九州大学学術情報リポジトリ
Kyushu University Institutional Repository
光学的バイオセンシングによる糖鎖高分子の分子認 識能解析
寺田, 侑平
https://doi.org/10.15017/1931885
出版情報:Kyushu University, 2017, 博士(工学), 課程博士 バージョン:
権利関係:
Molecular Recognition Analyses of Glycopolymer by Optical Biosensing
Kyushu University
Graduate School of Engineering
Department of Chemical Systems and Engineering
Yuhei Terada
Supervisor: Yoshiko Miura, Yu Hoshino
Contents
Chapter 1 Introduction……….………..1
1.1 Background of Sugar………...2
1.1.1 Role of Sugar chain...………...2
1.1.2 Glyco-cluster effect………..5
1.1.3 Glycomaterials………..6
1.2 Glycopolymer………..8
1.2.1 Glycopolymer for Biomaterial……….8
1.2.2 Synthesis of Glycopolymers……….……..10
1.2.3 Glycopolymer Interface………..………14
1.3 Biosensing and Analysis of Biomolecule Interaction using Optical Methods….….17 1.3.1 Biosensor using Optical Methods………...17
1.3.2 Structural Color for Biosensing………...19
1.3.3 SPR Technique for Biosensing………...23
References Chapter 2 Biosensing Protein Recognition by Glycopolymer using Structural Color produced from Two-dimensional Photonic Crystal………41
2.1 Introduction………...43
2.2 Experimental section………..………...45
2.2.1 Reagents………..…….…………..45
2.2.2 Apparatus………46
2.2.3 Synthesis of poly(NAS-r-PEGMA-r-TMSMA) ……….….……..47
2.2.4 Synthesis of mannose-incorporating hydrogel nanoparticle (ManNP)………..48
2.2.5 Synthesis of mannose homopolymer (ManHP)……….….49
2.2.6 Preparation of glycopolymer immobilized 2D-PhC films……….50
2.2.7 Measurement of protein binding onto the glycopolymer using QCM………...51
2.2.8 Detection of protein adsorption onto the glycopolymers via monitoring of the changes in the reflection intensity from the 2D-PhC film………...52
2.3 Results & Discussion……….53
2.3.1 Surface analysis of the 2D-PhC films using XPS………..53
2.3.2 Surface analysis of the 2D-Phc films using AFM………..55
2.3.3 Analysis of the interaction between the mannose-incorporating polymers and proteins using QCM……….………...58
2.3.4 Detection of protein adsorption on mannose-incorporating polymers via monitoring of the changes in the reflection intensity of the 2D-PhC film…………..60
2.4 Conclusion……….……64
References Chapter 3 Investigation of Polymer-brush Structure and Protein Recognition ability of Glycopolymer Interface using SPR Technique………...……69
3.1 Introduction……….…………..71
3.2 Experimental section………..………….…………..73
3.2.1 Reagents………..….………..73
3.2.2 Apparatus………74
3.2.3 Synthesis of thiol-terminated glycopolymers……….……74
3.2.4 SPR measurements……….76
3.2.5 Measurement of glycopolymer immobilization onto gold substrate using SPR method……….……76
3.2.6 Characteriation of glycopolymer layer………...77
3.2.7 Analysis of protein binding onto glycopolymer using SPR method…..………77
3.3 Results & Discussion……….78
3.3.1 Synthesis of glycopolymers………78
3.3.2 Immobilization of glycopolymers onto the gold substrate……….79
3.3.3 Charaterization of the glycopolymer layer using an SPR contrast variation technique………..82
3.3.4 Analysis of protein binding onto glycopolymer using SPR method…………..86
3.4 Conclusion……….………90
References Chapter 4 Screening of Protein Interaction against Array of Glycopolymers synthesized by glyco-module strategy………...……...94
4.1 Introduction………...96
4.2 Experimental section………..………...99
4.2.1 Reagents………..………...99
4.2.2 Apparatus………..100
4.2.3 Synthesis of sugar azide 1………100
4.2.4 Synthesis of sugar azide 2………101
4.2.5 Synthesis of sugar azide 3………102
4.2.6 Synthesis of sugar azide 4………103
4.2.7 Synthesis of polymer backbone by RAFT polymerization……….….104 4.2.8 Synthesis of glycopolymer by click chemistry……….104 4.2.9 SPRI measurement of protein binding onto glycopolymer array……….……105 4.3 Results & Discussion……….…..……106 4.3.1 Synthesis of glycopolymers by post-click chemistry………...106 4.3.2 Surface analysis of polymer immobilized gold substrate using XPS………...108 4.3.3 SPRI measurement of CTB binding to glycopolymer array……….110 4.3.4 Investigation of CTB interaction with glycopolymers synthesized by glyco-module method………111 4.3.5 Binding analysis of CTB binding to glycopolymer baring Gal and Neu5Ac...113 4.3.6 Analyzing the cooperativity of Gal unit and Neu5Ac unit for the interaction with CTB………...114 4.3.7 Binding analysis of CTB binding to glycopolymer synthesized by glyco-module method………...115 4.4 Conclusion……….…………..117 Reference
Appendix
Chapter 5 Summary………...………...………..…131
Acknowledgment
Chapter 1
Background
Background
1.1 Background of Sugar 1.1.1 Role of Sugar chain
In most case, sugars are known as sweet things or energy source in foods, or constructional material such as main component of cell wall (cellulose), and outer shell of crustacean and insects (chitin). These are certainly one of the important role that sugar possesses in biological system. On the other hand, it is known that many sugar chains exist on the cells that construct our bodies in forms of sugar clustered domains such as glycoprotein,1,2 glycolipid,2-4 and glycosaminoglycan.5,6 Furthermore, they are related to many important vital phenomena, and therefore they are attracting attention as the third biological chain next to protein and DNA. Those sugars are known in binding specifically with proteins, and the sugar-protein interaction grabs the key for the important vital phenomena such as virus infection, metastasis of cancer, cell adherence, signal transduction, etc. (Figure 1-1).4,7-11
Figure 1-1 Role of sugars on cell surface.
There are numerous kinds of sugars found in nature, and they each have different roles in biological system. When focused on physiologically active sugars, for instance, glycoproteins and glycolipids seen on our cell surfaces, number of monovalent sugars constructing those sugar cluster are confined to less than ten kinds: mannose (Man), glucose (Glc), galactose (Gal), N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), xylose (Xyl), fucose (Fuc), and N-acetylneuraminic acid (Neu5Ac) (Figure 1-2). Therefore, glycoproteins found are constructed by combination of these monovalent sugars. As seen in Figure 1-2, individual sugar has different framework structure and placement of hydroxyl groups, hence these differences are important for specific recognition of proteins. Some of known proteins and the sugars that recognize
Figure 1-2 Representatives of physiologically active sugars found in glycoproteins.
those proteins are shown in Table 1-1.12-20 It is described that different sugar recognizes different target, and are related to different biological phenomena dependent on the target they recognize. Additionally, it is shown that not only the difference in structure of monovalent sugar, but also the difference in combination of sugars (difference in oligosaccharide structure) is related to the lectin recognition. Although many about the lectin recognition of monovalent sugars had been revealed and their specific recognition ability is attractive, there are still unclear regions about the lectin recognition by sugar clusters, and the detail of the sugar-protein interaction is important. Therefore, development of sugar cluster materials and analyses of sugar-protein recognition is important in terms of developing useful biomaterials and understanding the biological phenomena.
Table 1-1 Examples of lectins and the corresponding sugars.12-20
1.1.2 Glyco-cluster effect
Sugars interact specifically to proteins due to its individual frame structure, and the main components of the interactions are hydrogen bond, van der Waals’ interaction, and hydrophobic stacking.21,22 Although interaction between sugar and protein is specific, the bond between monovalent sugar and protein is weak (binding constant Ka = 103 – 104 M-1).23 However, sugars exist as densely gathered domains such as glycoprotein, glycolipid, and glycosaminoglycan on the cell surface, resulting in achieving enhanced interaction to proteins. This is known as the glyco-cluster effect.21,24,25 Proteins that recognize sugars are called lectins,8 and they invariably have multiple sugar-binding pockets.26 For instance, concanavalin A (ConA) is known in recognizing mannose and glucose specifically, and it is tetrahedral shape consisted of four subunits with sugar binding point on the top of each subunits.14,27,28 Hence, the cluster effect is due to multipoint interaction between sugars and protein, which is an enthalpic contribution, and also entropic contribution arising from increment of the binding mode (Figure 1-3).
