃度が測定されている。本試験で実際に用いられた用量において,各患者 で測定された血中濃度の患者ごとの平均値を解析したところ,その平均値は 3.4 µg/mL (SD=1.8 µg/mL),中央値は 2.9 µg/mL(範囲 0.6 µg/mL∼8.4 µg/mL)であった。 c) 用量別有効性成績 国内第 3 相試験では,深在性真菌症治療において優れた有効性成績が確認されている。DRC により有効性評価対象例(PPS)とされた 65 例の有効率は 76.9% (50/65)であり,カンジダ症の 有効率は 91.7% (11/12),アスペルギルス症の有効率は 68.3% (28/41),クリプトコックス症の有 効率は 100% (8/8)であった。 PPS 65 例のうち,5∼7 日目の初期維持投与量決定までに治験を中止した 1 例を除く 64 例につ いて,5∼7 日目に決定された初期維持投与量別の有効性成績を表 11 に示す。なお,PPS のうち, 5∼7 日目の初期維持投与量決定までに投与を中止した 1 例の維持投与量は経口 1 回 200 mg 1 日 2 回が用いられた。本症例の DRC による総合効果判定は有効であり,因果関係を否定できない 有害事象のため 4 日目に投与が中止された。CYP2C19 遺伝子型は HEM であった。 初期維持投与量を決定した後,有効性・安全性・血中濃度に応じて治験責任医師の判断で投与 量を増減した結果,初期維持投与量及び CYP2C19 遺伝子型で有効性に明らかな違いは認められ Pl as ma c onc entr a ti on o f v o ri c onaz ol e (ug /mL ) 0 1 2 3 4 5 6 7 8 9 10 11 12 EM HEM PM Pl as ma c onc entr a ti on o f v o ri c onaz ol e (ug /mL ) 0 1 2 3 4 5 6 7 8 9 10 11 12 EM HEM PM 血漿中ボリコナゾール濃度( µg/ mL ) EM HEM PM Pl as ma c onc entr a ti on o f v o ri c onaz ol e (ug /mL ) 0 1 2 3 4 5 6 7 8 9 10 11 12 EM HEM PM Pl as ma c onc entr a ti on o f v o ri c onaz ol e (ug /mL ) 0 1 2 3 4 5 6 7 8 9 10 11 12 EM HEM PM 血漿中ボリコナゾール濃度( µg/ mL ) EM HEM PM
なかった。 表 11 初期維持投与量別有効性成績(PPS)a) 初期維持投与量 (1 回投与量) 有効率(%) EM HEM PM 不明 全体 経口 250 mg 0/0 0/0 0/0 1/1 1/1 経口 200 mg 3/3 10/10(100%) 1/1 1/1 15/15(100%) 経口 150 mg 7/8(87.5%) 4/4 6/7(85.7%) 0/2 17/21(81.0%) 経口 100 mg 1/2 3/4 0/0 0/1 4/7(57.1%) 経口 50 mg 0/0 0/1 0/0 0/0 0/1 注射 4.0 mg/kg 0/0 1/1 0/0 0/0 1/1 注射 3.5 mg/kg 0/0 1/1 0/0 0/0 1/1 注射 3 mg/kg 2/4 4/6(66.7%) 1/1 2/3 9/14(64.3%) 注射 2 mg/kg 0/0 1/1 0/1 0/1 1/3 合計 13/17(76.5%) 24/28(85.7%) 8/10(80.0%) 4/9(44.4%) 49/64(76.6%) 2.7.3.