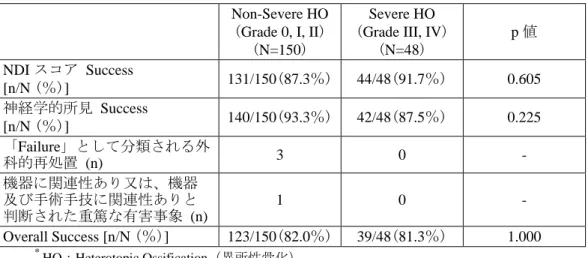
平 成 2 9 年 4 月 2 1 日
医
薬
・
生
活
衛
生
局
医 療 機 器 審 査 管 理 課
審議結果報告書
[類
] 医療用品 04 整形用品
別
[一般的名称] 人工椎間板
[販
] PRESTIGE LP CERVICAL Disc システム
売
名
[申
] メドトロニックソファモアダネック株式会社
請
者
[申
] 平成 28 年5月 30 日(製造販売承認申請)
請
日
【審 議 結 果
】
平成 29 年4月 21 日の医療機器・体外診断薬部会の審議結果は次のとおりであ
り、この内容で薬事分科会に報告することとされた。
本承認申請については、使用成績評価の対象として指定し、次の条件を付した
上で、承認することが適当である。生物由来製品及び特定生物由来製品には該当
しない。
なお、使用成績の調査期間は 5 年とすることが適当とされた。
本製造販売承認申請の承認条件
椎間板ヘルニア又は骨棘を主因とした頚椎の神経根症や脊髄症の治療及び頚椎
前方手術に対する十分な知識・経験を有する医師が、関連学会と協力して作成
された適正使用指針を遵守し、講習の受講により本品を用いた治療に関する技
術を得る等、本品が適切に用いられるよう、必要な措置を講じること。
1 審査報告書 平成 29 年 4 月 3 日 独立行政法人医薬品医療機器総合機構 承認申請のあった下記の医療機器にかかる医薬品医療機器総合機構での審査結果は、以 下のとおりである。 記 [ 類 別 ] : 医療用品 04 整形用品 [ 一 般 的 名 称 ] : 人工椎間板
[ 販 売 名 ] : PRESTIGE LP Cervical Disc システム
[ 申 請 者 ] : メドトロニックソファモアダネック株式会社 [ 申 請 年 月 日 ] : 平成 28 年 5 月 30 日
[ 特 記 事 項 ] : なし
2 審査結果
平成 29 年 4 月 3 日
[ 類 別 ] : 医療用品 04 整形用品 [ 一 般 的 名 称 ] : 人工椎間板
[ 販 売 名 ] : PRESTIGE LP Cervical Disc システム
[ 申 請 者 ] : メドトロニックソファモアダネック株式会社 [ 申 請 年 月 日 ] : 平成 28 年 5 月 30 日
審査結果
「PRESTIGE LP Cervical Disc システム」(以下「本品」という。)は、椎間板ヘルニアや骨 棘等の圧迫因子の除去後に頚椎の適応椎間の椎間板を本品に置換することで、適応椎間の 可動性を維持することを目的とした頚椎人工椎間板である。原則として、最低 3 か月の保存 療法が奏効しなかった患者で、画像所見(CT、MRI、X 線等)により確認される椎間板ヘル ニア又は骨棘を伴う神経根症又は脊髄症に用いることで、可動性の維持とともに除痛が期 待される。本品は、ボール部とトラフ部から構成され、ボール部とトラフ部が組み合わさり、 可動性を維持し、また、それぞれレール、チタンプラズマスプレーコーティング及びグリッ ドブラスト加工による粗面により椎体に固定される。 非臨床試験の成績として、機械的安全性、生物学的安全性及び MR 装置との相互作用に 関する試験の資料が提出され、特段の問題がないことが示された。
本品の臨床試験の成績として、米国での PMA(Premarket Approval Application)承認取得 の目的で実施された本品の 1 椎間使用における有効性及び安全性を評価するための IDE G040086 Pivotal 試験(以下「1 椎間治験」という。IDE:Investigational Device Exemption。)、 体内への金属イオン濃度の溶出等を評価するために追加実施された IDE G040086 Metal Ion 試験(以下「Metal Ion 治験」という。)