Figure 1-3 Schematic illustration of the binding modes in sugar-protein interaction.
1.1.3 Glycomaterials
Materials using sugars are attractive as useful biomaterial because of their specific interaction with proteins. Hence, applications using molecular recognition ability of the sugars are being challenged.29-31 For example, drug delivery system,32,33 biosensor,34-36 and immunoassay31,37 are the application areas where glycomaterials exert their performance effectively. Additionally, bringing out the glyco-cluster effect is necessary since those glycomaterials generally require not only specific, but also strong interaction against the target proteins. Hence, for the development of the bio-devices and other bio-related applications using sugars, materials exposing multiple sugar units on their surfaces are prepared. Furthermore, the strategy to exert glyco-cluster effect efficiently is an important topic in the development of glycomaterials.
Generally, the glyco-cluster effect is effective when dense sugars are exposed on the material surface (the more sugar exists, the more efficient cluster effect is possible).
Even on the cell surfaces, glycoproteins and glycolipids exist in micro-domain called raft38,39 and caveolae40,41 in order to form localized sugar-clusters. By mimicking these sugar-clusters and sugar dense surfaces, many kinds of glycomaterials were developed (Figure 1-4).
One simple way to prepare sugar-clustered surface is self-assembled monolayers (SAM).X By immersing material in a solution of organic molecules, those organic molecules self assemble and form a well-oriented monolayer. Most popular method is to immerse a gold substrate in a solution with alkanethiols or their derivatives. By modification of SAM with sugars, a surface with densely packed sugars can be prepared.42-45 Although this is a simple method, non-specific adsorption of proteins onto SAM surfaces by the hydrophobicity of the alkanethiol layer is a concern. However, this technique have broad utility, and can be applied to not only substrates but also
nanoparticles.47-49 Sugar-modified nanoparticles are useful, especially gold nanoparticles since the gold nanoparticles can absorb light in certain wavelength dependent on their sizes, and also the refractive index is high. Therefore, they are used for immunoassay,37 drug delivery reagent,50 and also to increase the detection limit of biosensors.47,48 Another example is a liposome, in which multiple sugars can be presented by incorporation of sugars into the lipid bilayer.51-53 The liposome has biocompatibility and biodegradability, and some materials are encapsulated inside, hence they are capable for biomarkers or drug delivery system. A dendrimer is also an attractive biomaterial because of its uniform chemical structure.54-57 The highly branched structure is suitable for presenting multiple sugars by modifying the terminal of each branch. The number of branches can be increased by additional synthesis step, and its nanostructure and number of sugar presented are controlled. Various glycomaterials have been developed, and more useful and practical glycomaterials are in demand.
Figure 1-4 Examples of glycomaterials.
1.2 Glycopolymer
1.2.1 Glycopolymer for Biomaterial
As mentioned in the former section, many kinds of glycomaterials had been developed, and glycomaterials exerting the glyco-cluster effect efficiently are required for the development of the practical bio-related applications. Another example, glycopolymer is an attractive candidate as a practical glycomaterial. Here, glycopolymer is defined as synthetic polymer which expose sugar units on the side chain just as ornaments of a pendant, and is not a tandem string of sugar chains like the polysaccharides.58-62
There are many merits for selecting glycopolymers in order to develop biomaterials.
First, not like antibodies, the glycopolymers are ordinarily stable to external stimuli such as heat and pH change. Therefore, storage for a long time is possible, and they could be applied for modification of materials in severe conditions such as high temperature, organic solvent, and high/low pH solvent. Secondly, facile synthesis is possible. The glycopolymers are obtained by polymerization of monomers containing sugar unit (glycomonomer), or incorporation of sugar units into a polymer backbone.
Glycomaterials which could be synthesized easily is valuable, especially in terms of mimicking nature products such as oligosaccharides and glycoporteins, because extraction or total synthesis of those sugar-clusters are difficult, and also require numbers of synthesis step and time.63-66 In addition, various polymer design is possible because of the progress in synthesis and polymerization technique (more detail is explained in the following section). The size, architecture and sequence of the monomers could be controlled.67 Another unique feature is that functions could be added by co-polymerization of other monomers with functional units. For instance, first glycopolymer with acrylamide was synthesized in 1975,68 and acrylamide co-polymerized glycopolymer is known as water-soluble polymer.69,70 In addition, our
group reported glycopolymer containing silane coupling reagent.71 This enables glycopolymers to be chemically bound to material surfaces, add flexibility or self-assembling ability, which expands the utility of the glycopolymers.
The synthesis of glycopolymers became much aggressive after 1980s.58,72-76 To date, with progress of polymerization and synthesis methods, many researchers have been reported glycopolymers with various design and polymer conformation for the enhanced interaction against proteins, for instance, simple linear polymer,77 block copolymer,78 star polymer,79 and polymer nanoparticles80 (Figure 1-5).
Figure 1-5 Examples of glycopolymers.
1.2.2 Synthesis of Glycopolymers
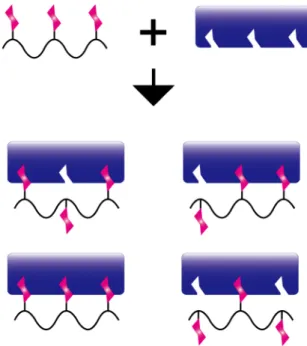
Synthetic glycopolymers are capable of mimicking clustered sugars in nature because of their various possible designs. For the synthesis of glycopolymer, there are two major strategies (Figure 1-6). One of the way is the polymerization of pre-synthesized glycomonomers (Figure 1-6a).81-82 In this method, compound with vinyl group is introduced into the anomeric position of the sugar. Although this is a simple method, generally, hydroxyl groups in sugar have to be protected before introducing the vinyl groups.36,37,71,83,84 The other method is to incorporate sugar unit into the pre-polymerized polymer backbone (Figure 1-6b). For the incorporation of sugar units, click chemistry85-87 such as thiol-ene reaction88 and Huisgen reaction (especially, the azide-alkyne cycloaddition)89,90 have been used. This method may reduce the protection step for the synthesis of glycomonomers.
Figure 1-6 Major strategies for the synthesis of glycopolymer. (a) Polymerization of glycomonomer, and (b) incorporation of sugar units into polymer backbone.
The simplest polymerization method for the synthesis glycopolymer is free radical polymerization.91 Monomers and initiator are dissolved in a solvent, and the polymer chains are propagated from radicals generated by exposure to heat or light. This is a simple and facile method, but the chain length of the polymers could not be controlled well. However, not only a linear polymer but also polymer nanoparticle can be synthesized with this method. Poly(N-isopropylacrylamide) (pNIPAm) is one of the representative gel particle material for its unique feature.92 It is known that the polymerization of NIPAm in water gives pNIPAm hydrogel particles with a diameter of micrometer order.93 This is called precipitation polymerization, and the grown polymer chain collapse upon itself by polymerization in high temperature, which is higher than the lower critical solution temperature (LCST). LCST is the point hydrophobic interaction between the polymer chains become dominant than the polymer chain interaction with the solvent. Furthermore, the free radical polymerization in water under the existence of surfactant lead to synthesis of hydrogel nanoparticle, which enables preparation of three-dimensional polymer chains with the diameter range of few ten nm to few hundred nm.94,95 This synthesis method is known as the pseudo precipitation polymerization (Figure 1-7).
Figure 1-7 Synthesis of hydrogel nanoparticle by pseudo-precipitation polymerization.
