Structural basis of the molecular
mechanisms underlying intracellular
quality control of glycoproteins mediated
by their glucosylation
Zhu, Tong
)octor of Philosophy
)epartment of Functional 2olecular Science
School of Physical Sciences
SO0EN)AI (The Graduate University for
Advanced Studies)
Structural basis of the molecular mechanisms
underlying intracellular quality control of
glycoproteins mediated by their glucosylation
Zhu
し ゅ
, Tong
と ん
朱 彤
Department of Functional Molecular Science,
School of Physical Sciences,
SOKENDAI (The Graduate University for Advanced Studies)
Abstract
A considerable number of proteins are modified with oligosaccharides that serve as the protein-quality tags. Enzymatic trimming of the oligosaccharides displayed on newly synthesized glycoproteins are coupled with exposure of the protein-fate determinants for interacting with a set of carbohydrate recognition proteins as guides for folding, secretory, and degradation processes. Despite of the biological importance, the physicochemical insights into the quality control system of glycoproteins remain unclear. For this reason, I was motivated to provide the structural basis for understanding the molecular mechanisms of the glycoprotein fate-determination process in my PhD thesis. It contains four chapters, including Chapter 1 “General introduction”, Chapter 2 “Elucidation of the structural basis of the sensing mechanism of the ER folding sensor enzyme UGGT”, Chapter 3 “Exploration of the conformational space occupied by the high-mannose-type oligosaccharide functioning as the folding signal”, and Chapter 4 “Conclusions and perspective”.
The endoplasmic reticulum (ER) in the eukaryotic cells is one of the main compartments for efficient protein folding. In the ER, a high-mannose-type oligosaccharide functions as folding signal for recruiting molecular chaperones to facilitate the folding of newborn glycoproteins. Correctly folded glycoproteins with a transportation tag are moved to the Golgi apparatus, while terminally misfolded ones are marked by extensive processing of the carbohydrate residues and thereby subjected to the degradation process.
The glycoprotein-fate determination system also involves a backup mechanism,
by which the folding intermediates losing the folding signal can be sorted out and their folding signal is regenerated to prolong the process for obtaining the correct three-dimensional (3D) structures. To accomplish this unique mechanism, a molecular
“gate keeper” that recognizes the folding intermediates conjugated to a certain type of oligosaccharide and labels them for bringing into additional folding pass plays a key role. An ER-located enzyme, UDP-glucose: glycoproteins glucosyltransferase (UGGT) is considered as the glycoprotein folding sensor. The incompletely folded glycoproteins with a high-mannose-type undecasaccharide are the potential substrates of UGGT which exclusively modifies them by selective glucosylation. The resulting product, a monoglucosylated high-mannose-type oligosaccharide, is responsible for recruiting the ER molecular chaperones to resume the folding maturation process.
To elucidate the molecular mechanisms underlying restoring the folding process mediated by glucosylation that involves specific protein-protein and protein-carbohydrate recognitions, it is essential to perform the structural analysis of the key enzyme UGGT itself as well as its substrate and product oligosaccharides. However, no structural information of UGGT had been available so far due to its huge size and instability. Furthermore, detailed conformational analyses of oligosaccharides remain challenging because of the heterogeneous and flexible properties.
In my thesis, I have overcome these obstacles and elucidated the structure of the glycoprotein folding sensor enzyme UGGT based on X-ray crystallographic analyses, as well as the homogeneous high-mannose-type oligosaccharide with terminal
glucosylation by using nuclear magnetic resonance (NMR) spectroscopy.
To clarify the mechanisms of the recognition towards the incompletely folded glycoproteins, I described the architectural study of UGGT by the combination of bioinformatics analysis and biophysical approaches. To solve the instability problem, UGGT originating from a thermophilic fungus was chosen for operating detailed structural analyses. Encouragingly, the fungal UGGT was successfully obtained in a milligram scale by using E. coli system. The bioinformatics analysis of fungal UGGT suggested that the N-terminal region possesses three-tandem thioredoxin (Trx) -like domains (termed Trx1, Trx2 and Trx3 respectively), followed by a domain rich in β sheet and C-terminal catalytic domain. I also performed architectural prediction of UGGT originating from other species and the results showed that the structural domain arrangement is quite conserved among species, indicating the significance of these domain characters for the function of UGGT. To understand the detailed structure at atomic resolution, crystallization of a series of UGGT constructs were performed. Consequently, I successfully resolved the crystal structure of Trx3 domain, which could give the first structural information of UGGT with atomic detail. The crystallographic study of Trx3 domain revealed that Trx3 contains an extensive hydrophobic patch that may serve as putative substrate-binding site. It is plausible that Trx1 and Trx2 also share the similar 3D structure and have the similar function, suggesting that UGGT recognizes the hydrophobic surface of the incompletely folded glycoprotein through the extensive hydrophobic patch harbored in its multiple Trx domains.
The recombinant UGGT thus obtained was also applied for the NMR study of high-mannose-type oligosaccharides by developing a method employing UGGT as catalyst for the terminal glucosylation. Considering the substrate specificity of this enzyme, denatured glycoprotein mixture derived from the genetically engineered yeast cells, which homogeneously expressed a specific high-mannose-type undecasaccharide, were employed as potential substrates. In order to conduct detailed conformational analyses of the oligosaccharides by stable isotope-assisted NMR measurements, UDP-[13C6]glucose as donor substrate of UGGT was chemically synthesized. By combining these techniques, the in vitro chemoenzymatic reaction catalyzed by UGGT successfully provided uniformly and selectively 13C-labeled monoglucosylated high-mannose-type oligosaccharides harboring intracellular glycoprotein folding signal.
Multidimensional NMR measurements of the high-mannose-type oligosaccharides indicated that attachment of one glucose residue induces little conformational changes, whereas the removal of one mannose residue results in significant modification of the dynamic behaviors of the carbohydrate chain. These results suggest that the 13C-labeled oligosaccharides could be a useful probe for NMR analyses of their conformational dynamics in solution and their interactions with the ER chaperones at the atomic level.
These studies would provide the structural basis for understanding the interaction mechanisms between UGGT and structurally imperfect glycoproteins, giving new insights into quality control of glycoproteins in cells.
Thesis contents:
Abstract
Thesis contents
Chapter 1. General introduction
1.1 Protein folding and quality control systems 1.2 Oligosaccharides on glycoproteins
1.3 Protein folding and quality control in the endoplasmic reticulum 1.4 The ER folding sensor enzyme UGGT
1.5 Conformational dynamics of oligosaccharides 1.6 Scope of this study
Chapter 2. Elucidation of the structural basis of the sensing mechanism of the ER folding sensor enzyme UGGT
2.1 Introduction 2.2 Results
2.3 Discussions and conclusions 2.4 Methods and materials
Chapter 3. Exploration of the conformational space occupied by the high-mannose-type oligosaccharide functioning as the folding signal
3.1 Introduction 3.2 Results
3.3 Discussions and conclusions 3.4 Methods and materials
Chapter 4. Conclusions and perspective
Acknowledgement
Chapter 1. General introduction
1.1 Protein folding and quality control systems
To exert their biological activities, proteins need to fold into thermodynamically stable three-dimensional (3D) structures within a short frame of time after biosynthesis. In the cells, the spontaneous structural maturation of proteins is often hindered because of the crowded, complex molecular environments. To prevent the possibility that aberrant proteins tend to aggregate and become toxic to the cells, transition from a random coil to a native structure requires the assistance of chaperones controlled in a sophisticated fashion so as to allow folding efficiency and avoid error1. Such mechanisms are best exemplified by an eukaryotic protein quality control system in which the oligosaccharides serve as protein-fate determinants via being monitored by a set of carbohydrate binding proteins collectively called intracellular lectins2.