、患者救済目的で Pivotal 試験(1 椎間治験)に追加 して実施された IDE G040086 Continued Access 試験(以下「Continued Access 治験」という。)、 ヒストリカルコントロールとして利用した Atlantis(前方固定用プレート)の 1 椎間の成績 を含む IDE G010188 試験、並びに本品の 2 椎間使用における有効性及び安全性を評価する ために実施された IDE G050202 試験(以下「2 椎間治験」という。)の成績が提出された。 本品の 1 椎間使用に係る主な評価資料である 1 椎間治験は、頚椎椎間板変性疾患が原因 で 1 椎間の外科的治療が必要な患者 280 例を対象とした多施設共同前向き非劣性既存対照 比較試験である。当該治験では、本品の旧モデル製品である PRESTIGE ST の承認取得のた めに実施した多施設共同前向き無作為割付非劣性比較試験(IDE G010188 試験)における比
3
較対照群である頚椎前方除圧固定術群(以下「ACDF 群」という。ACDF:Anterior Cervical Discectomy and Fusion。)がヒストリカルに比較対照群として用いられた。有効性については、 主要評価項目として設定された術後 24 か月時点の Overall Success(術後の Neck Disability Index(以下「NDI」という。)や神経学的所見等の 5 項目を組み合わせた評価項目)を達成 した被験者は、本品群 70.4%(159 例/226 例)、ACDF 群 63.2%(108 例/171 例)であった。 安全性については、有害事象等が評価され、本品との因果関係が否定できない有害事象は 34 例 61 件、8 事象であった。2 椎間治験は、頚椎椎間板変性疾患が原因で 2 椎間の外科的治療 が必要な患者 397 例(本品群 209 例、ACDF 群 188 例)を対象とした多施設共同前向き非劣 性比較対照試験であり、本申請においては 1 椎間治験に含まれていなかった高さ 5mm サイ ズの評価を行う目的で添付され、その他サイズと同等の有効性及び安全性が示された。 提出された資料について専門協議の議論を踏まえて総合的に評価した結果、本品は椎間 板ヘルニア又は骨棘に起因する頚椎の神経根症又は脊髄症で1椎間レベルの外科的治療が必 要な患者に対し、一定の有効性及び安全性が見込まれると判断した。しかしながら、本品は 国内における使用経験がないことから、国内における有効性及び安全性の評価は重要であ ると考え、製造販売承認後の使用成績調査が必要であると判断した。さらに、本品の特性を 考慮すると、適切な患者選択や手術手技等が本品の有効性及び安全性を担保する上で重要 であることから、本品の特性を理解し頚椎前方手術に習熟した医師が適応を遵守して使用 する必要があると考え、関連学会と連携して作成された適正使用指針の遵守及び講習の受 講等の必要な措置を講じることが適切と判断し、以下に示す承認条件を付すこととした。 以上、独立行政法人医薬品医療機器総合機構における審査の結果、以下の使用目的で本品 の製造販売を承認して差し支えないと判断し、医療機器・体外診断薬部会で審議されること が妥当と判断した。 使用目的 本品は、頚椎の椎間板を置換するために用いられる頚椎人工椎間板である。 適応部位 頚椎(C3~C7)の 1 椎間 適応疾患 本品の適応疾患は、画像所見(CT、MRI、X 線等)により確認された次のいずれかを伴 う 1 椎間レベルの神経根症又は脊髄症である。 1. 椎間板ヘルニア 2. 骨棘 なお、本品は最低 3 か月の保存療法が奏効しなかった患者に用いられる。ただし、進行性の 神経障害が確認された場合は、その限りではない。
4 承認条件 椎間板ヘルニア又は骨棘を主因とした頚椎の神経根症や脊髄症の治療及び頚椎前方手術 に対する十分な知識・経験を有する医師が、関連学会と協力して作成された適正使用指針を 遵守し、講習の受講により本品を用いた治療に関する技術を得る等、本品が適切に用いられ るよう、必要な措置を講じること。
