マンガン系正極活物質の合成と
電気化学特性
平成
27 年度
三重大学大学院 工学研究科
博士前期課程 分子素材工学専攻
エネルギー変換化学講座
高士 祐輔
目 次
1.
序論
1-1. 研究背景
1-2. リチウムイオン電池
1-3. 代表的なリチウムイオン電池用正極活物質
1-4. LiMn
1.5Ni
0.5O
41-5. Li
2MnO
31-6. 本研究の目的
2.
実験
2-1.
LiMn
1.5Ni
0.5O
4の合成
2-1-1. 固相法による LiMn1.5Ni0.5O4の合成 2-1-2. 低温合成法による八面体 LiMn1.5Ni0.5O4の合成2-2.
Li
2MnO
3の合成
2-2-1. 固相法による Li2MnO3の合成 2-2-2.LiH を用いた Li2MnO3-xの合成2-3. 各種正極活物質の評価法とその条件
2-3-1. X 線回折測定(XRD) 2-3-2. 粒度分布測定 2-3-3. 走査型電子顕微鏡(SEM) 2-3-4. 比表面積測定(BET 法) 2-3-5. 電気化学測定セルの試作と電気化学特性評価2-4. 使用試薬
3.
結果と考察
3-1. LiMn
1.5Ni
0.5O
4の結晶性制御
3-1-1. 合成の簡易化 3-1-2. 焼成時の原料の接触性による粒形状に対する影響 3-1-3. 低温合成時の窒素保持時間の粒形状に対する影響 3-1-4. 従来合成法との粒形状の比較 ・・・ 10 ・・・ 10 ・・・ 11 ・・・ 12 ・・・ 12 ・・・ 12 ・・・ 14 ・・・ 14 ・・・ 14 ・・・ 14 ・・・ 15 ・・・ 15 ・・・ 18 ・・・ 1 ・・・ 1 ・・・ 3 ・・・ 5 ・・・ 6 ・・・ 8 ・・・ 19 ・・・ 19 ・・・ 20 ・・・ 22 ・・・ 253-2.
低温合成
LiMn
1.5Ni
0.5O
4の電池特性評価
3-2-1. 低温合成 LiMn1.5Ni0.5O4の電気化学特性 3-2-2. 従来法との電気化学特性比較 3-2-3. 充放電後の電極表面観察3-3. Li
2MnO
3の合成
3-3-1. 焼成条件が活物質粒径に与える影響の評価 3-3-2. 出発物質が活物質粒径に与える影響の評価 3-3-3. 活物質粒径の違いによる電気化学特性の評価3-4.
Li
2MnO
3-xの合成
3-4-1. Li2MnO3-xの合成と相同定 3-4-2. Li2MnO3-xの電気化学特性4. 総括
参考文献
謝辞
・・・ 26 ・・・ 26 ・・・ 27 ・・・ 30 ・・・ 31 ・・・ 31 ・・・ 32 ・・・ 35 ・・・ 37 ・・・ 37 ・・・ 50 ・・・ 53 ・・・ 55 ・・・ 571-1. 研究背景
電気エネルギー蓄電デバイスには、キャパシタや二次電池などが挙げられるが、その中で もリチウムイオン電池は特に高いエネルギー密度を有する。そのため、リチウムイオン電池 は家電、通信機器、携帯電話、事務機器などの広い分野でその二次電池としてのシェアを拡 大してきた。さらに、近年では将来予測される化石燃料をはじめとする資源枯渇問題および 地球温暖化や酸性雨に代表される環境問題から、再生可能エネルギーを効率的に活用する ための手段や電気自動車用蓄電池としての需要の拡大が期待されている。 そのため、更なるリチウムイオン電池市場の拡大や実用化に向けて、リチウムイオン電池 の高容量化、高出力化、低コスト化、安全性の向上が求められている。1-2. リチウムイオン電池
リチウムイオン電池は正極にLiCoO2、負極にグラファイト、電解液としてリチウム塩を 溶かした有機溶媒を用いたものが最も普及している。Fig.1-1 にその概略図を示す。リチウ ムイオン電池では、リチウムをゲスト種、強固な結晶格子をホストとして可逆的に出し入れ する、いわゆるインターカレーション反応が起こる。その反応式を以下に示す。正極:Li1-xCoO2 + xLi+ + xe- ⇔ LiCoO2 (1.1)
負極:LixC6 ⇔ xLi + xe-+ C6 (1.2) リチウムイオン電池は、正極と負極の組み合わせによって発生電圧と容量が異なり、これ らにより入出力密度やエネルギー密度が決まる。正極にLiCoO2、負極にグラファイトを用 いた電池のエネルギー密度は150 Wh/kg 程度であり、携帯機器の電源としては適当な性能 を有しているが、大型機器用の電池として用いるには更なるエネルギー密度と出力密度の 向上が求められている。 Fig.1-2 にリチウムイオン電池の主な電極材料の容量と電位の関係を示す。負極材料は低 電位で高容量を示す化合物が存在するが、正極材料の容量はこれらに比べ低いことが分か る。このため、リチウムイオン電池の更なるエネルギー密度と出力密度の向上を達成するた めには、正極材料開発の研究が重要である。
1-3. 代表的なリチウムイオン電池用正極活物質
現在のリチウムイオン電池における代表的な材料をFig.1-2 に示す。層状岩塩型酸化物
最も代表的な層状岩塩型酸化物として、LiCoO2がある[3]。LiCoO2は、(111)面方向に形 成された酸素層の層間の八面体サイト(3a、3b)をそれぞれ遷移金属とリチウムが単独層を形 成する形で占有し、交互に積層している。LiCoO2のCo3+⇔Co4+の酸化還元反応が完全に達 成され、全てのリチウムが脱離すると仮定すれば、その理論容量は274 mAh/g である。し かし、構造内のリチウムをすべてひき抜くと構造が壊れ可逆性が失われるため、可逆的に得 られる充放電容量は半分程度のリチウムが脱離した約125 mAh/g となる[4]。LiNiO2はLiCoO2とよく似た構造を持つが、酸素の配列はCdCl2型である。電位はLiCoO2
と比べ0.25 V ほど低いが放電容量は 160 mAh/g 程度を示す[5]。LiNiO2は、コバルトに比 べニッケルは安価であるために、材料としては期待が高かったが、局所的に岩塩相が生成す ることで特性が急激に低下することや、ニッケルは高酸化状態においてコバルトより不安 定であり酸素を放出しやすいため、安全性に問題があり実用電池としては用いることは困 難とされている[6] [7] [8] [9]。これらの問題を解決するための材料として LiMn1/2Ni1/2O2 [10]や LiMn1/3Ni1/3Co1/3O2 [11]や LiNi0.8Co0.15Al0.05O2 [12]が現在注目されている。 高容量を示す材料としては、約 300 mAh/g の放電容量を得られる xLi2MnO3
-(1-x)LiMO2 (M : Mn, Co, Ni)で示されるリチウム過剰系材料が注目されている[13] [14]。
スピネル型酸化物
LiMn2O4は正スピネルと呼ばれる構造を持つ[15]。LiMn2O4の4 V 領域における Mn3.5+ ⇔Mn4+の酸化還元反応による理論容量は148 mAh/g と LiCoO2に比べやや小さいが、資源 的に豊富なマンガンをベースにしていて、安価で安全性も高いといった特徴を持ち、注目さ れている。この構造をAB2O4と表記した場合、32 個の酸素(32e)が面心立方格子を形成 し、その間隙において、Aは酸素の四面体4 配位の隙間の 1/8 を占め、Bは八面体6 配位の 隙間の1/2 を占める。遷移金属が八面体サイトを占める点では層状岩塩型構造と同様である が、リチウムが四面体サイトを占める点で異なる。充放電はリチウムの挿入脱離に伴いマン ガンが3 価と 4 価の間で価数変化することで進行する。欠点としてはサイクル特性が悪い といったことが挙げられる。放電状態においては 3 価のマンガンが多く存在する。そのた め不均化反応が起こり、2 価のマンガンが生成する。その 2 価のマンガンの溶出が起こるこ とが主な原因であると考えられている[16][17][18][19]。この LiMn2O4は、Mn イオンの一 部を他の低価数3d 遷移金属で置換することが可能で、サイクル特性の向上が報告されてい る。置換により、マンガンの平均価数が+3.5 より増大することで、ヤーン・テラー効果に よる劣化を低減でき、充放電に伴う放電容量維持率は向上すると報告されている[20]。また これらの3d 遷移金属を置換した材料は 5 V 級正極としても期待されている[21]。オリビン型酸化物
LiFePO4はオリビン型の構造を持つ[22]。充放電時の平均電圧は 3.4 V で、Fe2+⇔Fe3+の
酸化還元反応による理論容量は169 mAh/g である。LiFePO4はPO4の四面体とFeO6の八
面体が連なり、隙間の空間にリチウム原子が一次元方向に並んでいる。この構造で充放電を 行い、リチウムをひき抜くと四面体と八面体の構造を保持したままリチウムが抜けた構造
であるFePO4が生成し、LiFePO4とFePO4の二相共存状態で反応が進行する。そして、こ
れらの対称性が良いため充放電における劣化が少ない[23][24]。また、LiFePO4は資源的に 豊富な鉄を用いることができるという長所を持つ。 リチウムイオン電池の正極活物質は、酸化還元種と酸化物イオンで充放電に適した骨格 構造が形成され、かつ構造中に多量のリチウムを保持でき、高い電圧を発生させることがで きることが求められる。これらの条件を満たす電極材料として、高容量を示すリチウム過剰 層状岩塩型構造酸化物や高電位を示すスピネル型構造酸化物が最も有望であると考えられ る。そのため、これらの材料は次世代型のリチウムイオン電池用正極活物質として注目が集 まっている。 Fig.1-2 代表的なリチウムイオン電池用材料の電位-容量図[2].