Proteins, especially lectins vary in their structure, number of binding points, and molecular size.12-20,26 Especially, the size and the distance between binding points differ in nm order (Figure 1-8). For example, a lectin concanavalin A is consisted of 4 subunits with sugar binding point on the tip of each subunit, and the distance between each binding point is approximately 6.5 nm.14,27,28 Cholera Toxin is a toxin consisted of toxic A subunit and 5 binding B subunits positioned in distance of approximately 3 nm each.96,97 Therefore, synthesis of well-controlled glycopolymers in nm order are necessary for better specificity and binding strength against target lectins.
To achieve the synthesis of such tailored glycopolymers for the better affinity with proteins, living radical polymerization is an attractive method. A simple radical polymerization method for the synthesis of polymer is free radical polymerizaiton, however, the polymer obtained possess wide polydispersity. Living radical polymerization allows control of polymer length, and polymer with narrow
Figure 1-8 Examples of proteins and their sugar binding points.
polydispersity could be obtained. The living radical polymerization was developed in 1990s and since then, massive works aiming for better control of living polymerization were reported.98-101 The representative methods of living radical polymerization are reversible addition chain transfer (RAFT) polymerization,102 atom transfer radical polymerization (ATRP),103,104 and nitroxide-mediated living radical polymerization.105 RAFT polymerization and ATRP are shown again in the latter section. These controlled polymerization techniques enable synthesis of polymers with size variation in nm order.
For strong interaction of glycopolymer with proteins, to exert the glyco-cluster effect by multipoint interaction is necessary. Additionally, lectins vary in their structure and size, hence synthesis of glycopolymer for efficient interaction with the target by controlled polymerization is in demand. It is highly expected that these controlled polymers and synthesis method makes the glycopolymer attractive for the development of biomaterial.
1.2.3 Glycopolymer Interface
For the development of biomaterials using glycopolymers, surface of various materials such as membrane,106 particle,37,84 and substrate36 are grafted with glycopolymers and the interface grabs the key for the material function. In nature, sugars exist as clustered domains on cell surfaces. Hence, precise control of the glycopolymer interface and investigatin the detail of the interface are important matters for development of practicable biomaterial and also understanding biological functions.
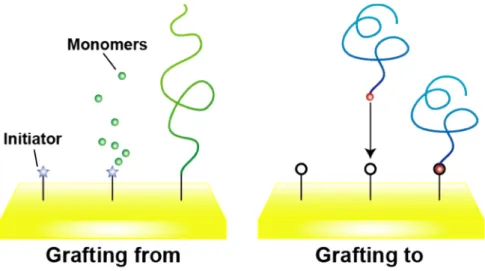
There are two representative methods for modifying polymers on material surfaces.107,108 One is “grafting to” method which polymer is synthesized before immobilizing onto the material surface. In this case, synthesized polymers are chemically bound to the surface by functional group in the polymer or physically adsorbed by adding polymer solution to the material. The other is “grafting from”
method which polymer is grafted by growing the polymer from the initiator previously immobilized on the material surface (Figure 1-9).
Figure 1-9 Illustration of representative methods for grafting polymers on a material surface. Grafting from (left) and grafting to (right) method.
These grafting methods have different features. For instance, in “grafting to”
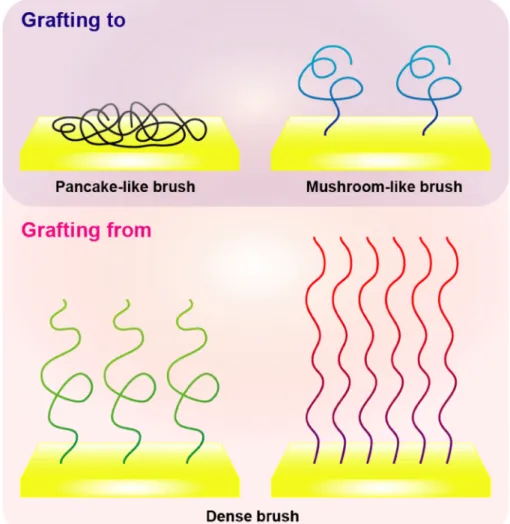
method, polymers could be immobilized onto various kinds of materials by changing the functional group in the polymer. The immobilization density of the polymer is affected by steric hindrance and solvation property of the polymer, and often result in pancake-like or mushroom-like polymer brushes.109 On the other hand, in “grafting from” method, immobilization density of the polymer could be controlled by the amount of initiator, and polymer brush with higher density could be achieved110 (Figure 1-10).
Figure 1-10 Illustration of polymer-brush architecture formed by the difference in grafting methods.
In order to develop useful biomaterial, well-controlled polymer interface is necessary. For precise control of the polymer interface, precise design of the polymer is important. As mentioned in the former section, living radical polymerization is the useful way for preparing well-controlled polymer interface. There are two major living radical polymerization used for the preparation of controlled polymer interface, RAFT polymerizationX and ATRP.X In RAFT polymerization, thiocarbonyl compound is used as the chain transfer agent and polymer with narrow molecular weight distribution could be achieved. In addition, obtained polymer may contain thiol at the terminal after reduction. The terminal thiol could be used to attach the polymer onto material surface, hence this method is often used in the “grafting to” method. ATRP is a method using transition metal complex as a catalyst and organic halogen compound as an initiator. To be more specific, surface initiated ATRP (SI-ATRP)X in which the initiator is immobilized on the material surface, could grow the polymer from the surface. This is a representative “grafting from” method.
1.3 Biosensing and Analysis of Biomolecule Interaction using Optical Methods 1.3.1 Biosensor using Optical Methods
Generally, biosensor is composed of two parts, molecular recognition part and detection part. The molecular recognition part is where ligand biomolecules interact with target biomolecules. For example, antigen-antibody interaction, sugar-protein interaction, and enzyme-substrate interaction are used.113 Those interaction is transduced to certain kind of signal such as heat, electrical signal, frequency, and refractive index change at the detection part. There are various kinds of detection methods that correspond to those signals such as isothermal titration calorimetry technique,114 field emission transistor,115,116 quartz qrystal microbalance method,117,118 and surface plasmon resonance (SPR) technique.119 For development of valuable biosensor, there are several important matter that need to be considered: improvement of detection limit, reduction of noise, design of ligand, surface modification, etc. Among various biosensing methods, optical methods are attractive because of high sensitivity, rapid detection method, versatility, and also require little amount of sample solution.120
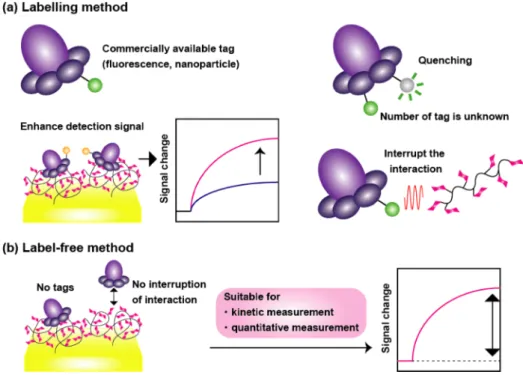
Optical biosensing can be divided into two categories, labelling detection and label-free detection (Figure 1-11). Well known example for the former method is fluorescent-labeled detection, where the biomolecule is labeled with fluorescent tags.121-123 This is widely used method since the equipment is in easy access, and there are many commercial reagents or biomolecules that are already labeled with fluorescent tag. Concern about the fluorescent-labeled detection is that fluorescent tags might affect the biomolecule interaction, and also quantitative analysis is difficult because the number of tags in each molecule is difficult to determine and quenching of fluorescence.
However, labelling detection is still useful for its sensitivity, and there are reports that show improvement of sensitive detection by labelling of molecules by fluorescent tags
and nanoparticles. In contrast, the label-free method enables quantitative analysis and kinetic measurement of biomolecule interaction. Especially in label-free detection using optical method, binding of the target molecule to a ligand is detected by refractive index change, which correlate with concentration of the target molecule. Hence, the measurement can be performed with little amount of sample. Examples of label-free optical biosensors are SPR sensors,124 photonic crystal sensors,36,125,126 and optical fiber sensors.127 In this thesis, it is focused on label-free biosensors.
