博 士 学 位 論 文
Development of Visible Light-driven Carbon-carbon
Bond Forming Reactions with Perylene-based
Photoredox Catalysts
近畿大学大学院
総合理工学研究科 物質系工学専攻
博 士 学 位 論 文
Development of Visible Light-driven Carbon-carbon
Bond Forming Reactions with Perylene-based
Photoredox Catalysts
平 成 30 年 1 月
近畿大学大学院
総合理工学研究科 物質系工学専攻
Contents
General Introduction………...1
Chapter 1 1-1. Introduction………...………....………...………17
1-2. Coupling Reaction of Benzaldehyde Using Perylene as a PRC....20
1-3. Coupling Reaction of Aldehydes and Ketones Using Perylene as a PRC...25
1-4. Possible Reaction Mechanism………..……….27
1-5. Conclusions………...………28 1-6. Experimental Details………...…………29 1-7. References………..………...…………35 1-8. Supplementary Data………...…..……...…………36 Chapter 2 2-1. Introduction………...………....………...………42 2-2. Synthesis of Urethane-perylene ……...…...………...……..43
2-3. Coupling Reaction of Ketones Using Urethane-perylene as a PRC...46
2-4. Coupling Reaction of Aldehydes Using Urethane-perylene as a PRC………..……….49
2-5. Possible Reaction Mechanism……...………...………51
2-6. Conclusions………..……...…………...………..………54
2-7. Experimental Details………...….………55
2-8. References……….………...….………61
2-10. Computational Methods………..………...….………83
Chapter 3 3-1. Introduction………...………....………...………88
3-2. Coupling Reaction of N-Benzylbenzaldimine Using Perylene as a PRC………...91
3-3. C o u p l i n g R e a c t i o n o f I m i n e s U s i n g P e r y l e n e a s a PRC...95
3-4. Possible Reaction Mechanism………..……….………99
3-5. Application to Crosslinking of Poly(imine)…….……….100
3-6. Conclusions………...……..…103 3-7. Experimental Details………....……..…104 3-8. References……….………...……….…116 3-9. Supplementary Data…………..………...………...……..118 General Conclusions………...130 Publication List……….………...132 Acknowledgments
1
General Introduction 1. Photoredox Catalyst
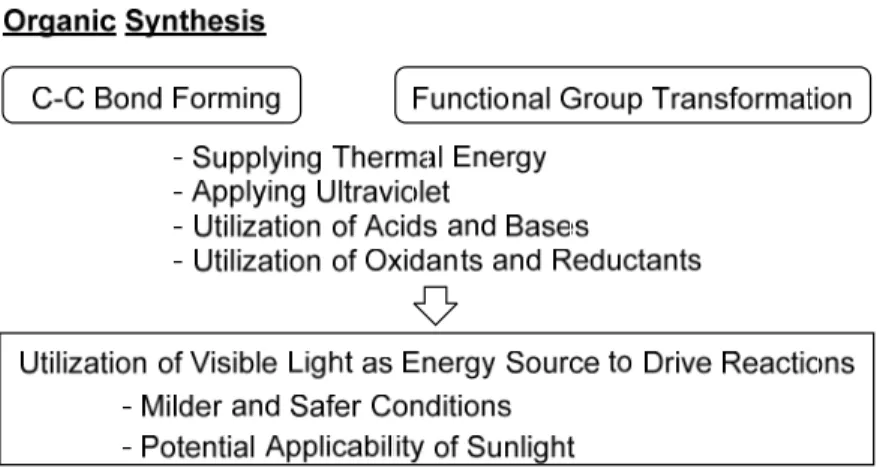
Development of efficient methods for carbon-carbon bond forming has been the core of organic chemistry. So far, those methods established through the tremendous efforts of researchers have brought about the current prosperity of the pharmaceutical, electronics, and aerospace industries. In those methods, enolates, organometallics containing main group elements, carbocations, carbenes, and radicals are used and thus the efficiency in their formations dominates the total reaction efficiency. Conventionally, the formation of such reactive species has required consumption of considerable amount of energy, i.e., applying heat, irradiating UV, and employing highly reactive reagents such as metallic oxidants and reductants (Figure 1-1). In contrast, the current important trend in the field of organic chemistry is to make the reactions safer, less expensive, and more sustainable by liberating them from thermal energy consumption and utilization of highly reactive metal reagents. In the context of such requirements growing unprecedentedly, utilization of visible light as an energy source for carbon-carbon bond forming is a promising concept that would fulfill the requirements.
2
The reasons why the utilization of visible light as an energy source in organic reactions is so attractive are 1) visible light is much less harmful than the other electromagnetic waves such as ultraviolet and 2) it may be supplied abundantly from the
sun light. h
powerful to activate organic molecules from its ground state to transition states. Therefore, there have been only a few reactions of organic molecules that can be driven directly by irradiation of visible light. Isomerization of photochromic molecules is a such example. From a couple of decades ago, it has been clarified that a variety of molecules with low band gaps can be activated upon absorption of visible light and then serve as reductants and oxidants during the quenching process. These molecules have been employed as “photoredox catalysts (PRCs)” in various reactions that can be driven by irradiation of visible light.1-5 Such molecules applicable as photo-redox catalysts includes transition metal complexes and organic dyes (Figure 1-2).
3
As shown in Figure 1-3, there are two routes for the excited PRC towards the ground state: 1) In route A, it can act as a reductant with releasing one electron to become a radical cation, then the radical cation can act as an oxidant. 2) In route B, it can act as an oxidant with accepting one electron to become a radical anion, then the radical anion can act as a reductant. In both the cases, two processes accompanying single electron transfer, which are orthogonal with each other, can be combined into a reaction system, giving rise to generation of two different species (radical anion of X and radical cation of Y). The resulting reactive species bearing different charges would undergo coupling into a new molecule X-Y or interconversion into molecules X’ and Y’.
4
Among a variety of the PRCs developed so far, ruthenium complexes and iridium complexes bearing plural pyridyl ligands have been most extensively studied and thus highly appreciated as promising PRCs for various reactions. Their photo-excited states have long-lives because of the intersystem crosslinking transforming singlets into triplets. In addition, the performance of these metal complexes can be tuned by selecting appropriate ligands.
Examples of photoredox catalysis using Ru(Ⅱ) as a PRC are shown in severl Schemes shown below. First, as shown in Figure 1-4, a Ru(II) complex was employed as a PRC for the dehalogenation of phenacyl bromide into acetophenone.6 Under irradiation
of visible light, Ru(bpy)32+ is excited into [Ru(bpy)32+]*, which oxidizes an aromatic
amine (= reductant) into its radical cation. At the same time, [Ru(bpy)32+]* is reduced into
Ru(bpy)3+. Then Ru(bpy)3+ reduces phenacyl bromide into phenacyl radical and bromide
anion, with recovering Ru(bpy)32+. From the radical cation of the amine, hydrogen radical
is transferred to phenacyl radical, achieving the reduction of phenacyl bromide into acetophenone.
Another example of photocatalysis, in which Ru(bpy)32+ was used as a PRC, was the
asymmetric α-alkylation of aldehydes (Figure 1-5).7 Under irradiation of visible light,
Ru(bpy)32+ is excited into [Ru(bpy)32+]*, which oxidizes “Radical A” formed by the
addition of “Radical B” to “Substrate A” into the corresponding iminium cation. Hydrolysis of the iminium cation gives the target compound, α-alkylated aldehyde. “Substrate A” formed by the dehydrative condensation of the substrate primary aldehyde and the chiral amine employed as a catalyst for the chiral induction in this reaction. The abovementioned oxidation step accompanies the reduction of [Ru(bpy)32+]* into
5
anion and “Radical B”, leading to the recovery of Ru(bpy)32+ in the ground state, which
then waits for excitation upon irradiation to drive the next catalysis cycle.