1.2 Oligosaccharides on glycoproteins
Glycosylation is one of the major covalent modifications of proteins in cells (Figure 1.2.1.)3. Comparing to other post-translational modifications like phosphorylation, which provides a digital two-state system4, glycosylation could produce a diverse, dynamic property to the proteins. The great variation of monosaccharide building blocks and possible glycosidic links as well as their high degrees of motional freedom give rise to various 3D structures. Consequently, the
oligosaccharides can act as signaling tags through interacting with the intracellular lectins (Figure 1.2.2.)5,6, thereby mediating various cellular events, including pathogen infection, cellular communication and immune response7,8. Recognition of oligosaccharides displayed on glycoproteins also plays central roles in protein quality control system.
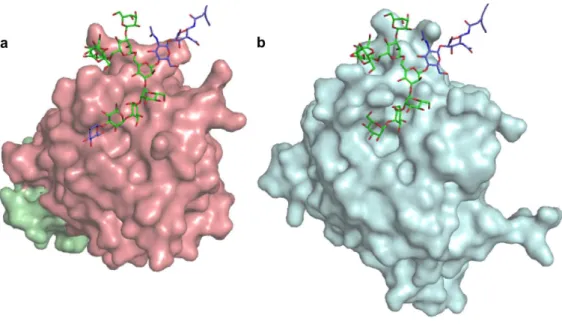
Figure 1.2.1. Representation of a glycoprotein in which oligosaccharide chains are covalently connected to the asparagine residues. Adapted from one subunit of tetrameric influenza neuraminidase with high-mannose-type oligosaccharides attached to Asn86, Asn146, Asn200 and Asn234 (PDB code: 1nn2) 3. Two glycans at Asn146 and Asn200 are highly ordered.
Figure 1.2.2. Representation of oligosaccharides interacting with intracellular lectins. Adapted from the crystal structures of (a) ERGIC53-CRD/MCFD2 (PDB code: 3WHT)5 and (b) VIP36-CRD (PDB code: 2DUO)6 co-crystalized with glycotope di-saccharide, Manα1-2Man. To model the entire ligand oligosaccharide, Glc1Man8GlcNAc2, non-glycotope sugar moiety originated from the Asn196-glycan of an insect arylphorin glycoprotein [PDB code: 3GWJ (molecule D)]9 was superposed with glycotope disaccharide Manα1-2Man based on the torsion angle energy estimated using the PDB-CARE program10.
1.3 Protein folding and quality control in the endoplasmic reticulum
The endoplasmic reticulum (ER) in the eukaryotic cells is an intracellular compartment for efficient protein folding. Proteins destined to the extracellular spaces and cell membranes through secretory pathway are co-translationally translocated into the ER, in which they attain their native structures before transport to their final
locations. A considerable proportion of these proteins are modified with oligosaccharides attached to the asparagine residue(s) in the ER. The oligosaccharides not only increase the solubility and stability of the polypeptides they modify11, but also control quality of the proteins, regulating their structural maturation by functioning as fate determinants2,12,13,14,15,16,17
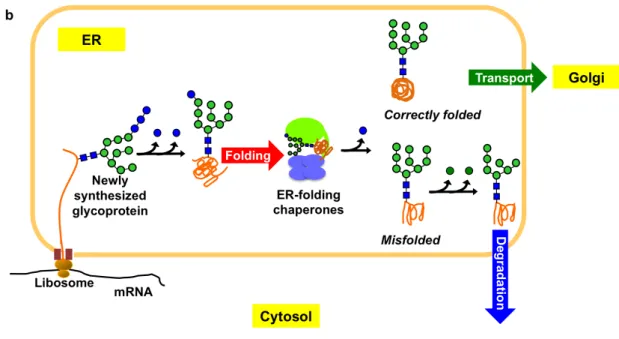
Initially, preassembled precursor high-mannose-type oligosaccharide (Glc3Man9GlcNAc2), that harbors three non-reducing terminal branches (termed D1, D2 and D3 branch respectively) with three glucose residues capping the D1 branch,is covalently attached to the emerging polypeptides (Figure 1.3.1.a). Immediately, stepwise processing of the oligosaccharides begins to transiently create the quality tags responsible for recruiting the ER-resident chaperones having lectin activity so that the fates of the newly synthesized glycoproteins, i.e. folding, transport and degradation, would be determined (Figure 1.3.1.a). Trimming of the first two glucose residues generates the folding signal, a monoglucosylated high-mannose-type oligosaccharide (Glc1Man9GlcNAc2), which is responsible for recruiting the ER chaperones to initiate the folding process12,14,18. The chaperoning process is terminated by removing the innermost glucose residue18,19,20. While correctly folded glycoproteins are transported to the Golgi apparatus for further processing2,5,6,10,11,12,13,14
, severely misfolded glycoproteins experience extensive removal of the terminal mannose residues leading to degradation process2,10,11,12,13,14,21,22
(Figure 1.3.1.b).
Figure 1.3.1. (a) Schematic representations of Glc3Man9GlcNAc2 oligosaccharide residues, branches, and the glycoprotein fate determinants encoded in the triantennary structure according to the custom stated by Vliegenthart J.F.G. et al.23 (b) Scheme of high-mannose-type oligosaccharide dependent fate determination of glycoproteins in the ER, which is coupled with oligosaccharide processing and interactions with a set of ER-resident chaperones with lectin activities.
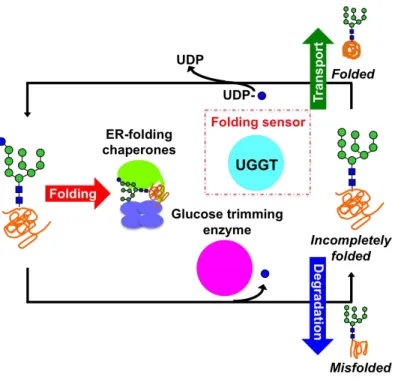
In some cases, one round association with the ER chaperones is not enough for nascent glycoproteins to obtain their native structures24. Indeed, they undergo several round interactions with the ER chaperones. This is achieved by an elaborate backup mechanism serving as molecular “gatekeeper” that can sort the deglucosylated folding intermediates out and regenerate the monoglucosylated glycoform for sending them back to the chaperone-assisted folding process.
Sorting incompletely folded glycoproteins out and labeling them by glucosylating their sugar chains are executed by one unique molecule, the folding sensor enzyme UDP-glucose: glycoproteins glucosyltransferase (UGGT). This enzyme functions as the molecular gatekeeper through its enzymatic specificity that exclusively recognizes the incompletely folded glycoproteins exhibiting high-mannose-type oligosaccharide(s) without terminal glucosylation as its potential substrates and thereby labels them by transferring a monoglucose residue to their oligosaccharide chains using uridine diphosphate (UDP)-glucose as glucose donnor18,19. The resulting product glycan, i.e. monoglucosylated high-mannose-type oligosaccharide, enables the incompletely folded glycoproteins to re-engage with the ER chaperones so that the folding process can be resumed18,19.
With the help of the gatekeeper UGGT, several rounds of folding cycle including re-glucosylation, association with ER chaperones, de-glucosylation, dissociation with ER chaperones are supposed to occur until the glycoproteins acquire their native structures, or until they are eventually marked for degradation18,19,20 (Figure 1.3.2.). Because the restoring process of folding maturation is mediated through specific protein-protein and protein-carbohydrate recognitions, it is essential for understanding the molecular mechanisms underlying this system to approach into the key enzyme UGGT along with its substrate and product oligosaccharides.
Figure 1.3.2. Scheme of the folding cycle initiated by the molecular gatekeeper UGGT.