付録(1)Table 4.7 より引用 a ) 初期維持投与量決定までに治験を中止した 1 例を除く d) 用量別安全性成績 国内第 3 相試験では安全性評価対象例 100 例中,99 例(99.0%)に因果関係を問わない有害事象 が,78 例(78.0%)に因果関係を否定できない有害事象が認められた。 5∼7 日目の初期維持投与量決定までに 100 例中,有害事象により 7 例(因果関係を否定でき ない:4 例,因果関係なし:3 例),死亡(因果関係なし)により 1 例が投与を中止した。 残りの 92 例について,5∼7 日目に決定された初期維持投与量別の初期維持投与量決定後に発 現した因果関係を問わない有害事象,及び因果関係を否定できない有害事象の発現頻度をそれぞ れ表 12,表 13 に示す。因果関係を問わない有害事象は CYP2C19 遺伝子型及び初期維持投与量 で明らかな差は認められなかった。しかしながら,因果関係を否定できない有害事象は PM にお いて EM/HEM より発現頻度が高かった。 また,治験期間中に因果関係を否定できない重篤な有害事象が 3 例に発現しているが,この 3 例における CYP2C19 遺伝子型と重篤な有害事象発現時の 1 日投与量を表 14 に示す。
なお,症例ごとの投与量の推移と,肝に関する臨床検査値(AST, ALT, Al-P, γ-GTP, 総ビリル ビン)の推移の関係を表す一覧表を 2.7.3 項付録に添付した。
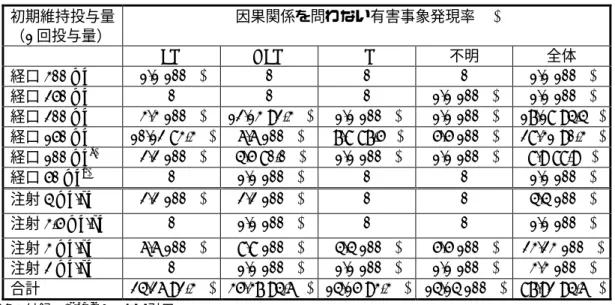
表 12 初期維持投与量別因果関係を問わない有害事象発現率 初期維持投与量 (1 回投与量) 因果関係を問わない有害事象発現率(%) EM HEM PM 不明 全体 経口 300 mg 1/1(100%) 0 0 0 1/1(100%) 経口 250 mg 0 0 0 1/1(100%) 1/1(100%) 経口 200 mg 3/3(100%) 12/13(92.3%) 1/1(100%) 1/1(100%) 17/18(94.4%) 経口 150 mg 10/12(83.3%) 6/6(100%) 7/8(87.5%) 5/5(100%) 28/31(90.3%) 経口 100 mga) 2/2(100%) 4/5(80.0%) 1/1(100%) 1/1(100%) 8/9(88.9%) 経口 50 mgb) 0 1/1(100%) 0 0 1/1(100%) 注射 4 mg/kg 2/2(100%) 2/2(100%) 0 0 4/4(100%) 注射 3.5 mg/kg 0 1/1(100%) 0 0 1/1(100%) 注射 3 mg/kg 6/6(100%) 8/8(100%) 4/4(100%) 5/5(100%) 23/23(100%) 注射 2 mg/kg 0 1/1(100%) 1/1(100%) 1/1(100%) 3/3(100%) 合計 24/26(92.3%) 35/37(94.6%) 14/15(93.3%) 14/14(100%) 87/92(94.6%) 2.7.3.