Figure 1-4. Reduction of phenacyl bromide under irradiation of visible light using Ru(bpy)32+ as a photoredox catalyst
6
Figure 1-5. Enantioselective α-alkylation of aldehydes under irradiation of visible light using Ru(bpy)32+ as a photoredox catalyst
7
2. Utilization of Organic Dyes as Photoredox Catalysts
As described above, the use of transition metal complex-based PRC is a useful and promising strategy to achieve efficient photocatalysis of various organic reactions. However, such transition metal complexes are precious and expensive even if their amounts required for the efficient progress of the reactions are small. Besides such metal complexes, organic dyes with extended π-conjugated systems are also attractive candidates for photoredox catalysts due to their structural diversity and lower cost and their non-dependence of the utilization of precious metals.8-13
Appropriately selected organic dyes catalyze reactions under irradiation of visible light. For example, enantioselective α-alkylation of aldehyde, which can be achieved by using Ru(bpy)32+, can be also achieved by using eosin Y, an organic dye that can absorb
visible light (λmax 539 nm) (Figure 2-1).14 Upon absorbing visible light, eosin Y (EY) can
be excited into photoexcited singlet state (1EY*). This singlet undergoes intersystem
crossing into triplet state (3EY*). The resulting triplet 3EY* oxidizes “Radical B” to give
a radical anion, EY·-, which then reduces secondary alkyl bromide (“Substrate B”) into
bromide anion and “Radical A”, leading to the recovery of EY in the ground state. Perylene diimides (PDIs) have been used as photoredox catalysts for arene-arene coupling (Figure 2-2).15 In the photocatalysis, PDI is photoexcited by absorbing visible light (λmax 455 nm). The resulting excited state PDI* oxidizes triethyl amine into its
radical cation, giving radical anion PDI·-. This radical anion absorbs visible light again to transform itself into the excited radical anion PDI·-*, which then reduces aryl chloride
readily into chloride anion and the corresponding aryl radical. The aryl radical undergoes coupling with N-methylpyrrole with releasing hydrogen radical to furnish the coupling product. This reduction process allows the recovery of PDI in the ground state.
8
Figure 2-1. Enantioselective α-alkylation of aldehydes under irradiation of visible light using eosin Y as a photoredox catalyst
9
Figure 2-2. Aryl-aryl coupling under irradiation of visible light using perylene diimide as a photoredox catalyst
10
3. Photooxidation and Photoreduction of Perylene
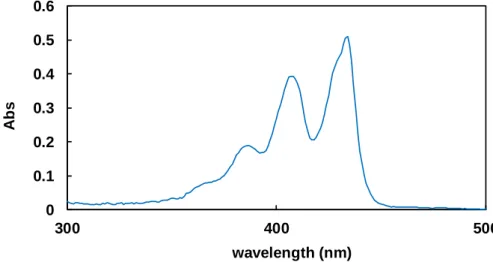
Perylene is a simple polyaromatic hydrocarbon that can absorb visible light. As shown in Figure 3-1, it absorbs visible lights in a region of wavelength from 350 to 450 nm. The absorption at 435 nm is attributable to the HOMO-LUMO gap of perylene, which was confirmed by DFT calculation (Figure 3-2).
The photoexcited perylene can be used as a reductant. Such reducing nature is efficiently used in the radical polymerization driven by irradiation of visible light (Figure 3-3).16 In this polymerization system, perylene is photoexcited upon irradiation by a white
LED. The excited perylene reduces α-bromoester moiety at the chain end to transform it into bromide anion and the corresponding radical, to which monomers reacts to permit the chain extension. At the same time, the excited perylene is oxidized into a cation radical of perylene. This radical cation oxidizes bromide anion into bromo radical, which undergoes recombination with the resulting propagating radical at the chain end.
Figure 3-1. UV-vis spectrum of perylene in acetonitrile
0 0.1 0.2 0.3 0.4 0.5 0.6 300 400 500 A b s wavelength (nm)
11
Figure 3-2. Energy levels of HOMO and LUMO of perylene (by DFT calculation, B3LYP/ 6-31 G (d, p))
Figure 3-3. Controlled radical polymerization of methyl methacrylate (MMA) under irradiation of visible light with using perylene as a photoredox catalyst
12
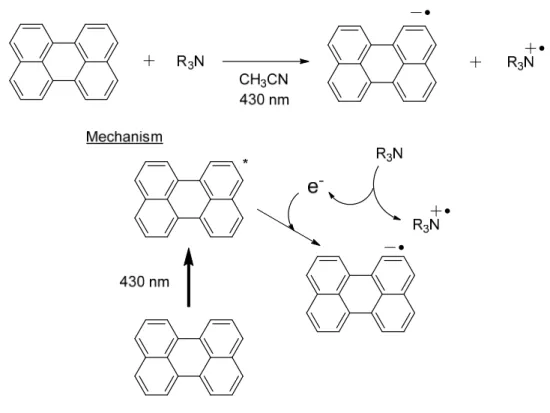
On the other hand, the photoexcited perylene can act also as an oxidant (Figure 3-4).17 Upon irradiating visible light (λmax 435 nm) to a solution of perylene and tertiary
amines in acetonitrile, the amines can be oxidized into the corresponding radical cations.
reduction process that uses the radical anion of perylene as a reductant, a new photocatalytic cycle would be achieved.
13
4. Target of This Work
As discussed above, perylene is a simple aromatic compound that is attractive as a photoredox catalyst. It can be excited upon irradiation of visible light, leading to the formation of photoexcited perylene that can be used as a reductant or an oxidant depending on redox potentials of substrates. In addition, perylene can be modified easily by reactions with electrophiles to afford a series of substituted perylenes.
What the author has focused on his research is the efficient use of the radical anion of perylene as a strong reductant. As shown in Figure 3-4, the radical anion can be prepared by the reduction of photoexcited perylene by a tertiary amine, in other words, a sacrificial electron donor that reduces the photoexcited perylene into the radical anion. As acceptors of electron from the radical anion, aldehydes, ketones, and imines were chosen (Figure 4-1). Upon accepting electron from the radical anion of perylene, these substrates can be transformed into the corresponding radical anions, which would undergo homocoupling to furnish the corresponding 1,2-diols and 1,2-diamines. This reduction process gives perylene in the ground state, which can be excited again by absorbing visible light. As a result, the combination of two different reactions, oxidation of amine and reduction of aldehydes, ketones, and imines, gives a complete cycle of a photoredox catalysis by perylene as a photoredox catalyst. If the radical anions formed by the reduction of the substrates undergo the homocoupling efficiently, it will give us a straightforward synthetic route for 1,2-diols and 1,2-diamines, which are highly useful compounds as monomers for polymer synthesis and ligands for metal complexes, via “C-C bond forming driven by visible light”.
14
Figure 4-1. Utilization of anion radical of perylene for reduction of aldehydes, ketones, and imines
This thesis deals with the investigations and achievements by the author in the course of his research toward his target, development of visible light-driven carbon-carbon bond forming reactions with peylene-based photoredox catalysts. This “General Introduction” is followed by three chapters:
In chapter 1, the development of visible light-driven reductive coupling of aromatic aldehydes and ketones using perylene as a photoredox catalyst is described. The author discovered that perylene worked as a photoredox catalyst efficiently in the reductive coupling of aromatic aldehydes and ketones under irradiation of visible light, leading to the successful formation of the corresponding 1,2-diols. The author postulated the mechanism for the photoredox catalysis that involves 1) the excitation of perylene by irradiation of visible light, 2) one electron transfer from a sacrificial tertiary amine to the photoexcited perylene to afford an anion radical of perylene, 3) single-electron-transfer
15
(SET) from the anion radical species to the substrates such as aromatic aldehydes and ketones to transform them into the corresponding ketyl radicals, with recovering perylene in the ground state.
In chapter 2, the development of new perylene derivative bearing urethane moiety (urethane-perylene) and its application as a photoredox catalyst to reductive coupling of ketones and aldehydes are described. The urethane-perylene was designed so that the urethane moiety would capture and activate ketones and aldehydes via hydrogen bonding, facilitating the single electron transfer from the radical anion of perylene to the captured substrates. The experiments clarified that the perylene urethanes were superior photoredox catalysts than pristine perylene, particular in the reductive coupling of ketones with higher LUMO levels. This finding proved the feasibility of the molecular design of photoredox catalysts with improved performance based on tethering hydrogen bond donors to photo-absorbing cores via covalent bonds.
In chapter 3, the development of visible light-driven reductive coupling of aromatic imines using perylene as a photoredox catalyst is described. The reductive coupling of imines is a straightforward synthetic approach to vicinal diamines
suffered from the consumption of low valent metal species. The authored investigation clarified that the photoredox system using perylene as the photoredox catalyst was highly efficient and appreciable from its metal-free nature. In addition, the author applied the visible light-driven reductive coupling of imines to crosslinking reaction of a polymer bearing imino groups in the main chain. The reaction proceeded readily to afford the corresponding networked polymer, demonstrating that the present coupling is applicable to polymer synthesis.
