目次
2.7.2
臨床薬理試験 ...3
2.7.2.1
背景及び概観...6
2.7.2.1.1
ヒト生体試料を用いた試験 ...6
2.7.2.1.2
臨床試験...10
2.7.2.2
個々の試験結果の要約 ...13
2.7.2.2.1
ヒト生体試料を用いた試験 ...13
2.7.2.2.1.1
血漿蛋白結合,血球移行性 ...13
2.7.2.2.1.2
代謝 ...14
2.7.2.2.1.3
薬物相互作用...16
2.7.2.2.1.4
排出トランスポータに関する検討 ...19
2.7.2.2.1.5
取り込みトランスポータに関する検討...20
2.7.2.2.1.6
その他...22
2.7.2.2.2
臨床薬理試験 ...23
2.7.2.2.2.1
日本人健康成人における第 I 相試験及び薬物動態試験 ...23
2.7.2.2.2.2
外国人健康成人における第 I 相試験及び薬物動態試験 ...33
2.7.2.2.2.3
特殊集団における薬物動態試験...49
2.7.2.2.2.4
薬物相互作用試験...73
2.7.2.2.2.5
薬力学試験 ...87
2.7.2.3
全試験を通しての結果の比較と解析 ...105
2.7.2.3.1
薬物動態学的な特徴 ...105
2.7.2.3.1.1
日本人健康成人における基本的な薬物動態パラメータ ...105
2.7.2.3.1.2
分布に関する評価...108
2.7.2.3.1.3
代謝に関する評価...109
2.7.2.3.1.4
排泄に関する評価... 111
2.7.2.3.2
内因性要因 ... 111
2.7.2.3.2.1
民族間差 ... 111
2.7.2.3.2.2
加齢の影響 ...117
2.7.2.3.2.3
性差 ...118
2.7.2.3.2.4
腎機能障害の影響...121
2.7.2.3.2.5
肝機能障害の影響...121
2.7.2.3.2.6
CYP2D6 の遺伝多型 ...121
2.7.2.3.2.7
OAB 患者...122
2.7.2.3.3
薬物間相互作用...125
2.7.2.3.3.1
CYP3A4 関連の相互作用(ケトコナゾール,リファンピシン,経口避妊薬,
ソリフェナシン)...126
2.7.2.3.3.2
CYP2C9 関連の相互作用(ワルファリン)...127
2.7.2.3.3.3
CYP2D6 関連の相互作用(メトプロロール,デシプラミン)...127
2.7.2.3.3.4
P-糖蛋白関連の相互作用(ジゴキシン) ...128
2.7.2.3.3.5
その他の相互作用(メトホルミン) ...128
2.7.2.3.4
薬力学作用 ...128
2.7.2.3.4.1
濃度-反応関係 ...128
2.7.2.3.4.2
用量/濃度-QT 関係 ...132
2.7.2.4
特別な試験 ...138
2.7.2
臨床薬理試験
本項で使用した略号及び用語の定義一覧を表 2.7.2-1 に示す。
表 2.7.2-1
略号及び用語の定義一覧
略号及び用語 定義 3H 質量数 3 の水素放射性同位元素 14C 質量数 14 の炭素放射性同位元素 α1-AGP α1-酸性糖蛋白 Ae 尿中排泄量 Aeinf 時間 0 から無限時間まで外挿した Ae Aelast 時間 0 から尿中濃度定量可能最終時点までの Ae Aet 時間 0 から時間 t までの Ae Ae% 尿中排泄率 Aeinf% 時間 0 から無限時間まで外挿した Ae% Aelast% 時間 0 から尿中濃度定量可能最終時点までの Ae% Aet% 時間 0 から時間 t までの Ae% AUC 血漿中濃度−時間曲線下面積AUCinf 時間 0 から無限時間まで外挿した AUC
AUClast 時間 0 から血漿中濃度定量可能最終時点までの AUC
AUCt 時間 0 から時間 t までの AUC
AUCtau 時間 0 から投与間隔までの AUC
BMI 体格指数
BOO bladder outlet obstruction:下部尿路閉塞
BW284c51 1,5-ビス(4-アリルジメチルアンモニウムフェニル)ペンタン-3-オン ジブロミ ド Caco-2 細胞 ヒト結腸腺癌由来細胞 CI 信頼区間 CL/F 経口クリアランス CLR 腎クリアランス Cmax 最高血漿中濃度 Css 定常状態における平均血漿中未変化体濃度 Ct 母集団薬物動態モデル(最終モデル)で推定した投与後経過時間 t 時間におけ る血漿中未変化体濃度 Ctrough 血漿中トラフ濃度 CV 変動係数 CYP チトクロム P450 ΔQTc 最終評価時における QTc のベースラインからの変化量 dQTcI 個体別補正因子を使って心拍数で補正した QT(QTcI)間隔のベースラインか らの変化量 ddQTcF Fridericia 式により補正した QTc(QTcF)間隔の同じ時刻のベースラインからの 変化量の実薬投与時とプラセボ投与時との差 ddQTcI 個体別補正因子を使って心拍数で補正した QT(QTcI)間隔の同じ時刻のベー スラインからの変化量の実薬投与時とプラセボ投与時との差 dRR RR のベースラインからの変化量 ECG 心電図 EE エチニルエストラジオール
略号及び用語 定義 eGFR 推定糸球体ろ過量 EM extensive metabolizer:ある薬物代謝酵素において,高い代謝能を有するヒト F 絶対バイオアベイラビリティ FMO フラビン含有モノオキシゲナーゼ Fr OAB 患者の 100 mg 群を基準にした相対バイオアベイラビリティ(母集団薬物 動態解析) fu 血漿蛋白非結合型分率 GFR 糸球体ろ過量 GMR 幾何平均比(最小二乗平均比も含む) HDL 高密度リポ蛋白 HPLC 高速液体クロマトグラフィー HPLC-UV 紫外光検出高速液体クロマトグラフィー HSA ヒト血清アルブミン IC50 50%阻害濃度 IR 即放性 Ki 阻害定数 Km 基質薬物のミカエリス定数 LC-MS 液体クロマトグラフィー-マススペクトロメトリー LC-MS/MS 液体クロマトグラフィー-タンデムマススペクトロメトリー LDL 低密度リポ蛋白 LNG レボノルゲストレル LOQ 定量下限値 LSC 液体シンチレーションカウンター
LUTS lower urinary tract symptom:下部尿路症状
M5 代謝物:ミラベグロン脱アシル体(M16)-Nw-アセチル体,YM-538852,M-5 M8 代謝物:ミラベグロン-N-a位酸化体(フェニル酢酸誘導体),YM-538853,M-8 M9 代謝物:4-アセチルアミノフェニル酢酸,YM-340790,M-9 M11 代謝物:ミラベグロン-O-グルクロニド,YM-382984,RB-3 M12 代謝物:ミラベグロンケトン酸化体(M18)-N-COO-グルクロニド,YM-538858, RB-6 M13 代謝物:ミラベグロン-N-COO-グルクロニド,YM-538859,RB-9 M14 代謝物:ミラベグロン-Nω-グルクロニド,YM-554028,RB-11 M15 代謝物:ミラベグロン-N-O-グルクロニド,YM-9636324,H1 M16 代謝物:ミラベグロン脱アシル体,YM-208876 M17 代謝物:ミラベグロン脱アシル体-o-O-グルクロニド,H2 MDR1 P-糖蛋白
MDRD modification of diet in renal disease
mRNA メッセンジャーRNA
MR 未変化体に対する代謝物の比
NA 適用せず
NADPH ニコチンアミドアデニンジヌクレオチドリン酸の還元型
NMR 核磁気共鳴スペクトロメトリー
OAB overactive bladder:過活動膀胱
OATP 有機アニオン輸送ポリペプチド
OCAS Oral Controlled Absorption System:持続吸収型経口徐放システム
OCT 有機カチオントランスポータ
略号及び用語 定義 PM poor metabolizer:ある薬物代謝酵素において,代謝能が欠損又は著しく低いヒ ト PT プロトロンビン時間 PTR ピークトラフ比 Qmax 最大尿流率 QTc corrected QT:補正 QT QTcF Fridericia 式により補正した QT QTcI 個体別補正因子を使って心拍数で補正した QT(=QT/RRβ) R2 決定係数 S9 9000 g 上清 SD 標準偏差 SE 標準誤差 t1/2 消失半減期 tlag 吸収ラグタイム tmax 最高血漿中濃度到達時間 Vss 定常状態での分布容積 Vz/F 経口投与時の消失相における分布容積 YM178 ミラベグロン
2.7.2.1
背景及び概観
2.7.2.1.1 ヒト生体試料を用いた試験
ヒト生体試料を用いた試験一覧を表 2.7.2-2 に示す。
In vitro 試験により,ミラベグロンの血漿蛋白結合,血球移行性,代謝,チトクロム P450(CYP)
阻害並びに誘導作用及び薬物間相互作用について検討した。また,種々のトランスポータによる
排出又は取り込み及びそれらのトランスポータに対する阻害活性についても検討した。
ミラベグロンの蛋白結合について,血漿蛋白結合率の測定[ME-021],主要結合蛋白の推定
[ME-044]並びに代謝物 M5 及び M16 の血漿蛋白結合率の測定[ME-128]を行った。また,ヒ
ト血液を用いてミラベグロンの血球移行性[ME-045]を検討した。
ミラベグロンの代謝について,肝ミクロソームを用いて代謝プロファイルの検討[ME-020],
肝ミクロソーム及びヒト CYP 発現系を用いて代謝に関与する CYP 分子種の同定[ME-002],ヒ
ト血漿を用いて加水分解代謝物の同定[ME-034],各種酵素源を用いて代謝に関与するエステラー
ゼ分子種の同定[ME-079]を行った。ミラベグロンの CYP 阻害作用について,肝ミクロソーム
又はヒト CYP 発現系を用いて検討した[ME-009,ME-068]。ミラベグロンの CYP 誘導作用につ
いて,ヒト初代肝培養細胞を用いて検討した[ME-074]。また,薬物間相互作用については,CYP3A4
の基質又は阻害剤,CYP2D6 の基質及びスルホニル尿素系血糖降下剤を用いて,ミラベグロンの
CYP 阻害作用並びにミラベグロンの代謝活性に及ぼす阻害作用を検討した[ME-015,ME-024]。
さらに,P-糖蛋白(MDR1)に対する基質性/阻害作用の検討[ME-031,ME-032],ヒト有機カチ