Figure 1-11 Illustration describing the feature of optical biosensing method by (a) labelling method, and (b) label-free method.
1.3.2 Structural Color for Biosensing
“Colors” are observed as light in phenomena representative in photoluminescence material or fluorescence, in which light was once absorbed in a material and emitted afterwards. On the other hand, there is a phenomenon that colors are observed by the interference of light controlled by nanometer order structure. The observed light is called structural color.128-130 Some examples seen in our daily life are back side of compact disc and rainbow color seen on bubble surface. On the other hand, a lot of structural colors are observed in nature, such as color of morpho butterfly wing, outer shell of insects, bird feather and squama of fish (Figure 1-12).131-133 Hence, many researchers have been trying to reveal the mechanism of structural color observed in nature, and also development of structural color material by biomimetic strategy are in challenge.
Figure 1-12 Examples of structural color found in nature.131-133
An opal134,135 constructed by colloidal crystal is a representative material producing structural color, and one of the basic principle for the expression of the structural color can be expressed by bragg’s law (Figure 1-13). Periodic nanostructure controls the
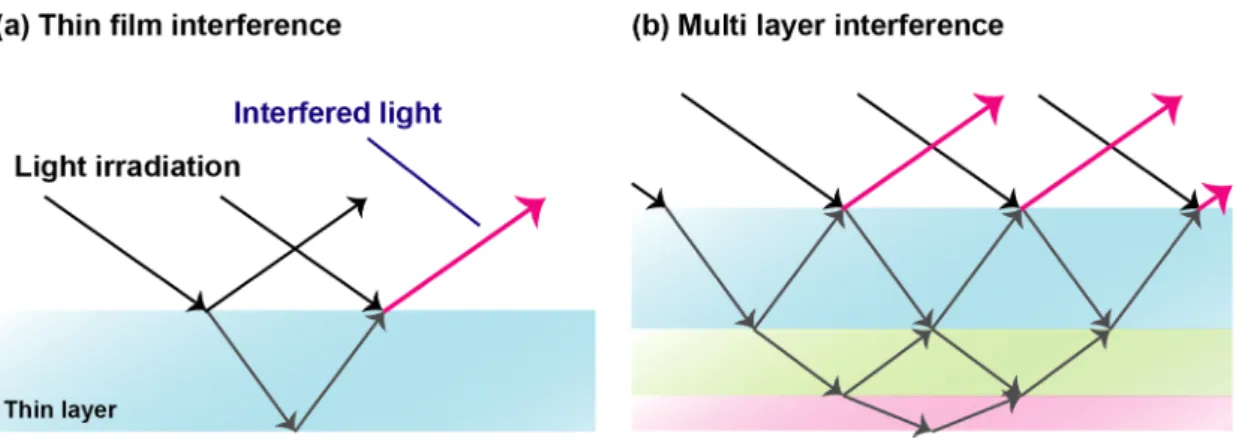
interference of light irradiated onto the structure material, and as a result, reflected (or scattered), and diffracted visible light is observed as color without using any pigments or fluorescence materials. Therefore, a lot of structural colors are observed in thin film materials or multi layered materials (Figure 1-14), and of course, imprinted materials.
Figure 1-13 (a) Illustration of structural color opal, and (b) explanation of bragg’s law.
Figure 1-14 Production of structural color by film materials. Schematic illustration of (a) thin film interference and (b) multi layer interference.
Furthermore, structural color can be observed by light scattering of particles. For instance, blue or red color of sky, and white color of cloud and milk are due to the scattering of light by small particles. The color of the sky is due to the light scattering from particles with a diameter smaller than wavelength of the light, which is an effect of Rayleigh scattering (Figure 1-15a). In contrast, white appearance of the clouds is due to the light scattering from particles with nearly a size of the incident light wavelength, so called Mie scattering (Figure 1-15b). Since the research about the mechanism and the preparation method of structural color materials have been progressed, it is expected to apply structural color for practical devices and materials. Structural color is being applied to materials and devices such as display,136 pigment,137,138 sensor139 and solar cell.140
Figure 1-15 Schematic illustration and explanation for two types of scattering: (a) Rayleigh scattering and (b) Mie scattering.
Among the application using structural color, biosensor is an attractive candidate as application using structural color, because detection using color is one of the simplest method and it does not require complex equipment or measurement system. In addition, labelling is not necessary like method using fluorescence, and there is no concern for color bleaching. For development of biosensor using structural color, photonic crystal36,125,126,141 is a powerful material. The photonic crystal is a periodic structure that has different refractive index from the surroundings aligned in nanometer order (Figure 1-16). Photonic crystal biosensor can detect the biomolecule interaction by change in structural color which occur by the structural change induced by adsorption of molecules on or into the material structure.
Figure 1-16 Images of two-dimensional photonic crystal material.
1.3.3 SPR Technique for Biosensing
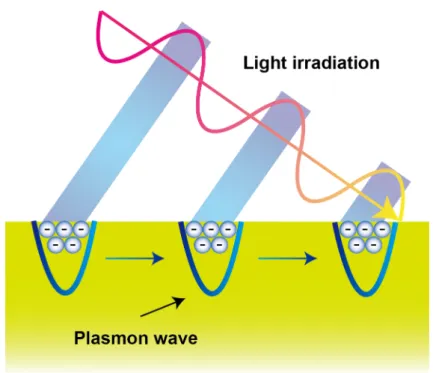
Metals such as gold, silver, and aluminium are delocalized electron rich materials.
Those electrons repel each other, and the electrons are difficult to become dense. When the electrons gather, they travel through the metal as a wave. This state resembles plasma, and those collective oscillation of electrons are called “plasmon” when they are quantized. Hence, surface plasmon is the delocalized electron oscillation at the interface of materials, especially metal-dielectric interface.142
When light is irradiated from one medium to another, both refracting light and reflecting light occur at small incident angle. The relation of the incident angle and the refraction angle can be explained by Snell’s law (Figure 1-17). Furthermore, the
refraction angle reach 90o by increasing the incident angle (this angle is called “critical angle”), and the refraction angle will no longer be observed when the incident angle became higher. This phenomenon is called total internal reflection (TIR). Under TIR condition, electromagnetic field that decay exponentially to the opposite side of the
Figure 1-17 Explanation of Snell’s law (law of refraction).
irradiated surface (“evanescent field” or “evanescent wave”) occurs (Figure 1-18).142 Here, the resonant oscillation of the electrons at the interface occurs by irradiating p-polarized light onto the surface under TIR condition. This physical process is called SPR, and this phenomena had been discovered and studied since 1902.143
To generate SPR on a metal thin film, irradiation of light onto the metal surface is necessary. However, SPR could not be generated simply by irradiating light because the velocity of light is faster than plasmon wave. The dispersion relation of wave can be described by equation (1).142
𝑣 = 2𝜋𝑓
𝑘 = 𝜔
𝑘)*+,- 𝜔 ⋯ 1
Here, f, k, v and w represent frequency, wavenumber, velocity and angular frequency respectively. Light speed c can be described c = 3 × 108 m/s under vacuum state, hence the dispersion relation shows linearity. By contrast, velocity of the SP wave changes by frequency change (Figure 1-19).142 The velocity of SP wave can be described as below:
144
𝑐 𝜀6 𝜔 + 𝜀8 𝜔
𝜀6 𝜔 𝜀8 𝜔 = 𝜔
𝑘89 𝜔 ⋯ 2
Fig. 1-18 Illustration of evanescent field that decay exponentially from the material interface.142
em(w) and es(w) represent the dielectric constant of metal and material on the metal respectively, and ksp(w) is the wavenumber component of SP. When a prism is equipped onto the metal and light was irradiated under TIR condition, wavenumber component of the wave that run along the boundary of evanescent wave kev(w) can be described as shown in equation (3).
𝑘:; 𝜔 = 𝑛9𝑘)*+,- 𝜔 sin 𝜃 ⋯ 3
Here, np and θ represent the refractive index of the prism and the angle of incident respectively, hence the wavenumber of the evanescent wave changes sinusoidally as a function of light incident angle. When ksp(w) shown in equation (2) and kev(w) are coincident, energy of the incident light is used for SPR (Figure 1-20).142 When generating SPR on a certain metal thin film, em(w) stays constant, therefore the irradiation angle for generating SPR θSP changes by the change in es(w).