16
5. References
1. Meggers, E. Chem. Commun., 2015, 51, 3290.
2. Peña- - Angew. Chem. Int. Ed., 2015, 54,
5006.
3. Chem. Rev., 2013, 113, 5322.
4. -J. Angew. Chem. Int. Ed., 2012, 51, 6828.
5. Chem. Soc. Rev., 2011, 40, 102.
6. Fukuzumi, S.; Mochizuki, S.; Tanaka, T. J. Phys. Chem., 1990, 94, 722.
7. Science, 2008, 322, 77.
8. Romero, N. A.; Nicewicz, D. A. Chem. Rev., 2016, 116, 10075.
9. Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Chem. Soc. Rev., 2016, 45, 6165.
10. , A. Chem. Soc. Rev., 2013, 42, 97.
11. - Chem. Rev.,
2012, 112, 1710.
12. Hoffmann, N. J. Photoch. Photobio. C., 2008, 9, 43.
13. Chem. Rev., 2007, 107, 2725.
14. Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed., 2011, 50, 951.
15. Ghosh, I.; Ghosh, T.; Bardagi, J. I.; König, B. Science, 2014, 346, 725. 16. Miyake, M. G. Theriot, C. J. Macromolecules, 2014, 47, 8255.
17. n, R. S. J. Chem. Soc. (C), 1971,
17
Chapter 1
Development of Visible Light-driven Reductive Coupling of
Carbonyl Compounds Using Perylene as a Photoredox Catalyst
1-1. Introduction
The reductive coupling of aldehydes and ketones is an attractive method for C-C bond forming, because it allows easy access to vicinal diols, which are an important class of synthetic building blocks used as monomers for polymer synthesis and chiral auxiliaries for asymmetric synthesis.1,2 Conventionally, highly efficient reductive coupling reactions have been achieved using stoichiometric amounts of metals or low-valent metal compounds with high reduction potentials such as Mg(0), Ti(II), Zn(0), and Sm(II), whose productions require significant amounts of energy.3 A typical example is shown in Scheme 1-1-1. In this example, the reductive coupling of aldehydes and ketones is achieved using samarium iodide and samarium metal in combination.3e In the coupling reactions using these low- valent metal species, addition of trimethylsilyl chloride is necessary to achieve high efficiency. Consequently, the products of the reactions are silyl ethers. By acidic treatment of the silyl ethers, the target diols can be obtained.
18
In contrast, as discussed in General Introduction, the successful use of visible light as an energy source for C-C bond forming reactions has attracted much attention, because it will lead to a much more cost-effective and environmentally friendly access to various useful chemicals. Recently, a visible-light-induced reductive coupling of aldehydes and ketones using an Ir (III) complex as a photoredox catalyst has been reported (Scheme 1-1-2).4 In the photocatalysis, the complex absorbs visible light to transform itself to the excited state. The resulting excited state receives one electron from a sacrificial tertiary amine. Then, the reduced Ir complex, which became sufficiently active to give an electron to C=O double bond, reduces aldehydes and ketones into the corresponding ketyl radical. The radicals thus formed undergo coupling to give the corresponding vicinal diols.
19
Another example of visible light photocatalysis of the reductive coupling of aldehyde is achieved by using poly(p-phenylene) as the photoredox catalyst (Scheme 1-1-3).5 The extended π-conjugated system of poly(p-phenylene) can
absorb visible light and thus is excited upon the absorption of visible light to induce the photoredox catalysis.
Scheme 1-1-3
Herein, the author describes a photoredox catalysis of reductive coupling of aldehydes and ketones, where perylene, a commercially available polycyclic aromatic hydrocarbon that can absorb visible light at ∼430 nm (Figure 1-1-1), was used as the photoredox catalyst (PRC). It has been reported that perylene can be excited upon absorbing visible light and the resulting excited perylene can be transformed into a radical anion species with oxidizing tertiary amine.6 As
described in General Introduction, the author envisaged that 1) perylene can be excited upon absorbing visible light, 2) the excited perylene would oxidize tertiary amine added as an electron donor, 3) the resulting anion radial of perylene would reduce aldehydes and ketones into the corresponding radical anions, with recovering perylene in the ground state, and 4) the radical anions would undergo homocoupling to afford the corresponding 1,2-diols.
20
Figure 1-1-1. UV-vis spectrum of perylene
1-2. Coupling Reaction of Benzaldehyde Using Perylene as a PRC
The initial experiment was performed using benzaldehyde 1a and triethylamine as the substrate and electron donor, respectively (Scheme 1-2-1, Table 1-2-1). Acetonitrile was selected as the solvent, because the successful photosensitised oxidation of tertiary amines using perylene in acetonitrile has been reported.6 To an acetonitrile solution of benzaldehyde 1a (initial concentration: 5
mM), perylene (8 mol%) and triethylamine (8 eq) were added, and visible light was irradiated using a commercially available 8.5 W white LED as the light source (entry 1). After the irradiation for 16 h, the reaction mixture was analysed by thin-layer chromatography (TLC), indicating the successful formation of the coupling product 2a. The reaction mixture was treated with acetic anhydride, pyridine, and 4-(N,N-dimethylamino)pyridine (DMAP) to transform diol 2a into the corresponding diacetate 3a, which was isolated by preparative TLC (PTLC). The yield was 39%.
21 Scheme 1-2-1
Table 1-2-1. Visible light-driven reductive coupling reaction of benzaldehyde. Entry [1a]0 (mM) R3N Amount of
perylene (mol%) Yield of 3a (%)a dl:meso of 3ab 1 5 Et3N 8 39 32:68 2c 5 Et 3N 8 0 - 3 5 Et3N 0 0 - 4 5 -d 8 0 - 5 20 Et3N 8 49 47:53 6 20 Et3N 12 52 49:51 7 20 i-Pr2NEt 12 66 52:48 8 20 PhNMe2 12 63 31:69 9 20 PhNEt2 12 37 50:50 10 20 12 7 47:53 11 20 12 4 12:88
a Yield of 3a isolated by silica gel column chromatography and preparative TLC (PTLC). b Determined by the 1H NMR analysis of 3a. c Without irradiation. d No amine was added.
22
Second, the necessity of visible light, perylene, and tertiary amine for the progress of the reaction was confirmed as follows: Without irradiation (entry 2), no reaction occurred. In the absence of perylene, no reaction occurred even if the solution was irradiated (entry 3). Without the addition of triethylamine, no reaction occurred (entry 4).
Third, the reaction was studied under varying reaction conditions. By increasing the initial concentration of benzaldehyde from 5 mM to 20 mM, the yield of 3a increased (entry 5 vs. entry 1). An increase in the amount of perylene was also effective in improving the yield of the product (entry 6).
Finally, tertiary amines were screened (entries 7–11). The results show that the reaction efficiency and diastereoselectivity significantly depend on the structure of amine. When triethylamine was replaced with diisopropylethylamine, the yield of 3a increased to 66% without influencing the diastereoselectivity. N,N-Dimethylaniline was also a suitable amine; its use afforded the coupling product in a good yield. Interestingly, the product was meso-rich. The reason for this selectivity is not clear at present; however, as reported by Nakajima et al., an attractive interaction occurs between aldehyde and the radical cation formed by the SET from tertiary amine to the excited photoredox catalyst;4 thus, the steric and electronic nature of amine would significantly affect the diastereoselectivity in the coupling reaction. The use of N,N-diethylaniline decreased the yield and diastereoselectivity. The use of cyclic amines such as methylpiperidine and N-methylpyrrolidine led to poor yields of the product. However, the use of latter led to exceptionally high meso-selectivity. These results indicate that the diastereoselectivity can be controlled by selecting an appropriate tertiary amine.
23
In Figure 1-2-1, the 1H-and 13C-NMR spectra of 3a obtained in entry 7 are shown.
In the 1H-NMR spectrum, signals attributable to the phenyl moiety, the methine moiety, and the acetyl moiety were observed in the relative intensity ratio that agreed with the chemical structure of 3a. In the 13C-NMR spectrum, signals at 169 ppm and 21 ppm were attributed to the carbonyl group and the methyl group of acetyl moiety, respectively. The four carbons composing the phenyl group were detected as eight signals from 136 to 127 ppm. In addition, the two carbons composing the formed methine group were detected as two signals at 77 ppm and 76 ppm. In the IR spectrum, a strong absorption peak was observed at around 1737 cm−1, confirming the presence of ester bonds.