1-4. LiMn
1.5Ni
0.5O
4 スピネル構造を持つLiMn2O4のMn の一部を 2 価の Ni で置換した LiMn1.5Ni0.5O4は、 その構造から全てのリチウムが脱離すると仮定すれば、148 mAh/g の理論容量を持ち、5 V 級正極活物質として注目されている[25]。Fig.1-3 にその結晶構造を示す。Ni2+のイオン半 径はMn3+と比べて大きいがNi2+の置換によりMn3+はMn4+となり、結果的にNi を置換す ることで格子定数は減少する。本材料はNi2+⇔Ni4+の酸化還元が主な反応として起こる。ま た、LiMn1.5Ni0.5O4はそれぞれP4332、Fd3̅mの空間群で表わされる、Ni と Mn が三次元 的に規則的に配列した超格子構造を持つ場合と、持たない場合が存在する。これら二種類は 酸化還元機構が異なり、Fig.1-4 に示すように充放電曲線の形も異なる[26]。超格子構造を 持たない試料では4 V 付近と 4.7 V 付近に電位平坦部が現れる。4 V 領域の電位平坦部は Mn の Mn3+⇔Mn4+の酸化還元反応によるもので、4.7 V 付近のプラトーは Ni の Ni2+⇔Ni4+ の酸化還元反応によるものである。一方で、超格子構造を持つ試料では4.7 V 付近に作動電 位 を示 すもの の、4 V 領域にはほとんど反応は見られない [27]。超格子構造を持つ LiMn1.5Ni0.5O4を合成するためには、その組成が重要となり、試料を化学量論組成にしなけ ればならない。本材料は、700ºC 以上で酸素の放出が起こるため、合成時の温度や雰囲気を 制御することが重要である[28][29]。また、本材料はその電位の高さから電解液の分解、導 電剤のカーボン分解、マンガンの溶出などが起こりサイクル特性に課題が残る。 超格子LiMn1.5Ni0.5O4 超格子を持たないLiMn1.5Ni0.5O41-5. Li
2MnO
3 Li2MnO3は、Li / M比が1 よりも大きいリチウム過剰系正極活物質であり、Mn4+⇔Mn6+ の酸化還元反応が完全に達成され、全てのリチウムが脱離すると仮定すれば、理論容量は 460 mAh/g となるため高容量インターカレーション材料として注目されている[30]。Fig.1-5 にその構造を示す。この構造は LiCoO2に代表されるような層状岩塩型の構造であり、Li イオン単独層とMn イオンと Li イオンを 2 : 1 の比で含む Mn-Li 層が酸化物イオンを介し て、交互に積層している。このMn-Li 層内において、Mn イオンと Li イオンが六角網目構 造を形成するように規則的に配列するという特徴を持つ。このMn イオンと Li イオンの規 則的な配列は、XRD 測定における超格子に由来する回折ピークから判断することが出来る。 またこの超格子構造を持つため、Li2MnO3はLiCoO2などが属する空間群R3̅mの六方晶系 ではなく、空間群C2/mの単斜晶系に属する[31]。このことにより、空間群R3̅mでは、陽 イオンが二種類、酸化物イオンは一種類の占有できる位置を持つことに対し、空間群C2/m では、陽イオンが四種類(4g、2b、2c、4h)、酸化物イオンは二種類(4i、8j)に増える。 また、Li2MnO3は4 価のマンガン酸化物であるため、Mn4+⇔Mn5+の酸化還元に伴うLi イオンの脱離が困難であることから、電気化学的に不活性であるとされてきた[32]。しかし 近年、低温焼成など試料合成方法の工夫や、4.5 V 以上まで充電することで電気化学的に活 性になることが報告されている[33]。Fig.1-7 にリチウム過剰系正極活物質の一つである、 Li2MnO3- LiMn1/3Ni1/3Co1/3O2材料の充放電曲線を示す。4.4 V までの充電では 100 mAh/g程度の容量しか得られていないのに対し、4.8 V まで充電したものは 350mAh/g 程度の高い 容量を示している[34]。4.8 V 充電では、4.5 V 付近に大きな電位平坦部を示している。こ の電位平坦部を経ることで高容量が得られている。この電位平坦部では、式(1.3)に示す形 で、Li と O が Li2O の形で脱離する反応が進行すると考えられている[35][36]。そして、引 き続きの放電において(1.4)に示す反応で MnO2にリチウムが挿入されることで高容量を示 す[37][38]。 Li2MnO3 → MnO2 + Li2O (1.3) MnO2 + Li+ + e- LiMnO2 (1.4) しかし、放電で取り出せる容量は250 mAh/g 程度であり、初回クーロン効率はとても低 い。初回不可逆容量の低減は本材料の課題の一つである。一方で、2 サイクル目以降では、 4.5 V 付近の電位平坦部は観測されない。このことは、2 サイクル目以降で Li2O の脱離反 応が起こらないことを示唆している。このことからLi2O の脱離反応が本材料の電気化学活 性過程でもあり、初回クーロン効率を低下させている原因であると考えられる。 サイクル特性も高電位で充電を行うことや充放電中の構造変化が起こるため良好でない とされている。サイクルを重ねるにつれて、電位低下が起こり、LiMn2O4に特徴的な3 V と 4 V 付近に電位平坦部が出現する。これは、Li2O の脱離反応の際に生成した、MnO2に関係
するものであると考えられる[39]。MnO2放電後に生成するLiMnO2はサイクルを重ねるこ
とで、スピネル相になることが知られており、非常に似た挙動が観察される[30]。
以上で示したように、本材料は充放電中において、非常に複雑な構造変化を伴うと考えら れており、現状ではその反応機構の全ては明らかになっておらず、今後より詳細な調査が求 められている。
Fig.1-5 Li2MnO3の結晶構造. Fig.1-6 Li2MnO3のMn-Li 層の結晶構造.