オントランスポータ(OCT)に対する基質性/阻害作用の検討[ME-092,ME-086]及びヒト凍結
肝細胞を用いた取り込みの検討[ME-109]を実施した。
Ex vivo の試験として,マスバランス試験[CL-007]で得られたヒト血漿及び尿試料を用いて,
ミラベグロンの光学異性体への変換について検討した[ME-041]。また,臨床試験[CL-003,CL-004,
CL-005 及び CL-007]で得られたヒト血漿及び尿試料を用いて,代謝物の同定及び構造推定を行っ
た[ME-046,ME-055 及び ME-083]。
表 2.7.2-2
ヒト生体試料を用いた試験一覧
試験項目 試験 番号 試験内容 ヒト組織,試験薬等 方法 ミラベグロン 添加濃度 添付資料 番号 ME-021 血漿蛋白結合率 血漿 限外濾過法 5000 ng/mL200, 1000, a 4.2.2.3-1 ME-044 血漿中主要結合蛋白 の推定 血清アルブミン (40 mg/mL) α1-酸性糖蛋白 (1 mg/mL) 高密度リポ蛋白 (3 mg/mL) 低密度リポ蛋白 (3 mg/mL) g-グロブリン (10 mg/mL) 限外濾過法 200, 1000, 5000 ng/mLa 5.3.2.1-1 In vitro 蛋白結合 ME-128 代謝物の 血漿蛋白結合率 血漿 限外濾過法 M5, M16: 10~250 ng/mL 5.3.2.1-2 In vitro 血球移行 ME-045 血球移行性 血液 インキュベー ション 100, 500, 2500 ng/mLa 4.2.2.3-2 ME-020 代謝プロファイルの 検討 肝ミクロソーム インキュベー ション 10 mmol/La 4.2.2.4-2 CYP 分子種同定 (発現系) CYP 発現系 FMO 発現系 インキュベー ション 0.2 mmol/L CYP 分子種同定 (親和性) 肝ミクロソーム CYP 発現系 インキュベー ション 0.01~25 mmol/L CYP 分子種同定 (相関) 肝ミクロソーム インキュベー ション 0.05,0.2, 1 mmol/L CYP 分子種同定 (阻害) 肝ミクロソーム インキュベー ション 0.2 mmol/L ME-002 CYP 分子種同定 (抗体) 肝ミクロソーム インキュベー ション 0.2 mmol/L 5.3.2.2-1 ME-034 加水分解代謝物の 同定 血漿 インキュベー ション 100 ng/mLa 5.3.2.3-4 エステラーゼ 分子種同定 (時間及び蛋白濃度 依存性) 血液 血漿 肝ミクロソーム 小腸ミクロソーム 肝 S9 小腸 S9 アセチルコリンエス テラーゼ(組換え体) の発現系 ブチリルコリンエス テラーゼ カルボキシルエステ ラーゼの発現系 インキュベー ション 5 mmol/L エステラーゼ 分子種同定 (基質濃度依存性) 血液 血漿 ブチリルコリンエス テラーゼ インキュベー ション 1~250 mmol/L In vitro 代謝 ME-079 エステラーゼ 分子種同定 (阻害) 血液 血漿 ブチリルコリンエス テラーゼ インキュベー ション 10 mmol/L 5.3.2.3-5ME-009 CYP 阻害作用 CYP 発現系 インキュベー ション 0.114~ 250 mmol/L 5.3.2.2-2 ME-068 CYP 阻害作用 肝ミクロソーム フェナセチン (60 mmol/L) ブプロピオン (50 mmol/L) アミオダロン (2.0 mmol/L) ジクロフェナック (7.5 mmol/L) S-メフェニトイン (40 mmol/L) デキストロメトル ファン(7.5 mmol/L) クロロゾキサゾン (30 mmol/L) テストステロン (100 mmol/L) ミダゾラム (5.0 mmol/L) インキュベー ション 0.1~100 mmol/L 5.3.2.2-5 ME-015 薬物相互作用 肝ミクロソーム デキストロメトル ファン(2.5~ 50 mmol/L) メトプロロール (2~100 mmol/L) ミダゾラム (2 mmol/L) ニフェジピン (0.5~50 mmol/L) インキュベー ション IC50算出時: 0.5~800 mmol/L Ki値算出時: 3.75~60 mmol/L 5.3.2.2-3 ME-024 薬物相互作用 肝ミクロソーム グリベンクラミド (0.01, 0.2, 1 mmol/L) トルブタミド (5, 100, 500 mmol/L) グリクラジド (0.5, 10, 50 mmol/L) インキュベー ション 0.03, 0.6, 3 mmol/L 5.3.2.2-4 In vitro 代謝 (相互作 用) ME-074 CYP 誘導作用 初代肝培養細胞 フェナセチン (80 mmol/L) テストステロン (250 mmol/L) インキュベー ション 0.1, 1, 10 mmol/L 5.3.2.2-6 ME-031 Caco-2 細胞による 経細胞輸送の評価 Caco-2 発現細胞 インキュベー ション 1~250 mmol/La 5.3.2.3-6 In vitro 膜透過 (排出) ME-032 MDR1 による ビンブラスチン輸送 に対する阻害 MDR1 発現細胞 MDR1 非発現細胞 3H-ビンブラスチン (1 mmol/L) インキュベー ション 16, 250 mmol/L 5.3.2.3-7
OCT1,OCT2,OCT3 による 細胞内取り込みの評 価 (時間依存性) OCT1 発現細胞 OCT2 発現細胞 OCT3 発現細胞 OCT 非発現細胞 (mock 細胞) インキュベー ション 10 mmol/La OCT1,OCT2,OCT3 による 細胞内取り込みの評 価 (基質濃度依存性) OCT1 発現細胞 OCT2 発現細胞 OCT3 発現細胞 OCT 非発現細胞 (mock 細胞) インキュベー ション 2~500 mmol/La ME-092 OCT1,OCT2,OCT3 による 細胞内取り込みの評 価(阻害) OCT1 発現細胞 OCT2 発現細胞 OCT3 発現細胞 OCT 非発現細胞 (mock 細胞) インキュベー ション 10 mmol/La 5.3.2.3-8 ME-086 OCT1,OCT2 による テトラエチルアンモ ニウム輸送に対する 阻害 OCT1 発現細胞 OCT2 発現細胞 OCT 非発現細胞 (mock 細胞) 14C-テトラエチルア ンモニウム (5 mmol/L) インキュベー ション 3~1000 mmol/L 5.3.2.3-9 肝細胞による 取り込みの評価 (時間依存性) 凍結保存肝細胞 インキュベー ション 1 mmol/La 肝細胞による 取り込みの評価 (基質濃度依存性) 凍結保存肝細胞 インキュベー ション 1~500 mmol/La In vitro 膜透過 (取り込 み) ME-109 肝細胞による取り込 みの評価(阻害) 凍結保存肝細胞 インキュベー ション 1 mmol/La 5.3.2.3-10 Ex vivo 異性体変換 ME-041 光学異性体への変換 血漿,尿 HPLC LC-MS 単回,空腹下,[CL-007] 経口,160 mga 5.3.2.3-1 ME-046 代謝物の検索 及び同定 血漿,尿 HPLC-UV LC-MS LC-MS/MS LSC [CL-007] 単回,空腹下, 経口,160 mga 5.3.2.3-2 ME-055 代謝物の構造推定 (H1, H2) 尿 HPLC-UV LC-MS LC-MS/MS NMR [CL-003] 12 週間(用量漸 増),食後,経口, 1 日 1 回投与, 60,130,200 mg [CL-004] 12 週間(用量漸 増),食後,経口, 1 日 1 回投与, 60,130,200 mg [CL-005] 単回,空腹下, 経口,160 mg 5.3.2.3-3 Ex vivo 代謝 ME-083 代謝物の同定 [ME-055]で尿試料 より精製した 代謝物試料(M15) LC-MS - 4.2.2.4-7 a:14C-ミラベグロン
2.7.2.1.2 臨床試験
ミラベグロンの薬物動態及び薬力学を検討した臨床試験の一覧を表 2.7.2-3 に示す。
ミラベグロンの臨床的な薬物動態特性を検討するため,「2.7.1 生物薬剤学試験及び関連する分
析法」に示す薬物動態試験の他に,健康成人を対象にした 6 試験を実施した(第 I 相単回及び反
復投与試験[CL-034],用量比例性試験[CL-066],単回投与及び食事の影響試験(IR カプセル),
[CL-001],反復投与試験(IR カプセル)
[CL-002],マスバランス試験[CL-007],反復投与,性
差及び高齢者試験[CL-031])。このうち,第 I 相単回及び反復投与試験[CL-034]と用量比例性
試験[CL-066]は国内で実施した。また,特別な集団を対象にした 3 試験を海外で実施した(性
差及び高齢者試験[CL-072],腎機能障害患者における薬物動態試験[CL-038],肝機能障害患者
における薬物動態試験[CL-039])。さらに,ミラベグロンの薬物相互作用を検討するため,健康
成人を対象にした 8 試験を海外で実施した(薬物相互作用試験(ケトコナゾール)[CL-036],薬
物相互作用試験(リファンピシン)[CL-070],薬物相互作用試験(ワルファリン)[CL-040],薬
物相互作用試験(メトプロロールと IR カプセル)[CL-005],薬物相互作用試験(デシプラミン)
[CL-058],薬物相互作用試験(ジゴキシン)[CL-059],薬物相互作用試験(メトホルミンと IR
錠)[CL-006],薬物相互作用試験(経口避妊薬)[CL-068])。以上の薬物動態試験の他に,国内
で実施した第 I 相単回及び反復投与試験[CL-034]の健康成人男性及び第 II 相試験[CL-045]の
過活動膀胱(OAB)患者から得られた血漿中未変化体濃度データを用いて,母集団薬物動態解析
を行った。また,国内で実施した第 III 相試験[CL-048]でミラベグロンの薬物動態を検討した。
ミラベグロンの薬力学試験として,下部尿路症状(LUTS)及び下部尿路閉塞(BOO)の患者を
対象にした尿流動態試験[CL-060]を海外で実施した。また,ICH E14 ガイドラインに基づき,