付録(1)Table 6.1 より引用 a) 体重 40 kg 未満 5 例,体重 40 kg 以上 4 例 b) 体重 40 kg 未満 表 13 初期維持投与量別因果関係を否定できない有害事象発現率 初期維持投与量 (1 回投与量) 因果関係を否定できない有害事象発現率(%) EM HEM PM 不明 全体 経口 300 mg 1/1(100%) 0 0 0 1/1(100%) 経口 250 mg 0 0 0 0/1(0%) 0/1(0%) 経口 200 mg 2/3(66.7%) 6/13(46.2%) 1/1(100%) 1/1(100%) 10/18(55.6%) 経口 150 mg 5/12(41.7%) 4/6(66.7%) 6/8(75.0%) 4/5(80.0%) 19/31(61.3%) 経口 100 mga) 1/2(50.0%) 3/5(60.0%) 1/1(100%) 1/1(100%) 6/9(66.7%) 経口 50 mgb) 0 0/1(0%) 0 0 0/1(0%) 注射 4 mg/kg 0/2(0%) 1/2(50.0%) 0 0 1/4(25.0%) 注射 3.5 mg/kg 0 1/1(100%) 0 0 1/1(100%) 注射 3 mg/kg 3/6(50.0%) 5/8(62.5%) 3/4(75.0%) 3/5(60.0%) 14/23(60.9%) 注射 2 mg/kg 0 0/1(0%) 1/1(100%) 1/1(100%) 2/3(66.7%) 合計 12/26(46.2%) 20/37(54.1%) 12/15(80.0%) 10/14(71.4%) 54/92(58.7%) 2.7.3.付録(1)Table 6.2 より引用 a) 体重 40 kg 未満 5 例,体重 40 kg 以上 4 例 b) 体重 40 kg 未満
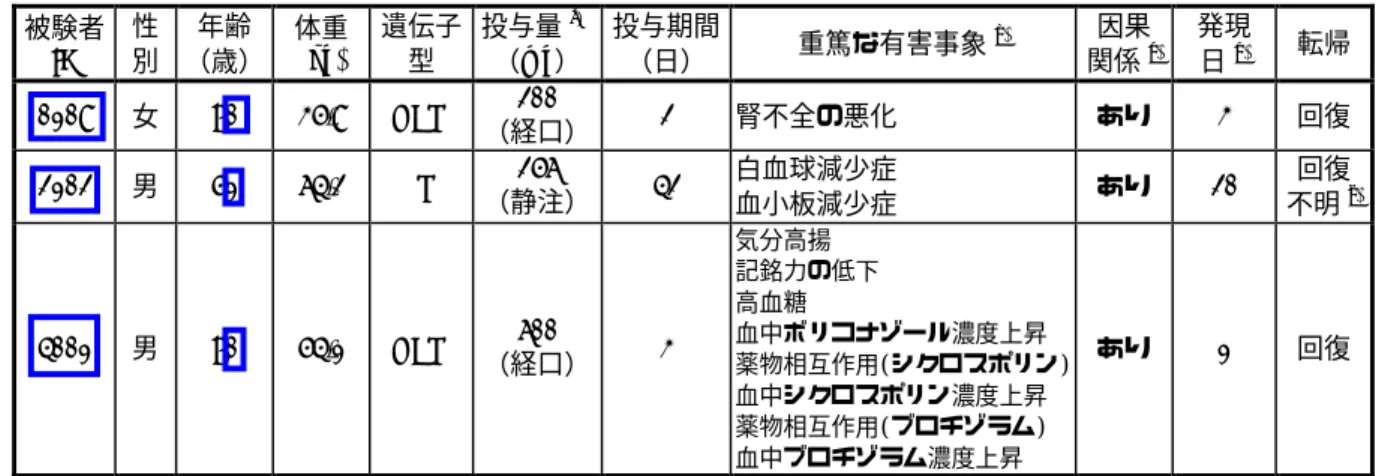
表 14 重篤な有害事象発現の原因として治験薬が「最も可能性が高い」と判断された症例 被験者 ID 性 別 年齢 (歳) 体重 (kg) 遺伝子 型 投与量a) (mg) 投与期間 (日) 重篤な有害事象 b) 因果 関係c) 発現 日d) 転帰 0108 女 70 35.8 HEM 200 (経口) 2 腎不全の悪化 あり 3 回復 2102 男 51 64.2 PM 256 (静注) 42 白血球減少症 血小板減少症 あり 20 回復 不明e) 4001 男 70 54.1 HEM 600 (経口) 3 気分高揚 記銘力の低下 高血糖 血中ボリコナゾール濃度上昇 薬物相互作用(シクロスポリン) 血中シクロスポリン濃度上昇 薬物相互作用(ブロチゾラム) 血中ブロチゾラム濃度上昇 あり 1 回復 a) 重篤な有害事象発現時の投与量又は発現前直近の1日投与量を示す。 b) 治験責任(分担)医師又は治験依頼者により重篤と判断された有害事象。 c) 治験薬と重篤な有害事象との因果関係。治験責任医師により,重篤な有害事象の原因として治験薬による可能 性が最も高いかどうかが判断された。 d) 投与開始日を 1 日目として算出した。 e) 基礎疾患の急性骨髄性白血病に対し化学療法を開始したため転帰は不明とされた。 e) 国内第 3 相試験成績で用いられた維持投与量に関する結論 国内第 3 相試験では,原則として初期維持投与量として経口剤 150 mg 又は 200 mg を 1 日 2 回 (体重 40 kg 未満の場合は 100 mg 1 日 2 回),注射剤 3 mg/kg 又は 4 mg/kg 1 日 2 回が用いられた。 