5 審査報告 平成 29 年 4 月 3 日 1. 審議品目 [ 類 別 ] : 医療用品 04 整形用品 [ 一 般 的 名 称 ] : 人工椎間板
[ 販 売 名 ] : PRESTIGE LP Cervical Disc システム
[ 申 請 者 ] : メドトロニックソファモアダネック株式会社 [ 申 請 年 月 日 ] : 平成 28 年 5 月 30 日 [ 申 請 時 の 使 用 目 的 ] : 本品は、頚椎の椎間板を置換するために用いられる頚椎人工 椎間板である。 適応部位 頚椎(C3~C7)の 1 椎間 適応疾患 本品の適応疾患は、画像所見(CT、MRI、X 線等)により 確認された次のいずれかを伴う 1 椎間レベルの神経根症 又は脊髄症である。 1. 椎間板ヘルニア 2. 骨棘 なお、本品は最低 3 か月の保存療法が奏効しなかった患者に 用いられる。ただし、進行性の神経障害が確認された場合は、 その限りではない。 [ 特 記 事 項 ] : なし 2. 審議品目の概要
「PRESTIGE LP Cervical Disc システム」(以下「本品」という。)(図 1)は、椎間板ヘル ニアや骨棘等の圧迫因子の除去後に頚椎の適応椎間の椎間板を本品に置換することで、適 応椎間の可動性を維持することを目的とした頚椎人工椎間板である。一定期間、保存療法が 奏効せず、画像所見(CT、MRI、X 線等)により確認される椎間板ヘルニア又は骨棘を伴う 神経根症又は脊髄症の患者に対し、本品を用いることで可動性の維持とともに除痛が期待 される。本品は、ボール部とトラフ部から構成され、ボール部とトラフ部が組み合わさるこ とで可動性を維持し、また、それぞれレール、チタンプラズマスプレーコーティング及びグ リッドブラスト加工による粗面により椎体に固定される。ボール部とトラフ部の原材料に
6 は、摩耗の低減と、埋植後、MR 画像によるインプラント周囲の評価を可能とするため、チ タン基複合材料(Ti-6Al-4V/10wt.%TiC、以下「チタン基複合材料」という。)が採用され ている。可動域は側屈±10°、屈曲・伸展±10°、トランスレーション±2mm となっており、ね じりの制限はない。椎間板高(5mm~8mm)と椎体の奥行(12mm~18mm)の組合せにより、 14 サイズの中から適切なサイズを選択して用いる。 図 1. 本品の外観図及び埋植イメージ 図 2. 本品の可動域 3. 提出された資料の概略及び総合機構における審査の概要 本申請において、申請者が提出した資料及び独立行政法人医薬品医療機器総合機構(以下 「総合機構」という。)からの照会事項に対する申請者の回答の概略は、以下のようなもの であった。 なお、本品に対して行われた専門協議の専門委員からは、「医薬品医療機器総合機構にお ける専門協議等の実施に関する達」(平成 20 年 12 月 25 日付 20 達第 8 号)第 5 項に該当し ない旨の申し出がなされている。 チタンプラズマスプレーコーティング レール インストゥルメント の把持部 ボール部 トラフ部
7 イ.開発の経緯及び外国における使用状況等に関する資料 【開発の経緯】 <提出された資料の概略> 頚椎の椎間板は線維軟骨で構成され、椎体と椎体をつなぐ、いわゆるクッション装置の役 割を果たしており、椎間板の中心部の髄核が脱出する状態を頚椎椎間板ヘルニアと呼ぶ。ヘ ルニアが後外側方向に脱出すると神経根が圧迫され、通常は一側上肢への疼痛と感覚・運動 障害が出現する(神経根症)。ヘルニアが後方に脱出すると脊髄が圧迫され、手指巧緻運動 障害、歩行障害及び排尿障害が生じる(脊髄症)1。また、前方からの骨棘による圧迫や後 方からの椎間関節等の肥厚による圧迫は、神経根症及び脊髄症の発症原因となり、これら静 的因子の他、前屈・後屈等の動的因子も脊髄症の原因となる2。 