1-6. 本研究の目的
LiCoO2は4 V 級正極活物質として約 140 mAh/g の実容量を示し、大量合成の簡便さや 容量・可逆性のバランスの良さから、近年までほとんどの実用リチウムイオン電池に採用さ れてきた。しかしながら、過充電時に生じる酸素の放出が電解液の燃焼反応を引き起こす問 題に加え、コバルトが希少金属でありコストが高いことや高環境負荷などの実用化におけ る不可避な課題が残っている。そのため、コバルト以外の元素を用いた代替材料開発が進め られている。そこで、資源的に豊富で安価かつ安全性も高いと言われているマンガンを用い たLi-Mn-O 系正極活物質が現在注目されている。 本研究では、Li-Mn-O 系正極活物質の中でも特に次世代正極活物質として注目されて いるスピネル型LiMn1.5Ni0.5O4および層状岩塩型Li2MnO3の二つの物質に着目した。これ らの材料の合成、構造、および電気化学特性を評価し、得られた知見からLi-Mn-O 系正極 活物質の電気化学特性向上の指針を見出すことを目指した。 以下に、本研究において行った各材料へのアプローチをまとめる。 ・LiMn1.5Ni0.5O4 電位平坦部が4.7 V 付近と非常に高く次世代の高出力正極材料として注目されてきたが、 その電位の高さから電解液の分解、導電剤のカーボン分解、マンガンの溶出などサイクル特 性に課題が残る。しかし近年、大阪市立大学の小槻らのグループにより、1000ºC の焼成に よって、活物質の表面形態は正八面体状へと変化し、特に優れたサイクル特性を示すことが 明らかになった[29]。さらに、本研究の先行研究によって、焼成時の酸素脱離反応がこの正 八面体状粒子の生成機構に関与し、焼成時の酸素分圧を下げることで低温において八面体 粒子の合成が可能であることが明らかにされた[40]。しかし、その低温合成での焼成条件と 電気化学特性に与える影響は明らかにされていない。そこで、本研究では、低温合成での焼 成条件の検討を行い、低温合成試料の粒形状の調査とその電気化学特性の評価を行った。 ・Li2MnO3 Li2MnO3は、4.5 V 付近に大きな電位平坦部を示し、この電位平坦部で Li2O の脱離反応 が起こることで高容量が得られている。そのため、この酸素脱離機構がリチウム過剰系正極 活物質の高容量化の鍵であると認識され、酸素脱離後の高容量を示す相の直接合成の試み が行われている。この領域の直接合成は難しくあまり開拓されてなかったが、近年、金属水 素化合物を用いた還元手法でLi2MnO3の酸素脱離相の合成に成功し、300 mAh/g 程度の容 量を示すことが報告されている[41]。リチウム過剰系正極活物質の高容量を示す相に関する 研究は現在も進められているが、依然として明らかにされていない部分が多く残っている。 そこで本研究では、金属水素化物を用いてLi2MnO3を還元し、その酸素脱離量を系統的に 変化させることで、酸素脱離反応での構造の変化を捉えることを試みた。そして、合成した 試料の構造解析および電気化学特性評価を行い、その相関を明らかにすることで、リチウム 過剰系正極活物質の複雑な反応機構および劣化機構の解明につながると考えた。そして、これらの粒形状や構造、電気化学特性の相関性を明らかにすることで、より優れ
2-1. LiMn
1.5Ni
0.5O
4の合成
2-1-1. 固相法による
LiMn
1.5Ni
0.5O
4の合成Fig.2-1 に示すような手順で LiMn1.5Ni0.5O4を合成した。
出発物質には[NiMn3](OH)8とLiOH・H2O を用いた。[NiMn3](OH)8はアルミナるつぼ
に入れ、電気炉で大気下において昇温速度5 ºC/min、500ºC で 8 h 仮焼した。仮焼の有無 による影響の考慮を行ったため、仮焼なしの試料も作製した。LiOH・H2O はボールミルに より粉砕を行ったものを用いた。ボールミル条件は、溶媒にヘキサンを用いて400 rpm で 1 h 粉砕とした。その後、それぞれをメノウ乳鉢により 30 min 混合し、アルミナるつぼに 入れ本焼した。本焼時は、出発物質同士の接触性を変えたものを3 種類用意した。通常通り 試料をるつぼに入れたもの、スパチュラによって粒子を分散させたもの、ふるいを用いて粒 子を分散させたもの、三つの方法を行った。その後、Fig.2-2 に示すように、電気炉で昇温 速度5 ºC/min、降温速度 1 ºC/min で 1000ºC または 800ºC (低温合成用),10 h、700ºC, 24 h、650ºC, 24 h、600ºC, 48 h 焼成した。得られた目的物質は、SEM 観察、充放電測定、 XRD 測定によって評価した。 Fig.2-1 固相法による LiMn1.5Ni0.5O4の合成フローチャート.
2-1-2. 低温合成法による八面体
LiMn
1.5Ni
0.5O
4の合成低温合成法でのLiMn1.5Ni0.5O4の合成は、*Fig.2-2 の 800ºC 処理を経て固相法で合成し
たLiMn1.5Ni0.5O4を原料として用いた。原料 LiMn1.5Ni0.5O4をアルミナるつぼに入れ焼成
した。焼成条件は電気炉でFig.2-3 に示すように、昇温速度 5 ºC/min、降温速度 1 ºC/min
で窒素雰囲気において850ºC,0 - 7 h、以後酸素雰囲気で 850ºC, 3 h、700ºC, 24 h、650ºC, 24 h、600ºC, 48 h 焼成した。窒素雰囲気での保持時間による粒形状への影響を調査するた めに、窒素雰囲気での保持時間を変化させた。得られた目的物質は、SEM 観察、充放電測 定、XRD 測定、比表面積測定によって評価した。 Fig.2-2 固相法での LiMn1.5Ni0.5O4焼成条件. Fig.2-3 低温合成法での八面体 LiMn1.5Ni0.5O4の焼成条件.
2-2. Li
2MnO
3の合成
2-2-1. 固相法による Li2MnO3の合成
Fig.2-4 に示すような手順で Li2MnO3を合成した。
出発物質にはMnO2または MnCO3とLiOH・H2O を用いた。出発物質による粒径への
影響を評価するためにマンガン源は二種類用いた。その後、それぞれをボールミルを用いて 混合した。ボールミル条件は、溶媒にアセトンを用いて400 rpm で 1 h 混合とした。その 後、ペレット状に加圧成形し、アルミナるつぼに入れ、電気炉で焼成した。焼成条件は昇温 速度5 ºC/min で・800ºC, 12 h ・700ºC, 12 h ・600ºC, 12 h の三つの条件で行った。焼成 条件による粒径への影響を評価するために三つの焼成温度を設定した。得られた目的物質 は、SEM 観察、充放電測定、XRD 測定、粒度分布測定によって評価した。 Fig.2-4 固相法による Li2MnO3の合成フローチャート.
2-2-2.LiH を用いた Li2MnO3-xの合成
Fig.2-5 に示すような手順で Li2MnO3-xを合成した。
出発物質には*Fig.2-4 の 600ºC 処理を経て固相法で合成した Li2MnO3と LiH を用い
た。それぞれをアルミナ乳鉢によりLi2MnO3 : LiH=1 : 2 (モル比)で混合した。混合物をペ レット状に加圧成形し、ガラス管に真空封入し、電気炉で焼成した。焼成条件は、昇温速度 2 ºC/min で・210ºC, 24 h ・210ºC, 72 h ・210ºC, 120 h の三つの条件で行った。焼成条 件による組成への影響を評価し、系統的に酸素欠損量を変化させるため三つの焼成時間を 設定した。その後、未反応の LiH および副生成物の Li2O をアンモニア水を用いて取り除 き、それを吸引濾過し、濾物を 120ºC 真空中において一晩乾燥させ目的物質を得た。得ら れた目的物質は、SEM 観察、充放電測定、XRD 測定によって評価した。
2-3. 各種正極活物質の評価法とその条件
2-3-1. X 線回折測定(XRD)
粉末X 線回折測定(X-ray diffraction; XRD)は理学電気株式会社製の「ロータレックス
RU-200B」回転対陰極形強力 X 線装置を使用した。X 線源には、CuKα 線を使用した。測定に はガラス製の試料ホルダーもしくは不活性雰囲気で測定可能な試料ホルダーを用い、管電
圧50 kV、管電流 250 mA、2θ 測定範囲 : 10°- 90°で測定した。
また、リートベルト解析用の測定にはBurker 製の D8-ADVANCE、Bragg Brentano 集
中光学系を使用した。X 線源には、CuKα 線を使用した。測定にはガラス製の試料ホルダー を用い、管電圧40 kV、管電流 40 mA、2θ測定範囲 : 10°- 90°で測定した。リートベルト 解析には Topas を用いて生成相の構造解析の精密化を試みた。 粉末法で解析を行うため試料は乳鉢で念入りに粉砕し、その粉末を詰めた試料ホルダー にX 線を照射し、発生する回折波をゴニオメーターで計測した。 また、不活性雰囲気での測定の際には、アルゴン雰囲気のグローブボックス内で試料を 試料ホルダーに充填し、測定を行った。 2-3-2. 粒度分布測定 粒度分布測定は、測定粒子にレーザビームを照射し、その粒子からの回折散乱光の強さの 分布を検出することでどれくらいの大きさの粒子がどれくらいの割合で含まれているか(粒 度分布) を求める測定である。本実験では、日機装製 Microtrac MT 3300 EXⅡを用いた。 正極活物質の屈折率η は未知であるため、無機固体で一般的に用いられる η=1.81 を使用し た。また、測定の際の溶媒には、蒸留水を用いた。 2-3-3. 走査型電子顕微鏡(SEM)
走査電子顕微鏡(Scanning electron microscope : SEM)により、試料形態に関する情報 を得た。電子銃から放出される電子線を細かく絞り、偏向コイルにより試料表面上の微小領 域に電子線を照射し、走査することにより形態を観察できる。本実験では、(株)日立製作所
製、走査型電子顕微鏡S-4800 を用いて、試料をカーボンテープで試料台に取り付けて観察
した。電極を観察する際には、Diethyl carbonate (DEC)によって電極を洗浄し、乾燥させ たものを用いた。また、電極など観察に不活性条件が必要なものは、アルゴン雰囲気のグロ ーブボックス内で不活性ホルダーに試料を取りつけて観察を行った。
2-3-4. 比表面積測定(BET 法) 正極活物質の粒径評価、電解液との接触面積を評価するため比表面積を測定した。各試料 の比表面積をガス吸着法 (BET : Brunauere-Emmette-Teller 法) を用いて行った。ガス吸 着法とは、粉体粒子の表面に吸着占有面積のわかったガス分子を吸着させ、その量から試料 の比表面積を求める方法である。本実験では、島津製作所製Tristar 3000 を使用した。各 試料の粉末をサンプル管に入れ、試料に付着した水分などを除去するため、90ºC, 3 h、200ºC, 2 h 減圧乾燥を行った。その後、試料を液体窒素で冷却し、N2分子を吹き込み、比表面積を 測定した。 2-3-5. 電気化学測定セルの試作と電気化学特性評価 Fig.2-6 に LiMn1.5Ni0.5O4に関する測定に用いた電気化学測定セル(有限会社日本トムセ ル製)の構造を示す。電池はアルゴン雰囲気のグローブボックス内で作製した。電池内部は、 下記図のように主に正極、セパレーター、電解液、負極で構成されている。またそれぞれの 電池構成物質については、下記に詳細を記す。ナガノBTS2004W を用いて下記条件にて定 電流充放電測定を行い電気化学特性を評価した。 LiMn1.5Ni0.5O4は3 サイクルのならし測定を行った後に本測定を行った。
正 極 : 活 物 質 に は 合 成 し た 試 料 、 導 電 剤 で あ る Acetylene black (AB) 、 N-methyl pyrrolidone (NMP)に溶かした Polyvinylidene difluoride (PVDF)を、活物質 : AB : PVDF = 80 : 10 : 10 (wt.%)で混合しアルミシートに塗布した。塗布後、空気中 80ºC, 2 h 乾燥、120ºC,3 h 真空乾燥行い、φ14 mm のポンチで切り取り正極とした。 負極 : リチウムシートをスペーサーに貼り付け、同じサイズに切り取ったものを使用した。 電解液 : 1 M LiPF6 in Ethylene carbonate (EC) : Dimethyl carbonate(DMC) (1:1 v/v%)
《ならし測定条件》 測定電位範囲 3.0 V - 5.1 V (vs. Li/Li+) 測定数 3 サイクル C レート 0.2C (1C = 147 mAh/g) 測定温度 60ºC 《本測定条件》 測定電位範囲 3.0 V - 5.1 V (vs. Li/Li+) 測定数 100 サイクル C レート 1C (1C = 147 mAh/g) 測定温度 60ºC Fig.2-6 LiMn1.5Ni0.5O4の測定に用いた電気化学測定セル. Table 2-1 LiMn1.5Ni0.5O4のならし測定条件. Table 2-2 LiMn1.5Ni0.5O4の本測定条件.