Fig. 1-19 Relation in the velocity of light and SP wave.142
There are two representative set ups for generating SPR using a prism and light irradiation, Otto configuration145 and Kretschmann configuration.146 In the former set up, evanescent field generated by TIR at the prism surface resonate the SP on the metal surface (Figure 1-21a). Because the evanescent field decay exponentially from the interface, the metal surface was placed close to the prism (~ 100 nm). In the latter set up, the metal was either coated on the prism, or metal deposited glass film was set on the prism. In this case, SP was resonated by the evanescent field from the prism which seeped through to the opposite side of the metal thin film (Figure 1-21b). This Kretschmann configuration is often used for SPR sensors. Figure 1-22 shows the reflectivity change of the light irradiated onto the gold surface (SPR curves) under air or using water as a solvent. The dip is due to the energy used for generating SPR, and the angle at the peak of this dip is θSP. Change of refractive index at the metal thin film surface produces the change in θSP, hence adsorption of molecules onto the metal
Fig. 1-20 SPR generated by irradiation of light onto a metal surface.142
surface, and concentration change of the molecule in the solvent could be measured.
Furthermore, by measureing the reflectivity change at the fixed light irradiation angle, refractive index change at the metal surface can be monitored in real-time. These techniques were used for biosensors, and the well-known BIACORE equipment was developed.147
Figure 1-21 Illustration of (a) Otto configuration, and (b) Kretschmann configuration.
Figure 1-22 Examples of SPR curve in (a) air and (b) water as a solvent.
By using SPR technique, analyses of biomolecule interactions,147 self-assembled monolayer membranes,148 and stimuli responsiveness of polymer brushes149 are possible.
For instance, by measuring θSP change and curve fitting of SPR curve, the thickness of the molecule layer can be obtained.148 By monitoring the reflectivity change of the light irradiated onto the gold thin film, binding analysis (calculation of binding/dissociation constants and binding/dissociation rate constants) of the target molecule to ligands immobilized on the gold surface is possible. Furthermore, using white light as the light source and CCD camera as a detector, measurement of interaction on multiple gold surface can be achieved. This technique is called SPR imaging (SPRI), and is known as a powerful method for screening of many biomolecule interactions (Figure 1-23).150
Figure 1-23 Schematic illustration of SPRI.
In recent years, imaging of single nanoparticle adsorption onto gold thin film was achieved by applying SPRI to a microscope (Figure 1-24).151 All of these SPR techniques can be performed with non-labeled biomolecules. Considering these various abilities of SPR techniques, SPR sensor is an attractive biosensor.
Fig. 1-24 Schematic illustration of SPRI microscope for the measurement of hydrogel nanoparticle adsorption onto a gold thin film.X
1.4 Aim of the Research
Sugars interact specifically with proteins, and the sugar-protein interaction is related to many important biological phenomena such as virus/pathogenic infection, metastasis of cancer, inflammation, etc. The interaction of monovalent sugar to a protein is relatively weak, however sugars generally exist in dense domains to enhance the interaction against protein. This glyco-cluster effect can be also brought out by synthesis of glycopolymer, which is a sugar-cluster presenting sugar units on the side chain. Hence, the synthesis and analysis of glycopolymer-protein interaction are notable topics for development of useful biomaterials. For the anlaysis of sugar-protein interaction, optical biosensing methods are attractive. Optical biosensors possess high sensitivity, and facile detection is possible. Furthermore, optical biosensors detect change in refractive index, and require a little amount of sample. As optical biosensing methods, photonic crystal and SPR techniques were focused in this thesis. The photonic crystals produce visible colors (structural color) by controlling the interference of light with its periodic nanostructure. The change in the intensity of structural color from the photonic crystal was used for the detection of protein. The SPR techniques are capable of detecting biomolecule adsorption onto a metal surface by the change in refractive index at the metal surface. By this technique, thickness of molecule layer on a metal surface, kinetic analysis and screening of biomolecule interaction are possible. In this thesis, various types of glycopolymers were synthesized, and their interaction with proteins were analyzed using optical biosensors.
In chapter 2, structural color biosensor immobilized with sugar-incorporating hydrogel nanoparticle was discussed. Hydrogel nanoparticle containing sugar units was synthesized by pseudo-precipitation polymerization. The hydrogel nanoparticles were immobilized onto a two-dimensional photonic crystal film by mediation of polymer and
protein. The adsorption of target lectin concanavalin A onto the sugar-incorporating hydrogel nanoparticle immobilized photonic crystal film was detected by the change in reflectivity change of the structural color.
In chapter 3, analysis of glycopolymer-brush interface on a gold film and its interaction with lectin using SPR technique were discussed. Glycopolymers with different sugar-incorporation ratio were synthesized by RAFT polymerization, and immobilized onto a gold surface. The thickness of the glycopolymer was estimated from the fitting against SPR curve of the glycopolymer immobilized gold film. Furthermore, the kinetic analysis of protein interaction to the glycopolymer-brush interface was perfomed by the time-dependent reflectivity change of the light irradiated onto the gold surface. Then the correlation between the glycopolymer-brush structure and protein recognition was discussed.
In chapter 4, screening protein interaction against glycopolymer array on a gold chip using SPRI method was discussed. Glycopolymers with different type of sugar units and also multiple sugar units were synthesized by RAFT polymerization and post-click chemistry. The interaction of the binding subunit of cholera toxin to the glycopolymer was enhanced by incorporating multiple types of sugar units into a polymer backbone.
References
1. A. Dell, H. R. Morris, Science 2001, 291, 2531-2356.
2. C. A. Bush, M. Martin-Pastor, Annu. Rev. Biophys. Struct. 1999, 28, 269-203.
3. T. Yamakawa, Y. Nagai, Trends in Biochemical Sciences 1978, 3, 128-131.
4. P. B. Savage, L. Teyton, A. Bendelac, Chem. Soc. Rev. 2006, 35, 771-779.
5. D. Sawitzky, Med Microbiol Immunol 1996, 184, 155-161.
6. R. E. Hileman, J. R. Fromm, J. M. Weiler, R. J. Linhardt, Bioessays 1998, 20, 156-167.
7. M. E. Taylor, K. Drickamer, Introduction to Glycobiology, Oxford University Press, Oxford, 2nd edn, 2002.
8. N. Sharon, H. Lis, Glycobiology 2004, 14, 53-62.
9. D. H. Dube, C. R. Bertozzi, Nature Reviews Drug discovery 2005, 4, 477-488.
10. C. Slawson, G. W. Hart, Nature Reviews Cancer 2011, 11, 678-684.
11. M. Worth, H. Li, J. Jiang, ACS Chem. Biol. 2017, 12, 326-335.
12. N. Shibuya, K. Tazaki, Z. Song, G. E. Tarr, I. J. Goldstein, W. J. Peumans, J.
Biochem. 1989, 106, 1098-1103.
13. N. M. Young, R. A. Z. Johnston, D. C. Watson, Eur. J. Biochem. 1991, 196, 631-637.
14. D. K. Mandal, N. Kishore, C. F. Brewer, Biochemistry 1994, 33, 1149-1156.
15. R. Banerjee, S. C. Mande, V. Ganesh, K. Das, V. Dhanaraj, S. K. Mahanta, K.
Suguna, A. Surolia, M. Vijayan, Proc. Natl. Acad. Sci. USA 1994, 91, 227-231.
16. H. Lis, N. Sharon, Chem. Rev. 1998, 98, 637-674.
17. E. E. Simanek, G. J. McGarvey, J. A. Jablonowski, C. H. Wong, Chem. Rev. 1998, 98, 833-862.
18. N. kaila, B. E. Thomas Ⅳ, Medicinal Research Reviews 2002, 22, 566-601.
19. H. E. Murrey, L. C. Hsieh-Wilson, Chem. Rev. 2008, 108, 1708-1731.
20. M. Aureli, L. Mauri, M. G. Ciampa, A. Prinetti, G. Toffano, C. Secchieri, S.
Sonnino, Mol Neurobiol 2016, 53, 1824-1842.