神経根症及び脊髄症の治療法として、一般的には疼痛が主訴の場合、初期療法として、装 具療法(頚椎カラーによる固定)や牽引療法、薬物療法等の保存療法が選択され、保存療法 が無効又は効果不十分な場合、手術療法が選択される。 神経根症の手術選択としては、頚椎前方除圧固定術(ACDF)又は後方からの椎間孔拡大 術が選択され、脊髄症については後方からの脊柱管拡大術(椎弓形成術)又は ACDF が選 択される。 ACDF は、前方進入により頚椎の椎間板及び圧迫因子を取り除いた後、移植骨や脊椎ケー ジ、頚椎前方固定プレート等を用いて罹患椎間を固定する手術で、国内外において広く行わ れている術式であり、その有効性及び安全性は確立されている。しかしながら、ACDF は、 除圧後に適応椎間を固定するため、固定術が行われた椎間に隣接している椎間に代償性に 過大な力学的負荷が加わり、椎間板の変性が進行するために生じる隣接椎間障害という合 併症を伴うことがあり、再手術が必要になることもある。 そのような背景から、適応椎間の除圧は行うが固定を行わない頚椎椎間板置換術を可能 とするため、1980年代後半より、本品の原型となる頚椎人工椎間板の開発が進められ、2007 年に本品の前世代品であるPRESTIGE STの1椎間使用が初めて米国においてPMA承認を取 得した(PMA Number:P060018)。 本品は、多椎間固定を可能とするため、PRESTIGE ST を改良し、スクリュー固定から主 にレール及びチタンプラズマスプレーコーティングにより椎体に固定させるデザインとす るとともに、摩耗の低減と MR 画像による評価を可能とするため、原材料をステンレスス チールからチタン合金に炭化チタンセラミックスを分散させたチタン基複合材料(Ti-6Al-4V/10wt.%TiC)へと変更し、2014 年 7 月、IDE 試験成績を添付し、1 椎間使用の PMA 承認 を取得している(PMA Number:P090029)。また、2016 年 7 月、2 椎間使用も IDE 試験成 績を添付し、PMA 承認を取得している(PMA Number:P090029/S003)。
本品の本邦における申請にあたっては、申請時点で米国において取得していた PMA 承認 が 1 椎間使用であったことから、まずは 1 椎間使用を目的とし、本品を用いた 1 椎間レベ ルの有効性及び安全性を評価するために実施された米国 IDE 試験(IDE G040086 Pivotal 試
8
験(以下「1 椎間治験」という。)、 IDE G040086 Metal Ion 試験(以下「Metal Ion 治験」と いう。)及び IDE G040086 Continued Access 試験(以下「Continued Access 治験」という。)) が添付された。また、本邦におけるニーズを考慮し、本申請において高さ 5mm サイズ(小 サイズ)を同時に承認取得する目的で、本品の 2 椎間レベルの有効性及び安全性を評価する ために実施された米国 IDE 試験(IDE G050202 試験(以下「2 椎間治験」という。))の成績 が添付された。 【外国における使用状況】 <提出された資料の概略> 本品は、56 か国で販売実績があり、2016 年 6 月までの期間で、計 XX,X 個販売されてい る。主要な諸外国における本品の承認・許可及び販売状況は、表 1 のとおりである。 なお、外国での本品の使用において、これまでに規制当局等に報告された不具合は 8 件 (インプラントの移動 7 件、不快感 1 件)であり、医療機器本体の変更、回収が必要になっ た重篤な不具合はなかった(2016 年 6 月時点)。 表 1. 主要な外国における認可状況 国名 等 使用目的又は効果 承認取得 年月日 累積販売 概数 米国 適応患者の年齢 -骨格が成熟していること。 適応部位 -C3-C7 の 1 又は 2 椎間 適応疾患 -難治性の神経根症(上肢痛及び/又は神経障害を伴 う。