1.4 The ER folding sensor enzyme UGGT
The folding sensor enzyme UGGT plays the key role in the glycoprotein quality control system by serving as molecular gatekeeper. Glycoproteins with high-mannose-type oligosaccharide(s), which are released from the ER chaperones, are inspected by UGGT before they go ahead in the early secretory pathway. Only those with structurally imperfect nature are sorted out and labeled by UGGT with glucose residue attached to the D1 branch of their carbohydrate chain as a signature of
“folding intermediates” 18,19. Whereas glycoproteins that have achieved native structures are supposed to be ignored by this enzyme25,26, suggesting that both protein
moieties and glycan moieties serve as determinants recognized by UGGT.
Extensive studies have been performed focusing on the substrate preference of UGGT. Regarding the N-glycan moieties, accumulating data indicate that UGGT recognizes the innermost GlcNAc residue in the N-glycan, which is supposed to be imbedded in the correctly folded glycoproteins25,27. Moreover UGGT exhibited highest enzymatic activity to high-mannose-type oligosaccharides containing nine mannose residues27,28. Considering the variations of glycoproteins, UGGT is expected to deal with a wide range of glycoprotein substrates in vivo, although very few endogenous substrates have so far been identified due to their transient existence29. In vitro, plenty of non-native glycoproteins with great variations in terms of size, shape, and distance between the structurally defective site and the N-glycosylation site for re-glucosylation, have been used as model substrates of UGGT25,26,28,29,30,31,32,33,34,35
. Interestingly, small synthetic hydrophobic fluorophore conjugated with a high-mannose-type oligosaccharide were used as defined probes for monitoring UGGT activity17,27, suggesting that UGGT recognizes non-protein aglycone determinant containing extensive hydrophobic stretch exposed to the solvent. However, the molecular basis for the recognition mechanism of UGGT responding to a wide range of incompletely folded glycoproteins as well as small analogues remains unknown.No structural information explaining the unique enzymatic specificity of UGGT, which only recognizes incompletely folded glycoproteins as its clients, has been provided till now due to its largeness and instability of this enzyme for structural study.
1.5 Conformational dynamics of oligosaccharides
To understand how the specific protein glycoform can be precisely recognized by molecular chaperons among all the possible counterparts containing homologous antennae and identical residues except for one glucose difference, it is essential to explore detailed conformational properties of the oligosaccharides.
The conformational variability exhibited by one certain glycoprotein glycan often inhibits it from being crystallized or makes it barely visible in X-ray crystallography12,14,36,37
. By contrast, nuclear magnetic resonance (NMR) spectroscopy is a powerful tool for providing detailed information about the conformational dynamics of carbohydrate chains in solution38,39,40. The utility of NMR spectroscopy is dramatically expanded by combining with stable isotope labeling of the carbohydrate chains12,41,42. However, this attempt was hindered by the limited availability of the stable-isotope labeled oligosaccharides that can be used as probes in adequate quality and quantity for NMR measurements. The transient existence makes it extremely difficult to obtain the glycans directly from natural sources. Moreover, their branched structures make the synthetic approach even harder. A feasible protocol for preparing the homogeneous monoglucosylated oligosaccharide is badly needed.
1.6 Scope of this study
Recently, the quality control system unique to eukaryotic glycoproteins has been
extensively studied. Despite of the biological and biophysical importance, the physicochemical insights into this system are still unclear. For this reason, I was motivated to provide the structural basis for understanding the molecular mechanisms for restoring the folding process. Since this process is mediated by glucosylation as a consequence of specific protein-protein and protein-carbohydrate recognitions, it is essential to perform the structural analyses of the key enzyme UGGT itself along with its substrate and product oligosaccharides. However, no structural information of this key enzyme had been available so far due to its huge size and thermodynamic instability. Furthermore, detailed conformational analyses of oligosaccharides remain challenging because of their heterogeneous and flexible properties.
In my thesis, I have overcome these obstacles and elucidated the structure of the glycoprotein folding sensor enzyme UGGT based on X-ray crystallographic analyses in conjunction with a bioinformatics approach and also performed structural characterization of the homogeneous high-mannose-type oligosaccharide with terminal glucosylation by using NMR spectroscopy. These results would provide physicochemical insights towards understanding the glycoprotein quality control system in cells.
References
1. Dobson CM. Protein folding and misfolding. Nature 426, 884-90 (2003).
2. Caramelo JJ, Parodi AJ. A sweet code for glycoprotein folding. FEBS Lett., (2015).
3. Varghese JN, Colman PM. Three-dimensional structure of the neuraminidase of influenza virus A/Tokyo/3/67 at 2.2 Å resolution. J Mol Biol. 221, 473-86 (1991). 4. Johnson LN. The regulation of protein phosphorylation. Biochem Soc Trans. 37, 627-41 (2009).
5. Satoh T, Suzuki K, Yamaguchi T, Kato K. Structural basis for disparate sugar-binding specificities in the homologous cargo receptors ERGIC-53 and VIP36. PloS One 9, e87963 (2014).
6. Satoh T, Cowieson NP, Hakamata W, Ideo H, Fukushima K, Kurihara M, et al. Structural basis for recognition of high mannose type glycoproteins by mammalian transport lectin VIP36. J Biol Chem. 282, 28246-55 (2007).
7. Brandley BK, Schnaar RL. Cell-surface carbohydrates in cell recognition and response. J Leukoc Biol. 40, 97-111 (1986).
8. Sharon N, Lis H. Carbohydrates in cell recognition. Scientific American 268, 82-9 (1993).
9. Ryu KS, Lee JO, Kwon TH, Choi HH, Park HS, Hwang SK, et al. The presence of monoglucosylated N196-glycan is important for the structural stability of storage protein, arylphorin. The Biochem J. 421, 87-96 (2009).
10. Lutteke T, von der Lieth CW. pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics 5, 69 (2004).
11. Shental-Bechor D, Levy Y. Folding of glycoproteins: toward understanding the biophysics of the glycosylation code. Curr Opin Struct Bio. 19, 524-33 (2009).
12. Satoh T, Yamaguchi T, Kato K. Emerging structural insights into glycoprotein quality control coupled with N-glycan processing in the endoplasmic reticulum. Molecules 20, 2475-91 (2015).
13. Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochemical Sci. 35, 74-82 (2010).
14. Kamiya Y, Satoh T, Kato K. Molecular and structural basis for N-glycan-dependent determination of glycoprotein fates in cells. Biochim Biophys Acta. 1820, 1327-37 (2012).
15. Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 4, 181-91 (2003).
16. Kato K, Kamiya Y. Structural views of glycoprotein-fate determination in cells. Glycobiology 17, 1031-44 (2007).
17. Takeda Y, Totani K, Matsuo I, Ito Y. Chemical approaches toward understanding glycan-mediated protein quality control. Curr Opin Chem Biol. 13, 582-91 (2009). 18. Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. 283, 10221-5 (2008).
19. D'Alessio C, Caramelo JJ, Parodi AJ. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin Cell Dev Biol. 21, 491-9 (2010).
20. Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci USA. 91, 913-7 (1994).
21. Lederkremer GZ. Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol. 19, 515-23 (2009).
22. Frenkel Z, Gregory W, Kornfeld S, Lederkremer GZ. Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6-5GlcNAc2. J Biol Chem. 278, 34119-24 (2003).
23. Vliegenthart J.F.G. DL, and Halbeek, H.V. High-resolution, 1H-nuclear magnetic resonance spectroscopy as a tool in the structural analysis of carbohydrates related to glycoproteins. Adv Carbohydr Chem Biochem. 41, 209-374 (1983).
24. Solda T, Galli C, Kaufman RJ, Molinari M. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol Cell. 27, 238-49 (2007).