Finally, perylene was replaced by other aromatic hydrocarbons to investigate their ability as photoredox catalysts for the coupling reaction (Scheme 1-2-2). Anthracene and naphthacene did not catalyse the reaction. Pyrene was also not an efficient photoredox catalyst. Its use afforded 3a only in a poor yield (14%).
24
Figure 1-2-1. 1H and 13C-NMR spectra of isolated diacetate 3a in CDCl
25
1-3. Coupling Reaction of Aldehydes and Ketones Using Perylene as a PRC
The protocol using perylene as a PRC for the reductive coupling of benzaldehyde 1a under visible light was extended to those of aromatic aldehydes and ketones (Scheme 1-3-1, Table 1-3-1). Notably, the less electron-deficient 4-halo-substituted benzaldehydes underwent the coupling more efficiently than those bearing electron-donating methyl and methoxy groups (entries 2-5). 1H- and 13C-NMR spectra of isolated diols 2b-e were shown in Supplementary Data (SD) shown in the last section in this chapter. In all cases, signals attributable to the newly formed methine groups were detected in both 1H- and
13C-NMR spectra. Interestingly, the presence of a methoxy group and fluorine atom in
the substrates led to the dominance of dl-products over meso-products, indicating that the introduction of heteroatoms with a higher electronegativity led to the controlled stereoselectivity.
Reductive coupling of aromatic ketones 1f-g were also investigated (entries 6-7). When benzophenone was irradiated with visible light under the optimized conditions for the reductive coupling reaction of benzaldehyde, the corresponding coupling product 2f was obtained in 38% yield. The yield was much lower than that expected from the lower reduction potential of benzophenone compared to benzaldehyde, making the SET to the former much easier than that to the latter.7 In addition to the formation of 2f, 1,1-diphenylmethanol was obtained in 28% yield, indicating that the coupling reaction of the anion radical formed by SET to benzophenone in the photoredox catalysis was competed by H radical abstraction from H radical donors by the anion radical. Another aromatic ketone, acetophenone, also underwent the coupling reaction. As expected from the higher reduction potential of acetophenone compared to benzaldehyde, the yield of the
26
corresponding coupling product 2g was not satisfactory, demonstrating that the photoredox catalysis of acetophenone using perylene is challenging. However, side reactions such as the reduction of acetophenone to 1-phenylethanol were not observed, indicating that the efficiency can be improved by further optimization of conditions. The 1H- and 13C-NMR spectra of 2f and 2g are shown in SD.
Scheme 1-3-1
Table 1-3-1. Visible light-driven reductive coupling of aldehydes and ketones.
Entry 1 X R Yield of 2 /%a dl : meso of 2b 1 1a H H 66 52 : 48 2 1b Me H 56 50 : 50 3 1c MeO H 56 81 : 19 4 1d F H 64 70 : 30 5 1e Cl H 67 53 : 47 6 1f H Ph 38 - 7 1g H Me 23 54 : 46
a Yield of 2 isolated by silica gel column chromatography and PTLC. b Determined by the 1H NMR analysis of 2 isolated by silica gel column chromatography and PTLC.
27
1-4. Possible Reaction Mechanism
A plausible reaction mechanism for the present photoredox catalysis by perylene is depicted in Scheme 1-4-1. First, the excited perylene (P*) upon absorbing visible light abstracts one electron from a tertiary amine. Second, from the resulting radical anion of perylene (P.-) to substrate 1, single electron transfer (SET) takes place. Third, the resulting
radical anions undergo a homo-coupling reaction to achieve carbon-carbon bond formation. Finally, target diol 2 can be obtained via abstraction of protons from proton donors such as the radical cation of the tertiary amine. Another possibility is that the radical anions abstract protons, and then the formed radicals undergo coupling reactions to form the corresponding diols 2.
28
1-5. Conclusions
Perylene, a simple polyaromatic hydrocarbon, was used as the PRC for the reductive coupling reactions of C=O double bond. The use of perylene enabled the efficient progress of the reactions using visible light emitted by an 8.5 W white LED as the energy source. In addition to the metal-free and energy-saving aspects, the present perylene-mediated photoredox system has the following advantages: 1) successful application to the reductive coupling reaction of 4-chlorobenzaldehyde; and 2) a dominant formation of the dl-diastereomers of the coupling products of 4-methoxybenzaldehyde and 4-fluorobenzaldehyde.
29
1-6. Experimental Details Materials
Acetic anhydride, acetophenone, acetonitrile, benzaldehyde, benzophenone, 4-chlorobenzaldehyde, deuterochloroform (CDCl3), N,N-diethylaniline,
dimethylaminopyridine (DMAP), dimethylaniline, 4-fluorobenzaldehyde, N,N-diisopropylethylamine, pyridine, methoxybenzaldehyde, methylbenzaldehyde, 4-methylpiperidine, 4-methylpyrrolidine, perylene, pyrene, pyridine and triethylamine were purchased from Wako Pure Chemical Industries Co., Ltd. Tetracene was purchased from TCI Co., Ltd. and used as received. Anthracene was purchased from Sigma Aldrich Co., Ltd. and used as received. Acetonitrile was distilled from calcium hydride under reduced pressure and stored over molecular sieve 4A in an argon atmosphere prior to use. The aldehydes and acetophenone were distilled under reduced pressure and stored in an argon atmosphere prior to use. The tertiary amines were also distilled under reduced pressure and stored over sodium hydroxide in an argon atmosphere prior to use. Other reagents were used as received.
Instruments
NMR spectra (400 MHz for 1H; 100 MHz for 13C) were recorded with a JEOL NMR
spectrometer (JNM-AL400) as solutions in CDCl3 using TMS as the internal standard.
Chemical shifts, δ, and coupling constants, J, are expressed in ppm and Hz, respectively. IR spectra were obtained using a JASCO FT/IR-470 spectrometer, and wavenumbers, ν, are given in cm−1. UV-vis spectra were obtained using a Shimadzu UV3101PC
spectrometer. The experimental setup for the reductive coupling reactions under visible-light irradiation is shown in Figure 1-6-1. For the irradiation, a HOZAN LED (model L – 711) was used. Its emission spectrum is shown in Figure 1-6-2.
30
Figure 1-6-1. Experimental setup
Figure 1-6-2. Emission spectrum of the LED used for irradiating visible light.
4 6 8 10 12 14 16 18 300 400 500 600 700 800 In te n s it y ( m W ) Wavelength (nm)
31
Coupling Reaction of Benzaldehyde 1a (Table 1-2-1, entry 7).
Typical procedure: Benzaldehyde 1a (53.0 mg, 0.500 mmol), perylene (15.1 mg, 60.0 μmol), i-Pr2EtN (0.70 mL, 4.0 mmol), and acetonitrile (25 mL) were added to a 100
mL flask under argon. The resulting solution was irradiated using a LED light under stirring at room temperature. After 16 h, the solution was concentrated under reduced pressure to obtain a mixture containing diol 2a.
To the mixture, acetic anhydride (5.0 mL), pyridine (10 mL), and DMAP (5.0 mg) were added, and the solution was left at room temperature for 4 h. Then, it was concentrated under reduced pressure. The resulting residue was chromatographed on silica gel (eluent: hexane / ethyl acetate / dichloromethane = 150 / 50 / 1) and preparative TLC (eluent: hexane / ethyl acetate / dichloromethane = 250 / 50 / 1) to afford diacetate
3a as a mixture of dl- and meso-diastereomers (49.2 mg, 0.165 mmol, 66% yield, dl :
meso = 52 : 48) as a pale yellow solid: 1H-NMR (CDCl
3, r.t.) δ 7.33-7.14 (m, 20H), 6.09
(s, 2H, meso), 6.05 (d, 2H, dl), 2.07 (s, 6H, dl) 2.00 (s, 6H, meso 13C-NMR (CDCl3, r.t.)
δ 169.9, 169.7, 136.2, 136.1, 128.5 (2C), 128.4, 128.3, 127.7, 127.6, 77.3, 76.5, 21.1,
IR (KBr) ν 3034, 2949, 1737, 1496, 1241 cm−1.
Coupling Reaction of 4-Methylbenzaldehyde 1b (Table 1-3-1, entry 2).
