2.7.6
個々の試験のまとめ
2.7.6.1
S4001 試験の概要(資料:5.3.5.1.2)
治験依頼者名:
Solvay Pharmaceuticals, Inc.
各治験の要約表
申請資料中該当箇所
(審査当局
使用欄)
商品名: LUVOX
錠
分冊番号:該当せず
有効成分名:
マレイン酸フルボキサミン
ページ:該当せず
治験の標題:
社会不安障害の治療におけるフルボキサミン:外来患者を対象とした
二重盲検プラセボ対照並行群間比較多施設共同試験
治験責任医師名
治験実施医療機関:
公表文献:
Am J Psychiatry 156: 5,May 1999
治験期間(年数) :
開発のフェーズ:
(最初の被験者の同意取得日): 19
年
月
日
(最後の被験者の最終観察日): 19
年
月
日
第
IV 相
目的:
<主要目的>
社会不安障害の治療におけるマレイン酸フルボキサミン(以下,フルボキサミン)(50
~300 mg/日)の有効性及び安全性をプラセボと比較し,検証することとした。主要有効性
評価項目は,Clinical Global Impression (CGI) global improvement とし、有効例は,二重盲検
薬の投与開始後に“much improved”又は“very much improved”と判定された者と定義した。
<副次目的>
Brief Social Phobia Scale(BSPS),Liebowitz Social Phobia Scale(LSPS,Liebowitz Social
Anxiety Scale と同一であるので以下 LSAS とする),Sheehan Disability Scale(SDS),Social
Phobia Inventory(SPIN),Patient Global Impression(PGI),Hamilton Rating Scale – Depression
(HAMD)及び CGI severity of illness scale を実施し,その結果に基づいて有効性を比較す
ることとした。
治験依頼者名:
Solvay Pharmaceuticals, Inc.
各治験の要約表
申請資料中該当箇所
(審査当局
使用欄)
商品名: LUVOX
錠
分冊番号:該当せず
有効成分名:
マレイン酸フルボキサミン
ページ:該当せず
治験方法:
本治験は,無作為割付二重盲検並行群間比較デザインに従って実施した。精神疾患の診
断・統計マニュアル第
4 版(DSM-IV)による社会不安障害の診断基準(300.23)を満たす
被験者をフルボキサミン(50~300 mg/日)又はフルボキサミンと外観が一致するプラセボ
(1~6 錠/日)のいずれかに無作為に割り付け,85 日間にわたり投与を行なった。本治験
では,4 施設で合計 92 例が組み入れられ,86 例(フルボキサミン群:42 例,プラセボ群:
44 例)が評価可能であった。共通の治験実施計画書及び症例報告書が各医療機関で使用さ
れた。
被験者数(計画時及び解析時):
92 名の患者がランダマイズされ,そのうち 86 名が評価対象となった。
診断及び主要な組入れ基準:
本治験の対象者は,DSM-IV による社会不安障害の診断基準を満たす 18 歳から 65 歳ま
での男性及び女性外来患者で,スクリーニング時及びベースライン時の
BSPS スコアが 20
以上,
HAM-D17 項目のスコアが 16 以下の者とした。ベースライン時の CGI global
improvement スコアが 1(very much improved)又は 2(much improved)であった場合,当
症例は除外した。
被験薬,ロット番号:
マレイン酸フルボキサミン
50 mg 錠(経口)
ロット番号:LUVOX
錠:E-032751060895,プラセボ錠:C-00539
治療期間:
12 週間
対照治療,用量及び投与方法:
7 日間の単盲検プラセボ観察期の後,各症例に対し,フルボキサミン(50~300 mg/日)又
はフルボキサミンと外観が一致するプラセボ(1~6 錠/日)のいずれかの投与を 85 日間実施
した。最初の
3 週間は漸増投与期とし,残りの 8 週間は維持用量期とした。維持用量期の平
均総
1 日用量は,フルボキサミン群 198.28 mg(3.86 錠),プラセボ群 4.85 錠であった。
治験依頼者名:
Solvay Pharmaceuticals, Inc.