頚部痛がない場合も含む) -脊髄症(椎間高位の異常による) -ただし、いずれの疾患についても、次の内、少なく とも一つの画像所見があること。 -椎間板ヘルニア -頚椎症(骨棘があること) -椎間板高の明らかな減少(隣接椎間に比べて) アプローチ -前方法 保存療法 -6 週間の保存療法に抵抗する患者が対象。ただし、 進行性の症状又は神経根/脊髄の圧迫が認められる場 合はその限りではない。 1 椎間: 2014 年 7 月 PMA 取得 (P090029) 2 椎間: 2016 年 7 月 PMA 取得 P090029/S003 XXX 個
9 その 他の 主要 な地 域 適応部位 -C2/C3 から C7/T1 適応疾患・適用する状況 -頚椎椎間板症及び不安定症 ・隣接椎間が骨癒合している患者(先天性及び医原性の 両方を含む) ・頚椎椎間板症に対する初回手術又は再手術(手術範囲 の延長) ・失敗した前方除圧術、術後の狭窄症又は術後の不安定 性に対する再手術 -偽関節又は失敗した椎体間固定術に対する再手術 EU: 2006 年 6 月 CE マーク取得 オーストラリ ア: 2004 年 4 月 中国: 2006 年 1 月 韓国: 2006 年 11 月 XXXX 個 (英、仏、 独 の 合 計) XX 個 XXX 個 XXX 個 ・その他の国における累積販売数:XXXX 個 ・XX 個以上の販売実績があるその他の国:イタリア、インド、オーストリア、カナダ、サウ ジアラビア、シンガポール、スイス、スペイン、台湾、チェコ、デンマーク、ニュージーラン ド、ブラジル、ベルギー、ポーランド、ポルトガル、南アフリカ、メキシコ。 【機器に関する情報】 <提出された資料の概略> 本品の性能に関する規格として、機械的安全性(亜脱臼試験、沈み込み試験、押し出し試 験、静的圧縮試験、圧縮疲労試験、静的圧縮剪断試験、圧縮剪断疲労試験、摩耗試験、チタ ンプラズマスプレーコーティングの静的引張試験、チタンプラズマスプレーコーティング の静的剪断試験、チタンプラズマスプレーコーティングの剪断疲労試験、チタンプラズマス プレーコーティングの摩耗試験及びチタンプラズマスプレーコーティングの厚さ測定)及 び生物学的安全性が設定され、各設定が妥当であることを説明する資料が提出された。 <総合機構における審査の概要> 総合機構は、後述するロ. 設計及び開発に関する資料【機械的安全性】及び【生物学的安 全性】における審査の結果、本品の規格設定に特段の問題はないと判断した。 ロ.設計及び開発に関する資料 【物理的、化学的特性】 <提出された資料の概略> 物理的、化学的特性について、本品のボール及びトラフ部の原材料であるチタン基複合 材料はチタン合金に炭化チタンセラミックスを分散させた新規原材料であるが、原材料規 格中に、物理的、化学的特性が規定されており、また、チタンプラズマスプレーコーティ ングの原材料である純チタンは既承認前例があるとして、試験が省略された。 <総合機構における審査の概要> 総合機構は新規原材料について、成分の種類及び配合比、気孔率、炭化チタンの粒径、機
10 械的性質等、物理的、化学的特性に関する情報が原材料規格として適切に規定されているこ とに加え、機械的安全性試験、生物学的安全性試験等の成績も踏まえると、物理的、化学的 特性に関する資料を省略することに対し、特段の問題はないと判断した。 【電気的安全性及び電磁両立性】 <提出された資料の概略> 本品は電気を使用しないため、電気的安全性及び電磁両立性に関する資料は省略された。 <総合機構における審査の概要> 総合機構は、電気的安全性及び電磁両立性に関する資料を省略することに対し、特段の問 題はないと判断した。 