QT/QTc 間隔に及ぼすミラベグロンの影響を海外の健康成人で検討した(QT/QTc 評価試験
[CL-037]及び QT/QTc 評価試験 2[CL-077]
(参考資料))。以上の薬力学試験の他に,国内で実
施した第 II 相試験[CL-045]の OAB 患者から得られた血漿中未変化体濃度データを用いて,OAB
患者における有効性評価項目及び脈拍数との濃度–反応関係,並びに濃度–QT 関係を検討した。な
お,QT/QTc 間隔に及ぼすミラベグロンの影響を国内外の OAB 患者で検討した結果については,
「2.7.4.4.3 QTc の評価」に示す。
表 2.7.2-3
ミラベグロンの薬物動態及び薬力学を検討した臨床試験の一覧
試験番号 (添付資料番号) 試験目的 実施地域 被験者種類 被験者数(男/女) ミラベグロンの剤型 用法・用量 (試験デザイン) 健康被験者での薬物動態試験 CL-034 (5.3.3.1-1) 単回及び反復投与時 の安全性,薬物動態 日本 健康成人 単回:40 (40/0) 反復:24 (24/0) OCAS 錠 単回:50,100,200,300,400 mg 又はプラセボを 空腹時に単回経口投与 反復:100,200 mg 又はプラセボを食後に単回経口 投与及び 1 日 1 回 7 日間経口投与 (無作為化,単盲検,プラセボ対照,用量漸増) CL-066 (5.3.3.1-2) 薬物動態の 用量比例性 日本 健康成人 12 (12/0) OCAS 錠 25,50,100 mg を低用量から順に空腹時に 単回経口投与 (非盲検,個体内増量) CL-001 (5.3.3.1-3) パート I:単回投与時 の安全性,忍容性, 薬物動態 パート II:食事の影響 英国 健康成人 パート I:85 (85/0) パート II:12 (12/0) IR カプセル パート I:0.1,0.3,1,3,10,30,100,160,240, 340 mg 又はプラセボを空腹時に単回経口投与 (無作為化,二重盲検,プラセボ対照,用量漸増) パート II:160 mg を食後,空腹時又は食前に 単回経口投与 (無作為化,非盲検,クロスオーバー) CL-002 (5.3.3.1-4) 反復投与時の安全性, 忍容性,薬物動態 英国 健康成人 40 (40/0) IR カプセル 40,80,160,240 mg 又はプラセボを空腹時に, 240 mg 又はプラセボを食後に 単回及び 1 日 1 回 7 日間経口投与 (無作為化,二重盲検,プラセボ対照,用量漸増) CL-007 (5.3.3.1-5) マスバランス オランダ 健康成人 4 (4/0) 14C-ミラベグロン溶液 160 mg を空腹時に単回経口投与 (非盲検) CL-031 (5.3.3.1-7) 反復投与時の薬物動 態,安全性,忍容性, 性差,年齢の影響 オランダ 健康非高齢者 64 (32/32) 健康高齢者 32 (16/16) OCAS 錠 非高齢者:50,100,200,300 mg 又はプラセボを 空腹時に単回及び 1 日 1 回 10 日間経口投与 高齢者:50,200 mg 又はプラセボを 空腹時に単回及び 1 日 1 回 10 日間経口投与 (無作為化,二重盲検,プラセボ対照) 特別な集団での薬物動態試験 CL-072 (5.3.3.3-1) 性差,年齢の影響 フランス 健康非高齢者 36 (18/18) 健康高齢者 39 (21/18) OCAS 錠 25,50 又は 100 mg を食後に 1 日 2 回 1 日間及び 1 日 1 回 6 日間経口投与 (無作為化,非盲検,クロスオーバー) CL-038 (5.3.3.3-2) 腎機能障害の影響 米国 健康成人 8 (4/4) 腎機能障害患者 25 (14/11) OCAS 錠 100 mg を空腹時に単回経口投与 (非盲検,群間比較) CL-039 (5.3.3.3-3) 肝機能障害の影響 スロバキア 健康成人 16 (9/7) 肝機能障害患者 16 (9/7) OCAS 錠 100 mg を空腹時に単回経口投与 (非盲検,群間比較)試験番号 (添付資料番号) 試験目的 実施地域 被験者種類 被験者数(男/女) ミラベグロンの剤型 用法・用量 (試験デザイン) 薬物相互作用試験 CL-036 (5.3.3.4-1) ケトコナゾールとの 薬物相互作用 米国 健康成人 24 (12/12) OCAS 錠 100 mg を空腹時に単回経口投与×2 回; ケトコナゾール 400 mg を 1 日 1 回 9 日間経口投与 (非盲検,クロスオーバー) CL-070 (5.3.3.4-2) リファンピシンとの 薬物相互作用 米国 健康成人 24 (13/11) OCAS 錠 100 mg を空腹時に単回経口投与×2 回; リファンピシン 600 mg を 1 日 1 回 11 日間経口投与 (非盲検) CL-040 (5.3.3.4-3) ワルファリンとの 薬物相互作用 フランス 健康成人 24 (12/12) OCAS 錠 100 mg を空腹時に 1 日 1 回 16 日間経口投与; ワルファリン 25 mg を単回経口投与×2 回 (非盲検,クロスオーバー) CL-005 (5.3.3.4-4) パート I:CYP2D6 PM と EM での薬物動態 パート II:メトプロ ロールとの薬物相互 作用 オランダ パート I:CYP2D6 PM 8 (8/0) EM 8 (8/0) パート II:CYP2D6 EM 12 (12/0) IR カプセル パート I:160 mg を空腹時に単回経口投与 (非盲検) パート II:160 mg を空腹時に 1 日 1 回 5 日間経口投与; 酒石酸メトプロロール 100 mg を単回経口投与×2 回 (非盲検,クロスオーバー) CL-058 (5.3.3.4-5) デシプラミンとの 薬物相互作用 フランス 健康成人 28 (14/14) OCAS 錠 100 mg を空腹時に 1 日 1 回 19 日間経口投与; デシプラミン 50 mg を単回経口投与×3 回 (非盲検,クロスオーバー) CL-059 (5.3.3.4-6) ジゴキシンとの 薬物相互作用 フランス 健康成人 25 (13/12) OCAS 錠 100 mg を空腹時に 1 日 1 回 14 日間経口投与; ジゴキシン 0.25 mg を単回経口投与×2 回 (非盲検,クロスオーバー) CL-006 (5.3.3.4-7) メトホルミンとの 薬物相互作用 オランダ 健康成人 32 (32/0) IR 錠 160 mg を空腹時に 1 日 1 回 16 日間又は 11 日間経口 投与;メトホルミン 500 mg を 1 日 2 回 5 日間 又は 16 日間経口投与 (クロスオーバー) CL-068 (5.3.3.4-8) 経口避妊薬との 薬物相互作用 フランス 健康成人 30 (0/30) OCAS 錠 100 mg 又はプラセボを空腹時に 1 日 1 回 10 日間経 口投与;経口避妊薬(Minidril:エチニルエストラジ オール 30 μg,レボノルゲストレル 150 μg)を 1 日 1 回 21 日間経口投与 (無作為化,二重盲検,クロスオーバー) 薬力学試験 CL-037 (5.3.4.1-1) QT/QTc 評価 米国 健康成人 48 (25/23)a OCAS 錠 100,200 mg 又はプラセボを空腹時に 1 日 1 回 7 日 間経口投与; モキシフロキサシン 400 mg 又はプラセボを 単回経口投与 (無作為化,二重盲検,プラセボ・実薬対照, クロスオーバー)
試験番号 (添付資料番号) 試験目的 実施地域 被験者種類 被験者数(男/女) ミラベグロンの剤型 用法・用量 (試験デザイン) CL-077 (5.3.4.1-2) (参) QT/QTc 評価 米国 健康成人 352 (176/176) OCAS 錠 50,100,200 mg 又はプラセボを空腹時に 1 日 1 回 10 日間経口投与; モキシフロキサシン 400 mg 又はプラセボを 単回経口投与 (無作為化,二重盲検,プラセボ・実薬対照, クロスオーバー) CL-060 (5.3.4.2-1) 下部尿路症状(LUTS) 及び下部尿路閉塞 (BOO)患者の 尿流動態への影響 米国とカナダ LUTS 及び BOO の 患者 200 (200/0) OCAS 錠 50,100 mg 又はプラセボを食後に 1 日 1 回 12 週間経口投与 (無作為化,二重盲検,プラセボ対照,群間比較) PM = poor metabolizer,EM = extensive metabolizer,a:無作為化後に治験薬を投与されなかった 1 例を除く
2.7.2.2
個々の試験結果の要約
2.7.2.2.1 ヒト生体試料を用いた試験
2.7.2.2.1.1
血漿蛋白結合,血球移行性
2.7.2.2.1.1.1 血漿蛋白結合率[ME-021]
··· 添付資料 4.2.2.3-1
14C-ミラベグロンの血漿蛋白結合率を限外濾過法により測定したところ,添加濃度 200~
5000 ng/mL における血漿蛋白結合率は日本人で 76.3%~76.9%,白人で 72.2%~73.3%であった。
検討した濃度範囲において血漿蛋白結合率は一定であった。
2.7.2.2.1.1.2 血漿中主要結合蛋白の推定[ME-044]
··· 添付資料 5.3.2.1-1
血漿中主要結合蛋白を推定する目的で,健康成人の血漿中蛋白含量を参考に調製した HSA
(40 mg/mL),a
1-酸性糖蛋白(a
1-AGP,1 mg/mL),HDL(3 mg/mL),LDL(3 mg/mL),g-グロブ
リン(10 mg/mL)に対する結合率を,限外濾過法により測定した。
14C-ミラベグロン(添加濃度
200~5000 ng/mL)と最も高い結合率を示したのは HSA(33.9%~37.4%)であり,次いでa
1-AGP
(24.0%~31.6%),LDL(9.9%~15.3%)及び HDL(4.2%~11.9%),g-グロブリン(2.1%~5.0%)
の順となった。検討した濃度範囲において,いずれの蛋白結合率においても明確な濃度依存性は
認められなかった。
2.7.2.2.1.1.3 代謝物の血漿蛋白結合率[ME-128]
··· 添付資料 5.3.2.1-2
M5 及び M16 の血漿蛋白結合率を限外濾過法により測定した。M5(添加濃度 10~250 ng/mL)