第 3 日目の血中濃度に応じた投与量の調整の結果,初期維持投与量として経口剤は 150 mg 1 日 2 回(体重 40 kg 未満の場合は 100 mg 1 日 2 回),注射剤の場合は 3 mg/kg 1 日 2 回が選択された症 例が多かった。5∼7 日目の初期維持投与量決定後,有効性・安全性に加え血中濃度も目安にし て治験責任医師の判断で投与量はさらに増減された。その結果 PM では減量される傾向にあり, HEM/EM では同量が投与あるいは増量される傾向にあった。 CYP2C19 遺伝子型及び初期維持投与量を問わず,有効性に明らかな差は認められなかった。 一方,初期維持投与量決定以降の因果関係を否定できない有害事象発現率は PM で EM/HEM よ り高かった。 (b) 安全性上の投与量上限に関する考察 深在性真菌症は多くの場合,生命に影響する重篤な疾患である。したがって,得られるベネフ ィットとそのリスクを考慮した上で,生命に影響する重篤な症例ではできる限り高い用量を早期 に用いて確実な治療効果を得る必要性がある。その後,慎重に安全性の観察をして忍容性に問題 があれば漸減していくべきである。 深在性真菌症患者において,ボリコナゾール投与時の安全性プロファイルは薬剤曝露のみなら ず,基礎疾患,全身状態,併用薬,患者の特異体質等,複数の要因が複雑に関与していると考え られる。したがって患者ごとに安全性上の投与量の上限には差があると考えられ,全ての深在性 真菌症患者について説明できる一律の投与量の上限を示すことは限界があると考えられる。しか しながら,本剤を安全に投与できる上限の目安は,本邦第 1 相試験成績から推測できる。 本邦における市販用製剤を用いた第 1 相反復経口投与試験(治験№ A1501022)では,400 mg 1
日 2 回を第 1 日目に負荷投与した後,2 日目以降 200 mg を 1 日 2 回反復投与した場合の忍容性 は全ての CYP2C19 遺伝子型で確認され,投与中止例は認められなかった。300 mg 1 日 2 回反復 投与は CYP2C19 遺伝子型が HEM/EM の被験者に対してのみ行われたが,10 例中 3 例で肝機能 検査値異常が認められ,また,そのうち肝機能検査値異常のため 2 例が治験薬の投与を中止した。 以上より「日本人において安全に経口剤を投与できる用量の上限は概ね 1 回 200∼300 mg 1 日 2 回」と推測される。 本邦における注射剤を用いた第 1 相反復静脈内投与試験から,3 mg/kg 1 日 2 回反復投与の忍 容性は CYP2C19 すべての遺伝子型で確認され,投与中止例は認められなかった。また,4 mg/kg 1 日 2 回反復投与の忍容性は CYP2C19 遺伝子型が HEM/EM の被験者で検討され,中止例は認め られなかった。以上より「日本人において安全に注射剤を投与できる用量の上限は概ね 1 回 3 mg/kg∼4 mg/kg 1 日 2 回」と推測される。 (c) CYP2C19遺伝子型と維持投与量及び安全性の考察 健康被験者では,CYP2C19 遺伝子型により薬剤曝露は数倍の差があり,CYP2C19 遺伝子型は 安全性プロファイルに影響を与えると可能性があると考えられた。