21. R. T. Lee, Y. C. Lee, Glycoconj. J. 2000, 17, 543-551.
22. J. Holgersson, A. Gustafsson, M. E. Breimer, Immunology and Cell Biology 2005, 83, 694-708.
23. T. K. Dam, C. F. Brewer, Chem. Rev. 2002, 102, 387-429.
24. Y. C. Lee, R. T. Lee, Acc. Chem. Res. 1995, 28, 321-327.
25. M. Mammen, S. K. Choi, G. M. Whitesides, Angew. Chem. Int. Ed. 1998, 37, 2754-2794.
26. T. B. H. Geitenbeek, S. I. Gringhuis, Nature Reviews Immunology 2009, 9, 465-479.
27. A. Jack, J. Weinzierl, A. J. Kalb, J. Mol. Biol. 1971, 58, 389-395.
28. Z. Derewenda, J. Yariv, J. R. Helliwell, A. J. Kalb, E. J. Dodson, M. Z. Papiz, T.
Wan, J. Campbell, The EMBO journal 1989, 8, 2189-2193.
29. G. Coullerez, P. H. Seeberger, M. Textor, Macromol Biosci. 2006, 6, 634-647.
30. S. Cecioni, Anne Imberty, S. Vidal, Chem. Rev. 2015, 115, 525-561.
31. A. Restuccia, M. M. Fettis, G. A. Hudalla, J. Mater. Chem. B 2016, 4, 1569-1585.
32. B. G. Davis, M. A. Robinson, Current Opinion in Drug Delivery & Development 2002, 5, 279-288.
33. S. Pearson, W. Scarano, M. H. Stenzel, Chem. Commun. 2012, 48, 4695-4697.
34. K. El-Boubbou, C. Gruden, X. Huang, J. Am. Chem. Soc. 2007, 129, 13392-13393.
35. S. Cunnigham, J. Q. Gerlach, M. Kane, L. Joshi, Analyst 2010, 135, 2471-2480.
36. Y. Terada, W. Hashimoto, T. Endo, H. Seto, T. Murakami, H. Hisamoto, Y. Hoshino, Y. Miura, J. Mater. Chem. B 2014, 2, 3324-3332.
37. M. Takara, M. Toyoshima, H. Seto, Y. Hoshino, Y. Miura, Polym. Chem. 2014, 5, 931-939.
38. K. Simons, D. Toomre, Nature Reviews Molecular cell biology 2000, 1, 31-39.
39. J. A. Allen, R. A. Halverson-Tamboli, M. M. Rasenick, Nature Reviews Neuroscience 2007, 8, 128-140.
40. B. Razani, S. E. Woodman, M. P. Lisanti, Pharmacological reviews 2002, 54, 431-467.
41. R. G. Parton, K. Simons, Nature Reviews Molecular cell biology 2007, 8, 185-194.
42. B. T. Houseman, M. Mrksich, Angew. Chem. Int. Ed. 1999, 38, 782-785.
43. M. Kleinert, N. Röckendorf, T. K. Lindhorst, Eur. J. Org. Chem. 2004, 18, 3931-3940.
44. Y. Miura, T. Yamauchi, H. Sato, T. Fukuda, Thin Solid Films 2008, 516, 2443-2449.
45. T. Weber, V. Chandrasekaran, I. Stamer, M. B. Thygesen, A. Terfort, T. K.
Lindhorst, Angew. Chem. Int. Ed. 2014, 53, 14583-14586.
46. J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo, G. M. Whitesides, Chem. Rev.
2005, 105, 1103-1169.
47. M. D. Malinsky, K. Lance, Kelly, G. C. Schatz, R. P. Van Duyne, J. Am. Chem. Soc.
2001, 123, 1471-1482.
48. S. Morokoshi, K. Ohhori, K. Mizukami, H. Kitano, Langmuir 2004, 20, 8897-8902.
49. C. R. Yozon, E. Jeoung, S. Zou, G. C. Schatz, M Mrksich, R. P. Van Duyne, J. Am.
Chem. Soc. 2004, 126, 12669-12676.
50. M. H. Stenzel, Chem. Commun. 2008, 30, 3486-3503.
51. N. Jayaraman, K. Maiti, K. Naresh, Chem. Soc. Rev. 2013, 42, 4640-4656.
52. J. Zhu, J. Xue, Z. Guo, L. Zhang, R. E. Marchant, Bioconjugate Chem. 2007, 18, 1366-1369.
53. Y. Ma, I. Sobkiv, V. Gruzdys, H. Zhang, X. L. Sun, Anal. Bioanal. Chem. 2012, 404, 51-58.
54. J. J. Lundquist, E. J. Toone, Chem. Rev. 2002, 102, 555-578.
55. M. Gingras, Y. M. Chabre, M. Roy, R. Roy, Chem. Soc. Rev. 2013, 42, 4823-4841.
56. T. Fukuda, S. Onogi, Y. Miura, Thin Solid Films 2009, 518, 880-888.
57. Y. Miura, S. Onogi, T. Fukuda, Molecules 2012, 17, 11877-11896.
58. V. Ladmira, E. Melia, D. M. Haddleton, European Polymer Jouranal 2004, 40, 431-449.
59. S. R. S. Ting, G. Chen, M. H. Stenzel, Polym. Chem. 2010, 1, 1392-1412.
60. Y. Miura, Polymer Journal 2012, 44, 679-689.
61. M. Ahmed, P. Wattanaarsakit, R. Narain, European Polymer Journal 2013, 49, 3010-3033.
62. Y. Miura, Y. Hoshino, H. Seto, Chem. Rev. 2016, 116, 1673-1692.
63. A. Kameyama, H. Ishida, M. Kiso, A. Hasegawa, Carbohydrate Research 1991, 209, c1-c4.
64. S. J. Danishefsky, M. T. Bilodeau, Angew. Chem. Int. Ed. Engl. 1996, 35, 1380-1419.
65. V. Kress, Nature 1997, 389, 587-591.
66. T. K. K. Mong, H. K. Lee, S. G. Durón, C. H. Wong, Proc. Nat. Acad. Sci. 2003, 100, 797-802.
67. Y. Abdouni, G. Yilmaz, C. R. Becer, Macromol. Rapid. Commun. 2017, in press.
68. R. L. Schnaar, Y. C. Lee, Biochemistry 1975, 14, 1535-1541.
69. V. Hořejší, P. Smolek, J. Kocourek, Biochimica et Biophysica Acta 1978, 538,
293-298.
70. R. Roy, F. D. Tropper, A. Romanowska, Bioconjugate Chem. 1992, 3, 256-261.
71. H. Seto, Y. Ogata, T. Murakami, Y. Hoshino, Y. Miura, ACS Appl. Mater. Interfaces 2012, 4, 411-417.
72. K. Kobayashi, H. Sumitomo, Y. Ina, Polymer Jouranal 1985, 4, 567-575.
73. R. Roy, F. O. Andersson, G. Harms, S. Kelm, R. Schauer, Angew. Chem. Int. Ed.
Engl. 1992, 31, 1478-1481
74. K. Nagata, T. Furuike, S. Nishimura, J. Biochem. 1995, 118, 278-284.
75. M. Mammen, G. Dahmann, G. M. Whitesides, J. Med. Chem. 1995, 38, 4179-4190.
76. G. B. Sigal, M. Mammen. G. Dahmann, G. M. Whitesides, J. Am. Chem. Soc. 1996, 118, 3789-3800.
77. B. D. Polizzotti, K. L. Kiick, Biomacromolecules 2006, 7, 483-490.
78. K. Jono, M. Nagao, T. Oh, S. Sonoda, Y. Hoshino, Y. Miura, Chem. Commun. 2017, in press.
79. Y. Chen, G. Chen, M. H. Stenzel, Macromolecules 2010, 43, 8109-8114.
80. Y. Hoshino, M. Nakamoto, Y. Miura, J. Am. Chem. Soc. 2012, 134, 15209-152121.
81. S. G. Spain, M. I. Gibson, N. R. Cameron, Journal of Polymer Science Part A Polymer Chemitry 2007, 45, 2059-2072.
82. C. R. Becer, Macromol. Rapid Commun. 2012, 33, 742-752.
83. K. Sasaki, Y. Nishida, T. Tsurumi, H. Uzawa, H. Kondo, K. Kobayashi, Angew.
Chem. Int. Ed. 2002, 41, 4463-4467.