各治験の要約表
申請資料中該当箇所
(審査当局
使用欄)
商品名: LUVOX
錠
分冊番号:該当せず
有効成分名:
マレイン酸フルボキサミン
ページ:該当せず
評価基準:
有効性:
治験期間中,CGI global improvement(主要有効性評価項目),BSPS,LSAS,SDS 及び
SPIN を用いて有効性の評価を行なった。更に,有効性を裏付ける評価として,CGI severity
of illness,HAM-D 及び PGI global improvement を実施した。
安全性:
被験者の初回来院時,病歴調査,身体的検査,尿検査を含む臨床検査などを行なって,
各被験者の健康状態を総合的に調査した。女性被験者に対しては妊娠検査(血清
β-HCG)
も実施した。臨床検査は第
2 来院時(ベースライン)及び投与終了時に,バイタルサイン
の測定は投与開始前,各来院時及び投与終了時にそれぞれ実施した。有害事象は,投与期
間を通して,被験者に特別な質問をするといった方法を取らずに調査した。
統計手法:
反応率の解析は,治験薬を少なくとも
1 回服用し,ベースライン時の評価可能な有効性
データをもつすべての症例について,Last Observation Carried Forward(LOCF:最終観察時
引 伸 ば し ) デ ー タ を 用 い て 実 施 し た 。 反 応 者 の 解 析 は , 施 設 を 層 と し た
Cochran
Mantel-Haenszel の検定を用いて行なった。有効性評価項目の各々によるベースラインスコ
アからの変化については,治療,施設及び施設-治療の交互作用を固定因子とした二元配置
分散分析モデルを用いて解析した。検定はすべて両側とし,
α=0.05 で行なった。
要約・結論:
92 例が無作為割り付けされ,症例番号が割り当てられた。実際に二重盲検薬を服用した
症例は
90 例(フルボキサミン群:46 例,プラセボ群:44 例)で,2 例は二重盲検薬を服
用する前に本治験を早期に中止した(1 例は治験実施計画書からの逸脱のため,他の 1 例
は同意の撤回のため)。本治験では,女性被験者(32 例)より男性被験者(58 例)が多数
を占め,フルボキサミン群においては男性被験者が有意に多かった(p=0.018)。被験者の
人口統計学的特性に関して,男女比以外で投与群間に差が認められた項目はなかった。
治験依頼者名:
Solvay Pharmaceuticals, Inc.
各治験の要約表
申請資料中該当箇所
(審査当局
使用欄)
商品名: LUVOX
錠
分冊番号:該当せず
有効成分名:
マレイン酸フルボキサミン
ページ:該当せず
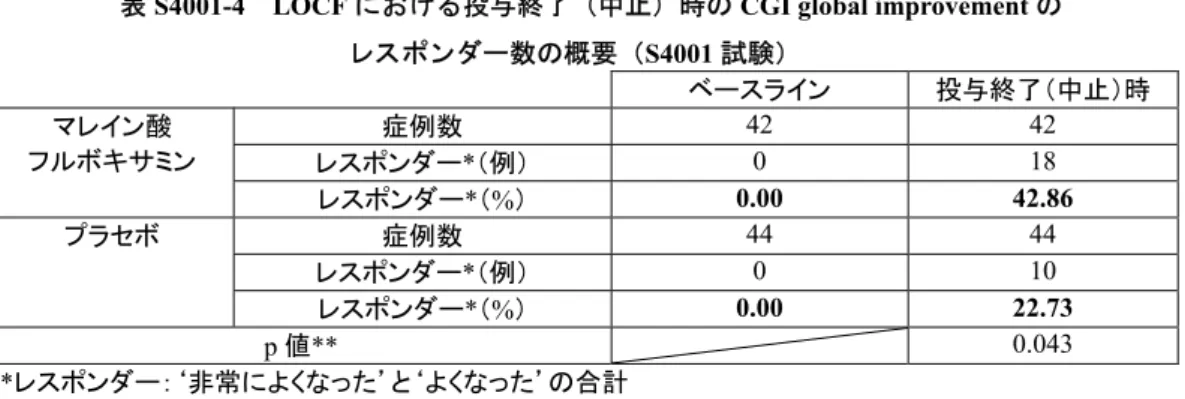
有効性の結果:
有効性の評価は,CGI global improvement(主要有効性評価項目),BSPS,LSAS,SDS
及び
SPIN を基に行なった。更に,有効性を裏付ける評価として,CGI severity of illness,
HAM-D17 項目の総スコア及び PGI global improvement を実施した。主要有効性評価項目に
よる評価の結果,治験終了時における反応率は,フルボキサミン群
43%,プラセボ群 23%
(p=0.043)であった。副次的評価項目を用いた評価の結果,SDS の 1 領域である“社会生