国内第 3 相試験でのポピュレ ーション PK 解析でも,CYP2C19 遺伝子型は患者の血中濃度に影響を与える重要な因子である ことが確認された(2.7.2.3 全試験を通しての結果の比較と解析参照)。深在性真菌症患者でも上 記の用量別安全性成績で述べたように CYP2C19 遺伝子型は曝露の差により安全性プロファイル に影響を与える因子のひとつと考えられる(2.7.4.5.1内因性要因参照)。 しかしながら,健康被験者と異なり,深在性真菌症患者の安全性プロファイルには薬剤曝露の みならず全身状態,基礎疾患,併用薬,患者の特異体質等の複数の因子が複雑に影響を与えるも のと考えられる。薬剤曝露についても健康被験者のように体重や CYP2C19 遺伝子型のみでなく, 患者の全身状態や併用薬などの複数の要因が影響すると考えられる。健康被験者でのシミュレー ションからは,国内第 3 相試験で第 3 日目のトラフ血中濃度は EM,HEM では 2.5 µg/mL を下回 ると推定されていたが,実際は 1 µg/mL を下回る者から 6 µg/mL を上回る者まで大きなばらつき が認められた(図 1)。したがって,深在性真菌症患者における安全性は,PM では他の遺伝子 型より血中濃度が高くなる可能性があり,また,PM では因果関係を否定できない有害事象の発 現頻度が高い傾向が認められたものの,CYP2C19 遺伝子型のみから一律に予測できるものでは なく,個々の患者ごとに違いがありうると考えられる。初期維持投与量を開始後,EM 及び HEM の患者についても PM の患者と同様に安全性モニタリングに加え血中濃度もモニタリング し,必要に応じて維持投与量の調整が必要と考えられる。 (d) 維持投与量に関する結論 患者の重篤度及び経口剤については体重も考慮して,初日の負荷投与後,初期維持投与量は, 経口剤は 1 回 150 mg 又は 1 回 200 mg を 1 日 2 回(体重 40 kg 未満の場合は 100 mg 1 日 2 回), 注射剤は 1 回 3 mg/kg 又は 1 回 4 mg/kg を 1 日 2 回が推奨される。初期維持投与量を開始後,有 効性及び安全性を十分に高めるためには,さらに患者ごとの安全性及び有効性のモニタリングに 加え,血中濃度も目安にして,投与量を担当医の判断で必要に応じて適宜増減することが必要で
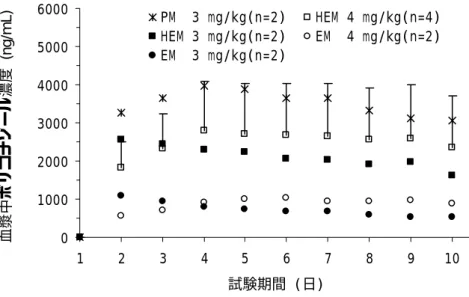
ある。ただし,増量時の経口剤の最高投与量は 1 回 300 mg 1 日 2 回,注射剤の最高投与量は 1 回 4 mg/kg 1 日 2 回までとする。なお,維持投与量の調整については後述の 5) 初期維持投与量 開始後の投与量の調整法についての検討で論じる。 2) 初日の負荷投与について 深在性真菌症は多くの場合,生命に影響し緊急性を要する重篤な疾患であり,できるだけ早く 薬剤血中濃度が定常状態に達することが有効性の観点から重要である。 国内第 1 相試験(治験№ A1501022)では,負荷投与量として第 1 日目に経口剤 400 mg 1 日 2 回,その後維持投与量として経口剤 200 mg を 1 日 2 回投与した。その結果,血中濃度は第 2∼3 日目にほぼ定常状態に達した(図 2)。本用法・用量での忍容性は良好であった。 図 2 治験№ A1501022 での血中濃度トラフ値の推移(経口反復) また,国内第 1 相試験(治験№ VRC-JP-97-501)では,負荷投与量として第 1 日目に注射剤 6 mg/kg 1 日 2 回,その後維持投与量として 3 mg/kg 又は 4 mg/kg を 1 日 2 回投与した。