25. Sousa M, Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 14, 4196-203 (1995).
26. Izumi M, Makimura Y, Dedola S, Seko A, Kanamori A, Sakono M, et al. Chemical synthesis of intentionally misfolded homogeneous glycoprotein: a unique approach for the study of glycoprotein quality control. J Am Chem Soc. 134, 7238-41 (2012).
27. Totani K, Ihara Y, Matsuo I, Koshino H, Ito Y. Synthetic substrates for an endoplasmic reticulum protein-folding sensor, UDP-glucose: glycoprotein glucosyltransferase. Angew Chem Int Ed Engl. 44, 7950-4 (2005).
28. Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein
glucosyltransferase. Biochemistry 31, 97-105 (1992).
29. Labriola C, Cazzulo JJ, Parodi AJ. Retention of glucose units added by the UDP-GLC:glycoprotein glucosyltransferase delays exit of glycoproteins from the endoplasmic reticulum. J Cell Biol. 130, 771-9 (1995).
30. Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Sci USA. 100, 86-91 (2003).
31. Taylor SC, Thibault P, Tessier DC, Bergeron JJ, Thomas DY. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 4, 405-11 (2003).
32. Ritter C, Helenius A. Recognition of local glycoprotein misfolding by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase. Nat Struct Biol. 7, 278-80 (2000).
33. Ritter C, Quirin K, Kowarik M, Helenius A. Minor folding defects trigger local modification of glycoproteins by the ER folding sensor GT. EMBO J. 24, 1730-8 (2005).
34. Pearse BR, Gabriel L, Wang N, Hebert DN. A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. The Journal of cell biology 181, 309-20 (2008).
35. Taylor SC, Ferguson AD, Bergeron JJ, Thomas DY. The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to
local changes in glycoprotein conformation. Nat Struct Mol Biol. 11, 128-34 (2004). 36. Taylor ME, Drickamer K. Convergent and divergent mechanisms of sugar recognition across kingdoms. Curr Opin Struct Biol. 28, 14-22 (2014).
37. Weis WI, Drickamer K. Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem 65, 441-73 (1996).
38. Yagi-Utsumi M, Kato K. Structural and dynamic views of GM1 ganglioside. Glycoconj J. 32, 105-12 (2015).
39. Peters T, Pinto BM. Structure and dynamics of oligosaccharides: NMR and modeling studies. Curr Opin Struct Biol. 6, 710-20 (1996).
40. Yamaguchi Y, Yamaguchi T, Kato K. Structural analysis of oligosaccharides and glycoconjugates using NMR. Adv Neurobiol. 9, 165-83 (2014).
41. Kamiya Y, Satoh T, Kato K. Recent advances in glycoprotein production for structural biology: toward tailored design of glycoforms. Curr Opin Struct Biol. 26, 44-53 (2014).
42. Kato K, Yamaguchi Y, Arata Y. Stable-isotope-assisted NMR approaches to glycoproteins using immunoglobulin G as a model system. Prog Nucl Magn Reson spectrosc. 56, 346-59 (2010).
Chapter 2. Elucidation of the structural basis of the
sensing mechanism of the ER folding sensor enzyme
UGGT
This chapter is adapted and modified from Zhu T, Satoh T, and Kato K, Structural insight into substrate recognition by the endoplasmic reticulum folding-sensor enzyme: crystal structure of third thioredoxin-like domain of UDP-glucose:glycoprotein glucosyltransferase. Scientific Reports 4, Article number: 7322 (2014).
2.1 Introduction
In the ER glycoprotein quality control system, a backup mechanism to sort the folding intermediates lost the folding signal and prolong the process for obtaining the correct three-dimensional structures is provided. The folding sensor enzyme UGGT functions as the molecular gatekeeper which regenerates the monogulcosylated glycoforms exclusively on the de-glucosylated glycoproteins yet to attain the native structures1-10, providing them additional opportunities to fold by interacting with ER chaperones.
UGGT is a large enzyme, comprising approximately 1,500 amino acid residues. It has been putatively divided into two functional regions: the N-terminal region accounting for 80% of the enzyme with low similarity to any known structural families is responsible for sensing the folding state, the C-terminal region which comprised the remaining 20% of the enzyme which shows great similarity to the glycosyltransferase 8 family11, 12, is responsible for the enzymatic activity. However, no further structural information is available on this key enzyme to date.
To provide the molecular insights towards understanding the folding cycle, the structural basis of the working mechanisms of the folding sensor enzyme UGGT was explored. Considering the availability and stability of this enzyme, Chaetomium thermophilum, a thermophilic fungus, which survives at temperatures of up to 60°C13, was selected as the source organism for the structural study of UGGT. The bioinformatics and crystallographic data demonstrated that the folding-sensor region of UGGT contains three tandem thioredoxin (Trx)-like domains. Moreover, the 3D
structure of a Trx domain of UGGT was determined, thereby providing structural insights into the mechanism of substrate recognition of the folding-sensor.
2.2 Results
Folding sensor region of UGGT possesses three tandem Trx-like domains
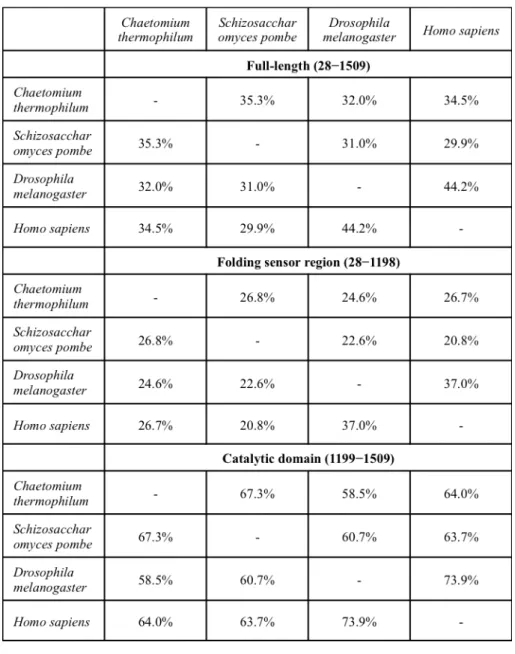
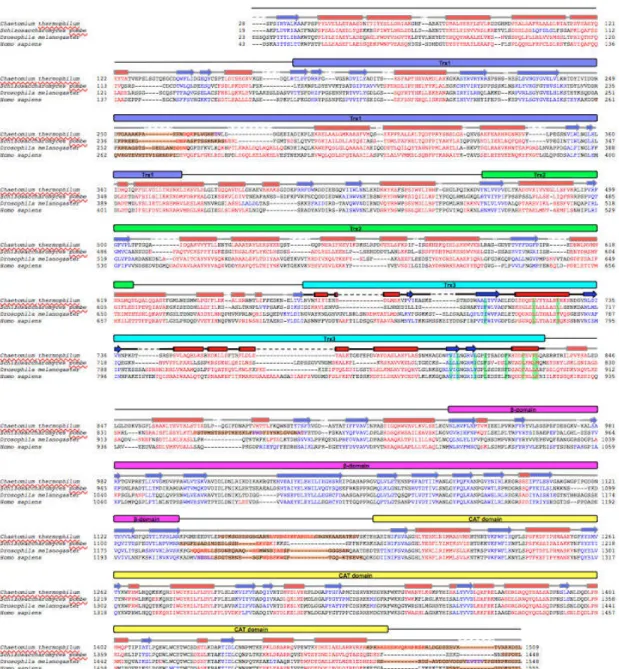
To investigate the structure of the N-terminal folding-sensor region of UGGT, I subjected its amino acid sequence (residues 28–1198) to bioinformatics analysis using the programs PSIPRED14 and DISOPRED215. The results indicate that the folding-sensor region of UGGT exhibits well-formed secondary structures; a mixed α/β region in the N-terminal part (residues 28–939) and a β-strand-rich region (termed the β-domain, residues 940–1140) around the C-terminus (Figure 2.2.1.). Although sequence homology of UGGT was modestly low (32.0%–34.5% identities) between the thermophilic fungus and humans (Table 2.2.1.), the secondary structure distributions appeared highly conserved across species. A remarkably disordered segment was identified at the connection between the β- and C-terminal catalytic domains (Figure 2.2.1.). This structural feature is consistent with previously reported results of limited proteolysis12.