活”では有意傾向が示されたのみであったが,他のすべての評価項目で投与群間に統計学
的な有意差が認められた。
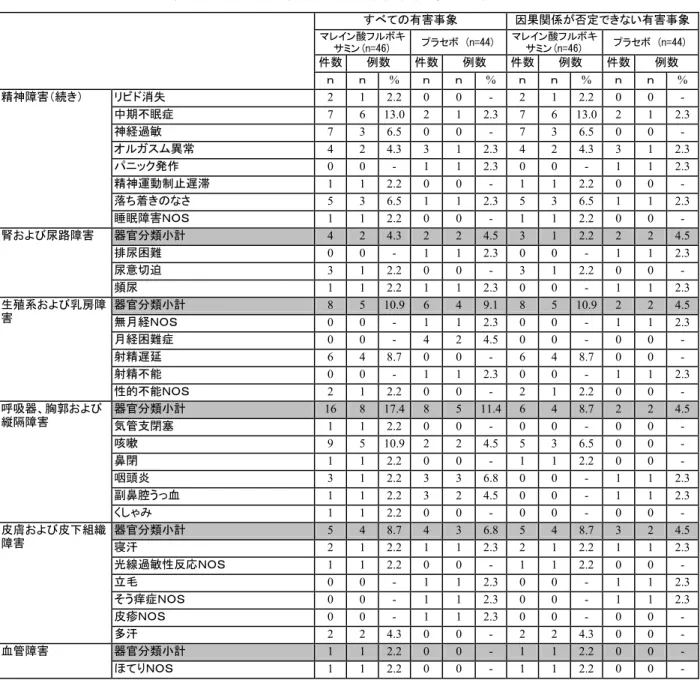
安全性の結果*:
有害事象及びバイタルサインの記録は各来院時に行ない,臨床検査は無作為割り付け前
及び治験終了時に実施した。
ITT 安全性被験者群(N=90)における TESS の発現率に関し,
フルボキサミン群(87%)は,プラセボ群(86%)とほぼ同値を示した。本治験の期間中,
重篤な有害事象が
3 件(フルボキサミン群:2 件,プラセボ群:1 件)報告された。治験
責任医師により重篤と報告されていた
3 件のうち,2 件の有害事象は重篤な有害事象の標
準的な定義に該当せず,Solvay Pharmaceuticals, Inc.の判断により重篤として報告する必要
はないとされた。重篤な有害事象のため本治験を早期に中止した症例は
1 例(骨折 NOS)
のみであった。また,これらの重篤な有害事象と治験薬との因果関係は,いずれも“関連
なし”と判断された。フルボキサミン群において最も頻繁(発現率>30%)に報告された
有害事象は,頭痛,嘔気,不眠(症)及び傾眠であった。有害事象のため本治験を早期に
中止した症例は,プラセボ群
4 例,フルボキサミン群 12 例とフルボキサミン群に多く認
められた。フルボキサミン群において,治験中止に至った症例で最も多く報告された有害
事象名は,嘔気:5 例及び不眠(症):5 例であった。
結論:
フルボキサミンに関する本プラセボ対照試験の結果,次の結論を得た。
• フルボキサミンは,社会不安障害症状や機能障害の改善に有効である。
• フルボキサミンは,社会不安障害患者において,1 日 50~300 mg で安全で,忍容性
に優れている。
報告書作成日: 20
年
月
日
*:副作用用語は COSTART で集計(和訳は J-ART による)
概要補遺:
S4001 試験(資料:5.3.5.1.2)
社会不安障害の治療におけるマレイン酸フルボキサミン:外来患者を対象とした二重盲
検プラセボ対照並行群間比較多施設共同試験
(1)試験方法の概略
項 目
内 容
治験の標題
社会不安障害の治療におけるマレイン酸フルボキサミン(以下,フルボキサミン):
外来患者を対象とした二重盲検プラセボ対照並行群間比較多施設共同試験
目的
<主要目的>
社会不安障害の治療におけるフルボキサミン(
50~300 mg/日)の有効性及び安全性を
プラセボと比較し,検証することとした。主要有効性評価項目は, CGI global
improvement とし,有効例は,二重盲検薬の投与開始後に“much improved”又は“very
much improved”と判定された者と定義した。
<副次目的>
BSPS,LSAS,SDS,SPIN,PGI,HAMD 及び CGI severity of illness を実施し,その結
果に基づいて有効性を比較することとした。
被験薬:マレイン酸フルボキサミン
50 mg 錠(経口)
ロット番号:LUVOX®-032751060895,プラセボ錠:C-00539
被 験 薬 , 対 照
薬,投与方法
本治験は,無作為割付二重盲検並行群間比較デザインに従って実施した。
精神疾患の診断・統計マニュアル第
4 版(DSM-IV)による社会不安障害の診断基準