84. M. Toyoshima, T. Oura, T. Fukuda, E. Matsumoto, Y. Miura, Polymer Journal 2010, 42, 172-178.
85. J. E. Moses, A. D. Moorhouse, Chem. Soc. Rev. 2007, 36, 1249-1262.
86. W. H. Binder, R. Sachsenhofer, Macromol. Rapid Commun. 2007, 28, 15-24.
87. V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh, X. Chen, Chem.
Rev. 2016, 116, 3086-3240.
88. C. E. Hoyle, C. N. Bowman, Angew. Chem. Int. Ed. 2010, 49, 1540-1573.
89. R. Huisgen, Angew. Chem. Int. Ed. 1963, 2, 565-598.
90. J. E. Hein, V. V. Fokin, Chem. Soc. Rev. 2010, 39, 1302-1315.
91. K. Ohno, T. Fukuda, H. Kitano, Macromol. Chem. Phys. 1998, 199, 2193-2197.
92. H. G. Schild, Prog. Polym. Sci. 1992, 17, 163-249.
93. R. H. Pelton, P. Chibante, Colloids and Surfaces 1986, 20, 247-256.
94. S. Nayak, L. A. Lyon, Angew. Chem. Int. Ed. 2005, 44, 7686-7708.
95. B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu, R. Stepto, Advances in Colloid and Interface Science 2009, 147-148, 251-262.
96. E. A. Merritt, S. Sarfaty, F. Van Den Akker, C. L’Hoir, J. A. Martial, W. G. J. Hol, Protein Sci. 1994, 3, 166-175.
97. R. G. Zhang, D. L. Scott, M. L. Westbrook, S. Nance, B. D. Spangler, G. G.
Shipley, E. M. Westbrook, J. Mol. Biol. 1995, 251, 563-573.
98. K. Matyjaszewski, J. Xia, Chem. Rev. 2001, 101, 2921-2990.
99. C. J. Hawker, A. W. Bosman, E. Harth, Chem. Rev. 2001, 101, 3661-3688.
100. M. Kamigaito, T. Ando, M. Sawamoto, Chem. Rev. 2001, 101, 3689-3745.
101. B. M. Rosen, V. Percec, Chem. Rev. 2009, 109, 5069-5119.
102. J. Chiefari, Y. K. B. Chong, F. Ercole, J. Kristina, J. Jeffery, T. P. T. Le, R. T. A.
Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559-5562.
103. M. Kato, M. Kamigaito, M. Sawamoto, T. Higashimura, Macromolecules 1995, 28, 1721-1723.
104. J. S. Wang, K. Matyjaszewski, J. Am. Chem. Soc. 1995, 117, 5614, 5615.
105. R. D. Puts, D. Y. Sogah, Macromolecules 1996, 29, 3323-3325.
106. Y. Ogata, H. Seto, T. Murakami, Y. Hoshino, Y. Miura, Membranes 2013, 3, 169-181.
107. B. Zhao, W. J. Brittain, Prog. Polym. Sci. 2000, 25, 677-710.
108. W. J. Brittain, S. Minko, Journal of Polymer Science Part A Polymer Chemistry 2007, 45, 3505-3512.
109. A. Bousquet, H. Awada, R. C. Hiorns, C. Dagron-Lartigau, L. Billon, Prog. Polym.
Sci. 2014, 39, 1847-1877.
110. T. Wu, K. Efimenko, J. Genzer, J. Am. Chem. Soc. 2002, 124, 9394-9395.
111. C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu, S. Perrier, Chem. Rev. 2009, 109, 5402-5436.
112. J. B. Kim, M. L. Bruening, G. L. Baker, J. Am. Chem. Soc. 2000, 122, 7616-7617.
113. S. P. Mohanty, E. Kougianos, Ieee Potentials 2006, 25, 35-40.
114. I. Jelesarov, H. R. Bosshard, J. Mol. Recognit. 1999, 12, 3-18.
115. L. Torsi M. Magliulo, K. Manoli, G. Palazzo, Chem. Soc. Rev. 2013, 42, 8612-8628.
116. D. Sarkar, W. Liu, X. Xie, A. C. Anselmo, S. Mitragotri, K. Banerjee, ACS Nano 2014, 8, 3992-4003.
117. K. A. Marx, Biomacromolecules 2003, 4, 1099-1120.
118. M. A. Cooper, V. T. Singleton, J. Mol. Recognit. 2007, 20, 154-184.
119. X. Guo, J. Biophotonics 2012, 5, 483-501.
120. X. Fan, I. M. White, S. I. Shopova, H. Zhu, J. D. Suter, Y. Sun, Analytica Vhimica Acta 2008, 620, 8-26.
121. U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke, T. Nann, Nature
Methods 2008, 5, 763-775.
122. W. R. Algar, A. J. Tavares, U. J. Krull, Analytica Chimica Acta 2010, 673, 1-25.
123. X. Feng, L. Liu, S. Wang, D. Zhu, Chem. Soc. Rev. 2010, 39, 2411-2419.
124. K. Inamori, M. Kyo, Y. Nishiya, Y. Inoue, T. Sonoda, E. Kinoshita, T. Koike, Y.
Katayama, Anal. Chem. 2005, 77, 3979-3985.
125. T. Endo, M. Sato, H. Kajita, N. Okuda, S. Tanaka, H. Hisamoto, Lab Chip 2012, 12, 1995-1999.
126. T. Endo, C. Ueda, H. Kajita, N. Okuda, S. Tanaka, H. Hisamoto, Microchim Acta 2013, 180, 929-934.
127. M. Wakao, S. Watanabe, Y. Kurahashi, T. Matsuo, M. Takeuchi, T. Ogawa, K.
Suzuki, T. Yumino, T. Myogadani, A. Saito, K. Muta, M. Kimura, K. Kajikawa, Y.
Suda, Anal. Chem. 2017, 89, 1086-1091.
128. S. Kinoshita, S. Yoshioka, ChemPhysChem 2005, 6, 1442-1459.
129. O. Sato, S. Kubo, Z. Z. Gu, Accounts of Chemical Research 2009, 42, 1-10.
130. Y. Zhao, Z. Xie, H. Gu, G. Z. Zhu, Z. Gu, Chem. Soc. Rev. 2012, 41, 3297-3317.
131. S. Kinoshita, S. Yoshioka, J. Miyazaki, Rep. Prog. Phys. 2008, 71, 076401.
132. E. Shevtsova, C. Hansson, D. H. Janzen, J. Kjaerandsen, Proc. Nat. Acad. Sci.
2011, 108, 668-673.
133. X. Yang, Z. Peng, H. Zuo, T. Shi, G. Liao, Sensors and Actuators A 2011, 167, 367-373.
134. O. L. J. Pursiainen, J. J. Baumberg, H. Winkler, B. Viel, P. Spahn, T. Ruhl, Optics Express 2007, 15, 9533-9561.
135. Y. Takeoka, J. Mater. Chem C 2013, 1, 6059-6074.
136. D. Ge, E. Lee, L. Yang, Y. Cho, M. Li, D. S. Gianola, S. Yang, Adv. Mater. 2015, 27, 2489-2495.
137. Y. Takeoka, S. Yoshioka, A. Takano, S. Arai, K. Nueangnoraj, H. Nishihara, M.
Teshima, Y. Ohtsuka, T. Seki, Angew. Chem. Int. Ed. 2013, 52, 7261-7265.
138. S. H. Kim, J. G. Park, T. M. Choi, V. N. Manoharan, D. A. Weitz, Nature Commun.
2014, 5.