その結果, ボリコナゾールの血中濃度は第 2∼4 日目には定常状態に達した(図 3)。本用法・用量での忍容 性は良好であった。 0 2000 4000 6000 8000 1 2 3 4 5 6 7 試験期間(日) 血 漿中ボリ コナゾー ル濃度 (ng/ m L ) 200mg PM (n=10) 200mg HEM (n=5) 200mg EM (n=5)
図 3 治験№ VRC-JP-97-501 での血中濃度トラフ値の推移(静注反復) 国内第 3 相試験(治験№ A1501001)では経口剤 300 mg 1 日 2 回,注射剤 6 mg/kg 1 日 2 回の 負荷用量が投与第 1 日目に用いられたが,第 1 日目に治験薬を中止あるいは中断した症例は認め られなかった。第 1 日目に認められた主な有害事象は視覚に関する有害事象であり,また,維持 投与期と異なる特別な有害事象は認められず,忍容性上問題はなかった。 経口剤 300 mg を 1 日 2 回(症状に応じて増量できるが,初日投与量の上限は忍容性の確認さ れている 400 mg を 1 日 2 回までとする),注射剤 6 mg/kg を 1 日 2 回負荷投与した際の忍容性は 良好と考えられる。かつ,負荷投与を行わない場合は血中濃度が定常状態に達するまでに約 5 日 以上かかると考えられるが,初日の負荷投与により血中濃度は早期に定常状態に達する。したが って,対象疾患である深在性真菌症は生命にかかわる重篤な疾患であり緊急に治療効果を得るこ とが必要であることを考慮すると,負荷投与により早期に十分な血中濃度が得られるベネフィッ トは安全性上のリスクを上回ると考えられる。 3) 注射剤から経口剤へのスイッチ投与について ボリコナゾール経口剤のバイオアベイラビリティーは,健康被験者を対象としたポピュレーシ ョン PK 解析より 96%と推定され,CV 値は 13%と吸収率のばらつきは少なかった。また,国内 第 3 相試験でのポピュレーション PK 解析では,バイオアベイラビリティーはほぼ 100%と推定 された。さらに類薬であるイトラコナゾールで知られている胃内 pH の上昇による吸収の低下も 本剤では認められない。したがって,経口剤でも注射剤と同様,安定して十分な血中濃度が得ら れると考えられる。 本剤の対象疾患は経口剤を投与できない全身状態のことも多く,その際は注射剤が必要となる。 しかしながら全身状態が安定し,経口投与可能となった場合,経口剤にスイッチすることにより 0 1000 2000 3000 4000 5000 6000 1 2 3 4 5 6 7 8 9 10 試験期間 (日) 血漿中ボ リコナゾ ール濃度 (ng/ m L ) PM 3 mg/kg(n=2) HEM 4 mg/kg(n=4) HEM 3 mg/kg(n=2) EM 4 mg/kg(n=2) EM 3 mg/kg(n=2)