の血漿蛋白結合率は日本人で 64.5%~67.1%,白人で 44.4%~47.2%であり,M16(添加濃度 10~
250 ng/mL)の血漿蛋白結合率は日本人で 47.4%~48.5%,白人で 32.4%~33.7%であった。検討し
た濃度範囲において血漿蛋白結合率は一定であった。
2.7.2.2.1.1.4 血球移行性[ME-045]
··· 添付資料 4.2.2.3-2
14C-ミラベグロン(添加濃度 100~2500 ng/mL)の赤血球移行率,赤血球-血漿間分配比及び血
液-血漿間分配比は,それぞれ 60.39%~60.82%,1.94~1.98,1.41~1.43 であった。検討した濃度
範囲において濃度依存性は認められなかった。
2.7.2.2.1.2
代謝
2.7.2.2.1.2.1 代謝プロファイルの検討[ME-020]
··· 添付資料 4.2.2.4-2
ヒト肝ミクロソームを用いて
14C-ミラベグロン(添加濃度 10 mmol/L)の代謝プロファイルを検
討したところ,NADPH 存在下で
14C-ミラベグロンは代謝され,5 種類の第一相代謝物が生成した。
しかしながら,ミラベグロンの代謝速度は遅く,肝ミクロソーム蛋白濃度 1 mg/mL においても代
謝物の生成量はわずかであった。また,NADPH 非存在下では明らかな代謝は見られなかった。
2.7.2.2.1.2.2 代謝に関与するチトクロム P450(CYP)分子種の同定[ME-002]
··· 添付資料 5.3.2.2-1
ヒト肝ミクロソーム及びヒト CYP 発現系を用いてミラベグロンの代謝に関与する CYP 分子種
を検討した。
ヒト CYP 発現系を用いてミラベグロンの代謝酵素を検討したところ,ミラベグロン(添加濃度
0.2 mmol/L)に対する明確な代謝活性を示した分子種は CYP2D6 及び CYP3A4 であった。ミラベ
グロンの代謝による減少量から推定した K
m値はヒト肝ミクロソーム(ミラベグロン添加濃度 0.01
~10 mmol/L),CYP2D6(ミラベグロン添加濃度 0.01~5 mmol/L)及び CYP3A4(ミラベグロン添
加濃度 0.01~15 mmol/L)に対してそれぞれ 8.5,0.23 及び 39.9 mmol/L であった。
ヒト肝ミクロソームによるミラベグロン(添加濃度 0.2 mmol/L,n=3)の代謝活性はテストステ
ロン 6β 水酸化活性(CYP3A4/5)と有意な相関(R
2= 0.39775~0.58863,P = 0.00052~0.00524)が
あった。また,3 回中 1 回の実験で,ミラベグロン(添加濃度 0.2 mmol/L)の代謝活性はデキスト
ロメトルファン O-脱メチル化活性(CYP2D6)とも有意な相関(R
2= 0.32251,P = 0.02174)があっ
た。ミラベグロンの異なる添加濃度(0.05 及び 1 mmol/L,n=1)における相関を追加で検討したと
ころ,ミラベグロンの代謝活性はテストステロン 6β 水酸化活性(CYP3A4/5)と有意な相関
(R
2= 0.49075~0.59417,P = 0.00102~0.00251)が認められたが,デキストロメトルファン O-脱
メチル化活性(CYP2D6)とは有意な相関は認められなかった(R
2= 0.15797~0.21723,P = 0.06881
~0.12739)。
ヒト肝ミクロソームによるミラベグロン(添加濃度 0.2 mmol/L)の代謝は CYP3A4 の特異的阻
害剤であるケトコナゾール及びトロレアンドマイシンの存在下で阻害され,それぞれの阻害の程
度は 1 mmol/L のケトコナゾールで 52%,100 mmol/L のトロレアンドマイシンで 20%であった。ま
た,ヒト肝ミクロソームによるミラベグロン(添加濃度 0.2 mmol/L)の代謝は抗 CYP2D6 抗体及
び抗 CYP3A4 抗血清の存在下で阻害されたが,抗 CYP3A4 抗血清が最大 80%の阻害を示したのに
対し,抗 CYP2D6 抗体による阻害は 10%であった。
以上の結果から,ミラベグロンの CYP 代謝には CYP3A4 が主として関与していると考えられた。
CYP2D6 が関与する可能性も除外できなかったが,その後の臨床試験において CYP2D6 の extensive
metabolizer(EM)群と poor metabolizer(PM)群を比較したところ,ミラベグロンを 160 mg 単回
経口投与後の血漿中ミラベグロン濃度の C
max及び AUC
infは PM の方が EM に比べ高値であったも
のの,両者の差は小さかった(2.7.2.2.2.3.4 薬物相互作用試験(メトプロロールと IR カプセル)
-CYP2D6 PM/EM における試験[CL-005])。
2.7.2.2.1.2.3 加水分解代謝物の同定[ME-034]
··· 添付資料 5.3.2.3-4
ミラベグロンはヒト血漿中において分解される。ヒト血漿中において生じるミラベグロンの分
解物(代謝物)を同定した。
14C-ミラベグロンをヒト血漿とインキュベートした in vitro 試料を放射能検出 HPLC 分析するこ
とにより代謝物を検索した。クロマトグラム上に 1 種の代謝物ピークが認められ,HPLC 保持時
間及び MS スペクトルを合成標品と比較することにより,分解物(代謝物)の構造を M16(ミラ
ベグロン脱アシル体,YM-208876)と同定した。ミラベグロンはヒト血漿中に存在するエステラー
ゼによりアミド部位で加水分解を受けることが推察された。
2.7.2.2.1.2.4 代謝に関与するエステラーゼ分子種の同定[ME-079]
··· 添付資料 5.3.2.3-5
ヒト血液,血漿,肝ミクロソーム,小腸ミクロソーム,肝 S9,小腸 S9,アセチルコリンエステ
ラーゼ(組換え体)の発現系,ヒト血清から精製したブチリルコリンエステラーゼ及びカルボキ
シルエステラーゼ 1 並びに 2 の発現系を用いて,ミラベグロンの加水分解(代謝)に関与するエ
ステラーゼ分子種を検討した。
各種酵素源を用いてミラベグロン(添加濃度 5 mmol/L)の加水分解活性を検討した結果,ヒト
血液,血漿及びブチリルコリンエステラーゼが活性を示したが,ヒト肝ミクロソーム,小腸ミク
ロソーム,肝 S9,小腸 S9,アセチルコリンエステラーゼの発現系及びカルボキシルエステラーゼ
1 並びに 2 の発現系は活性を示さなかった。
加水分解代謝物 M16 の生成から推定した K
m値(ミラベグロン添加濃度 1~250 mmol/L)はヒト
血液,血漿及びブチリルコリンエステラーゼに対してそれぞれ 14.5,15.2 及び 13.4 mmol/L とほぼ
同等であり,ヒト血液及び血漿中のミラベグロン加水分解活性はいずれもブチリルコリンエステ
ラーゼによって媒介されると考えられた。
ヒト血液,血漿及びブチリルコリンエステラーゼによるミラベグロン(添加濃度 10 mmol/L)
の加水分解は,いずれもブチリルコリンエステラーゼ阻害能を有するフッ化フェニルメチルスル
ホニル(セリン水解酵素阻害剤),エゼリン(コリンエステラーゼ阻害剤),ジイソプロピルフル
オロリン酸(コリンエステラーゼ及びカルボキシルエステラーゼ阻害剤)及びエトプロパジン(ブ
チリルコリンエステラーゼ阻害剤)によりほぼ完全に阻害された。一方,アセチルコリンエステ
ラーゼ阻害剤である 1,5-ビス(4-アリルジメチルアンモニウムフェニル)ペンタン-3-オン ジブロ
ミド(BW284c51)による阻害は中等度で,またパラオキソネース/アリールエステラーゼ阻害剤
(スルフヒドリル試薬)である 5,5'-ジチオビス(2-ニトロ安息香酸),カルボキシルエステラーゼ
阻害剤であるビス-p-ニトロフェニルリン酸及びパラオキソネース/アリールエステラーゼの阻害
剤であるエチレンジアミン四酢酸によってはほとんど阻害されなかった。各種阻害剤を用いた検
討は,ヒト血液及び血漿中のミラベグロン加水分解活性がブチリルコリンエステラーゼによるも
のであることを支持した。
2.7.2.2.1.3
薬物相互作用
2.7.2.2.1.3.1 CYP 分子種に対する阻害作用(CYP 発現系)[ME-009]
···· 添付資料 5.3.2.2-2
ヒトの主要 CYP 分子種代謝活性に及ぼすミラベグロンの阻害作用をヒト CYP 発現系ミクロ
ソームを用いて検討した。ミラベグロン(添加濃度 0.114~250 mmol/L)は CYP2D6 に対する阻害
作用は強かった(IC
50値:0.67 mmol/L)が,CYP2C19(IC
50値:227 mmol/L)及び CYP3A4(IC
50値:42.5 mmol/L)に対する阻害作用は弱かった。ミラベグロンは CYP1A2 及び CYP2C9 に対して
は阻害作用をほとんど示さなかった (IC
50値:> 250 mmol/L)(表 2.7.2-4)。
表 2.7.2-4
ヒト CYP 分子種の代謝活性に対するミラベグロンの阻害作用
CYP 分子種 IC50(mmol/L) CYP1A2 > 250 CYP2C9 > 250 CYP2C19 227 ± 13 CYP2D6 0.67 ± 0.10 CYP3A4 42.5 ± 4.7 4 回の測定値の平均値 ± 標準偏差2.7.2.2.1.3.2 CYP 分子種に対する阻害作用(肝ミクロソーム)[ME-068]
··· 添付資料 5.3.2.2-5
ヒト肝ミクロソームを用いて更に詳細にミラベグロンのヒト主要 CYP 分子種代謝活性に及ぼ
す阻害作用を検討した。ミラベグロン(添加濃度 0.1~100 mmol/L)は CYP2D6 に対して中等度の
直接阻害作用(IC
50値:13 mmol/L)を示したが,CYP1A2,CYP2B6,CYP2C8,CYP2C9,CYP2C19,
CYP2E1 及び CYP3A4/5 に対する直接阻害作用は弱かった(IC
50値:> 100 mmol/L)(表 2.7.2-5)。
CYP2D6 阻害作用には時間依存性が認められ,30 分プレインキュベーション時に IC
50値は
4.3 mmol/L に低下した。しかしながら,それ以外の CYP 分子種に対する時間依存的阻害作用は弱
く,30 分プレインキュベーション時の IC
50値はいずれも 100 mmol/L 以上であった。ミラベグロン
による CYP2D6 の時間依存的阻害には NADPH 依存性が認められ,ヒト肝ミクロソームによるミ