Table 2.2.1. Sequence identity of UGGT among species.
Figure 2.2.1. Bioinformatics study of full-length UGGT. (a) Domain structure of C. thermophilum UGGT. The Trx3 domain (residues Asn671-Ala831) was crystalized in this study. (b) Structure-based sequence alignment of full-length UGGT among species. The predicted secondary structure elements are highlighted as in Figure 2.2.1.a. Predicted segments and residues involving the C-terminal α6 or detergent interactions are highlighted in orange and green, respectively.
Next, I attempted to identify structural domain(s) within the N-terminal folding-sensor region using InterPro16 and Phyre217. Regarding the β-domain, no significantly homologous domains were identified. On the other hand, the folding-sensor region of UGGT was found to harbor three tandem Trx-like domains: Trx1 (residues 168–379), Trx2 (residues 467–624) and Trx3 (residues 671–831) (Figure 2.2.1.). The arrangement of these domains is essentially identical across species, suggesting that the 3D structural architecture of UGGT is evolutionarily conserved, which is convinced by the previous report that the chimeric UGGTs combining the C and N terminal regions that were originated from two species were active in vivo12.
Crystal structure of the third Trx-like domain of UGGT
Based on the finding that folding-sensor region of UGGT possesses three tandem Trx-like domains, I performed bacterial expression, purification and crystallization of a series of Trx domains. First, I expressed each of the three Trx domains. Although Trx3 domain was able to be expressed as a soluble protein, the Trx1 and Trx2 domains formed inclusion bodies in E. coli cells. Therefore, I made tandem constructs for their expression. Consequently, I was able to express Trx1-Trx2, Trx2-Trx3 and Trx1-Trx2-Trx3 proteins in their soluble form. Of these constructs, I successfully crystallized the Trx3 domain with the optimization of its N- and C-terminal sequences (residues 671–831), based on the identification of proteolytically stable fragments. However, despite extensive trials, I could not obtain crystals of the tandem constructs
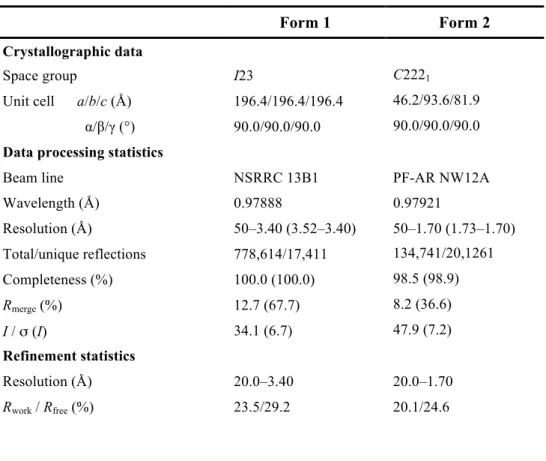
Two forms of the crystal structure of Trx3 domain were determined at 3.4 and 1.7 Å resolutions. The final model of Form 1, refined to a resolution of 3.40 Å, had an Rwork of 23.5% and Rfree of 29.2% (Table 2.2.2.). The crystal belonged to space group I23 with six molecules per asymmetric unit. The structures of molecules A–F were highly similar to each other with an RMSD value of 0.11–0.37 Å for superimposed Cα atoms 94–155. Molecule A in the crystal structure, which had the lowest average B value (Table 2.2.2.), was used for the comparative analysis and will be primarily described hereafter. On the other hand, Form 2 of the Trx3 domain of UGGT belonged to space group C2221 and diffracted up to 1.70-Å resolution. In the crystal structure, one molecule was contained per asymmetric unit. The final model of Form 2 had an Rwork of 20.1% and Rfree of 24.6% (Table 2.2.2.).
Table 2.2.2. Data collection and refinement statistics for UGGT-Trx3 domain.
Form 1 Form 2
Crystallographic data
Space group I23 C2221
Unit cell a/b/c (Å) 196.4/196.4/196.4 46.2/93.6/81.9 α/β/γ (°) 90.0/90.0/90.0 90.0/90.0/90.0 Data processing statistics
Beam line NSRRC 13B1 PF-AR NW12A
Wavelength (Å) 0.97888 0.97921
Resolution (Å) 50–3.40 (3.52–3.40) 50–1.70 (1.73–1.70) Total/unique reflections 778,614/17,411 134,741/20,1261
Completeness (%) 100.0 (100.0) 98.5 (98.9)
Rmerge (%) 12.7 (67.7) 8.2 (36.6)
I / σ (I) 34.1 (6.7) 47.9 (7.2)
Refinement statistics
Resolution (Å) 20.0–3.40 20.0–1.70
Rwork / Rfree (%) 23.5/29.2 20.1/24.6
R.m.s. deviations from ideal
Bond lengths (Å) 0.010 0.011
Bond angles (°) 1.28 1.47
Ramachandran plot (%)
Favored 96.5 98.3
Allowed 3.5 1.7
Number of atoms
Protein atoms (A/B/C/D/E/F) 1239/1246/1127/
1231/738/871 1166
Water molecules - 120
Detergent molecule - 37
Average B-values (Å2)
Protein atoms (A/B/C/D/E/F) 79.7/80.6/92.6
95.2/135.1/139.8 23.8
Water molecules - 30.1
Detergent molecule - 64.9
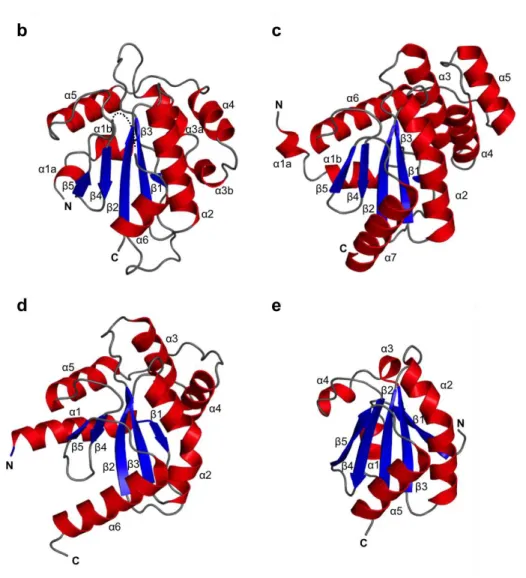
As expected from the bioinformatics analysis, the crystal structure displayed a typical Trx-like fold, i.e. a five-stranded β-sheet with a β1–β3–β2–β4–β5 arrangement surrounded by six α-helices (Figure 2.2.2.a and b). In the crystal structure, the β5–α6 loop (residues 816–818) was disordered. The C-terminal α6-containing segment showed a higher crystallographic B-factor (87.7 Å2) than the average value (79.7 Å2; Table 2.2.2.). Comparison of the structure of the Trx3 domain of UGGT with known protein structures using the DALI server revealed that the protein disulfide bond isomerase (DsbA/C) homologue, Salmonella enterica ScsC18, was the most structurally similar protein (Z-score = 9.4; RMSD = 2.9; PDB code: 4GXZ, Figure 2.2.2.c). As representative of the DsbA/C structure, the well-characterized crystal structure of E. coli DsbC (PDB code: 1EEJ)19 is also shown in Figure 2.2.2.d. The overall folds were very similar between the Trx3 domain of
UGGT and DsbC, except for the N-terminal α1 helix, which directly follows the dimerization domain in DsbC, and variable α3/α4 helices (Fig. 2.2.2.d). Compared with the crystal structure of the E. coli thioredoxin trxA20 (PDB code: 2TRX; Figure 2.2.2.e), which exhibits typical Trx fold, three contiguous helical insertions, α3, α4 and α5, were identified between β3 and β4, as observed in DsbC19. Furthermore, an N-terminal segment containing α1 and β1 regions of the Trx3 domain of UGGT was significantly different from that of E. coli trxA20 in terms of topological arrangement. In the folds shared by the Trx3 domain of UGGT and DsbC, α1 precedes β1, which makes anti-parallel β-strands with β3 (Figure 2.2.2.b-d). In contrast, α1 was inserted between β1 and β2, both of which were parallel with respect to β3 (Figure 2.2.2.e).