不要な侵襲を軽減でき,また,点滴を終了して入院から外来管理に移行できるため医療費の削減 につながる。したがって,注射剤で治療を開始後,経口投与可能となった時点で,医師の判断で 経口剤へスイッチすることが適切であると考えられる。 このような利便性の高い投与方法ができる抗真菌剤は本邦では本剤とフルコナゾールのみであ る。 4) 体重 40 kg 未満の患者での経口剤の投与量について 初期の外国第 2 相試験(治験№ 150-303)において,体重 44 kg の被験者に 1 回 200 mg(カプ セル剤)を 1 日 2 回経口投与したところ,ボリコナゾール血中濃度が 10 µg/mL を超えて高い血 中濃度となったため,治験実施計画書を改訂し,低体重の基準を 40 kg とし,体重 40 kg 未満の 場合は,投与量を半量とすることとした。また,その後の外国臨床試験では体重 40 kg 未満の場 合の経口剤の投与量は半量とした。 その後の健康成人におけるポピュレーション PK 解析より,体重は経口剤投与時の血中濃度を 規定する重要な因子と考えられた。経口剤は注射剤と異なり,体重あたりの投与量ではないため, 体重が低い被験者ではそうでないものと比較して血中濃度が上昇することが予想された。したが って国内第 3 相試験でも体重 40 kg 未満の場合に 40 kg 以上の場合と比較して血中濃度が高くな らないように経口剤の投与量を補正した。体重 40 kg 未満の患者では初日の負荷投与量は 150 mg 1 日 2 回,2 日目以降の維持投与量は 100 mg 1 日 2 回とした。 その結果,国内第 3 相試験で第 3 日目の時点で経口剤が投与された 61 例の第 3 日目のトラフ 血中濃度は表 15 のようであった。 表 15 経口剤投与時の体重別第 3 日目トラフ血中濃度 血漿ボリコナゾール濃度(µg/mL) 体重 例数 中央値 25%点-75%点 最小-最大 40 kg 未満 8 2.6 1.4 - 3.4 0.6 - 5.2 40 kg 以上 53 3.8 2.4 – 5.5 0.7 - 8.0 2.7.3.付録(1)Table 3.3 より引用 経口剤の投与量を 40 kg 未満の場合に調整したことにより,体重 40 kg 未満の被験者で高い曝 露となることを避けることができたと考えられる。 さらに,国内第 3 相試験で,有効性評価対象例 65 例のうち,体重 40 kg 以上の群での有効率 は 76.8%(43/56),体重 40 kg 未満の群での有効率は 77.8%(7/9)であり,明らかな差は認めら れなかった。また安全性評価対象例 100 例において,因果関係を否定できない有害事象の発現率 は体重 40 kg 以上の群では 80.2%(69/86),体重 40 kg 未満の群では 64.3%(9/14)であり明らかな 差は認められなかった。 以上から,体重 40 kg 未満の場合に,初日の負荷投与量は 150 mg 1 日 2 回,2 日目以降の維持 投与量は 100 mg 1 日 2 回とするのは妥当と考えられる。
5) 初期維持投与量開始後の投与量の調整法についての検討
一般的に,将来の有効性及び安全性を予測できる血中濃度閾値が存在する場合は,薬剤投与量 調整の判断材料として通常の有効性及び安全性モニタリングに加え,血中濃度に基づく投与量の 調整,つまり Therapeutic Drug Monitoring (TDM)は臨床上有益な手段と考えられている。
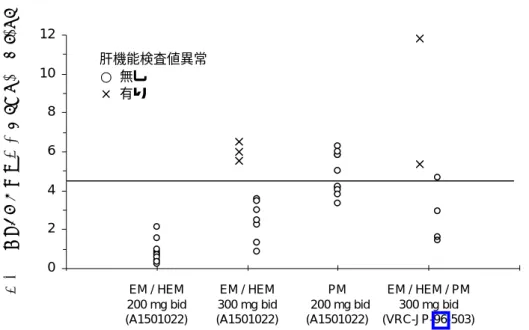