ラベグロンの代謝に起因するものと推察された。また,ミラベグロンの時間依存的阻害により減
少した CYP2D6 の代謝活性(コントロール活性の 28.0%)は,その大半が反応液を希釈すること
によりコントロールレベルに戻った(コントロール活性の 76.2%)ことから,ミラベグロンによ
る CYP2D6 阻害の大半は可逆的阻害であると推察された。
表 2.7.2-5
ヒト CYP 分子種の代謝活性に対するミラベグロンの阻害作用
IC50(mmol/L) CYP 分子種 CYP 反応 直接阻害 (0 分-プレインキュベーション) 時間依存的阻害 (30 分-プレインキュベーション) CYP1A2 フェナセチン O-脱エチル > 100 > 100 CYP2B6 ブプロピオン 水酸化 > 100 > 100 CYP2C8 アモジアキン N-脱アルキル > 100 > 100 CYP2C9 ジクロフェナック 4’-水酸化 > 100 > 100 CYP2C19 S-メフェニトイン 4’-水酸化 > 100 > 100 CYP2D6 デキストロメトル ファン O-脱メチル 13 4.3 CYP2E1 クロルゾキサゾン 6-水酸化 > 100 > 100 CYP3A4/5 テストステロン 6b-水酸化 > 100 > 100 CYP3A4/5 ミダゾラム 1’-水酸化 > 100 > 100 2 回の測定値の平均値2.7.2.2.1.3.3 薬物相互作用の検討(CYP2D6 の基質及び CYP3A4 の基質又は阻害剤)
[ME-015]
··· 添付資料 5.3.2.2-3
ヒト肝ミクロソームを用いて,各種 CYP2D6 基質(デキストロメトルファン並びにメトプロロー
ル)及び CYP3A4 基質(ミダゾラム並びにニフェジピン)の代謝に及ぼすミラベグロンの阻害作
用を検討した。デキストロメトルファン(添加濃度 15 mmol/L),メトプロロール(添加濃度
20 mmol/L),ミダゾラム(添加濃度 2 mmol/L)及びニフェジピン(添加濃度 10 mmol/L)の代謝に
及ぼすミラベグロンの IC
50値は,それぞれ 14.0,4.7~8.5,479~>500 及び 34 mmol/L であった(表
2.7.2-6)。また,デキストロメトルファン(添加濃度 2.5~50 mmol/L),メトプロロール(添加濃度
2~100 mmol/L)及びニフェジピン(添加濃度 0.5~50 mmol/L)の代謝に及ぼすミラベグロンの K
i値は,それぞれ 10.5,3.7~7.9 及び 13.3 mmol/L であった。デキストロメトルファン及びメトプロ
ロールの代謝に及ぼすミラベグロンの K
i値とミラベグロンの臨床推奨用量(50 mg)における C
maxとの比(K
i/C
max)は 25~70 であり,ミラベグロンは in vivo において CYP2D6 基質(デキストロ
メトルファン及びメトプロロール)の代謝に対して軽度の阻害をする可能性が示された。一方,
CYP3A4 基質のミダゾラムの代謝に及ぼすミラベグロンの IC
50値はミラベグロンの臨床推奨用量
における C
maxの 3000 倍以上で,治療域濃度をはるかに上回っており,ミラベグロンは in vivo に
おいてミダゾラムの代謝を阻害しないと考えられた。そのため K
i値の算出は行わなかった。同じ
く CYP3A4 の基質であるニフェジピンの代謝に及ぼすミラベグロンの K
i値とミラベグロンの臨床
推奨用量における C
maxとの比は 89 であり,ミラベグロンはニフェジピン代謝に対しては弱い阻
害作用を示した。
ヒト肝ミクロソームを用いて,ミラベグロンの代謝に及ぼす CYP3A4 阻害剤(ケトコナゾール
及びリトナビル)の阻害作用を検討した。ミラベグロン(添加濃度 5 mmol/L)の代謝に及ぼすケ
トコナゾールの IC
50値及び IC
50値と治療域濃度との比(IC
50/C
p)はそれぞれ 0.47 mmol/L 及び 0.022
~0.094,リトナビルの IC
50値及び IC
50値と治療域濃度との比はそれぞれ 0.065 mmol/L 及び 0.0040
~0.016 といずれも低く,CYP3A4 阻害剤(ケトコナゾール及びリトナビル)はミラベグロンの代
謝を in vivo においても阻害すると考えられた(表 2.7.2-7)。しかし,ミラベグロンの代謝に及ぼ
す CYP3A4 阻害剤(ケトコナゾール及びリトナビル)の阻害作用は,添加した濃度範囲(ケトコ
ナゾール:0.1~100 mmol/L,リトナビル:0.01~10 mmol/L)において最大でも約 65%にとどまっ
たことから,K
i値は算出しなかった。
表 2.7.2-6
各種 CYP2D6 基質及び CYP3A4 基質の代謝に及ぼすミラベグロンの阻害作用
基質 代謝物 IC50(mmol/L) IC50/Cmaxa Ki(mmol/L) Ki/Cmaxa
デキストロメト ルファン デキストロル ファン 14.0 93 10.5 70 メトプロロール メトプロロール a-水酸化体 4.7 31 3.7 25 メトプロロール O-脱メチル体 8.5 57 7.9 53 ミダゾラム ミダゾラム 1’-水酸化体 >500 >3333 – – ミダゾラム 4-水酸化体 479 3193 – – ニフェジピン デヒドロニフェ ジピン 34 227 13.3 89
a:ミラベグロンの Cmaxを 0.15 mmol/L として算出。
–:算出せず
表 2.7.2-7
ミラベグロンの代謝に及ぼす CYP3A4 阻害剤の阻害作用
阻害剤 IC50(mmol/L) 阻害剤の治療域濃度(Cp)(mmol/L) IC50/Cpa ケトコナゾール 0.47 5–21b 0.022–0.094 リトナビル 0.065 4–16c 0.0040–0.016 a:阻害剤の治療域濃度 b:Jamis-Dow ら(1997)[1]及び Daneshmend ら(1988)[2]の報告 c:Danner ら(1995)[3]及び Barry ら(1997)[4]の報告2.7.2.2.1.3.4 薬物相互作用の検討(スルホニル尿素系血糖降下剤)[ME-024]
··· 添付資料 5.3.2.2-4
ヒト肝ミクロソームを用いて,ミラベグロンとスルホニル尿素系血糖降下剤との in vitro 薬物代
謝相互作用を検討した。ミラベグロン(添加濃度 0.03~3 mmol/L)はグリベンクラミド(添加濃
度 0.01~1 mmol/L)及びトルブタミド(添加濃度 5~500 mmol/L)の代謝に影響を与えなかった。
また,グリベンクラミド(添加濃度 0.01~1 mmol/L),トルブタミド(添加濃度 5~500 mmol/L)
及びグリクラジド(添加濃度 0.5~50 mmol/L)はミラベグロン(添加濃度 0.03~3 mmol/L)の代謝
に影響を与えなかった。In vivo においてミラベグロンとこれらスルホニル尿素系血糖降下剤とが
代謝阻害により薬物相互作用を引き起こすことはないものと考えられた。
2.7.2.2.1.3.5 CYP 分子種に対する誘導作用(初代肝培養細胞)[ME-074]
··· 添付資料 5.3.2.2-6
CYP1A2 及び CYP3A4/5 分子種に及ぼすミラベグロンの酵素誘導作用をヒト初代肝培養細胞を
用いて検討した。ミラベグロン(添加濃度 0.1,1 及び 10 mmol/L)は CYP1A2 及び CYP3A4 の mRNA
量をそれぞれ最大 1.82 倍及び 1.13 倍に上昇させたが,CYP1A2 及び CYP3A4/5 分子種代謝活性に
はほとんど影響を及ぼさなかった(表 2.7.2-8)。以上の結果から,ミラベグロンは併用薬剤に対し
て CYP 酵素誘導に基づく薬物相互作用を引き起こす可能性は低いと考えられた。
表 2.7.2-8
ヒト CYP 分子種の代謝活性に対するミラベグロンの誘導作用
コントロール群に対する活性の上昇 (処理群/コントロール群) 処理 濃度 フェナセチン O-脱アルキル (CYP1A2) テストステロン 6b-水酸化 (CYP3A4/5) DMSO 0.1% (v/v) 1.00 ± 0.80 1.00 ± 0.45 ミラベグロン 0.1 mmol/L 1.01 ± 0.05 1.06 ± 0.06 1 mmol/L 1.04 ± 0.02 1.08 ± 0.04 10 mmol/L 1.08 ± 0.13 1.23 ± 0.18 オメプラゾール 100 mmol/L 18.2 ± 2.2a 1.84 ± 0.28a リファンピン 10 mmol/L 1.76 ± 0.57 4.74 ± 2.35a 3 例の平均値 ± 標準偏差 a:Vehicle コントロール群(DMSO, 0.1% (v/v))に対して有意差あり2.7.2.2.1.4
排出トランスポータに関する検討
2.7.2.2.1.4.1 P-糖蛋白(MDR1)に対するミラベグロンの基質性の検討[ME-031]
··· 添付資料 5.3.2.3-6
MDR1 を発現する Caco-2 細胞を介したミラベグロンの経細胞輸送活性を測定し,ミラベグロン
の MDR1 基質性を検討した。
14C-ミラベグロン(添加濃度 1~250 mmol/L)の基底膜側から頂側膜
側への輸送は頂側膜側から基底膜側への輸送に対して高い値を示した(表 2.7.2-9)。この基底膜側
から頂側膜側への
14C-ミラベグロンの経細胞輸送は飽和性のものであり,その K
m値は 250 mmol/L
以上であった。また,この経細胞輸送は MDR1 の特異的阻害剤であるベラパミルによりほぼ完全
に阻害された。以上の結果から,ミラベグロンは MDR1 の低親和性基質であると考えられた。
表 2.7.2-9
Caco-2 細胞単層膜を用いたミラベグロンの経細胞輸送の評価
ミラベグロン濃度 (mmol/L) A→Ba (×10-6cm/s) B→Ab (×10-6cm/s) A→Ba + ベラパミル (×10-6cm/s) B→Ab + ベラパミル (×10-6cm/s) 1 1.48 ± 0.17 13.22 ± 0.50 3.12 ± 0.22 4.01 ± 0.25 3 1.25 ± 0.11 13.72 ± 0.41 3.37 ± 0.23 4.37c 10 1.85 ± 0.15 14.87 ± 0.47 3.01 ± 0.30 4.21 ± 0.21 30 1.77 ± 0.30 13.13 ± 0.63 3.34 ± 0.34 4.14 ± 0.40 100 2.12c 9.27 ± 1.26 3.11c 3.98 ± 0.60 250 1.87 ± 0.11 7.65 ± 0.63 2.84 ± 0.05 3.79 ± 0.30 3 例の平均値 ± 標準偏差 a:頂側膜側から基底膜側への輸送 b:基底膜側から頂側膜側への輸送 c:n=22.7.2.2.1.4.2 P-糖蛋白(MDR1)に対するミラベグロンの阻害作用の検討[ME-032]