The reductive coupling reaction of 4-methylbenzaldehyde 1b (53.2 mg, 0.501 mmol) was performed under the same conditions as those for benzaldehyde. The mixture was purified by silica gel column chromatography (eluent: hexane/ethyl acetate = 3:1), furnishing diol 2b as a mixture of dl- and meso-diastereomers (33.9 mg, 0.140 mmol, 56% yield, dl : meso = 50 : 50) as a pale yellow solid: 1H-NMR (CDCl
3, r.t.) δ 7.13 (dd,
32
2.95 (br, s, 4H), 2.33 (s, 6H, meso), 2.28 (s, 6H, dl); 13C-NMR (CDCl
3, r.t.) δ 137.9, 137.5,
137.1 (2C), 129.1, 128.9, 127.2, 127.0, 78.9, 78.1, 21.3 (2C); IR (KBr) ν 3346, 3028, 2914, 2857, 1516 cm−1.
Coupling Reaction of 4-Methoxybenzaldehyde 1c (Table 1-3-1, entry 3).
The reductive coupling reaction of 4-methoxybenzaldehyde 1c (68.0 mg, 0.500 mmol) was performed under the same conditions as those for benzaldehyde. The mixture was purified by silica gel column chromatography (eluent: hexane/ethyl acetate =3/1), producing diol 2c as a mixture of dl- and meso-diastereomers (38.2 mg, 0.139 mmol, 56% yield, dl : meso = 81 : 19) as a pale yellow solid: 1H-NMR (CDCl3, r.t.) δ 7.18 (d, J = 8.8
Hz, 4H, meso), 7.02 (d, J = 8.8 Hz, 4H, dl), 6.85 (d, J = 8.8 Hz, 4H, meso), 6.75 (d, J = 8.8 Hz, 4H, dl), 4.72 (s, 2H, meso), 4.60 (s, 2H, dl), 3.79 (s, 6H, meso), 3.75 (s, 6H, dl), 2.90 (br, 13C-NMR (CDCl
3, r.t.) δ 159.5, 159.3, 132.2 (2C), 128.5, 128.3, 113.8,
113.6, 78.9, 77.9, 55.4, 55.3 IR (KBr) ν 3345, 2916, 2360, 1516, 1491 cm−1.
Coupling Reaction of 4-Fluorobenzaldehyde 1d (Table 1-3-1, entry 4).
The reductive coupling reaction of 4-fluorobenzaldehyde 1d (62.1 mg, 0.500 mmol) was performed under the same conditions as those for benzaldehyde. The mixture was purified by silica gel column chromatography (eluent: hexane/ethyl acetate = 3:1), affording diol 2d as a mixture of dl- and meso-diastereomers (39.9 mg, 0.159 mmol, 64% yield, dl : meso = 70 : 30) as a pale yellow solid: 1H-NMR (CDCl
3, r.t.) δ 7.15–7.11 (m,
4H), 7.05–6.88 (m, 12H), 4.81 (s, 2H, meso), 4.60 (s, 2H, dl), 2.71 (br, 13C-NMR (CDCl3, r.t.) δ 163.8,
33
Coupling Reaction of 4-Chlorobenzaldehyde 1e (Table 1-3-1, entry 5).
The reductive coupling reaction of 4-chlorobenzaldehyde 1e (70.3 mg, 0.500 mmol) was performed under the same conditions as those for benzaldehyde. The mixture was purified by silica gel column chromatography (eluent: hexane/ethyl acetate = 3:1), furnishing diol 2e as a mixture of dl- and meso-diastereomers (47.4 mg, 0.168 mmol, 67% yield, dl : meso = 53 : 47) as a pale yellow solid: 1H-NMR (CDCl3, r.t.) δ 7.27 (d, J = 8.8
Hz, 4H, meso), 7.21 (d, J = 8.3 Hz, 4H, dl), 7.11 (d, J = 8.8 Hz, 4H, meso), 7.03 (d, J = 8.3 Hz, 4H, dl), 4.84 (s, 2H, meso), 4.63 (s, 2H, dl), 2.89 (s, 2H, dl), 2.33 (s, 2H, meso
13C-NMR (CDCl
3, r.t.) δ 138.1, 137.9, 134.0, 128.5 (3C IR (KBr) ν 3329,
2918, 2355, 1598, 1491 cm−1.
Coupling Reaction of Benzophenone 1f (Table 1-3-1, entry 6).
The reductive coupling reaction of benzophenone 1f (91.1 mg, 0.500 mmol) was performed under the same conditions as those for benzaldehyde. The mixture was purified by silica gel column chromatography (eluent: hexane/ethyl acetate = 8:1), furnishing diol
2f (35.2 mg, 0.0955 mmol, 38% yield) as a pale yellow solid: 1H-NMR (CDCl3, r.t.) δ
7.31–7.28 (m, 8H), 7.18–7.16 (m, 12H), 3.03 (s, 2 13C-NMR (CDCl
3, r.t.) δ 144.3,
128.8 IR (KBr) ν 3558, 3054, 2965, 2360, 1493 cm−1.
Coupling Reaction of Acetophenone 1g (Table 1-3-1, entry 7).
The reductive coupling reaction of acetophenone 1g (60.2 mg, 0.501 mmol) was performed under the same conditions as those for benzaldehyde. The mixture was purified by silica gel column chromatography (eluent: hexane/ethyl acetate = 3:1) and by preparative TLC (eluent: hexane/ethyl acetate = 10:1) furnishing diol 2g as a mixture of
34
dl- and meso-diastereomers (14.0 mg, 0.0575 mmol, 23% yield, dl : meso = 54 : 46) as a pale yellow solid: 1H-NMR (CDCl3, r.t.) δ 7.26–7.19 (m, 20H), 2.55 (s, 2H, dl), 2.25 (s,
2H, meso), 1.59 (s, 6H, meso), 1.51 (s, 6H, dl 13C-NMR (CDCl
3, r.t.) δ 143.9, 143.6,
IR (KBr) ν 3491, 3056, 2983, 2361, 1446 cm−1.
35
1-7. References
1 (a) Yokota, K.; Uesaka, T.; Obata, M.; Shimomura, H.; Kakuchi, T. Polym. J.,
1999, 31, 1037. (b) Okano, K. Tetrahedron, 2011, 67, 2483. (c) Domínguez-Pérez,
B.; Ferrer, É.; Figueredo, M.; Maréchal, J.-D.; Balzarini, J.; Alibés, R.; Busqué, F. J. Org. Chem., 2015, 80, 9495.
2 (a) Archer, R. M.; Hutchby, M.; Winn, C. L.; Fossey, J. S.; Bull, S. D. Tetrahedron,
2015, 71, 8838. (b) Kleinnijenhuis, R. A.; Timmer, B. J. J.; Lutteke, G.; Smits, J.
M. M.; de Gelder, R.; van Maarseveen, J. H.; Hiemstra, H. Chem. Eur. J., 2016, 22, 1266.
3 (a) Enders, D.; Ullrich, E. C. Tetrahedron: Asym., 2000, 11, 3861. (b) Uchiyama, M.; Matsumoto, Y.; Nakamura, S.; Ohwada, T.; Kobayashi, N.; Yamashita, N.; Matsumiya, A.; Sakamoto, T. J. Am. Chem. Soc., 2004, 126, 8755. (c) Wen, J.; Zhao, J.; Wang, X.; Dong, J.; You, T. J. Mol. Cat. A: Chem., 2006, 245, 242. (d) Sun, J.; Dai, Z.; Li, C.; Pan, X.; Zhu, C. J. Organomet. Chem., 2009, 694, 3219. (e) Yoshimura, A.; Saeki, T.; Nomoto, A.; Ogawa, A. Tetrahedron, 2015, 71, 5347. 4 Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Angew. Chem. Int.
Ed., 2015, 54, 8828.
5 Zhang, M.; Rouch, W. D.; McCulla, R. D. Eur. J. Org. Chem., 2012, 31, 6187. 6 Bartholomew, R. F.; Brimage, D. R. G.; Davidson, R. S. J. Chem. Soc. (C), 1971,
3482.