【生物学的安全性】 <提出された資料の概略> 生物学的安全性について、チタンプラズマスプレーコーティングを含む本品全体に関す る評価として、本品を試験検体に用いた細胞毒性試験及び抽出試験、並びに新規原材料に対 する評価として本品と同一のチタン基複合材料からなる検体を試験に用いた感作性試験、 刺激性試験(皮内反応)、急性全身毒性試験、遺伝毒性試験(復帰突然変異試験及び染色体 異常試験)及び筋肉内埋植試験の成績が提出され、局所及び全身に対する毒性がないことが 示された。また本品の使用に際し、発生する可能性のある摩耗粉の生物学的安全性について、 ウサギ腰椎硬膜外腔への微粒子投与試験の成績が提出され、投与に関連する神経毒性、全身 毒性及び局所的影響の兆候は認められず、生体適合性に特段の懸念がないことが示された。 <総合機構における審査の概要> 総合機構は、抽出試験の一部において本品特異的に検出された物質の特定及び毒性リス ク評価を求めた。申請者は、各試験において検出された物質を特定し、当該物質の累積の毒 性リスク評価を行い、患者への健康リスクに特段の懸念はないと説明した。 総合機構は、提出された各試験成績及び上記説明に基づき、審査した結果、本品の生物学 的安全性に特段の問題はないと判断した。 【放射線に関する安全性】 <提出された資料の概略> 本品は、放射線を放出する機器ではないとして、放射線に関する安全性に関する資料は省 略された。
11 <総合機構における審査の概要> 総合機構は、放射線に関する安全性に関する資料を省略することに対し、特段の問題はな いと判断した。 【機械的安全性】 <提出された資料の概略> 本品の機械的安全性について、亜脱臼試験、沈み込み試験、押し出し試験、静的圧縮試験、 圧縮疲労試験、静的圧縮剪断試験、圧縮剪断疲労試験及び摩耗試験が実施され、チタンプラ ズマスプレーコーティングに関する評価として、静的引張試験、静的剪断試験、剪断疲労試 験及び摩耗試験、並びにチタンプラズマスプレーコーティングの厚さ、気孔率及び気孔径の 測定が実施された。いずれの試験においても設定された規格値を満たすことが示された。ま た、チタンプラズマスプレーコーティングの骨に対する固定性能については、本品を用いた 動物試験は省略されたが、同等のチタンプラズマスプレーコーティングを有する本邦既承 認品に関する資料が提出され、生体内における骨とコーティングを介したインプラントの 結合能が説明された。 <総合機構における審査の概要> 総合機構は、本品の摺動面はメタルオンメタル構造であるが、提出された摩耗試験の成績 から、海外既承認品である PRESTIGE ST と比較して摩耗量が少ないことが示されており、 ウサギの脊椎(硬膜外)に投与した微粒子投与試験の結果も踏まえると、本品の摩耗粉につ いて臨床上許容できないリスクはないと判断した。また、その他の機械的安全性試験の成績 及びチタンプラズマスプレーコーティングに関する資料を審査した結果、本品の機械的安 全性に特段の問題はないと判断した。 【安定性及び耐久性】 <提出された資料の概略> 本品の原材料については、特定の貯蔵方法によらなくとも品質を確保でき、経時的にその 安定性及び耐久性が低下しないと考えられるとして、本品の安定性及び耐久性に関する資 料は省略された。 <総合機構における審査の概要> 総合機構は、チタン基複合材料を構成するチタン合金及び純チタンは広く医療用材料と して使用されており、それらの安定性及び耐久性は既知であること、また、炭化チタンの特 性や本品の臨床使用実績を踏まえても、本品の安定性及び耐久性に臨床上許容できないリ スクはないと考えられたことから、安定性及び耐久性に関する資料を省略することに対し、 特段の問題はないと判断した。