Fig.2-7 に Li2MnO3および Li2MnO3-xに関する測定に用いた電気化学測定セルの構造を 示す。測定には、2032 型コインセルを用いた。電池は乾燥空気雰囲気下で作製した。電池 内部は、下記図のように主に正極、セパレーター、電解液、負極で構成されている。またそ
れぞれの電池構成物質については、下記に詳細を記す。ナガノBTS2004W を用いて下記条
件にて定電流充放電測定を行い電気化学特性を評価した。
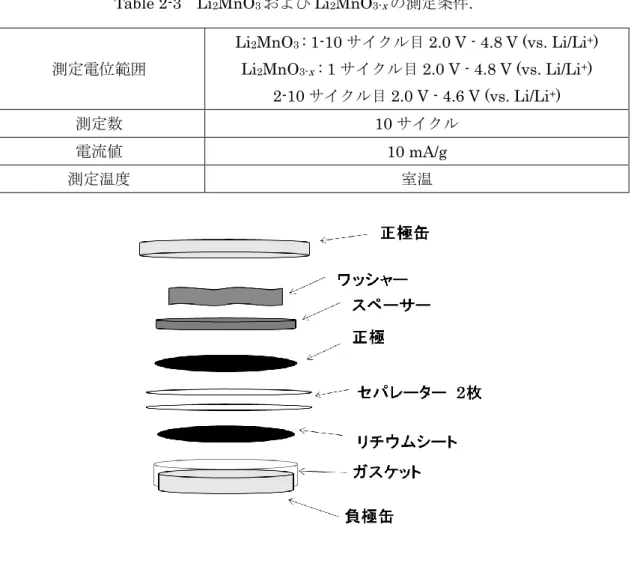
正 極 : 活 物 質 に は 合 成 し た 試 料 、 導 電 剤 で あ る Acetylene black (AB) 、 N-methyl pyrrolidone (NMP)に溶かした Polyvinylidene difluoride (PVDF)を、活物質 : AB : PVDF = 80 : 10 : 10 (wt.%)で混合しアルミシートに塗布した。塗布後、空気中 80ºC, 2 h 乾燥、120ºC,3 h 真空乾燥行い、φ12 mm のポンチで切り取ったものを正極と した。 負極 : リチウムシートをφ14 mm のポンチで切り取ったものを使用した。 電解液 : 1 M LiPF6 in EC : DEC (1:1 v/v%) セパレーター : ポリエチレンフィルター SETELA F20 BMU 測定電位範囲 Li2MnO3 : 1-10 サイクル目 2.0 V - 4.8 V (vs. Li/Li+) Li2MnO3-x : 1 サイクル目 2.0 V - 4.8 V (vs. Li/Li+) 2-10 サイクル目 2.0 V - 4.6 V (vs. Li/Li+) 測定数 10 サイクル 電流値 10 mA/g 測定温度 室温
2-4. 使用試薬
[NiMn3](OH)8 田中化学研究所(株) LiOH・H2O (LiMn1.5Ni0.5O2用) 和光純薬工業(株) LiOH・H2O (Li2MnO3用) ナカライテスク(株) MnO2 (株)高純度化学 MnCO3 Sigma-Aldrich LiH Sigma-Aldrich アンモニア水 ナカライテスク(株) ヘキサン ナカライテスク(株) アセトン ナカライテスク(株) AB STREM CHEMICALS NMP ナカライテスク(株) PVDF アルケマ(株) 1 M LiPF6 in EC : DEC(1:1 v/v%) キシダ化学(株) 1 M LiPF6 in EC : DMC(1:1 v/v%) キシダ化学(株) ポリエチレンフィルター 東レバッテリーセパレータ フィルム(株) ガラスフィルター Whatman Li シート 本城金属(株) Al シート(厚さ=18 μm) サンクメタル(株)3-1. LiMn
1.5Ni
0.5O
4の結晶性制御
3-1-1. 合成の簡易化
Fig.2-1 に示すフローチャートにより LiMn1.5Ni0.5O4の合成を行った。本研究の先行研究
において、出発物質である[NiMn3](OH)8は500ºC で 8 h 仮焼を行ったものを合成に用いて
いた[40]。仮焼を行うことで粒成長が起きること、合成の手順が複雑になることを考慮し、
本研究では[NiMn3](OH)8の仮焼を行わずLiMn1.5Ni0.5O4の結晶性の制御が行えるのか検証
した。
Fig.3-1 に仮焼有無による合成した試料の X 線回折図形、Fig.3-2 に SEM 画像を示す。 Fig.3-1 より、両試料の各回折ピークがリファレンスピークと一致し、目的物質が得られ たことが確認された。Fig.3-2 より、両試料の粒形状を比較すると同様の八面体粒子が得ら れていることが分かった。以上の結果から、両試料の間には大きな差はなく同様の目的物質 が得られていることを確認した。このことから、本研究では以後、[NiMn3](OH)8の仮焼は 行わずLiMn1.5Ni0.5O4の合成を行った。 仮焼有 仮焼無
Fig.3-1 [NiMn3](OH)8の仮焼の有無による合成試料のX 線回折図形.