7 Izutsu, K. Electrochemistry in Nonaqueous Solutions, Wiley-VCH, Weinheim,
36
1-8. Supplementary Data
1H-NMR spectrum of isolated diol 2b in CDCl
3 (Table 1-3-1, entry 2)
13C-NMR spectrum of isolated diol 2b in CDCl
37
1H-NMR spectrum of isolated diol 2c in CDCl
3 (Table 1-3-1, entry 3)
13C-NMR spectrum of isolated diol 2c in CDCl
38
1H-NMR spectrum of isolated diol 2d in CDCl
3 (Table 1-3-1, entry 4)
13C-NMR spectrum of isolated diol 2d in CDCl
39
1H-NMR spectrum of isolated diol 2e in CDCl
3 (Table 1-3-1, entry 5)
13C-NMR spectrum of isolated diol 2e in CDCl
40
1H-NMR spectrum of isolated diol 2f in CDCl
3 (Table 1-3-1, entry 6)
13C-NMR spectrum of isolated diol 2f in CDCl
41
1H-NMR spectrum of isolated diol 2g in CDCl
3 (Table 1-3-1, entry 7)
13C-NMR spectrum of isolated diol 2g in CDCl
42
Chapter 2
Development of a Perylene Derivative Bearing Urethane Moiety and
Its Utilization as a Photoredox Catalyst with Substrate-capture Ability
2-1. Introduction
As described in Chapter 1, the author demonstrated the use of perylene as a photoredox catalyst (PRC) enabled photocatalysis of reductive coupling of aromatic aldehydes and ketones under visible light. In the reaction, a single electron transfer from the radical anion of perylene to the substrate gives the corresponding anion radicals, and their homocoupling affords the corresponding 1,2-diols. The photoredox catalysis using perylene is simple and metal free; however, the yields of the products are moderate, particularly in the reactions of ketones and aldehydes with relatively high LUMO levels.
To improve the visible light-driven coupling of carbonyl compounds, the author envisaged catalysis assisted by hydrogen bonds, which could tether the substrates to the PRCs non-covalently. Capture and activation of carbonyl compounds by appropriately designed organocatalysts with hydrogen bond donors have been efficiently utilized to achieve enantioselective reactions1,2 and controlled ring-opening polymerizations.3 Recently, an well-constructed asymmetric reaction
environment was achieved by designing chiral catalysts bearing a planar π-conjugated wall and acidic arm, which cooperatively captured and facilitated enantioselective reactions.4 In addition, in the visible light-driven reductive coupling of aldehydes and ketones using iridium(III) complexes, hydrogen bond
43
activation of the substrates by ammonium salts is postulated.5 Moreover, the
efficiency of the coupling of acetophenone was improved by adding Brϕnsted acids.5
These relevant studies motivated the author to design a PRC with a hydrogen bond donor that could capture substrates and facilitate electron transfer from the photo-reduced moiety of the catalyst to the substrate. The author designed a perylene-derivative bearing a urethane moiety as a hydrogen bond donor (Figure 2-1-1). The pKa of urethanes is estimated to be approximately 13,6 which would be enough low to act as a hydrogen bond donor. In this chapter, the author describes the visible light-driven reductive coupling of ketones and aldehydes using the urethane-perylene catalyst.
Figure 2-1-1. Design of perylene derivatives bearing urethane moiety
2-2. Synthesis of Urethane-perylene
Urethane-perylene 1 was synthesized by the reaction of its precursor 3-(hydroxymethyl)perylene7 with tert-butylisocyanate (Scheme 2-2-1). 1 was soluble in a wide range of organic solvents, presumably owing to the presence of the tert-butyl group. The UV-vis spectrum of 1 is shown in Figure 2-2-1. It was almost
44
same as the spectrum of pristine perylene. The 1H- and 13C-NMR spectra of 1 are
shown in Figure 2-2-2
Scheme 2-2-1
45
Figure 2-2-2. 1H and 13C-NMR spectra of urethane-perylene 1 in CDCl 3.
46
2-3. Coupling Reaction of Ketones Using Urethane-perylene as a PRC
The reductive coupling of acetophenone 2a and benzophenone 2b was performed using perylene or urethane-perylene 1 as PRCs (Scheme 3-1, Table 2-3-1). As the sacrificial electron donor and solvent, N,N-diisopropylethylamine and acetonitrile were used, respectively, because this combination exhibited the best efficiency in the reductive coupling of benzaldehyde using perylene. Visible light was applied for 16 h, and then the reaction mixture was analysed via 1H-NMR to
determine the conversion of 2, yield of diol 3, and diastereomer ratio (dl : meso). The yields of alcohols 4 were calculated based on the relative signal intensity of the methine proton in the 1H-NMR spectra. Next, diol 3 was isolated by silica gel column chromatography andpreparative thin layer chromatography (PTLC). The
1H-NMR spectra of the crude products and the 1H- and 13C-NMR spectra of the
isolated products are shown in the Supplementary Data (SD) shown in the last section in this chapter.
In chapter 1, the use of a large amount of perylene (12 mol%) in the reductive coupling of acetophenone was described (Table 2-3-1, entry 1). Therefore, the author explored suitable reaction conditions to reduce the amount of perylene. However, when the amount of perylene was reduced to 1 mol%, the conversion of acetophenone was as low as 25%, even when the initial concentration of acetophenone 2a was increased to 100 mM (Table 2-3-1, entry 2). Next, urethane-perylene 1 was employed in place of urethane-perylene in the coupling of 2a, expecting that the urethane moiety would capture 2a via hydrogen bonds and activate 2a (entry 3). The conversion of 2a was higher than that achieved by using perylene. The yield of diol 3a was improved to approximately 40%. The diastereomers, dl- and
47
meso-ones, formed in an almost 1:1 ratio. In entry 4, in place of the white LED, a 420 nm LED was used. The coupling proceeded efficiently, confirming that 1 absorbed blue light to catalyse the coupling, as was expected from its absorption maximum at around 420 nm.
To confirm the validity of the design of urethane-perylene 1 as a PRC, reductive coupling of acetophenone 2a was carried out using perylene and benzyl tert-butylcarbamate (BTBC), where these two components act as the PRC and hydrogen bond donor independently (entry 5). Compared to the reaction without
BTBC (Table 2-3-1, entry 2), the conversion of 2a was improved from 25 to 37%,
implying that the urethane moiety of BTBC activated 2a by forming a hydrogen bond. However, this improvement was much less than that achieved using urethane-perylene 1 (Table 2-3-1, entry 3), confirming that tethering the perylene and urethane moieties promoted the reaction.
48
Table 2-3-1. Visible light-driven reductive coupling of ketones.
Entry 2 PRC Amount of PRC/ mol% Additive [2]0/ mM Conversion of 2 /%a Yield of 3/%a dl : meso of 3a Yield of 4/%a 1b 2a Perylene 12 None 20 N.D (23)c (46:54)d - 2 2a Perylene 1 None 100 25 20 (16)c 56:44 (49:51)d 0 3 2a 1 1 None 100 57 43 (38)c 52:48 (50:50)d 0 4e 2a 1 1 None 100 61 41 59:41 0 5 2a Perylene 1 BTBC (1 mol%) 100 37 27 (22)c 51:49 (49:51)d 0 6 2b Perylene 1 None 100 49 28 (23)c - 21 7 2b 1 1 None 100 57 45 (38)c - 12 a Determined by 1H-NMR analysis of the reaction mixture. b Reported in Chapter
1. c Yield of 3 isolated by silica gel column chromatography and PTLC. d Determined by the 1H-NMR analysis of 3a isolated by silica gel column
chromatography and PTLC. e A 420 nm blue LED (6 W) was used for the irradiation of visible light.
The use of 1 in place of perylene resulted in the improved coupling of benzophenone 2b (entry 6 vs. entry 7). The photoredox reaction of 2b gave coupling product 3b and alcohol 4b. The use of perylene resulted in the formation of 4b in 21%, while 1 resulted in suppressed formation of 4b, allowing the yield of diol 3b to increase to approximately 40%.
49
2-4. Coupling Reaction of Aldehydes Using Urethane-perylene as a PRC
Upon confirming the higher performance of 1, as compared with perylene, as a PRC for the coupling of ketones, its ability to catalyse the coupling of aldehydes
5 was investigated (Scheme 2-1, Table 2-1). In Chapter 1, the coupling of
4-methoxybenzaldehyde 5a, benzaldehyde 5b, and 4-fluorobenzaldehyde 5c using 12 mol% of perylene was described. Thus, prior to using urethane-perylene 1 as a PRC, the coupling of 5 was reinvestigated using 1 mol% perylene and 100 mM instead of 20 mM. As a result, the corresponding diols 6 were obtained in good yields (64%~78%) (entries 1, 3, and 6). The conversions of 5b and 5c
however, the conversion of 5a was lower than those of 5b and 5c, presumably owing to its more electron rich nature.
50
Table 2-4-1. Visible light-driven reductive coupling of aldehydes.