12 【性能】 <提出された資料の概略> 本品の性能は、機械的安全性試験及び臨床試験の成績から説明可能として、性能に関する 資料は省略された。 <総合機構における審査の概要> 総合機構は、性能に関する資料を省略することに対し、特段の問題はないと判断した。 【使用方法】 <提出された資料の概略> 臨床試験の試験成績により説明され、使用方法を裏付ける試験に関する資料の提出が省 略された。 <総合機構における審査の概要> 総合機構は、本品の使用方法を裏付ける試験に関する資料については、臨床試験の試験成 績により確認可能であるため、特段の問題はないと判断した。 【MR 装置との相互作用】 <提出された資料の概略> 本品と MR 装置との相互作用について、磁気誘導性偏位力測定試験、磁気誘導性トルク 測定試験、アーチファクト測定試験及び発熱測定試験の成績が提出され、1.5T 又は 3.0T の MR 環境にある患者に対して重大なリスクや危険を与える可能性は低いことが示された。ま た、アーチファクトについては、撮像部位が本品の埋植部位付近又は同一部位にある場合、 画質が損なわれる可能性があるが、測定されたシグナルボイドは本品のサイズ及び形状に 対して低いレベルであることが示された。 <総合機構における審査の概要> 総合機構は、提出された資料を審査した結果、特定の条件下での MR 装置の使用は可能 であると判断した。また、添付文書上に適切に情報提供がなされており、特段の問題はない と判断した。 ハ.法第 41 条第 3 項に規定する基準への適合性に関する資料 <提出された資料の概略> 医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律第 41 条第 3 項に基 づき厚生労働大臣が定める医療機器の基準(平成 17 年厚生労働省告示第 122 号。以下「基 本要件」という。)への適合性を宣言する適合宣言書が提出された。
13 <総合機構における審査の概要> 総合機構は、本品に関する基本要件への適合性について審査した結果、特段の問題はない と判断した。 ニ.リスクマネジメントに関する資料 <提出された資料の概略>
本品のリスクマネジメントに関する資料については、ISO 14971:2012(Medical Devices – Application of risk management to medical devices)及び JIS T 14971:2012(医療機器-リスクマ ネジメントの医療機器への適用)を参照し実施したリスクマネジメントとその実施体制及 び実施状況の概要を示す資料が提出された。 <総合機構における審査の概要> 総合機構は、リスクマネジメントに関する資料について審査した結果、特段の問題はない と判断した。 ホ.製造方法に関する資料 <提出された資料の概略> 本品の製造方法に関する資料として、製造工程及び製造施設に関する資料、品質管理に関 する資料並びに滅菌方法に関する資料が提出された。 <総合機構における審査の概要> 総合機構は、製造方法に関する資料を審査した結果、特段の問題はないと判断した。 ヘ.臨床試験の試験成績に関する資料又はこれに代替するものとして厚生労働大臣が認め る資料 添付資料として、本品の 1 椎間使用における有効性及び安全性を評価するために実施さ れた IDE G040086 Pivotal 試験(1 椎間治験)、血中の Metal Ion 濃度等を評価するために追加 実施された IDE G040086 Metal Ion 試験(Metal Ion 治験)、患者救済目的で 1 椎間治験に追加 して実施された IDE G040086 Continued Access 試験(Continued Access 治験)、ヒストリカル コントロールとして利用した Atlantis(前方固定用プレート:本邦既承認品である「滅菌済 ATLANTIS スパイナルシステム、承認番号:21600BZY00581000、承認年月日:平成 16 年 10 月 28 日」又は「滅菌済 ATLANTIS VISION スパイナルシステム、承認番号:21800BZY10031000、 承認年月日:平成 18 年 2 月 8 日」i。ただし治験では未滅菌品を用時滅菌して使用。)の 1 椎 間の成績を含む IDE G010188 試験、及び本品の 2 椎間使用における有効性及び安全性を評 i 医療機器・体外診断薬部会終了後に追記(又は「滅菌済 ATLANTIS VISION スパイナルシステム、承認番号: 21800BZY10031000、承認年月日:平成 18 年 2 月 8 日」)