3-1-2. 焼成時の原料の接触性による粒形状に対する影響 Fig.3-2 から、合成した八面体粒子は非常に凝集していることが分かった。凝集している ことにより、二次粒子の粒径が大きくなり活物質として利用できる部分が減少すること、そ の一次粒子間の粒界では八面体粒子が成長せず、粒子表面に酸素の最密面が形成されない ことにより電池性能の劣化が起こることが懸念された。そのため、この凝集を防ぐ方法とし て、焼成時に出発物質の粒子同士の接触性を変化させることを考えた。従来法、スパチュラ によって粒子を分散、ふるいを用いて粒子を分散させる、三つの方法を行った。それぞれ粒 子間の接触性は、従来法>スパチュラ>ふるいの順に大きくなる。 Fig.2-1 に示すフローチャートにより LiMn1.5Ni0.5O4の合成を行った。
Fig.3-3 に出発物質の接触性を変化させ合成した試料の X 線回折図形、Fig.3-4 に SEM 画 像を示す。 Fig.3-3 より、接触性が下がると不純物相が現れ、ピーク位置もずれていることが分かる。 このことは、水酸化リチウム一水和物と遷移金属水酸化物の接触性が下がりすぎ、十分に反 応が進行せず、合成試料はリチウム不足な組成になってしまったことが原因であると考え られる。Fig.3-4 より、全ての試料の粒形状を比較すると、接触性が悪くなるにつれて粒形 状は丸みをおびた形になり、八面体粒子へと成長していないことが分かった。このことから、 八面体粒子への成長は材料の組成も重要であることが分かった。また、凝集も起こっており、 この方法では凝集を防げないことが明らかになった。凝集を防ぐためには、出発物質である 遷移金属水酸化物の粒形状の制御や焼成温度を下げることが必要であると考えた。
仮焼有
仮焼無
ふるい
スパチュラ
従来法
Fig.3-3 出発物質の接触性を変化させた合成試料の X 線回折図形. 従来法 ふるい スパチュラ Ref. LiMn1.5Ni0.5O43-1-3. 低温合成時の窒素保持時間の粒形状に対する影響 本研究の先行研究において、八面体粒子の成長機構には酸素脱離反応が関与しており、合 成時の酸素分圧を変化させることで、低温で八面体粒子を合成出来ることが明らかになっ ている [40]。先行研究において、Fig.2-3 に示すように、窒素中 850ºC で焼成し、その後の 再酸化過程を経ることで八面体粒子の合成を行っていた。その際、窒素中での保持時間の影 響が調査されていなかった。そのため、本研究において、窒素保持時間の粒形状に対する影 響を調査し、その最適値の調査を行った。 Fig.3-5 に窒素中保持時間を変更し合成した試料の X 線回折図形、Fig.3-6 に再酸化を行 わず合成した試料のX 線回折図形、Fig.3-7 に窒素中保持時間を変更し合成した試料の SEM 画像、Fig.3-8 に再酸化を行わず合成した試料の SEM 画像を示す。 Fig.3-5 より、全ての試料の各回折ピークがリファレンスピークと一致し、目的物質が得 られたことが確認された。Fig.3-7 より、試料の粒形状を比較すると窒素中での保持時間が 長くなるにつれ、一次粒子の粒径が大きくなり綺麗な八面体粒子が形成されていないこと が分かった。0 h 保持では酸素脱離量が少なく遷移金属の拡散が大きく起こらないことから、 原料スピネルと同様の形状であったと考えられる。3 h 保持では酸素脱離量が適当量で、よ り小さな八面体一次粒子が形成されていることが確認できた。7 h 保持では、酸素脱離量が 適切量より多すぎ、より大きな一次粒子に成長したことで、八面体粒子が形成されていない 部分ができたと考えられる。 また、Fig.3-6 から、再酸化を行わずに合成した試料は、ニッケルとマンガンの相分離が 起こっていた。Fig.3-8 から、再酸化を行っていない試料は大きく粒成長が起こり、球状の 粒形態になっていた。このことから、酸素脱離量が適切量より多すぎ、大きく相分離が起こ ったことにより、酸素欠損に応じた粒形態に粒成長が起こったと考えられる。そのため、7 h 保持試料では、大きく粒成長したことにより、再酸化過程において八面体粒子が形成され るまでの時間を多く必要とし、そのために八面体粒子が形成されなかったと考えられる。加 えて、再酸化前では八面体粒子が形成されていないことから、再酸化過程において八面体粒 子が生成することが示唆された。
Ref. LiMn1.5Ni0.5O4 Ref. Li0.3Ni1.7O2 Fig.3-6 再酸化を行わなかった合成試料の X 線回折図形. Ref. Mn3O4 850ºC 7 h 再酸化無 Fig.3-5 窒素中での保持時間を変化させた合成試料の X 線回折図形. 0 h 原料 LiMn1.5Ni0.5O4 3 h 5 h Ref. LiMn1.5Ni0.5O4 7 h
10
μ
m
10
μ
m
10
μ
m
10
μ
m
10
μ
m
原料 LiMn
1.5Ni
0.5O
40 h
3 h
5 h
7 h
Fig.3-7 窒素中での保持時間を変化させた合成試料の SEM 画像. Fig.3-8 再酸化を行わなかった合成試料の SEM 画像.10
μ
m
3-1-4. 従来合成法との粒形状の比較
低温合成試料と従来法試料の比較のため、SEM による粒形状の観察と BET 法による比 表面積測定を行った。
Fig.3-9 に合成した試料の SEM 画像、Table 3-1 に比表面積測定結果を示す。SEM 画像 から一次粒子の大きさは低温合成試料の方が小さいことが分かった。従来法では一次粒子 の粒径が1 μm - 5 μm 程度であるのに対し、低温合成試料では 1 μm - 3 μm の一様な八面 体粒子であった。比表面積測定結果から低温合成試料は従来法と比較して、約17%比表面 積が大きかった。このことから、従来法より一次粒子の小さな八面体粒子が合成されてい ることを確認した。
従来法
(空気中 1000ºC 10 h)
低温合成法
(窒素中 850ºC 3 h)
Fig.3-9 合成試料の SEM 画像比較. Table 3-1 比表面積測定結果.3-2.
低温合成
LiMn
1.5Ni
0.5O
4の電池特性評価
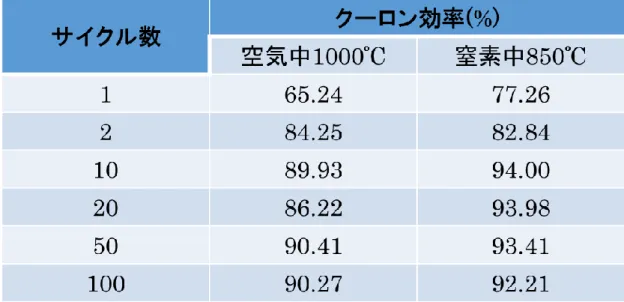
3-2-1. 低温合成 LiMn1.5Ni0.5O4の電気化学特性 Fig.3-10 に低温合成試料(窒素中 850ºC 3 h)の充放電曲線、Fig.3-11 にサイクルごとの容 量の推移を示す。測定は、Table 2-2 に示す条件で行った。 Fig.3-10 より、低温合成試料は LiMn1.5Ni0.5O4の充放時の特徴である、4 V 領域の充電容 量が見られなかった。このことから、低温合成試料はマンガンとニッケルが規則的に並んだ 超格子を持つLiMn1.5Ni0.5O4であることが示唆された。またLiMn1.5Ni0.5O4の特徴である、 4.7 V 付近での電位平坦部も確認された。Fig.3-11 から低温合成試料の 100 サイクル後の放 電時の容量維持率は90%程度であった。容量維持率は 1 サイクル目の放電容量を 100%と して 100 サイクル目の値から算出したものである。次項で従来法により合成を行った試料 との電気化学特性の比較を行い、低温合成の有意性について議論する。 Fig.3-11 低温合成試料のサイクル毎の充放電容量. Fig.3-10 低温合成試料の充放電曲線.3-2-2. 従来法との電気化学特性比較 Fig.3-12 に従来法合成試料(空気中 1000ºC 10 h)の充放電曲線、Fig.3-13 にサイクルごと の容量の推移、Table 3-2 に測定結果のまとめ、Table 3-3 に各サイクルでのクーロン効率の 比較を示す。 Fig.3-12 より、従来法合成試料も低温合成試料と同様に LiMn1.5Ni0.5O4の充放時の特徴 である、4 V 領域の充電容量が見られず、超格子を持つ LiMn1.5Ni0.5O4であることが示唆さ れた。Fig.3-13 から従来法合成試料の 100 サイクル後の放電時の容量維持率は 96 %程度で あった。容量維持率は1 サイクル目の放電容量を 100%として 100 サイクル目の値から算 出したものである。また、1 サイクル目の充電容量が大きくなっているが、これは本測定前 のならし測定から、本測定へ移行する際に恒温槽の温度が変化し、新たな被膜の形成や電解 液の分解等が起こったことによるものだと考えている。 Fig.3-10 と Fig.3-12 の比較を行うと、同様の形状の充放電曲線を示していることが分か っ た 。 こ の こ と か ら 、 低 温 合 成 試 料 に お い て も 従 来 法 と 同 様 の 目 的 と す る 八 面 体
LiMn1.5Ni0.5O4が合成できていることが示唆された。また、Fig.3-11 と Fig.3-13 と Table
3-3 の比較から、100 サイクル通じて低温合成試料は従来法試料と比べて 10%程度大きな放 電容量を示し、クーロン効率も2 - 7%良いことが分かった。それに対して、低温合成試料で は放電容量維持率が6%程度低い値を示した。 これらの原因は、低温合成試料が従来法合成試料と比較して、一次粒子の粒径が小さく、 比表面積が大きいことにあると考える。一次粒子の粒径が小さく、比表面積が大きいことか ら、活物質として利用できる割合が従来法と比較して増加するため、その放電容量が大きく なり、クーロン効率も向上したと考えられる。一方で、一次粒子の粒径が小さく、その比表 面積が大きいことによって、一次粒子間の粒界の面積も増加し、酸素の最密面が最表面に形 成されていない部分が増えている。そして、その部分が電解液と接触することで、電解液の 酸化分解や遷移金属の溶出を引き起こし、従来法と比較して放電容量維持率が低下したと 考えられる。また、電解液の分解反応は電解液と活物質との界面で進行するため、比表面積 が大きいことが、容量維持率として不利に働くと考えられる。
Fig.3-13 従来法合成試料のサイクル毎の充放電容量. Fig.3-12 従来法合成試料の充放電曲線.