139. M. A. Haque, T. Kurokawa, G. Kamita, Y. Yue, J. P. Gong, Chem. Mater. 2011, 23, 5200-5207.
140. D. Colonna, S. Colodrero, H. Lindström, A. D. Carlo, H. Míguez, Energy Environ.
Sci. 2012, 5, 8238-8243.
141. S. M. Shamah, B. T. Cunningham, Analyst 2011, 136, 1090-1102.
142. 永田和宏、半田宏、生体物質相互作用のリアルタイム解析実験法:BIACORE
を中心に、1998年、シュプリンガー。
143. R. W. Wood, Philysophical magazine 1902, 4, 396-402.
144. M. Puiu, C. Bala, Sensors 2016, 16, 870.
145. A. Otto, Phys. Stat. Sol. 1968, 26, K99-K101.
146. E. Kretschmann, H. Reather, Z. Naturf. 1968, 23A, 2135-2136.
147. B. Liedberg, C. Nylander, I. Lundström, Biosensors & Bioelectronics 1995, 10, i-ix.
148. S. Balamurugan, S. Mendez, S. S. Balamurugan, M. J. O’Brien II, G. P. López, Langmuir 2003, 19, 2545-2549.
149. Y. Yokota, A. Miyazaki, K. Fukui, T. Enoki, K. Tamada, M. Hara, J. Phys. Chem. B 2006, 110, 20401-20408.
150. E. A. Smith, W. D. Thomas, L. L. Kiessling, R. M. Corn, J. Am. Chem. Soc. 2003, 125, 6140-6148.
151. A. M. Maley, Y. Terada, S. Onogi, K. J. Shea, Y. Miura, R. M. Corn, J. Phys. Chem.
C 2016, 120, 16843-16849.
Chapter 2
Biosensing Protein Recognition by Glycopolymer using Structural Color produced from Two-dimensional
Photonic Crystal
Biosensing Protein Recognition by Glycopolymer using Structural Color produced from Two-dimensional Photonic Crystal
Abstract
A two-dimensional photonic crystal (2D-PhC) biosensor immobilized with hydrogel nanoparticle containing sugar units capable of detecting protein adsorption was developed (Figure 2-1). Glycopolymers with two different polymer conformations, linear sugar homopolymer and sugar-incorporating hydrogel nanoparticle were synthesized by free radical polymerization, and were immobilized onto the nano-imprinted 2D-PhC film using intermediate polymer and lectin. The surface modification of 2D-PhC film was analyzed in detail using atomic force microscope (AFM) and x-ray photoelectron spectroscopy (XPS), and the glycopolymer-protein interaction was detected by changes in the reflection intensity of structural color produced by the 2D-PhC film. The synthesized glycopolymers were immobilized onto the 2D-PhC film successfully, and specific interactions of protein with the glycopolymers were detected successfully. Enhanced interaction was present between the glycopolymers and the target proteins because of the glyco-cluster effect. The sugar-incorporating hydrogel nanoparticles showed larger binding capacity compared to
Figure 2-1 Schematic illustration of 2D-PhC biosensor with sugar-incorporating hydrogel nanoparticle.
the sugar homopolymers because of its three-dimensional polymer network, which contributed to the detection of low concentrated protein (6.0 ng / mL). The protein detection limit of the developed 2D-PhC biosensor was lower than that of the high sensitive SPR biosensor (1.43 µg / mL). The results indicated the potential of the optical biosensor using sugar-incorporating hydrogel nanoparticle as the molecular recognition part for the high-sensitive and easy detection of proteins.
2.1 Introduction
There is an increasing need for high-sensitive and accurate biosensors that can detect the infection disease, cancer, pathogens, or toxins, and those biosensors are also useful for the basic biological science. Biosensors typically consist of two parts; the detection part and the molecular recognition part. Various detection methods are utilized for the biosensor such as optical analysis methods (e.g. fluorescence method1, surface plasmon resonance technique2, structural color3 etc), electrochemistry4, quartz crystal microbalance5 and field emission transistor.6 Among them, optical analysis methods are promising due to its high sensitivity and also the recent development of the laser technology. Structural colors are one of the optical analyzing methods that enable simple and rapid detection of the target molecules.3 Because of dye-free color creation, the production of durable colors with long-term retention properties is possible. There have been several ambitious reports for biosensor using structural colors of photonic crystals (PhC). 7-17 Asher et al. reported PhC biosensors using crystalline colloidal arrays,18 and Takeoka et al. reported colorimetric glucose sensors using inverse opal hydrogels.19 In the former reports, they prepared the PhC which was successful in the biosensing, however, at the same time, preparing the PhC is tedious for preparation of versatile sensing devices.
On the other hand, Endo et al. previously reported PhC biosensors based on the structural color of nano-imprinted two-dimensional PhC (2D-PhC), where the PhC with two-dimensional patterns could be produced easily and utilized by a simple modification step such as nano-imprinting.20 They have also reported the detection of antigen-antibody reactions using the 2D-PhC.21 Therefore, the nano-imprinted 2D-PhC is a novel and convenient materials for biosensing based on the structural color. One important thing in PhCs is the appropriate surface modification for immobilization of biomacromolecules. So far, Miura group reported the surface modification using polymers, where they have achieved the protein detection using glycopolymers.22 In the reports, the modification of various materials with glycopolymers such as gold (nanoparticles22 and substrates23) and siliceous materials (glass,24 silica,25,26 and silicon wafers27) were achieved.
In the view of molecular recognition for biosensors, the glycopolymers are attractive, because sugars play important roles in vivo. It is known that sugars on cell surfaces interact specifically with proteins.28,29 It has been discovered that sugar-protein interactions are involved in various biological phenomena, including pathogenic infections, metastasis of cancer, cell adherence.30 In human bodies, sugars exist in clustered domains such as glycoproteins, glycolipids, and glycosaminoglycans. These compact, dense sugar structures result in enhanced interactions of the sugars and proteins; this is known as the gluco-cluster effect.31 The glyco-cluster effect can be achieved via the synthesis of glycopolymers. Because glycopolymers are capable of binding with proteins strongly and specifically, they have the potential as highly effective molecular recognition part for biosensors. We have previously reported glycopolymers and sugar-incorporating hydrogel nanoparticles that interact with target molecules strongly and specifically.22-27
In this study, we developed the sugar-incorporating hydrogel nanoparticle immobilized 2D-PhC biosensor for the detection of proteins. In the previous work, Endo et al immobilized polymers containing succinimide group and silane coupling agent on 2D-PhC.20,21 In this study, the surface modification of 2D-PhC was analyzed in detail. A lectin concanavalin A (ConA) was used as a model target protein. Mannose was used as sugar units in glycopolymers to achieve the specific recognition of ConA. It is known that ConA binds specifically to mannose.32,33 Two different types of polymers—specifically, mannose homopolymer (ManHP), and mannose-incorporating hydrogel nanoparticle (ManNP)—were synthesized. These glycopolymers were immobilized onto the nano-imprinted cyclo-olefin polymer film using an intermediary consisting of the succinimide-containing polymer and ConA successfully. The strong and specific interactions between the mannose-incorporating polymers and ConA were detected by monitoring the changes in the reflection intensity of the 2D-PhC film.
Because sugars act as ligands for various proteins, bacteria, and viruses, sugar-incorporating polymers can be used for the recognition of various kinds of biomolecules.34-36 The results of this study suggested that the developed biosensor has great potential, and could be applied for the development of 2D-PhC biosensors.
2.2 Experimental section 2.2.1 Reagents
The following chemicals were purchased from commercial sources, and were used as received: N-acryloxysuccinimide (NAS), N,N’-methylenebisacrylamide (BIS), and 3,3’-dithiodipropionic acid were obtained from Tokyo Chemical Industry Co., Ltd.
(Tokyo, Japan); (trimethoxysilyl) propyl methacrylate (TMSMA), poly ethylene glycol ethyl ether methacrylate (PEGMA), and bovine serum albumin (BSA) were obtained