Figure 2.2.2. Crystal structure of the Trx3 domain of UGGT comparing to homologous structures. (a) Structure-based alignment of the Trx3 domain of C. thermophilum UGGT (form 1). (b) Ribbon models of Trx3 domain of C. thermophilum UGGT (form 1). The secondary structures are highlighted (α-helix, red; β-sheet, blue) and the linker regions are shown in grey. The positions of N- and C-termini are also indicated. Dotted line indicates disorder segment. (c) DsbA/C homologue, Salmonella enterica ScsC (PDB code: 4GXZ). (d) E. coli DsbC (PDB code: 1EEJ). For clarity, the N-terminal dimerization domain (residues 1–60) is not shown in the model. (e) E. coli thioredoxin trxA (PDB code: 2TRX). The secondary
In addition, it is plausible that Trx1 and Trx2 also exhibit Trx-folds similar to Trx3 and their structural homologs as suggested by homolog modelling, except for the N-terminal and variable α helical segments between 3 and 4 as well as an insertion loop (residues 226-293) in Trx1 (Figure 2.2.3.).
Figure 2.2.3. Comparison of the Trx-like domains in UGGT. (a) Homology model of
Trx1 domain (residues 168-379) of C. thermophilum using Neisseria gonorrhoeae disulfide interchange protein (PDB code: 3GV1) as template. An insertion loop (residues 226-293) is shown in dash line. (b) Homology model of Trx2 domain (residuues 467-624) of C. thermophilum using Neisseria meningitidis Thiol:disulfide interchange protein DsbA (PDB code: 3DVW) as template. (c) Trx3 domain (residues 671-831) of C. thermophilum UGGT (form 1). The secondary structures are colored as in Figure 2.2.2.
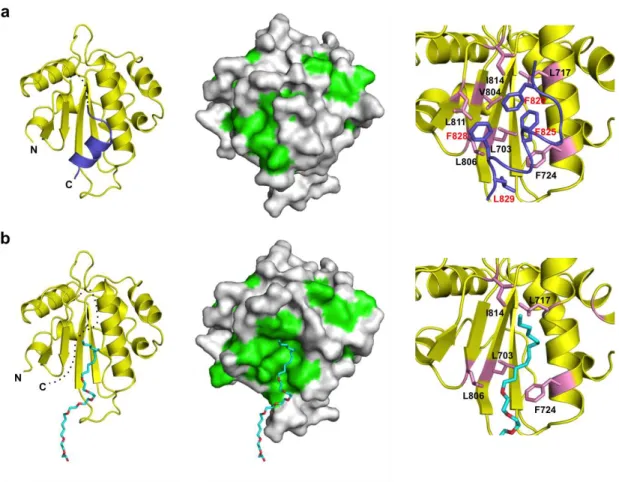
The C-terminal α6 helix, which is followed by a putatively flexible linker region in UGGT, was completely disordered in the crystal structure of Form 2, suggesting the instability of this helix (Figure 2.2.3.b, left). Because of the absence of the α6 helix, an extensive hydrophobic patch was exposed on the surface of the Trx3 domain (Figure 2.2.3.b, centre). The detergent ANAPOE C12E8 was accommodated on this exposed hydrophobic patch. The α6 helix was stabilized mainly through its hydrophobic surface, containing Phe820, Phe825, Phe828 and Leu829, which made contact with the hydrophobic patch, including Leu703 (β2), Leu717, Phe724 (α2), Val804, Leu806 (β4), Leu811 (β5) and Ile814 (β5–α6 loop) (Figure 2.2.4.a, right). Most of these hydrophobic residues were involved in the interaction with the detergent in Form 2. Thus, the C-terminal α6 helix and detergent molecule occupy the common hydrophobic surface of the Trx3 domain. These hydrophobic residues are highly conserved among species (Figure 2.2.1.).
Figure 2.2.4. An extensive patch of the Trx3 domain is concealed by a flexible C-terminal helix. The crystal structures of the Trx3 domain in Form1 and Form2 are indicated in (a) and (b), respectively. The ribbon and surface models are shown in the left and centre. Dotted lines indicate disordered segments. In the surface model (centre), the hydrophobic residues are shown in green. Close-up views of the C-terminal helix or detergent-interacting regions are represented on the right. Residues involved in these interactions are highlighted in the pink stick model. In Form1 (a), the C-terminal α6 helix is highlighted in slate. In Form2 (b), the detergent ANAPOE C12E8 is shown as a stick model.
2.3 Discussions and conclusions
In this research, through comprehensive bioinformatics studies, I revealed the
architectural structure of the N terminal folding sensor region of UGGT, in which three tandemly lined-up Trx-like domains are arranged and followed by a β-sheet rich domain. Moreover, I successfully resolved the crystal structure of the third Trx-like (Trx3) domain, which could give the first structural information of UGGT with atomic detail. As expected, this domain shows a typical Trx-like fold with the highest similarity to bacterial DsbA/C: a central five-stranded β-sheet flanked by six α helices. Two crystal forms were resolved: an open form in which the hydrophobic patch was exposed with the attachment of a substrate-mimicking detergent and a closed form in which the hydrophobic patch was concealed by the flexible C-termianl alpha helix, indicating the extensive hydrophobic patch may function as the putative substrate binding site as well as the involvement of a regulation mechanism.
The Trx-like domain was commonly found in protein disulfide isomerase (PDI) family members that are responsible for assisting protein folding21. The bacterial PDI members are expressed as monomer containing a redox active CXXC motif21,22. However higher PDI members are evolved as multi-domain proteins containing both redox-active and inactive Trx-like domains in different arrangment and actively involved in protein folding maturation process in the ER21-23. In UGGT, none of the three Trx-like domains contain the CXXC motif, excluding the possibility that UGGT is directly involved in the thiol/disulfide modification reaction. In this context, the cis-Pro loop adjacent to the CXXC motif, a hallmark of redox-active Trx-fold proteins21 and involved in substrate recognition in DsbA24, is also not present in the Trx3 domain of UGGT. Accumulating evidence indicated that noncatalytic Trx-like
domains are often involved in substrate recognitions25-27. The crystallographic study I performed indicated that Trx3 domain may bind to incompletely folded protein through its extensive hydrophobic patch. In addition, homolog modeling predicted similar or even larger hydrophobic patches in the other two Trx-like domains (Figure 2.3.1.), suggesting Trx1 and Trx2 domain may have the similar structures and functions to that of Trx3 domain.
Figure 2.3.1. Surface hydrophobicity comparison of the Trx-like domains in UGGT. Homology model of (a) Trx1 and (b) Trx2 domain are the same with Figure 2.2.3.a and b. Crystal structure of (c) Trx3 is the same with Figure 2.2.4.a Form1, The hydrophobic residues are colored green.