3-2-3. 充放電後の電極表面観察 Fig.3-14 に低温合成試料(窒素中 850ºC 3 h)と従来法合成試料(空気中 1000ºC 10 h)の 100 サイクル充放電後の電極のSEM 画像を示す。 SEM 画像から、どちらも充放電前では綺麗な八面体粒子が形成されていることが確認で きた。充放電後の従来法合成試料においては、粒子に割れが存在していることが分かった。 一方で、低温合成試料ではその割れは確認できず綺麗な八面体粒子が残ったままであった。 これは、低温合成試料は一次粒子の粒径が小さいため、リチウムの挿入脱離に伴う粒子の歪 みの影響が小さく、そのことによって割れが起こりにくいことが理由であると考えられる。 粒子の割れの有無と放電容量維持率に相関性があまり見られないことから、従来法合成試 料における割れの影響が少ないと推測される。しかしながら、酸素の最密面を保つという意 味で低温合成において一次粒子の粒径を小さくすることの有用性は確認された。
5
μ
m
5
μ
m
5
μ
m
5
μ
m
充放電前
空気中
1000°C
10 h
窒素中
850°C
3 h
Fig.3-14 充放電後の電極表面 SEM 画像.充放電後
3-3. Li
2MnO
3の合成
本研究では、LiH を用いて Li2MnO3を還元させることでLi2MnO3-xの合成を目指した。
Li2MnO3の還元反応を均一かつ円滑に進めるためには、Li2MnO3の微粒子を還元反応の出
発物質として用いる必要がある。そのため、本項では、Li2MnO3合成の出発物質および焼
成温度とLi2MnO3の粒径への影響を検討した。
3-3-1. 焼成条件が活物質粒径に与える影響の評価
本項では、Li2MnO3の微粒子を得るために、Li2MnO3の焼成条件を検討した。焼成は、
600, 700, 800ºC で 12 h 行った。Fig. 2-3 に Li2MnO3の合成手順をに示す。
Fig. 3-15 に異なる温度で合成した Li2MnO3の X 線回折図形、Fig.3-16 に合成試料の
SEM 画像を示す。 いずれの試料も、空間群C2/mで帰属される層状岩塩相の反射が観測され、目的物質の単 一相の生成が確認された。焼成温度が上昇するにつれて各ピークの半値幅は、狭くなってい た。試料の結晶子サイズは、600ºC 試料で 174.2 Å、700ºC 試料で 218.5 Å、800ºC 試料 で400.4 Åであった。このことから、焼成温度の増加により、Li2MnO3の結晶成長が確認 された。600ºC で焼成した試料と 800ºC で焼成した試料の粒形状を比較すると、800ºC で 焼成した試料では、Li2MnO3粒子同士の焼結による粒成長が確認された。これらの結果は、 焼成温度が高くなるにつれて粒成長が起こり、結晶性が増加するという一般的な結果とも 一致していた。微細なLi2MnO3を得るには、600ºC での合成がよいと考えられる。 Ref. Li2MnO3 600ºC 800ºC 700ºC
3-3-2. 出発物質が活物質粒径に与える影響の評価
出発物質にMnCO3またはMnO2を用いてLi2MnO3の合成を行い、出発物質の違いによ
るLi2MnO3の粒径の違いを検討した。Fig.3-17 に出発物質として用いた MnCO3とMnO2
のSEM 画像、Fig. 3-18 にこれらの試料の粒度分布を示す。MnO2は1 μm 程度の球形の一
次粒子が凝集し、数 μm 程度の二次粒子を形成している。これに対して、MnCO3は、1 μm
程度の粒子が凝集せず、各々独立した形状であった。Fig.2-3 に Li2MnO3の合成手順を示
す。
Fig. 3-19 に、異なる出発物質を用いて空気中 600ºC, 12 h の条件で合成した Li2MnO3の
X 線回折図形、Fig. 3-20 に合成した Li2MnO3のSEM 画像、Fig. 3-21 に合成した試料の粒
度分布測定結果を示す。 いずれの試料も、空間群C2/mで帰属される層状岩塩相の反射が観測され、目的物質の生 成が確認された。試料の結晶子サイズは、MnCO3を用いた試料で174.2 Å、MnO2を用い た試料で162.0 Åであった。合成した試料の粒は、いずれの試料も、1 μm 以下の不規則な 形状だった。Li2MnO3の粒形状は、混合時や焼成過程での粒成長等で出発物質から変化し たと考えられる。粒度分布測定では、いずれの試料も1 μm 以下の粒子が最も多く、MnCO3 を出発物質として用いたLi2MnO3の方が、3 μm 以下の粒子が多かった。MnCO3の脱炭酸 反応が合成中の粒子の凝集を防ぎ、Li2MnO3の粗大粒子が減少したのと考えられる。出発 物質の粒径の違いは、Li2MnO3 の粒径に顕著な影響を与えなかったが、出発物質として MnCO3を用いることで粗大粒子の少ないLi2MnO3が合成得られた。
2
μ
m
600ºC
800ºC
2
μ
m
Fig.3-16 焼成温度を変化させ合成した試料の SEM 画像.2
μ
m
10
μ
m
2
μ
m
10
μ
m
MnCO
3MnO
2 Fig.3-17 出発物質の SEM 画像. Fig.3-18 出発物質の粒度分布測定結果.MnCO
3MnO
22
μ
m
2
μ
m
Fig.3-20 出発物質を変化させ合成した試料の SEM 画像.MnCO
3MnO
2 MnCO3 MnO2 Ref. Li2MnO3 Fig.3-19 出発物質を変化させ合成した試料の XRD 測定結果.3-3-3. 活物質粒径の違いによる電気化学特性の評価
ここまでの項において、Li2MnO3合成の出発物質、焼成条件のLi2MnO3の粒径への影響
を検討してきた。本項では、Li2MnO3の粒径の電気化学特性への影響を調査した。
MnCO3を出発物質として用い、600 および 800ºC で合成した Li2MnO3、MnO2を出発物
質として用い600ºC で合成した Li2MnO3の電気化学特性を評価した。活物質の一次粒子の
粒径は、それぞれ、MnCO3 600ºC < MnO2 600ºC < MnCO3 800ºC である。
Fig.3-22 にこれらの試料の初回充放電曲線を示す。すべての試料の初回充電過程に Li2MnO3の特徴である4.5 V 付近での電位平坦部が確認された。MnCO3を用いて600ºC で 焼成した試料は、300 mhA/g を超える初回充電容量を示し、初回放電過程で 280 mhA/g の 高容量を示した。Li2MnO3の粒径が大きくなるにつれて、初回充電過程で観測された電位 平坦部の電位が高くなり、放電容量が減少した。MnCO3を用いて800ºC で焼成した試料の 初回充電容量は、80 mhA/g まで減少し、初回放電容量は、50 mhA/g だった。 以上より、Li2MnO3の充放電容量が、一次粒子の粒径に大きく依存することが確認され た。特にLi2MnO3一次粒子の粒成長が確認された800ºC で合成した試料の容量の低下は顕 著であり、低温合成した Li2MnO3のみ高容量を示すという報告と一致する結果を示した
[33]。これは、Li2MnO3の初回充電における反応が大きく影響している。Li2MnO3の初回
充電では酸素脱離反応が起こり、その反応を経ることで高容量を示す [34]。その反応は活 物質表面で起こるため、活物質の一次粒子が小さくその表面積が大きい方が有利である。そ のため、一次粒子の大きな試料では、その反応が円滑に起こらず、初回充電の電位平坦部の 電位が高く、容量が少ない結果になったと考えられる。 これらの結果から、本研究では以後、出発物質としてMnCO3を用いて空気中600ºC, 12 h 焼成を行い Li2MnO3-xの合成を行った。 Fig.3-21 合成試料の粒度分布測定結果.
MnCO
3MnO
2MnCO
3800ºC
MnO
2600ºC
MnCO
3600ºC
3-4.