Entry 5 PRC Conversion of 5 /%a Yield of 6 /%a dl : meso of 6a Yield of 7 /%a 1 5a Perylene 78 64 (62)b 83:17 (85:15)c 0 2 5a 1 98 69 (66)b 80:20 (83:17)c 9 3 5b Perylene 100 78 (74)d 56:44 (55:45)e 0 4f 5b Perylene 100 71 (69)d 54:46 (52:48)e 0 5 5b 1 100 88(86)d 56:44 (53:47)e 0 6 5c Perylene 100 69 (64)b 58:42 (63:37)c 0 7 5c 1 100 77(71)b 64:36 (69:31)c 0
a Determined by 1H-NMR analysis of the reaction mixture. b Yield of 6 isolated by
silica gel column chromatography and PTLC. c Determined by 1H-NMR analysis
of 6 isolated by silica gel column chromatography and PTLC. d Yield of 8b, diacetate of 6b, isolated by silica gel column chromatography and PTLC. e Determined by 1H-NMR analysis of 8b isolated by silica gel column
chromatography and PTLC. f The reaction was carried out in the presence of BTBC (1 mol%).
51
Next, the use of 1 was investigated in the coupling of 5a (entry 2). Upon replacing perylene with 1, the conversion of 5a was improved from 78% to 98%, and the yield of diol 6a was improved from 64% to 69%, suggesting that 5a would be efficiently captured by the urethane moiety and activated. On the other hand, the conversions of 5b and 5c were 100% even by using pristine perylene, and thus it seemed there was no chance to improve the efficiency further by using 1. However, it turned out that 1 gave increased yields of product diols 6b and 6c (entries 5 and 7), suggesting that unclarified side reactions accompanying the perylene-catalyzed reductive coupling of aldehydes were successfully suppressed in the 1-catalyzed reactions. One possible side reaction is the coupling of the anion radical formed by the one-electron reduction of aldehyde and the radical formed via the one-electron oxidation of the tertiary amine.8 Using 1, the coupling reaction was promoted over the competing side reaction, leading to high yields of 6b (88%) and 6c (77%). Such a high yield of 6b was not achieved by the perylene-catalyzed coupling of benzaldehyde 5b by adding BTBC (entry 4), confirming that the tethering of the urethane and perylene moieties in 1 was indispensable for the achievement of the higher yield in entry 5 than that in entry 3.
2-5. Possible Reaction Mechanism
Figure 2-5-1 describes possible mechanisms for the photocatalysis. As described in Chapter 1, perylene can be excited by visible light, and one electron can be transferred from tertiary amines added as sacrificial electron donors to excited perylene. Analogously, the perylene moiety of 1 excited by visible light abstracts an electron from the tertiary amine, leading to the formation of 1.-, a
52
strong reductant. From the radical anion, a single electron transfer (SET) to substrate takes place, giving the corresponding radical anion of the substrate, which undergoes a homocoupling reaction. The radical cations of the tertiary amine act as proton donors. As discussed above, the use of 1 led to the higher conversions of some of the substrates such as acetophenone and 4-methoxybenzaldehyde with
thus, capturing them through hydrogen bonds lowered their LUMO levels.
Figure 2-5-1. Photocatalysis of reductive coupling of ketones and aldehydes by urethane-perylene 1.
Another significant aspect of the use of 1 as a PRC, is that it facilitated higher yields of the coupling products in reactions of substrates with lower LUMO levels such as benzophenone, benzaldehyde, and 4-fluorobenzaldehyde. It would be
53
reasonable to postulate a faster proton transfer from the urethane moiety to the radical anions formed from these substrates. Upon protonation, the radical anions can be transformed into radical species without a negative charge, which can undergo the coupling reaction more easily. Another explanation can be based on the presence of hydrogen bonds between the urethane moieties of 1. Such interactions facilitate the aggregation of 1, which would lead to the increased probability of coupling of radical anions.
54
2-6. Conclusions
A new PRC bearing a visible light-responsive perylene moiety and a urethane moiety was synthesized and employed in the visible light-driven reductive coupling of ketones and aldehydes. The urethane moiety was introduced to capture and activate the substrates through hydrogen bonds for efficient electron transfer from the radical anion of the perylene moiety to the substrates. The improved conversions of substrates with higher LUMOs such as acetophenone and 4-methoxybenzaldehyde prove that the urethane moiety captured these substrates as expected. Further investigations into the role of the urethane moiety will be continued to achieve appropriate molecular designs of perylene derivatives as PRCs.
55
2-7. Experimental Details Materials
Acetic anhydride, acetophenone, acetonitrile, benzaldehyde, benzophenone, benzyl alcohol, tert-butylisocyanate, deuterochloroform (CDCl3), dibutyltin dilaurate (DBTDL),
o-dichlorobenzene, N,N-dimethylamino pyridine (DMAP), N,N-dimethylformiamide (DMF), 4-fluorobenzaldehyde, N,N-diisopropylethylamine, phosphoryl chloride, pyridine, 4-methoxybenzaldehyde, perylene, pyridine, sodium borohydride and tetrahydrofuran (THF) were purchased from Wako Pure Chemical Industries Co., Ltd.
Acetonitrile, DMF and THF was distilled from calcium hydride under reduced pressure and stored over molecular sieve 4A in an argon atmosphere prior to use. The aldehydes, acetophenone, benzyl alcohol and tert-butylisocyanate were distilled under reduced pressure and stored in an argon atmosphere prior to use. N,N-diisopropylethylamine were also distilled under reduced pressure and stored over sodium hydroxide in an argon atmosphere prior to use. Other reagents were used as received.
Instruments
NMR spectra (400 MHz for 1H; 100 MHz for 13C) were recorded with a JEOL NMR spectrometer (JNM-AL400) using tetramethylsilane (TMS) as the internal standard. Chemical shifts, δ, and coupling constants, J, are expressed in ppm and Hz, respectively. IR spectra were obtained using a JASCO FT/IR-470 spectrometer, and wavenumbers, ν, are given in cm−1. High-resolution mass spectroscopic analyses were performed using a JEOL mass spectrometer (JMS-T100LP AccuTOF LC-plus) in a direct analysis in real time (DART) mode and detected by the time-of-flight (TOF) method. UV-vis spectra were obtained using a Shimadzu UV3101PC spectrometer. For irradiation, a HOZAN LED (model L–711) or a OptoCode LED (model EX-420) was used.
56
Synthesis of Perylen-3-ylmethyl tert-Butylcarbamate 1 (Scheme 2-2-1) Synthesis of Perylen-3-ylmethanol.
o-dichlorobenzene (2.0 mL),
reaction mixture was stirred at 100 °C for 16 h, and then neutralized by adding 5M sodium hydroxide (2.0 mL). The resulting mixture was poured into 5% sodium acetate (200 mL), and filtered with suction to obtain a brown solid. The solid was dissolved in THF (226
were added successively. The mixture was stirred at room temperature for 4 h, and then concentrated under reduced pressure. The resulting residue was chromatographed on silica gel (eluent: chloroform) to obtain perylen-3-ylmethanol
as a yellow solid: 1H-NMR (CDCl3, r.t.) δ 8.25-8.15 (m, 4H), 7.95 (d, J = 8.3 Hz, 1H),
7.69 (d, J = 7.8 Hz, 2H), 7.58-7.47 (m, 4H), 5.11 (s, 2H), 1.76 (br, s, 1H). 13C-NMR
(CDCl3, r.t.) δ 136.0, 134.8, 133.4, 132.7, 131.9, 131.7, 131.4, 131.2, 130.7, 130.4, 129.6,
HRMS m/z: [M+H]+
Calcd for [C21H15O]+ 283.1123 Found 283.1119.
Synthesis of Perylen-3-ylmethyl tert-Butylcarbamate 1.
To a solution of perylen-3-ylmethanol
tert-The solution was heated at 100 °C for 16 h, and concentrated under reduced pressure. tert-The resulting residue was chromatographed on silica gel (eluent: chloroform) and further purified by recrystallization (chloroform) to afford perylen-3-ylmethyl tert-butylcarbamate 1 as yellow powder (262.1 mg ol, 69%): m.p. 197.9-199.4 °C.