··· 添付資料 5.3.2.3-7
MDR1 の代表的な基質であるビンブラスチンを用いて,ミラベグロン(添加濃度 16 及び
250 mmol/L)存在下におけるヒト MDR1 発現細胞単層膜における
3H-ビンブラスチン(添加濃度
1 mmol/L)の輸送を検討した。ミラベグロンは MDR1 発現細胞における基底膜側から頂側膜側へ
のビンブラスチン輸送に対して 250 mmol/L の高濃度においても影響を及ぼさなかった。一方,頂
側膜側から基底膜側へのビンブラスチン輸送は,ミラベグロン添加濃度 16 mmol/L では明白な影
響は認められなかったが,250 mmol/L の高濃度において 3.4 倍に上昇した。この結果から,ミラ
ベグロンが高濃度において MDR1 を介した薬物輸送を阻害する可能性を除外できなかったが,基
底膜側から頂側膜側(MDR1 を介した排出方向)へのビンブラスチン輸送には影響がみられなかっ
たことから,その阻害の程度は軽度であると推察された。
2.7.2.2.1.5
取り込みトランスポータに関する検討
2.7.2.2.1.5.1 有機カチオントランスポータ(OCT)に対するミラベグロンの基質性の検討
[ME-092]
··· 添付資料 5.3.2.3-8
ヒト OCT1,OCT2 及び OCT3 の発現細胞を用いて,
14C-ミラベグロンの細胞内取り込み活性を
測定し,ミラベグロンが OCT の基質か否かを検討した。
14C-ミラベグロン(添加濃度 10 mmol/L)
の細胞内取り込み活性は,いずれの OCT 発現細胞においても OCT を発現していないコントロー
ル細胞に比べて 1.3~2.0 倍高い値を示した。OCT1 及び OCT3 を介した
14C-ミラベグロン(添加濃
度 2~500 mmol/L)の細胞内取り込み輸送は飽和性を示し,その K
m値はそれぞれ 108 mmol/L 及び
439 mmol/L であった(表 2.7.2-10)。一方,OCT2 を介した
14C-ミラベグロンの細胞内取り込み輸
送はミラベグロン濃度 500 mmol/L まで飽和性を示さなかった。また,OCT の代表的阻害剤である
0.5 mmol/L のイミプラミン及びデシプラミンによりいずれの OCT 細胞内取り込み活性もほぼ完全
に阻害された。以上の結果から,ミラベグロンは OCT1,OCT2 及び OCT3 の低親和性基質である
と考えられた。
表 2.7.2-10
ヒト OCT 発現細胞を用いたミラベグロンの細胞内取り込みの基質濃度依存性
トランスポータ Km (mmol/L) Vmax (pmol/min/mg protein) Vmax/Km (mL/min/mg protein) OCT1 108 1480 13.7 OCT2 ミラベグロン濃度 500 mmol/L まで飽和性を示さなかった。a OCT3 439 1980 4.51a:ミカエリス・メンテン(S-V)プロットにおける直線回帰式の傾き: 4.75 mL/min/mg protein
2.7.2.2.1.5.2 有機カチオントランスポータ(OCT)に対するミラベグロンの阻害作用の検討
[ME-086]
··· 添付資料 5.3.2.3-9
ヒト OCT 発現細胞を用いて,同トランスポータの典型的基質であるテトラエチルアンモニウム
の細胞内取り込み活性に対するミラベグロンの阻害作用を検討した。OCT1 による
14C-テトラエチ
ルアンモニウム(添加濃度 5 mmol/L)取り込み活性に対するミラベグロン(添加濃度 3~
1000 mmol/L)の IC
50値は 47.2 µmol/L であった。OCT2 に対する阻害作用は比較的弱く,ミラベグ
ロンが 1 mmol/L の高濃度においても OCT2 による
14C-テトラエチルアンモニウム取り込み活性は
コントロールの 55.8%を維持していた。OCT1 によるテトラエチルアンモニウム取り込み活性に及
ぼすミラベグロンの IC
50値とミラベグロンの臨床推奨用量(50 mg)における C
max(0.15 mmol/L)
との比は 300 倍以上であり,ミラベグロンの IC
50値はミラベグロンの治療域濃度をはるかに上回っ
ていることから,ミラベグロンは in vivo において OCT1 及び OCT2 による輸送を阻害しないと考
えられた。OCT1 及び OCT2 の代表的基質であるメトホルミンとの薬物相互作用試験において,
ミラベグロン(160 mg q.d. 反復経口投与)はメトホルミンの薬物動態に影響を及ぼさなかった
(2.7.2.2.2.4.8 薬物相互作用試験(メトホルミンと IR 錠)[CL-006])。
2.7.2.2.1.5.3 ヒト凍結肝細胞への取り込みの検討[ME-109]
···添付資料 5.3.2.3-10
ヒト凍結肝細胞を用いて,
14C-ミラベグロンの細胞内取り込み活性を測定した。
14C-ミラベグロ
ン(添加濃度 1 mmol/L)のヒト肝細胞への取り込みの時間及び温度依存性を検討したところ,試
験に使用したすべての肝細胞ロット(3 ロット)で時間及び温度依存性が観察され,ミラベグロ
ンのヒト肝細胞への取り込みに能動的な機構(薬物トランスポータ)が関与することが示唆され
た(表 2.7.2-11)。また,
14C-ミラベグロンの肝細胞取り込みの基質濃度依存性(添加濃度 1~
500 mmol/L)を検討したところ,3 ロット中 2 ロットで飽和傾向が認められ,薬物トランスポータ
の関与の可能性を支持した。
14
C-ミラベグロン(添加濃度 1 mmol/L)の細胞内取り込みに対する各種阻害剤の影響を検討した
結果,有機アニオン輸送ポリペプチド(OATP)の阻害剤であるシクロスポリン A(0.1 及び 1 mmol/L)
及び OCT1 の阻害剤であるキニジン(25 及び 250 mmol/L)が
14C-ミラベグロンの細胞内取り込み
を阻害した。しかしながら,阻害の程度はいずれも 50%未満であり,OATP 及び OCT1 の寄与率
はいずれも低いと推察され,ミラベグロンの肝細胞取り込みに主として関与する薬物トランス
ポータを同定することはできなかった。有機アニオントランスポータ 2(OAT2)及び OCT1 の阻
害剤であるプロベネシド(1 mmol/L)並びにプロスタグランジン F
2a(30 mmol/L),OATP 及び胆
汁酸輸送担体(NTCP)の阻害剤であるタウロコール酸(1 mmol/L),OATP の阻害剤であるエス
トラジオール 17-グルクロニド(100 mmol/L)はミラベグロンの肝細胞取り込みを阻害しなかった。
OCT1 の阻害剤であるメチルフェニルピリジニウム(1 mmol/L)は 3 ロット中 1 ロットでミラベ
グロンの肝細胞取り込みを 25%阻害したが,残りの 2 ロットでは阻害しなかった。
表 2.7.2-11
ヒト凍結肝細胞を用いた
14C-ミラベグロンの細胞内取り込みの時間及び濃度依存性
細胞内取り込み量(mL/106cells) 化合物 温度 終濃度 (mmol/L) インキュ ベーション 時間(min) ロット 1 ロット 2 ロット 3 14C-ミラベグロン 氷上 1 1 21.2 12.7 10.4 3 24.4 16.0 14.6 5 30.8 14.6 16.9 10 31.1 19.7 13.9 37 °C 1 1 31.4 17.6 22.1 3 70.0 31.9 48.5 5 79.2 40.2 57.3 10 118.8 41.7 65.9 3H-E2Ga 氷上 0.1 0.5 8.9 7.4 4.0 2 14.0 6.6 6.6 37 °C 0.1 0.5 24.8 7.7 12.2 2 65.5 15.0 28.8 3H-MPPb 氷上 1 0.5 7.4 3.7 4.6 2 7.5 5.4 4.4 37 °C 1 0.5 44.2 11.9 21.9 2 80.2 25.8 45.8 2 回の実験の平均a:β-Estradiol 17-(β-D-glucuronide) sodium salt(OATP の基質)
b:1-Methyl-4-phenylpyridinium iodide(OCT1 の基質)
2.7.2.2.1.6
その他
2.7.2.2.1.6.1 光学異性体への変換[ME-041]
··· 添付資料 5.3.2.3-1
マスバランス試験[CL-007]において得られたヒト血漿及び尿サンプルを用いて,ミラベグロ
ンの光学異性体(YM-181687)への変換の有無を検討した。
14C-ミラベグロンはヒト血漿及び尿中
2.7.2.2.1.6.2 代謝物の同定及び構造推定[ME-046]
··· 添付資料 5.3.2.3-2
マスバランス試験[CL-007]において得られたヒト血漿及び尿サンプルを用いて,ミラベグロ
ン代謝物の同定及び構造推定を行った。ヒト血漿中において M5,M8,M11 及び M16 が,ヒト尿
中において M5,M8,M9,M11 及び M16 が検出され,それらの構造が同定された(図 2.7.2-21)。
更にヒト血漿及び尿中に M12 及び M13 の存在が確認された。また,ヒト血漿及び尿中に存在す
る M14 及び M15,並びにヒト尿中に存在する M17 の分子量が明らかとなった。
2.7.2.2.1.6.3 代謝物の構造推定[ME-055]
··· 添付資料 5.3.2.3-3
未治療の 2 型糖尿病患者を対象とした試験[CL-003],メトホルミン療法を実施中の 2 型糖尿病
患者を対象とした試験[CL-004]及び薬物相互作用試験(メトプロロールと IR カプセル)
[CL-005]
において得られたヒト尿サンプルを用いて,ミラベグロン代謝物 M15 及び M17 の構造推定を行っ
た。M15 は,二級アミンに酸素が結合し,更にグルクロン酸が抱合した化学構造と推定された(図