(
300.23)を満たす被験者をフルボキサミン(50~300 mg/日)又はフルボキサミンと
外観が一致するプラセボ(1~6 錠/日)のいずれかに無作為に割り付け,85 日間にわ
たり投与を行った。最初の
3 週間は漸増投与期とし,残りの 8 週間は維持用量期とした。
治療期間
12 週間
対象 DSM-IV による社会不安障害の診断基準を満たす 18 歳から 65 歳までの男性及び女性
外来患者で,スクリーニング時及びベースライン時の
BSPS スコアが 20 以上,
HAM-D17 項 目のスコアが 16 以下の者とした。ベ ースライン時 の CGI global
improvement スコアが 1(very much improved)又は 2(much improved)であった場合,
当症例は除外した。
選択基準
1)
年齢
18~65 歳の男女。妊娠の可能性のある女性又は閉経後 1 年未満の女性は,医
学的に適切な方法で避妊していること[
3 ヵ月間以上経口避妊薬を服用している
者,
2 ヵ月間以上 medroxyprogesterone(Depo-Provera
®)を服用している者,二重盲
検投与期開始前に
2 ヵ月間以上子宮内避妊法を実施している又は levonorgestrel
(
Norplant
®)の埋植を行っている者,若しくは避妊処理法を行なっている者,コ
ンドームと殺精子剤を併用している者]。二重盲検期開始前に,血清
βヒト絨毛性
ゴナドトロピン(
β-HCG)妊娠検査が陰性でなければならない。
2) DSM-IV の構造的問診(SCID)改訂版に従って調査するとき,DSM-IV の社会不安
障害の診断基準(
300.23)を満たす者
3) BSPS 総スコアが 20 以上の者
4) 臨床症状及び臨床検査所見から判断して,臨床的に異常ではないと判断された者
5) 読み書きの点で英語又はスペイン語が流暢である者
6) 文書による同意が得られる者
除外基準
1)
過去
1 年間に次のいずれかに診断(DSM-IV)された者
・大うつ病性障害(296.2 又は 296.3)
・強迫性障害(
300.3)
・精神分裂病(295.xx)の主診断
・社会不安障害以外の不安障害(
300.xx)の主診断
・適応障害(309.xx)
・双極性障害(
296.4x,296.5x,296.6x,296.7,296.80 又は 296.89)
・境界性人格障害(301.83)
2) HAM-D 17 項目のスコアが 16 以上の者
3) 第 1 日の CGI global improvement スコアが 1 又は 2 の者
4) 過去 6 ヵ月以内に,ニコチンを除く物質乱用障害(DSM-IV 305.xx)の既往を有す
る者
項 目
内 容
5) 臨床的に重要な造血系疾患,自己免疫疾患又は心・血管障害の現病歴を有する者
6) 次のいずれかの現病歴・既往歴を有する者
・治験薬の吸収・分布・代謝・排泄に影響を及ぼす可能性のある胃腸管障害,肝障
害,腎障害又は他の障害
・発作障害(小児性熱性発作の単一エピソードを除く),脳血管障害又は脳外傷
・臨床的に重要な不安定内分泌障害(甲状腺機能低下症等)又は不安定インスリン
依存性糖尿病
・いずれかの型の悪性疾患の現病歴・既往歴
7) 本治験開始前の 30 日以内に,主要臓器に対して毒性を発現する可能性があること
が確認されている薬剤による治療を受けた者