UGGT consists of multi-domains with hydrophobic patches connected by
flexible linkers in between, suggesting that UGGT may exhibit architectural flexibility during substrate recognizing process instead of a rigid conformation, which enables UGGT bind to the solvent exposed extensive hydrophobic region on the glycoproteins with multi-hydrophobic patches in its Trx-like domains, so that UGGT can catalyze the glucosylation reaction towards diverse non-native glycoproteins in variations of shape, size and distance between the structural imperfect and its target N-glycans1-10.
Moreover, a newly identified ER-resident 15 kDa selenoprotein (Sep15) has been found to form a tight complex with UGGT at a 1:1 ratio28,29 (Figure 2.3.2.). Sep15 contains a Trx-like domain in which a selenocystein is involved in the redox-active motif. Although the in vivo enzymatic activity of this enzyme is still not clear, it is proposed to catalyze the isomerization or reduction of disulfide bonds according to its redox potential30. In vitro data indicated that Sep15 is able to enhance the glucosyltransferase activity of UGGT31. It is plausible that Sep15 serves as a structural extension of UGGT with a complementary function, plays cooperative role through binding with UGGT in the quality control system.
Figure 2.3.2. Schematic drawing of the Sep15 cooperate with the folding sensor UGGT.
In summary, my bioinformatics and crystallographic analyses revealed that the folding-sensor region of UGGT harbours three tandem Trx-like domains. Moreover, I provided 3D structural snapshots of the third Trx-like domain, in which a putative substrate-binding hydrophobic patch is intramolecularly masked or involved in an intermolecular interaction. These findings offer a key breakthrough toward understanding of the molecular recognition mechanisms of this ER folding-sensor enzyme.
2.4. Methods and materials Protein expression and purification
C. thermophilum var. thermophilum La Touche (DSM 1495) was obtained from
DSMZ, Braunschweig, Germany. Total RNA was isolated using TRIzol® reagent (Life Technologies). The cDNA was synthesized using SuperScript® III Reverse Transcriptase (Life Technologies) with oligo d(T) primers according to the manufacturer’s instructions. Full-length UGGT cDNA was cloned by PCR using a C. thermophilum genomic DNA database13. Recombinant UGGT proteins were expressed as glutathione S-transferase (GST)-fused proteins. The Trx1 (residues 168– 379), Trx2 (residues 467–624), Trx3 (residues 671–831), Trx1-Trx2 (residues 168– 624), Trx2-Trx3 (residues 467–831) and Trx1-Trx2-Trx3 (residues 168–831) domains were amplified by PCR and subcloned into the BamHI and XbaI sites of a modified pCold-GST vector (Takara Bio Inc.)32, in which the factor Xa site was replaced with the tobacco etch virus (TEV) protease recognition site. Recombinant proteins were expressed in E. coli BL21 Star™ cells (Life Technologies) according to the manufacturer’s protocols (Takara Bio Inc.). GST-fused proteins were purified using glutathione-Sepharose™ columns (GE Healthcare). Subsequently, the GST tag was removed by adding TEV protease to the resin for 12 h at 277 K, leaving two additional residues Gly-Ser at the N terminus. The resultant proteins were further purified by size-exclusion chromatography (Superdex-200; GE Healthcare) using a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.1 mM EDTA. The selenomethione (SeMet)-labelled Trx3 domain was expressed in E. coli B834 (DE3) using M9 minimal medium with SeMet. Expression and purification were performed following the same protocol as that for the native protein. Purified proteins were dialyzed against a buffer containing 10 mM Tris-HCl (pH 7.5) and 100 mM NaCl.
The integrity of the protein samples was validated by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS) analysis using an AXIMA-CFR™ spectrometer (Shimadzu) and N-terminal Edman sequencing with a Procise 494HT protein sequenator (ABI/Life Technologies).
Protein crystallization, X-ray data collection and structure determination
The crystals of the Trx3 domain of UGGT (Form 1, 10 mg/ml) were grown in a buffer containing 60% Tacsimate (pH 7.0) for 2 weeks at 289 K. The crystals of the Trx3 domain of UGGT (Form 2) were obtained by equilibrating a solution of 8 mg/ml protein with 1.2 mM ANAPOE C12E8 (polyoxyethylene[8]dodecyl ether • 3,6,9,12,15,18,21,24-octaoxahexatriacontan-1-ol) mixed with an equal volume of precipitant solution containing 23% PEG3350, 0.1 M Tris-HCl (pH 7.0) and 0.2 M ammonium acetate for 6 days at 289 K. The crystals were transferred into the reservoir solution and flash-cooled in liquid nitrogen. Data sets for Forms 1 and 2 were collected using synchrotron radiation at 13B1 of the National Synchrotron Radiation Research Center (Hsinchu, Taiwan) and AR-NW12A of the Photon Factory (Tsukuba, Japan), respectively. All diffraction data were processed using HKL200033. Crystal parameters are summarized in Table 2.2.2.
The 1.70 Å-resolution crystal structure of the Trx3 domain of UGGT (Form 2) was solved using the SAD method. The initial phase was determined using the SHELX C/D/E program34. The initial model was automatically built using ARP/wARP35. Further manual model building into the electron density maps and
Å-resolution structure of the Trx3 domain of UGGT (Form 1) was solved by molecular replacement using the program Phaser38 with the crystal structure of Form 2 as a search model. The stereochemical quality of the final model was assessed by RAMPAGE39. The final refinement statistics are summarized in Table 2.2.2. Graphic figures were prepared using PyMOL (http://www.pymol.org/).
Additional information
The co-ordinates and structural factors of the crystal structures of the Trx3 domain of C. thermophilum UGGT (Forms 1 and 2) have been deposited in the Protein Data Bank under the accession numbers 3WZT and 3WZS, respectively.
References
1. Izumi M, Makimura Y, Dedola S, Seko A, Kanamori A, Sakono M, et al. Chemical synthesis of intentionally misfolded homogeneous glycoprotein: a unique approach for the study of glycoprotein quality control. J Am Chem Soc. 134, 7238-41 (2012).
2. Sousa M, Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 14, 4196-203 (1995).
3. Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc
Natl Acad Sci of USA. 100, 86-91 (2003).
4. Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry 31, 97-105 (1992).
5. Taylor SC, Thibault P, Tessier DC, Bergeron JJ, Thomas DY. Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 4, 405-11 (2003).
6. Ritter C, Helenius A. Recognition of local glycoprotein misfolding by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase. Nat Struct Biol. 7, 278-80 (2000).
7. Ritter C, Quirin K, Kowarik M, Helenius A. Minor folding defects trigger local modification of glycoproteins by the ER folding sensor GT. EMBO J. 24, 1730-8 (2005).
8. Pearse BR, Gabriel L, Wang N, Hebert DN. A cell-based reglucosylation assay demonstrates the role of GT1 in the quality control of a maturing glycoprotein. The J Cell Biol. 181, 309-20 (2008).
9. Taylor SC, Ferguson AD, Bergeron JJ, Thomas DY. The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat Struct Mol Biol. 11, 128-34 (2004). 10. Labriola C, Cazzulo JJ, Parodi AJ. Retention of glucose units added by the UDP-GLC:glycoprotein glucosyltransferase delays exit of glycoproteins from the endoplasmic reticulum. J Cell Biol. 130, 771-9 (1995).
11. Arnold SM, Kaufman RJ. The noncatalytic portion of human UDP-glucose: glycoprotein glucosyltransferase I confers UDP-glucose binding and transferase function to the catalytic domain. J Biol Chem. 278, 43320-8 (2003).
12. Guerin M, Parodi AJ. The UDP-glucose:glycoprotein glucosyltransferase is organized in at least two tightly bound domains from yeast to mammals. J Biol Chem. 278, 20540-6 (2003).
13. Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, et al. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146, 277-89 (2011).
14. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 292, 195-202 (1999).
15. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 337, 635-45 (2004).
16. Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40, D306-12 (2012).
17. Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 4, 363-71 (2009).