Li
2MnO
3-xの合成
Li2MnO3およびその固溶系は、初回充電の 4.5 V 付近の電位平坦部において酸素脱離反
応を経ることで、250 mAh/g 以上の高容量を示す[34]。しかし、Li2MnO3をはじめとする
リチウム過剰系正極活物質の充放電反応機構は複雑であり、充放電過程での電位の低下や 容量の減少といった課題の解決が困難となっている[39]。近年、kubota 等は、LiH を用い
た還元合成により、Li2MnO3-xの直接合成に成功した[41]。しかし、酸素脱離反応による
Li2MnO3の組成、構造の変化と電気化学特性との相関について、更なる研究が必要である。
そこで本研究では、LiH を用いた Li2MnO3-xの還元合成条件を検討し、Li2MnO3-xの酸素
欠損量を変化させた試料を合成し、その結晶構造と電気化学特性の変化について系統的な 調査を行った。
3-4-1. Li2MnO3-xの合成と相同定
Li2MnO3-xは、還元剤であるLiH と Li2MnO3を混合したペレットを210ºC で反応時間を
24、72、120 h と変化させて合成した。Fig.2-4 に Li2MnO3-xの合成手順を示す。還元反応
後の試料に存在した不純物は、アンモニア水を用いて洗浄し、目的物質を得た。
Fig.3-23 に還元反応後の Li2MnO3-xのX 線回折図形、Fig.3-24 に 2 = 43 - 46°の拡大図
を示す。還元反応の時間を変化させて合成した試料は、いずれも、空間群C2/mで帰属され る反射が確認された。還元反応時間が長くなるにつれて、2 = 45°に観測された(131)反射 の位置は、低角度側へ移動した。Li2MnO3-xの還元反応は、層状岩塩構造を維持したまま反 応が進行したと考えられる。各回折ピークは、還元反応時間が長くなるにつれてブロードに なり、試料の結晶性の低下が確認された。また、2 = 34, 56°に Li2O の反射が観測され、 還元反応の進行に伴い、そのピーク強度は増加した。Li2MnO3の還元反応は、(1)に示す反 応式で進行すると考えられる。
Li2MnO3 +2 LiH → Li2MnO3-x + xLi2O + (2-x) LiH + x H2 ・・・(1)
Fig.3-25に洗浄後試料のLi2MnO3-xのX 線回折図形、Fig.3-26 に 2 = 43° - 46°の拡大
図を示す。観測された反射は全て、空間群 C2/m の層状岩塩型構造で帰属され、2 = 34, 56°に観測された Li2O のピークは消失した。洗浄により不純物が除去され、目的物質の単 一相が得られた。Fig.3-27 に反応時間 120 h 試料の洗浄前後の X 線回折図形、Fig.3-28 に 2 = 43 - 46°の拡大図を示す。洗浄前後の Li2MnO3-xの回折図形を比較すると、洗浄後の 試料のピークは、高角度側へ僅かに移動し、その形状もシャープとなった。不純物の洗浄過 程で、Li2MnO3-xとH2O や OH-との反応が示唆された。
Fig.3-29 に洗浄後試料、Fig.3-30 に還元反応前試料の SEM 画像を示す。還元反応前後の
Li2MnO3-xは、いずれも1 - 2m 程度の粒子で、還元反応による粒成長や粒形状の変化は、
Fig.3-23 還元反応後の Li2MnO3-xのX 線回折図形. Fig.3-24 還元反応後の Li2MnO3-xの X 線回折図形(43°-46°を拡大). Fig.3-25 洗浄後試料の Li2MnO3-xのX 線回折図形. Li2O Ref. Li2MnO3 24 h 72 h 120 h 還元反応前 Ref. Li2MnO3 24 h 72 h 120 h 還元反応前 Fig.3-26 洗浄後試料の Li2MnO3-xの X 線回折図形(43°-46°を拡大).
(131)
(131)
10
μ
m
Fig.3-29 洗浄後試料の SEM 画像.10
μ
m
2
μ
m
2
μ
m
Ref. Li2MnO3 洗浄前 還元反応前 洗浄後Fig.3-27 洗浄前後の Li2MnO3-xのX 線回折図形. Fig.3-28 洗浄前後の Li2MnO3-xの X 線回折図形(43°-46°を拡大).
Li2MnO3-xの還元時間に対する構造の変化を明らかとするため、各試料の粉末 X 線リー トベルト解析を行った。空間群C2/m の層状岩塩構造を用いて[41]、格子定数、原子位置、 占有率の精密化を行った。還元前試料は、4i位置と8j位置の酸素の占有率に変化が無かっ たため、これらの値を1 に固定して解析を行った。還元反応後試料は、4i位置と8j位置の 酸素の占有率を変化させて行ったが、8j 位置の酸素の占有率のみ変化が確認された。この ため、4i位置の酸素の占有率は固定して解析を行った。Fig.31 - 34 及び Table 4 - 3-7 に還元前試料、還元反応時間 24 h、3-72 h、120 h 試料のリートベルト解析結果を示す。解 析結果は、全ての試料において、Rwp = 7.5%, Rp = 5.3%, RB = 2.3%と低い値を示し、計算 値は測定値と良い一致を示した。
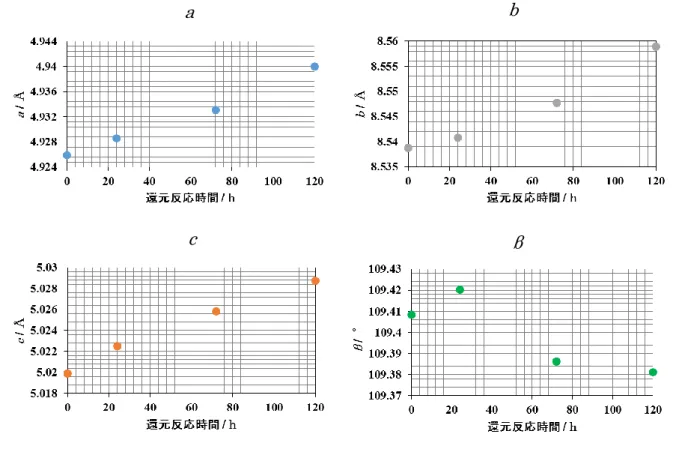
Fig.3-35 及び Table 3-8 に還元時間に対する Li2MnO3-xの格子定数の変化を示す。Table
3-9 に各サイトの占有率から算出した Li2MnO3-xの組成を示す。a, b, cいずれの軸も還元反 応時間の増加とともに格子が伸張した。各サイトの占有率から算出した Li2MnO3-xの組成 は、Li2MnO3の還元反応の進行に伴い、酸素欠損量の増加と共に、Li / Mn 比の減少も確認 された。解析結果からマンガンの価数を算出すると、還元時間の増加と共にマンガンの価数 は減少した。還元時間に対するLi2MnO3-xの格子の伸張は、還元によるマンガンイオンの半 径の増加が原因と考えられる。
Fig.3-36 及び Table 3-10 に還元時間に対する Li2MnO3-xの各サイトの占有率の変化を示
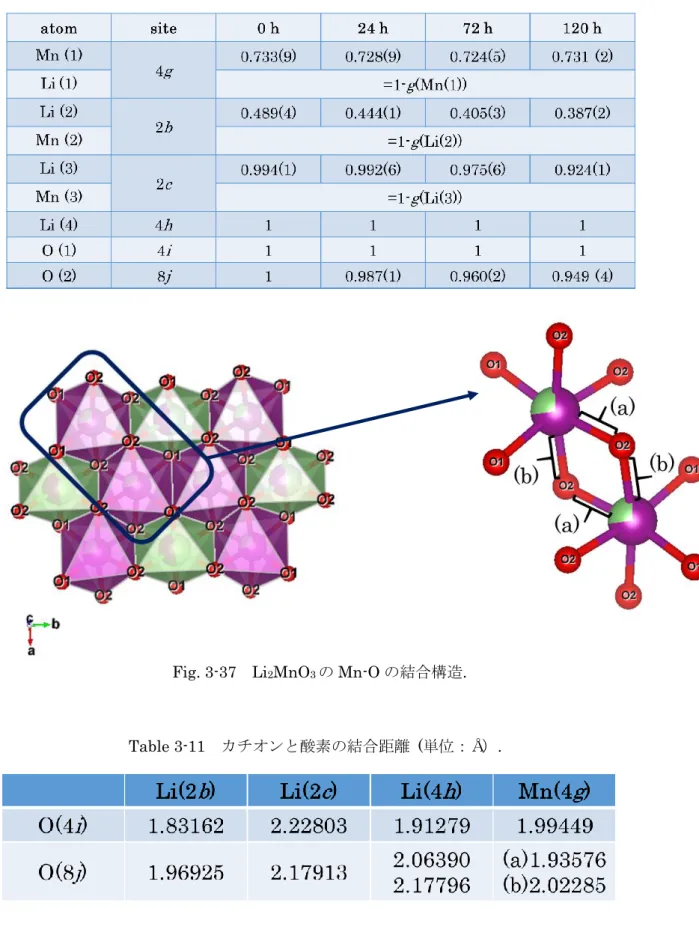
す。Li2MnO3の構造中で、4g八面体位置、2b八面体位置は、ハニカム状の遷移金属層内の Mn の占有する八面体と Li の占有する八面体にそれぞれ対応する。2c, 4h位置は、Li 層内 の八面体位置に対応する。Li2MnO3-xの8j位置の酸素の占有率は、還元時間の増加に伴い、 1.0 から 0.95 まで減少し、構造中への酸素欠損の導入が確認された。4g八面体位置のマン ガンの占有率は、還元時間に対して一定であったのに対して、2b八面体位置のマンガンの 占有率は、0.511 から 0.613 まで増加した。リチウム層では、Li2MnO3の遷移金属層内のリ チウム位置の下に位置する2c位置の遷移金属の占有率が増加した。 Fig.3-37 に Mn と O の結合構造、Table 3-11 に各カチオンと酸素の結合距離を示す。Mn は、2 つの 4i位置のO と 4 つの 8j位置のO と結合し、八面体を形成する。Mn と 8j位置 のO の結合は二種類ある。(a)は、1.93576 Åと短く、(b)は 2.02285 Åと長い。4i位置の O と Mn の結合は 1.99449 Åであり、8j位置のO との結合(b)と比較すると短い。また、 2b、4h位置のLi と 8j位置のO の結合は、4i位置のO との結合より長い。そのため、還 元反応の進行に伴い、8j位置の酸素が優先的に脱離したと考えられる。しかし、稜共有する Mn 八面体間での、Mn と 8j位置のO の結合は長いものと短いものが連結しているため、 両方の結合が切れることが難しいと考えられる。そのため、酸素欠損が起こりにくく、その 電荷を補償する形で、マンガンリッチな構造へと変化していくことが考えられる。
測定値 計算値 T ab le. 3-4 還元 反応 前 試 料のリ ート ベル ト解 析結 果 . モデル 構造 : C 2/ m a = 4. 9258 (8) Å , b = 8. 53 87(0) Å c = 5. 0198 (8) Å , β = 109. 408(4) ° R wp = 7. 52 % , R p= 5. 30 % , R wp /R e = 1. 68 % , R B = 2. 33 0% F ig. 3-31 還元 反応 前試 料のリ ート ベル ト解 析 結果 .