8) セロトニン再取込み阻害薬に対して過敏症を示す者,Seldane ®(terfenadine),
Hismanal®(astemizole)又は Propulsid®(cisapride)を併用している者
9) 医学的に適切な方法で避妊していない妊娠の可能性のある女性,スクリーニング
期の妊娠検査が陽性である女性,妊娠している女性又は授乳中の女性
10) 治験実施計画書において併用可能とした薬剤を除き,治験期間中,電気痙攣療法
又は他の向精神薬治療を必要とする者
11) 治験期間中,構造的精神療法(認知療法等)を必要とする者
12) 次のいずれかの薬剤又は治療をそれぞれ一定の期間内に受けた者
・二重盲検投与期開始前
3 ヵ月間以内に電気痙攣療法
・二重盲検投与期開始前
30 日間以内に他の治験薬
・二重盲検投与期開始前
14 日間以内にモノアミン酸化酵素阻害薬又は長時間作
用型フェノチアジン系薬剤
・二重盲検投与期開始前
5 週間内に fluoxetine
・二重盲検投与期開始前
1 週間以内に terfenadine 又は cisapride,2 ヵ月以内に
astemizole
・二重盲検投与期開始前
7 日間以内に他の精神活性剤
13) 器質性又は全身性疾患を有する者,若しくは,他に規定のない限り,本治験薬の
有効性及び安全性の評価に影響を及ぼすと考えられる他の治療の介入を必要とす
る者
14) 事前に同意を得ることが困難と考える者
15) 過去に治療に対するコンプライアンスが不良であった者
被験者数
92 名の患者がランダマイズされ,そのうち 86 名が評価対象となった。
有 効 性 評 価 項
目
治験期間中,CGI global improvement(主要評価項目),BSPS,LSAS,SDS 及び SPIN
を用いて有効性の評価を行った。更に,有効性を裏付ける評価として,
CGI severity of
illness,HAMD 総スコア及び PGI improvement を実施した。
安全性評価
被験者の初回来院時,病歴調査,身体的検査,尿検査を含む臨床検査などを行なって,
各被験者の健康状態を総合的に調査した。女性被験者に対しては妊娠検査(血清
β-HCG)も実施した。臨床検査は第 2 来院時(ベースライン)及び投与終了時に,バ
イタルサインの測定は投与開始前,各来院時及び投与終了時にそれぞれ実施した。有
害事象は,投与期間を通して,被験者に特別な質問をするといった方法を取らずに調
査した。
統計手法
反応率の解析は,
治験薬を少なくとも
1 回服用し,投与開始時の評価可能な有効性デー
タが評価可能であるすべての症例について,
LOCF を用いて実施した。反応者の解析
は,施設を層とした
Cochran Mantel-Haenszel の検定を用いて行い,有効性評価項目の
各々による投与開始時のスコアからの変化については,治療,施設及び施設
-治療の交
互作用を固定因子とした二元配置分散分析モデルを用いて解析した。検定はすべて両
側とし,
a=0.05 で行った。
症例数の
設定根拠
プラセボに反応する被験者の比率を
25%と仮定し,症例数を以下のように見積もった。
プラセボ マ レ イ ン 酸 フ ルボキサミン 有効性評価項目 P1 P2 α 検出力 症例数2xN
CGI Global Improvement 0.25 0.65 0.05 80% 32