国内第 3 相試験では,第 3 日目の血中濃度に応じて初期維持投与量を決定した後,維持投与量 の調整方法としてその後さらに有効性及び安全性のみならず,血中濃度も考慮して治験責任医師 の判断で必要に応じて維持投与量は増減された。血中濃度の目安としては,トラフ値が 4.5 µg/mL を超える場合は頻回に肝機能検査を実施し,必要に応じて投与量を調節することとした。 外国では,深在性真菌症患者における PK/PD 関係と,有効性及び安全性を予測できる血中濃 度閾値の有無について外国第 2/3 相試験成績から検討されたうえで申請され,本剤は承認されて いる。 外国での検討結果を参考にした上で,本邦において,通常の有効性・安全性モニタリングに基 づく投与量の調整に加え,さらに TDM を実施することの意義について検討した。 (a) 血中濃度による安全性の予測 ボリコナゾールを減量する原因あるい投与中止に至る因果関係を否定できない有害事象は多く の場合,肝に関する有害事象である。以下では,血中濃度と肝に関する有害事象の関係を検討し た。 a) 国内第 1 相試験における検討 国内反復投与試験では,2 試験で計 5 例に肝機能検査値異常が認められており,そのうち 2 例 が肝機能検査値異常により投与を中止している。国内第 1 相試験において,肝機能検査値異常が 認められた被験者のトラフ血中濃度を図 4 に示す。図に示した内訳は,経口単回・反復投与試 験(治験№ A1501022)において,反復投与時に因果関係を否定できない肝機能検査値異常(AST, ALT,γ-GTP)が認められた 3 例(全例 HEM:1 回 300 mg 1 日 2 回反復投与群)と認められなかっ た被験者,及び経口反復投与試験(治験№ VRC-JP-96-503)において因果関係を否定できない肝 機能検査値異常(AST,ALT,γ-GTP)が認められた 2 例(PM 1 例,HEM 1 例:1 回 300 mg 1 日 2 回反復投与群)である。肝機能検査値異常が認められた 5 例の血中濃度(×で表示)は認められ なかった被験者と比較して高い傾向にあった。また,5 例とも血中濃度トラフ値は国内第 3 相試 験で減量を考慮する目安としていた 4.5 µg/mL を超えていた。しかしながら,例数が少なく,ま た,トラフ値が 4.5 µg/mL を超えた被験者で全て肝機能検査値異常が出現したわけではないため, 4.5 µg/mL が明確な閾値とは言えず,国内第 1 相試験から閾値は確認できなかった。
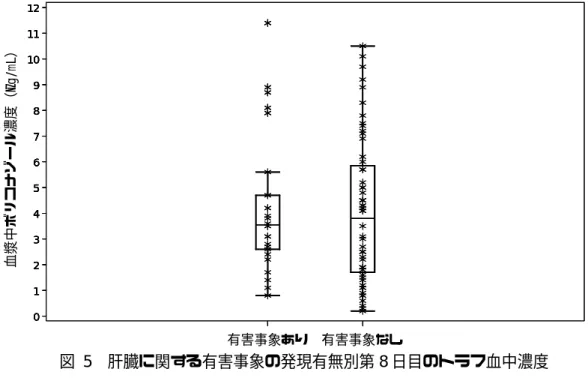
図 4 国内第 1 相試験における肝機能検査異常の有無とトラフ血中濃度 b) 国内第 3 相試験における検討 国内第 3 相試験では,第 3 日目の血中濃度に応じて 5∼7 日目に投与量を調整して初期維持投 与量を決定した。初期維持投与量を決定後は最長 84 日目まで,有効性・安全性に加え血中濃度 も目安にして治験責任医師の判断で投与量を調整できるものとした。各被験者は第 8 日目のトラ フ時の血中濃度が測定されている。肝臓に関する因果関係を否定できない有害事象の発現の有無 と,第 8 日目のトラフ血中濃度の関係を図 5 に示す。 0 2 4 6 8 10 12 血漿中 ボ リ コ ナ ゾ ー ル濃度 のト ラ フ 値 ( μ g/ m L ) EM / HEM 200 mg bid (A1501022) EM / HEM 300 mg bid (A1501022) PM 200 mg bid (A1501022) EM / HEM / PM 300 mg bid (VRC-JP-96-503) 肝機能検査値異常 ○ 無し × 有り