モデル 構造 : C 2/ m a = 4. 9285 (8) Å , b = 8. 54 08(2) Å c = 5. 0224( 8) Å , β = 109. 420( 3) ° R wp = 7. 20 % , R p = 5. 14 % , R wp /R e = 1. 56 % , R B = 2. 074 % F ig. 3-32 24 h 還元 反応 後試料 のリ ート ベル ト解 析結果 . T ab le 3-5 24 h 還元 反応 後試料 のリ ート ベル ト解 析結果 . 測定値 計算値
モデル 構造 : C 2/ m a = 4. 9330 (6) Å , b = 8. 54 76(9) Å , c = 5. 0258( 0) Å , β = 109. 386( 2) ° R wp = 6. 80 % , R p= 4. 97 % , R wp /R e= 1. 59 % , R B= 2. 027 % F ig. 3-33 72 h 還元 反応 後試料 のリ ート ベル ト解 析結果 . T ab le 3-6 72 h 還元 反応 後試料 のリ ート ベル ト解 析結果 . 測定値 計算値
測定値 計算値 モデル 構造 : C 2/ m a = 4. 9399( 9) Å , b = 8. 55 58( 9) Å c = 5. 0287 (5) Å , β = 109.381( 1) ° R wp = 6. 69 % , R p= 4. 99 % , R wp /R e = 1. 52 % , R B = 1. 63 8% T ab le 3-7 12 0 h 還元 反応 後試 料の リー トベ ルト 解析結 果 . 測定値 計算値 Fi g.3 -34 120 h 還元 処理 後試 料 のリ ート ベル ト解 析結 果 .
a
b
c
β
Fig.3-35 各軸の格子定数の変化.
Fig.3-36 リートベルト解析から得た各サイトの占有率の変化.
4g
2b
8j
2c
Table 3-10 各サイトの占有率の変化.
Fig. 3-37 Li2MnO3のMn-O の結合構造.
Fig.3-38 に Li2MnO3 - Li2MnO2 - LiMnO2三成分系状態図、Fig.3-39 に還元反応による
Li2MnO3-xの構造の変化、Fig.3-40 に Li2MnO3のMn-Li 層の構造を示す。Li2MnO3から
酸素脱離反応が進行すると、生成物の組成は、Li2MnO3 - Li2MnO2連結線に示される。一
方、Li/Mn 比の減少による還元反応が進行すると、生成物の組成は、Li2MnO3 - LiMnO2
連結線上に示される。これら2 つの還元反応の進行により得られた本研究の還元相は、
Li2MnO3 - Li1.5MnO2連結線上で組成の変化が確認された。
還元反応の進行に伴い、Li2MnO3-xは、構造中の酸素欠損量の増加とLi/Mn 組成比の変
化による構造中のMn 量が増加した。構造中の Mn の増加は、Li 位置への Mn の置換とし
て記述される。Li2MnO3は、立方最密充塡下した酸化物イオンの空隙の八面体位置をマン
ガンとリチウムが交互に積層した層状岩塩構造を持つ。構造中の遷移金属層は、LiO6八面
体が6 つの MnO6八面体と連結したハニカム構造をとる。Li2MnO3-xのMn が、遷移金属
層のリチウム位置を占有する場合、還元反応後も層状岩塩構造は維持されるが、リチウム
層の八面体位置をMn が占有すると、Mn 八面体が不規則に配列した岩塩やスピネルに近
い構造となる。Li2MnO3-xは、還元時間の増加と共に、遷移金属層とリチウム層でマンガ
ンの増加が確認され、還元反応の進行と共に、層状岩塩構造が不安定となることが分かっ た。
Fig.3-38 Li2MnO3 - Li2MnO2 - LiMnO2 3 成分系相図での組成プロット.
Fig.3-39 還元反応による構造内での変化の模式図.
Fig.3-40 Li2MnO3のMn-Li 層の構造.
.
3-4-2. Li2MnO3-xの電気化学特性
Fig.3-41 に Li2MnO3-x の初回充放電曲線、Table 3-12
に初回充放電結果のまとめ、Fig.3-42 に電位平坦部までに注目した初回充電曲線、Fig.3-43 に各試料の 10 サイクル目までの 充放電曲線を示す。 Table 2-3 に示す条件、電流値 : 10 mA/g 、測定電位範囲 : 1 サイクル目 4.8 V - 2.0 V 2 - 10 サイクル目 4.6 V - 2.0 V で測定を行った。 Fig.3-41 から、還元反応時間の増加と共に、充電時においては 4.5 V 付近の電位平坦部で の容量の減少、電位平坦部に至るまでの容量、充放電時においては3 V 領域での容量が増 加していることが分かる。また、これらの結果は、還元反応時間が増加すると共に還元反応 が進行していることを示唆している。Table 3-12 より、還元前と比較して、還元反応後の試 料はクーロン効率が改善していることが分かる。このことから、還元反応によってリチウム 過剰系正極活物質の課題である初回不可逆容量が低減した。しかし、120 h 試料は 72 h 試 料と比べ、容量も小さくクーロン効率が改善しなかった。このことは、120 h 試料ではマン ガンリッチな相が72 h 試料と比べ多く存在することや、リチウム層へマンガンが移動した ことによって、リチウムの移動が妨げられ、活物質として機能していない部分が増えたこと によると考えられる。また、これらの現象がリチウム過剰系正極活物質における劣化を引き 起こす原因である可能性が示唆された。 Table 3-9 のリートベルト解析結果より得られたマンガンの価数から、それぞれ 4 価のマ ンガンまで酸化還元反応が進行する仮定すると、得られる容量はそれぞれ、24 h 試料 : 33 mAh/g 、72 h 試料 : 58 mAh/g、120 h 試料 : 92 mAh/g である。その結果と、Fig.3-42 か ら得られた電位平坦部までの容量を比較すると、良い値の一致を示した。このことから、電 位平坦部までの容量はマンガンの還元によるものであることが示唆された。Fig.3-43 から、 各試料の充放電曲線を比較すると、還元反応時間が長いものほど、10 サイクルまでの充放 電時における 3 V 領域の容量が大きいことが分かった。還元による層状岩塩構造の乱れの 増加と共に、充放電過程での 3 V 領域での容量が増加し、放電過程の電位の低下が顕著と なることが分かった。また、サイクル特性は改善されていないことを確認した。120 h 試料 においても、まだ4.5 V 付近の電位平坦部が存在することから、初回充電において構造変化 が起こっていることが考えられる。そして、それに引き続く充放電による構造変化が起こる ことにより、電位低下及び容量の低下が起こるため、サイクル特性が改善しないと考えられ る。
24 h 72 h 120 h 還元反応前 Fig.3-41 Li2MnO3-x の初回充放電曲線. Table 3-12 初回充放電結果のまとめ. 24 h 72 h 120 h
24 h
還元反応前
120 h
72 h
Fig.3-43 Li2MnO3-x の10 サイクル目までの充放電曲線. 2-10 サイクル目 10 サイクル目 1 サイクル目本研究では、Li-Mn-O 系正極活物質の中でも特に次世代正極活物質として注目されてい るスピネル型LiMn1.5Ni0.5O4および層状岩塩型Li2MnO3の二つの物質に着目した。これ らの材料の合成、構造、および電気化学特性を評価し、得られた知見からLi-Mn-O 系正極 活物質の電気化学特性向上の指針を見出すことを目指した。 得られた結果と、それに基づくLi-Mn-O 系正極活物質の材料設計指針を以下に示す。
・LiMn
1.5Ni
0.5O
4の結晶制御
・出発物質である[NiMn3](OH)8の仮焼なく八面体LiMn1.5Ni0.5O4が合成でき、合成の簡易
化が可能であることを明らかにした。 ・低温合成において窒素保持時間を変更、すなわち酸素脱離量を変化させることで粒形状が 大きく変化した。 ・酸素脱離量が大きくなると、遷移金属であるニッケルとマンガンの相分離が顕著に起こ り、それに伴い粒成長が起こった。 ・また、再酸化を行わない試料との比較結果から、再酸化過程において八面体粒子の生成が 起こっていることが明らかになった。 ・窒素保持850ºC,3 h の条件が、低温での八面体粒子の合成条件として最も適当であった。 ・低温合成試料は従来法と比較して、17%程度比表面積が大きくなった。 ・低温合成が一次粒子の小さな八面体粒子の合成に有効であることを明らかにした。