2.7.2-21)。M17 は,M16(ミラベグロン脱アシル体)のアニリン側のベンゼン環に酸素が結合し,
更にグルクロン酸が抱合した化学構造と推定された。
2.7.2.2.1.6.4 代謝物の同定[ME-083]
··· 添付資料 4.2.2.4-7
ミラベグロンを経口投与したラットの胆汁サンプルから精製した M12[ME-018],M13[ME-018]
及び M14[ME-052]並びにミラベグロンを経口投与したヒトの尿サンプルから精製した M15
[ME-055]を用いて,合成標品と比較することにより,それらの構造を同定した。M13 は,二級
アミンに二酸化炭素が結合し,更にグルクロン酸が抱合した化学構造(カルバモイルグルクロニ
ド)と同定された(図 2.7.2-21)。M12 は,M13 の二級水酸基がケトンに酸化されたカルバモイル
グルクロニドと同定された。M14 は,末端(チアゾール環)の一級アミンにグルクロン酸が直接
抱合した代謝物と同定された。M15 は,二級アミンに酸素が結合し,更にグルクロン酸が抱合し
た化学構造と同定された。
2.7.2.2.2 臨床薬理試験
2.7.2.2.2.1
日本人健康成人における第 I 相試験及び薬物動態試験
2.7.2.2.2.1.1 第 I 相単回及び反復投与試験[CL-034] ··· 添付資料 5.3.3.1-1
1.
方法
本試験は,日本人健康成人男性における,ミラベグロン OCAS 錠(50,100 及び 200 mg)の単
回並びに反復経口投与時の安全性及び薬物動態を検討するための,第一部及び第二部からなるプ
ラセボを対照とした単盲検試験である。第一部では 50,100,200,300,400 mg を空腹時単回投
与し,第二部では 100,200 mg を朝食後に単回経口投与し,2 日間の休薬後,更に朝食後 7 日間
反復投与した。薬物動態の評価では,血漿中及び尿中未変化体濃度から薬物動態パラメータを算
出し,第一部では単回投与による用量依存性を,第二部では反復投与による蓄積性を検討した。
第一部では 40 例(プラセボ群 10 例,各実薬群 6 例),第二部では 24 例(プラセボ群 8 例,各
実薬群 8 例)の被験者が無作為化され,すべての被験者が脱落することなく,試験を完了した。
2.
結果
(1) 第一部(単回投与)
血漿中ミラベグロン濃度推移を図 2.7.2-1 に,血漿中及び尿中薬物動態パラメータを表 2.7.2-12
及び表 2.7.2-13 に示す。
ミラベグロンを 50~400 mg の範囲で単回投与したときの血漿中濃度は,経口投与後速やかに上
昇し,最高血漿中濃度(以下,C
max)到達後 2 相性に消失した。C
max及び時間 0 から無限時間ま
で外挿した血漿中濃度-時間曲線下面積(以下,AUC
inf)の平均値はそれぞれ 31.01~720.14 ng/mL
及び 292.24~4142.50 ng·h/mL であり,いずれも用量に依存して増加した。最高血漿中濃度到達時
間(以下,t
max)の平均値は 2.8~4.0 時間と用量に依らず一定であり,消失半減期(以下,t
1/2)の
平均値は 23.9~36.4 時間であった。また,経口クリアランス(以下,CL/F)と経口投与時の消失
相における分布容積(以下,V
z/F)の平均値は,いずれも用量増加に伴い減少する傾向がみられ
た。腎クリアランス(以下,CL
R)の平均値は 9.91~15.21 L/h といずれの用量でも同程度であり,
時間 0 から投与 72 時間までの尿中排泄率(以下,Ae
72h%)の平均値は用量増加に伴い上昇する傾
向がみられた。
図 2.7.2-1
日本人健康成人男性における単回投与時の血漿中ミラベグロン濃度推移(第一部)
[CL-034]
表 2.7.2-12
日本人健康成人男性における単回投与時のミラベグロンの
血漿中薬物動態パラメータ(第一部)[CL-034]
パラメータ (単位) 投与群 平均値 SD 最小値 最大値 CV Cmax 50 mg 31.01 18.06 9.17 57.40 58.25% (ng/mL) 100 mg 130.67 43.79 88.94 196.27 33.52% 200 mg 164.51 82.99 43.01 301.77 50.45% 300 mg 548.52 92.50 374.10 640.81 16.86% 400 mg 720.14 264.40 380.33 1095.94 36.72% tmax 50 mg 3.5 1.4 2.0 5.0 39.38% (h) 100 mg 3.3 0.8 2.0 4.0 24.49% 200 mg 2.8 1.3 2.0 5.0 46.91% 300 mg 3.7 1.0 2.0 5.0 28.17% 400 mg 4.0 1.3 2.0 5.0 31.62% AUClast 50 mg 223.99 78.96 103.95 335.13 35.25% (ng·h/mL) 100 mg 773.02 215.55 529.90 1162.36 27.88% 200 mg 1251.58 417.16 721.66 1908.28 33.33% 300 mg 3053.27 300.18 2484.43 3358.64 9.83% 400 mg 3917.41 694.76 2685.05 4678.42 17.74% AUCinf 50 mg 292.24 76.93 173.44 373.49 26.32% (ng·h/mL) 100 mg 882.40 234.53 619.57 1308.81 26.58% 200 mg 1382.68 441.45 853.78 2085.90 31.93% 300 mg 3285.08 333.94 2635.76 3574.22 10.17% 400 mg 4142.50 735.89 2814.32 4868.01 17.76% t1/2 50 mg 36.4 11.8 22.6 49.6 32.48% (h) 100 mg 30.8 3.4 25.9 34.0 11.11% 200 mg 26.4 3.6 21.9 32.7 13.68% 300 mg 25.1 4.3 19.5 31.8 17.03% 400 mg 23.9 4.9 17.1 30.1 20.33% CL/F 50 mg 183.49 58.11 133.87 288.29 31.67% (L/h) 100 mg 119.34 28.11 76.41 161.40 23.55% 200 mg 157.61 50.64 95.88 234.25 32.13% 300 mg 92.24 10.89 83.93 113.82 11.80% 400 mg 99.79 22.03 82.17 142.13 22.08% Vz/F 50 mg 9817.82 4879.64 4544.11 17573.53 49.70% (L) 100 mg 5404.52 1741.99 2999.93 7908.90 32.23% 200 mg 5934.29 1878.55 3784.66 8919.47 31.66% 300 mg 3299.98 373.14 2859.52 3988.43 11.31% 400 mg 3346.70 465.45 2866.10 4070.40 13.91% 各群 n=6表 2.7.2-13
日本人健康成人男性における単回投与時のミラベグロンの
尿中薬物動態パラメータの要約統計量(第一部)[CL-034]
パラメータ (単位) 投与群 平均値 SD 最小値 最大値 CV Ae72h% 50 mg 7.20 2.32 3.89 10.54 32.16% (%) 100 mg 7.61 3.62 3.49 12.67 47.58% 200 mg 9.01 2.66 5.17 12.15 29.50% 300 mg 14.57 2.48 11.28 16.96 17.01% 400 mg 11.81 2.55 8.29 14.34 21.63% CLR 50 mg 15.21 1.85 13.01 18.19 12.14% (L/h) 100 mg 9.91 4.45 5.20 16.39 44.86% 200 mg 14.61 1.96 11.47 17.63 13.43% 300 mg 14.29 1.80 11.16 16.41 12.58% 400 mg 12.14 2.07 8.66 14.66 17.09% 各群 n=6用量調整した C
max(C
max/Dose)及び AUC
inf(AUC
inf/Dose)の各群間の幾何平均比(以下,GMR)
及びその 95%信頼区間(以下,CI)を推定した結果,C
max及び AUC
infは 100 mg 以下の用量では
用量比を超えて上昇し,300 mg 以上の高用量では用量比に伴って上昇することが示唆された(図
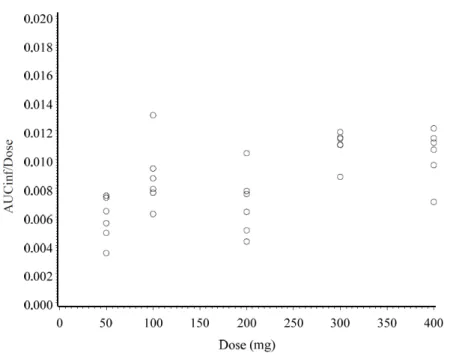
2.7.2-2,図 2.7.2-3,表 2.7.2-14)。しかし,200 mg 群で得られた濃度値は全体的に低かったため,
100~300 mg 付近の薬物動態と用量との関係は明らかでなかった。
また,C
max及び AUC
infの対数変換値を用量の対数変換値に対してプロットした回帰直線の傾き
とその 95%CI を検討した結果,C
maxでの傾きは 1.501[95%CI:1.222-1.781],AUC
infでの傾きは
1.260[95%CI:1.120-1.400]であり,いずれの 95%CI も 1 を含まなかった(表 2.7.2-15)。
表 2.7.2-14
日本人健康成人男性における単回投与時の用量調整した C
max,AUC
infの
用量間の GMR(第一部)[CL-034]
95%CI 項目 比較 GMR 下限 上限 Cmax/Dose 100 mg / 50 mg 2.412 1.345 4.328 200 mg / 50 mg 1.380 0.769 2.475 300 mg / 50 mg 3.483 1.941 6.248 400 mg / 50 mg 3.272 1.824 5.870 200 mg / 100 mg 0.572 0.319 1.026 300 mg / 100 mg 1.444 0.805 2.590 400 mg / 100 mg 1.356 0.756 2.433 300 mg / 200 mg 2.524 1.407 4.528 400 mg / 200 mg 2.371 1.322 4.254 400 mg / 300 mg 0.940 0.524 1.686AUCinf/Dose 100 mg / 50 mg 1.519 1.134 2.034