57 1H-NMR (CDCl 3, r.t.) δ 8.21-8.15 (m, 3H), 8.11 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.68 (d, J = 7.8 Hz, 2H), 7.55-7.45 (m, 4H), 5.43 (s, 2H), 4.76 (br, s, 1H), 1.34 (s, 13C-NMR (CDCl 3, r.t.) δ 154.9, 134.7, 133.2, 132.1, 131.9, 131.3, 131.1, 129.2, HRMS m/z: [M+H]+ Calcd for [C 26H23NO2]+ 381.1729 Found 381.1738. 2974, 2363, 1698, 1602, 1521 cm-1.
Synthesis of Benzyl tert-Butylcarbamate (BTBC) (Scheme 2-3-1).
To a solution of benzyl alcohol (1.08 g, 10.0 mmol) and DBTDL (63.2 mg, 0.100 mmol) in DMF (10 mL), tert-butylisocyanate (1.53 mL, 13.0 mmol) was added. The solution was stirred at 100 °C for 20 h, then concentrated under reduced pressure. The resulting residue was chromatographed on silica gel (eluent: hexane/ethyl acetate = 5/1) to obtain BTBC (1.95 g, 9.40 mmol, 94%) as a colorless liquid: 1H-NMR (CDCl
3, r.t.) δ
7.35-7.25 (m, 5H), 5.04 (s, 2H), 4.76 (br, s, 1H), 1.32 (s, 9H); 13C-NMR (CDCl3, r.t.) δ
154.7, 136.9, 128.6, 128.2, 128.1, 66.1, 50.5, 29.0; HRMS m/z: [M+H2O]+ Calcd for
[C12H19NO3]+ 225.1365; Found 225.1361. IR (KBr); 3353, 3033, 2970, 2363, 1712, 1269,
1213 cm-1.
Coupling Reaction of Acetophenone 2a Using 1 (Table 2-3-1, entry 3).
In a test tube, 2a (60.1 mg, 0.500 mmol), 1 (1.9 mg, 5.0 μmol), and diisopropylethyamine (0.70 mL, 4.0 mmol) were dissolved in acetonitrile (5 mL) under argon. The resulting solution was irradiated using a LED light under stirring at room temperature. After 16 h, the solution was concentrated under reduced pressure. The resulting residue was chromatographed on silica gel (eluent: hexane / ethyl acetate /
58
dichloromethane = 200 / 50 / 1) and preparative TLC (eluent: hexane / ethyl acetate / dichloromethane = 500 / 50 / 1) to afford diol 3a as a mixture of dl- and meso-diastereomers (23.2 mg, 0.0957 mmol, 38% yield, dl : meso = 50 : 50) as a pale yellow solid: 1H-NMR (CDCl3, r.t.) δ 7.26–7.19 (m, 20H), 2.57 (s, 2H, dl), 2.27 (s, 2H, meso),
1.59 (s, 6H, meso), 1.51 (s, 6H, dl); 13C-NMR (CDCl
3, r.t.) δ 143.9, 143.6, 127.5, 127.4,
127.3, 127.2, 127.0, 79.0, 78.7, 25.3, 25.1; IR (KBr) ν 3491, 3056, 2983, 2361, 1446 cm−1.
Coupling Reaction of Acetophenone 2a Using Perylene in the Presence of BTBC (Table 2-3-1, entry 5).
According to general procedure, the reductive coupling reaction of 2a (60.1 mg, 0.500 mmol) using perylene (1.2 mg, 5.0 μmol) and BTBC (1.0 mg, 5.0 μmol) was performed to afford diol 3a as a mixture of dl- and meso-diastereomers (13.2 mg, 0.0545 mmol, 22% yield, dl : meso = 49 : 51) as a pale yellow solid.
Coupling Reaction of Benzophenone 2b Using 1 (Table 2-3-1, entry 7).
According to the procedure given for the coupling reaction of acetophenone 2a using
1, the reductive coupling reaction of benzophenone 2b (91.1 mg, 0.500 mmol) using 1
(1.9 mg, 5.0 μmol) was performed. The resulting mixture was purified by silica gel column chromatography (eluent: hexane / ethyl acetate / dichloromethane = 500 / 50 / 1) and preparative TLC (eluent: hexane / ethyl acetate / dichloromethane = 500 / 50 / 1) to afford diol 3b (34.9 mg, 0.0952 mmol, 38% yield) as a pale yellow solid: 1H-NMR
(CDCl3, r.t.) δ 7.31–7.28 (m, 8H), 7.18–7.16 13C-NMR (CDCl3,
59
Coupling Reaction of 4-Methoxybenzaldehyde 5a Using 1 (Table 2-4-1, entry 2).
According to the procedure given for the coupling reaction of acetophenone 2a using
1, the reductive coupling reaction of 4-methoxyasbenzaldehyde 5a (68.2 mg, 0.501
mmol) using 1 (1.9 mg, 5.0 μmol) was performed. The resulting mixture was chromatographed on silica gel (eluent: hexane / ethyl acetate = 2 / 1 to 1 / 1) and preparative TLC (eluent: hexane / ethyl acetate / dichloromethane = 200 / 50 / 1) to afford diol 6a as a mixture of dl- and meso-diastereomers (45.1 mg, 0.164 mmol, 66% yield, dl : meso = 83 : 17) as a pale yellow solid: 1H-NMR (CDCl3, r.t.) δ 7.16 (d, J = 8.8 Hz, 4H,
meso), 7.00 (d, J = 8.8 Hz, 4H, dl), 6.83 (d, J = 8.8 Hz, 4H, meso), 6.73 (d, J = 8.8 Hz, 4H, dl), 4.71 (s, 2H, meso), 4.58 (s, 2H, dl), 3.79 (s, 6H, meso), 3.75 (s, 6H, dl), 3.06 (br
13C-NMR (CDCl
3, r.t.) δ 159.5, 159.2, 132.2, 132.1, 128.4, 128.3, 113.8, 113.6,
78.9, 77.8, 55.4, 55.3 IR (KBr) ν 3345, 2916, 2360, 1516, 1491 cm−1.
Coupling Reaction of Benzaldehyde 5b Using 1 (Table 2-4-1, entry 5).
According to the procedure given for the coupling reaction of acetophenone 2a using
1, the reductive coupling reaction of benzaldehyde 5b (52.8 mg, 0.498 mmol) using 1 (1.9
mg, 5.0 μmol) was performed. The formation of diol 6b was confirmed by 1H-NMR. The
solution was concentrated under reduced pressure, and to the resulting residue, acetic anhydride (5 mL), DMAP (5 mg), and pyridine (10 mL) were added. The resulting solution was stirred at room temperature for 16 h and concentrated under reduced pressure. The resulting residue was chromatographed on silica gel (eluent: hexane / ethyl acetate / dichloromethane = 150 / 50 / 1) and preparative TLC (eluent: hexane / ethyl acetate / dichloromethane = 250 / 50 / 1) to afford diacetate 8b as a mixture of dl- and meso-diastereomers (64.1 mg, 0.215 mmol, 86% yield, dl : meso = 53 : 47) as a pale yellow
60 solid: 1H-NMR (CDCl 3, r.t.) δ 7.33-7.14 (m, 20H), 6.09 (s, 2H, meso), 6.05 (d, 2H, dl), 2.07 (s, 6H, dl) 2.00 (s, 6H, meso 13C-NMR (CDCl3, r.t.) δ 169.9, 169.7, 136.2, 136.1, IR (KBr) ν 3034, 2949, 1737, 1496, 1241 cm−1.
Coupling Reaction of 4-Fluorobenzaldehyde 5c Using 1 (Table 2-4-1, entry 7).
According to the procedure given for the coupling reaction of acetophenone 2a using
1, the reductive coupling reaction of 4-fluorobenzaldehyde 5c (61.9 mg, 0.499 mmol)
using 1 (1.9 mg, 5.0 μmol) was performed. The resulting mixture was purified by silica gel column chromatography (eluent: hexane / ethyl acetate = 2 / 1 to 1 / 1) and preparative TLC (eluent: hexane / ethyl acetate / dichloromethane = 200 / 50 / 1) to afford diol 6c as a mixture of dl- and meso-diastereomers (44.0 mg, 0.176 mmol, 71% yield, dl : meso = 69 : 31) as a pale yellow solid: 1H-NMR (CDCl
3, r.t.) δ 7.14–6.88 (m, 16H), 4.80 (s, 2H,
meso), 4.59 (s, 2H, dl), 3.06 (br, 13C-NMR (CDCl3, r.t.) δ 163.8 (2C), 161.4, 161.3,
135.5 (2C), IR (KBr) ν 3322, 2898,