18. Shepherd M, Heras B, Achard ME, King GJ, Argente MP, Kurth F, et al. Structural and functional characterization of ScsC, a periplasmic thioredoxin-like protein from Salmonella enterica serovar Typhimurium. Antioxid Redox Signal. 19,
1494-506 (2013).
19. McCarthy AA, Haebel PW, Torronen A, Rybin V, Baker EN, Metcalf P. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat Struct Biol. 7, 196-9 (2000).
20. Katti SK, LeMaster DM, Eklund H. Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J Mol Biol. 212, 167-84 (1990).
21. Heras B, Kurz M, Shouldice SR, Martin JL. The name's bond...disulfide bond. Curr Opin Struct Biol. 17, 691-8 (2007).
22. Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochemical Sci. 31, 455-64 (2006).
23. Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta 1783, 535-48 (2008).
24. Freedman RB, Klappa P, Ruddock LW. Protein disulfide isomerases exploit synergy between catalytic and specific binding domains. EMBO Rep. 3, 136-40 (2002).
25. Klappa P, Ruddock LW, Darby NJ, Freedman RB. The b' domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927-35 (1998).
26. Serve O, Kamiya Y, Maeno A, Nakano M, Murakami C, Sasakawa H, et al. Redox-dependent domain rearrangement of protein disulfide isomerase coupled with exposure of its substrate-binding hydrophobic surface. J Mol Biol. 396, 361-74
(2010).
27. Serve O, Kamiya Y, Kato K. Redox-dependent chaperoning, following PDI footsteps. Protein Folding (ECWalters ed), NOVA Science Publishers (New York), 489-500 (2011).
28. Korotkov KV, Kumaraswamy E, Zhou Y, Hatfield DL, Gladyshev VN. Association between the 15-kDa selenoprotein and UDP-glucose:glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J Biol Chem. 276, 15330-6 (2001).
29. Labunskyy VM, Ferguson AD, Fomenko DE, Chelliah Y, Hatfield DL, Gladyshev VN. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem. 280, 37839-45 (2005). 30. Ferguson AD, Labunskyy VM, Fomenko DE, Arac D, Chelliah Y, Amezcua CA, et al. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J Biol Chem. 281, 3536-43 (2006).
31. Takeda Y, Seko A, Hachisu M, Daikoku S, Izumi M, Koizumi A, et al. Both isoforms of human UDP-glucose:glycoprotein glucosyltransferase are enzymatically active. Glycobiology 24, 344-50 (2014).
32. Hayashi K, Kojima C. pCold-GST vector: a novel cold-shock vector containing GST tag for soluble protein production. Protein Expr Purif 62, 120-7 (2008).
33. Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307-26 (1997).
34. Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A, Found
Crystallogr. 64, 112-22 (2008).
35. Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 3, 1171-9 (2008).
36. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 66, 486-501 (2010).
37. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 53, 240-55 (1997).
38. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 40, (2007).
39. Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437-50 (2003).
Chapter 3. Exploration of the conformational space
occupied by the high-mannose-type oligosaccharide
functioning as the folding signal
This chapter is adapted and modified from Zhu T, Yamaguchi T, Satoh T, and Kato K, A hybrid strategy for the preparation of 13C-labeled high-mannose-type oligosaccharides with terminal glucosylation for NMR study, Chemistry Letters 44, 1744-1746 (2015).
3.1 Introduction
During glycoprotein biosynthesis, the branched structures of N-linked oligosaccharides displayed on the newly synthesized polypeptides play key roles. The high-mannose-type oligosaccharides serve as protein quality tags, which indicate the folding states of the glycoproteins and are recognized by a series of intracellular lectins, and thereby contributing to intracellular fate determination of the glycoproteins1,2,3,4,5,6,7,8,9
. The monoglucosylated high-mannose-type dodecasaccharide (abbreviated as GM9) (Figure 3.1.1.) is displayed on proteins as a tag indicating the incompletely folded status of glycoproteins, which is recognized by the ER lectins operating as chaperones, i.e. calnexin and calreticulin1,2,3,4,5,6,7,8,9
.
Figure 3.1.1. Representation of the monoglucosylated high-mannose-type dodecasaccharide GM9.
For a better understanding of the mechanisms underlying these molecular processes, it is essential to investigate the conformational properties of carbohydrate
crystallographic studies have provided many conformational snapshots of carbohydrate-protein complexes, most of the studies were performed using smaller di- or tri-saccharides as ligands instead of the entire oligosaccharides because the intrinsic flexibility of the carbohydrate chains hampers the crystallographic approach1,3,10,11. Moreover, such flexible properties of carbohydrate chains are vitally relevant to carbohydrate-mediated biomolecular interactions12. Thus, the static views given by X-ray crystallography may provide limited insight into the functional mechanisms of the sugar chain recognition. In contrast, NMR is a one of the potentially valuable tool for detailed evaluation of the conformational dynamics of oligosaccharides12,13,14. Stable isotope-labeling techniques of the carbohydrate chains would provide the great advantages of this method15,16,17.
For detailed conformational analyses, chemically synthesized di- or trisaccharides were prepared with 13C-labeling at specific positions18,19,20,21
, although such synthetic approaches have been difficult to apply to larger, branched oligosaccharides with a few prominent exceptions22,23. In contrast, metabolic labeling methods have been developed using a variety of production vehicles to produce isotopically labeled glycoproteins using immunoglobulin G as a model system16,17,24,25,26
. In the biosynthetic methods, control of glycoprotein glycoforms remains challenging due to their transient existence. To address this issue, previously my group employed genetically engineered Saccharomyces cerevisiae strains that lack specific genes involved in processing of high-mannose-type oligosaccharides27,28,29. Using this method, overexpression systems of homogenous
high-mannose-type oligosaccharides Manα1-2Manα1-6(Manα1-2Manα1-3)Manα1-6(Manα1-2Manα1-2Manα1-3)Manβ1- 4GlcNAcβ1-4GlcNAc (abbreviated as M9) and its derivative (abbreviated as M8B) that lacks the nonreducing-terminal mannose residue at the central D2 branch was established. These oligosaccharides carry glycoprotein fate determinants in their triantennary structures, directing the glycoproteins to vesicular transport to the Golgi or ER-associated degradation1,2,3,5,7. By cultivating the yeast cells using 13C-labeled glucose as the sole carbon source, these oligosaccharides in 13C-labeled form could be prepared for detailed NMR analyses of their dynamic conformations27,28.
For the exclusive production of GM9 by the yeast engineering technique, at least six genes have to be deleted including alg8, gls2, mnn1, mnn4, mns1, and och1. However, multiple gene knock-out is generally cumbersome and may result in a low production yield. To approach the conformational space occupied by GM9 oligosaccharide functioning as the folding signal in the glycoprotein quality control system, I developed a hybrid strategy that combines the yeast engineering with chemoenzymatic synthesis to produce the uniformly as well as selectively 13C-labeled monoglucosylated high-mannose-type dodecasaccharide GM9 (Figure 3.1.2.).
Figure 3.1.2. The scheme depicting the hybrid approach mediated by UGGT for GM9 oligosaccharide production.
The ER glycoprotein quality control system involves a backup mechanism for regenerating the folding signal of the folding intermediates, in which the glucose residue is transferred to the nonreducing terminus of the D1 branch of the high-mannose-type oligosaccharides (mainly M9) displayed on the proteins with conformational defect by the action of a folding sensor enzyme, UGGT8,30. In my studies, I have successfully obtained this protein in milligram scale. Therefore, I attempted to use this enzyme as catalyst for the terminal glucosyalation of M9. In this chapter, I applied this method for exploration of the dynamic conformations of the high-mannose-type oligosaccharides by NMR techniques in conjunction with molecular simulation.
3.2 Results