200 mg / 50 mg 1.171 0.874 1.568 300 mg / 50 mg 1.927 1.439 2.580 400 mg / 50 mg 1.803 1.347 2.415 200 mg / 100 mg 0.771 0.576 1.033 300 mg / 100 mg 1.269 0.947 1.699 400 mg / 100 mg 1.187 0.887 1.590 300 mg / 200 mg 1.645 1.229 2.203 400 mg / 200 mg 1.540 1.150 2.062 400 mg / 300 mg 0.936 0.699 1.253
図 2.7.2-2
日本人健康成人男性における単回投与時の用量調整した C
maxの分布(第一部)
[CL-034]
図 2.7.2-3
日本人健康成人男性における単回投与時の用量調整した AUC
infの分布(第一部)
[CL-034]
表 2.7.2-15
日本人健康成人男性における単回投与時の用量に対する C
max,AUC
infの
対数変換回帰分析[CL-034]
95%CI パラメータ 傾き 下限 上限 Cmax 1.501 1.222 1.781 AUCinf 1.260 1.120 1.400(2) 第二部(反復投与)
血漿中ミラベグロン濃度推移を図 2.7.2-4 に,血漿中及び尿中薬物動態パラメータを表 2.7.2-16
及び表 2.7.2-17 に示す。
血漿中ミラベグロン濃度推移から,100 及び 200 mg 群ともに初回投与後第 7 日目(反復投与開
始後第 4 日目)以降トラフ値はほぼ一定となった。第 10 日目(反復投与開始後第 7 日目)の C
maxは,100 mg 群で第 1 日目(初回投与時)と比較して上昇し,第 10 日目の時間 0 から投与後 24 時
間までに血漿中濃度-時間曲線下面積(以下,AUC
24h)は 100 及び 200 mg 群ともに第 1 日目と比
較して上昇した。また,第 10 日目の t
max,t
1/2,CL
Rは,いずれの用量でも第 1 日目と比較してほ
ぼ同じであり,第 10 日目の Ae%は,いずれの用量でも第 1 日目と比較してやや上昇した。
100mg (n=8) 200mg (n=8) Mean±SDPlasma YM178 Concentration (ng/mL)
0.00 100.00 200.00 300.00 400.00 500.00 Time (h) 0.0 24.0 48.0 72.0 96.0 120.0 144.0 168.0 192.0 216.0 240.0 264.0 288.0
![表 2.7.2-14 日本人健康成人男性における単回投与時の用量調整した C max ,AUC inf の 用量間の GMR(第一部)[CL-034] 95%CI 項目 比較 GMR 下限 上限 C max /Dose 100 mg / 50 mg 2.412 1.345 4.328 200 mg / 50 mg 1.380 0.769 2.475 300 mg / 50 mg 3.483 1.941 6.248 400 mg / 50 mg 3.272 1.824 5.870 200 mg / 1](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/27.892.172.730.731.1110/日本人男性おける単回投与用量調整用量間CLCI項目比較下限上限.webp)
![表 2.7.2-26 外国人健康成人男性におけるミラベグロン IR カプセル投与時の 尿中薬物動態パラメータ[CL-002] 投与群 パラ 投与時期 メータ (単位) 40 mg 初回 40 mg最終 80 mg初回 80 mg最終 160 mg初回 160 mg最終 240 mg初回 空腹時 240 mg最終空腹時 240 mg初回食後 240 mg最終食後 Ae a (mg) 1.92±0.5428% 1.3 - 2.7 3.37±1.0431%2.3 - 4.7 6.29±1.9431%3.4 -](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/39.892.94.804.178.449/おけるミラベグロンカプセル動態パラメータCLパラメータ初回最終.webp)
![表 2.7.2-39 外国人健康高齢被験者における尿中薬物動態パラメータ(最終投与時)[CL-031] 投与群 Ae 24h (mg) Ae 24h %(%) CL R (L/h) 50 mg 女性 (n=6) 2.29±0.6026%1.6 - 3.0 4.59±1.2126%3.3 - 6.0 8.72±3.2037%5.0 - 13 200 mg 女性 (n=6) 20.8±5.225%12 - 27 10.4±2.625%5.9 - 13 10.5±2.624%7.8 - 15 50 mg 男性 (](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/49.892.221.673.192.439/齢被おける尿中薬物動態パラメータ最終投与投与CL女性女性男性.webp)
![表 2.7.2-41 外国人健康高齢,非高齢男女における反復投与時(7 日目)の 薬物動態パラメータ(25 mg)[CL-072] 非高齢者 高齢者 パラメータ (単位) 男性 (n=11) 女性 (n=11) 男性 (n=13) 女性 (n=12) C max (ng/mL) 21.6±10.549% 4.8 - 39 20.1±5.628%13 - 27 11.7±4.639%5.4 - 20 19.7±5.629%14 - 31 t max (h) 4.14±0.842.5 - 5.0 3.86±0.](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/52.892.174.721.183.683/おける投与パラメータ高齢高齢パラメータ単位男性女性男性女性C.webp)
![表 2.7.2-42 外国人健康高齢,非高齢男女における反復投与時(7 日目)の 薬物動態パラメータ(50 mg)[CL-072] 非高齢者 高齢者 パラメータ (単位) 男性 (n=12) 女性 (n=12) 男性 (n=11) 女性 (n=11) C max (ng/mL) 54.4±24.545% 23 - 102 58.1±15.827%31 - 84 43.5±18.943%23 - 71 66.3±27.341%16 - 108 t max (h) 3.92±0.872.5 - 5.0 4.58](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/53.892.172.720.184.683/おける投与動態パラメータ高齢高齢パラメータ単位男性女性女性C.webp)
![表 2.7.2-45 外国人健康高齢,非高齢男女における反復投与時(7 日目)の M11 の薬物動態パラメータ[CL-072] 非高齢者群 高齢者群 用量 パラメータ (単位) 男性 女性 男性 女性 25 mg C max (ng/mL) 4.45±2.4511 4.70±2.4711 3.49±1.5013 5.62±1.2012 AUC tau (ng·h/mL) 47.1±21.911 51.0±23.111 46.1±18.813 76.5±22.512 Ae tau % (%) 0.614±0.](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/55.892.172.724.194.660/おける日目パラメータCL者群用量パラメータ単位男性女性男性女性.webp)
![表 2.7.2-46 外国人健康高齢,非高齢男女における反復投与時(7 日目)の M12 の薬物動態パラメータ[CL-072] 非高齢者群 高齢者群 用量 パラメータ (単位) 男性 女性 男性 女性 25 mg C max (ng/mL) 3.87±3.0111 2.81±1.8711 2.08±1.6613 2.71±1.1912 AUC tau (ng·h/mL) 40.2±29.411 27.5±19.011 29.1±23.913 38.3±23.312 Ae tau % (%) 0.153±0.](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/56.892.172.724.194.659/おける日目パラメータCL者群用量パラメータ単位男性女性男性女性.webp)
 非高齢者群 高齢者群 代謝物 用量 パラメータ (単位) 男性 女性 男性 女性 M13 25 mg C max (ng/mL) 1.067±0.6187 1.037±0.6393 0.783±0.1963 0.681±0.1159 AUC tau (ng·h/mL) 3.36±3.627 3.17±3.853 1.47±0.793 2.06±1.809 Ae tau % (](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/58.892.139.759.175.1068/おける薬物パラメータCL続き高齢者群高齢者群代謝物用量パラメータ.webp)
 非高齢者群 高齢者群 代謝物 用量 パラメータ (単位) 男性 女性 男性 女性 M15 25 mg C max (ng/mL) 1.61±0.599 1.63±0.8911 1.20±0.6211 2.06±0.7212 AUC tau (ng·h/mL) 6.88±3.119 8.61±7.1011 6.04±5.3811 18.2±10.912 Ae tau % (%)](https://thumb-ap.123doks.com/thumbv2/123deta/6519780.664684/59.892.140.760.173.1068/おける代謝パラメータCL続き高齢者群高齢者群代謝物用量パラメータ.webp)