14 価するために実施された IDE G050202 試験(2 椎間治験)の成績が提出された。 表 2. 米国で実施された臨床試験一覧 試験名 被験機器・ 対照機器 症例数 1 椎間の症候性頚椎椎間板変性疾患を対象とした Artificial disc LP の前向き多施設共同比較試験 (IDE G040086 Pivotal 試験(1 椎間治験)) 本品 280 1 椎間の症候性頚椎椎間板変性疾患を対象とした Artificial disc LP の前向き多施設共同比較試験
(IDE G040086 Metal Ion 試験(Metal Ion 治験))
本品 30
1 椎間の症候性頚椎椎間板変性疾患を対象とした Artificial disc LP の前向き多施設共同比較試験
(IDE G040086 Continued Access 試験(Continued Access 治験))
本品 24 1 椎間の症候性頚椎椎間板変性疾患を対象とした Artificial disc の 前向き多施設共同比較試験 (IDE G010188 試験) PRESTIGE ST 276 Atlantis 265 2 椎間の症候性頚椎椎間板変性疾患を対象とした Artificial disc LP の前向き多施設共同比較試験 (IDE G050202 試験(2 椎間治験)) 本品 209 Atlantis 188 <提出された資料の概略> (1)IDE G040086 Pivotal 試験(1 椎間治験)(2005 年 1 月~2008 年 1 月) 1 椎間治験は、頚椎椎間板変性疾患が原因で 1 椎間の外科的治療が必要な患者に被験機器 を埋植し、本品による治療群(以下、「本品群」という。)の術後 24 か月時の Overall Success rate(有効性及び安全性を組み合わせた全般的な成功率)が既存療法である ACDF(以下 「ACDF 群」という。)との比較において劣らないこと(非劣性)を検証する目的で実施さ れた多施設共同前向き非劣性既存対照比較試験である。なお、本品群の ACDF 群に対する 非劣性が認められた場合には、副次的に優越性の検証も行う計画とされていた。 対照群には、本品の旧モデル製品である PRESTIGE ST の承認取得のために実施した多施 設共同前向き無作為割付非劣性比較試験(IDE G010188 試験、2007 年 7 月 16 日 PMA 承認 取得、PMA 番号:PMA P060018)における比較対照群(ACDF 群)がヒストリカルに用い られた。 なお、本治験及び G010188 における対象患者(選択基準及び除外基準)、観察項目(主要 評価項目、副次的評価項目及び安全性評価項目)及び観察時期は同一であった。対照群は、 頚椎椎間板変性疾患患者の 1 椎間の標準的手術手法である ACDF において評価され、ACDF には、同種移植骨及び前方固定用プレート Atlantis を用いる術式が採用された。 主な患者選択基準、除外基準の概要は表 3 のとおりである。
15 表 3. 主な患者選択基準・除外基準及び目標症例数の設定根拠 選択基準 以下の選択基準の全てに合致する患者: 1. 下記に定義された頚椎変性椎間板疾患: - 画像診断(CT、MRI、X 線など)にて椎間板ヘルニアもしくは骨棘形成が 確認され、いずれか、あるいは両方の圧迫因子が原因で、症候性で難治 性の神経根症あるいは脊髄症と診断された患者 - 病歴にこれら症状(疼痛、機能障害、神経障害等のいずれか)が記載され ている患者 2. 1 椎間に外科的治療を必要とする患者 3. C3/C4 から C6/C7 の椎間に疾患のある患者 4. 約6週間の保存療法に奏効しない患者、又は保存療法にも関わらず、進行性 の症状又は神経根及び脊髄の圧迫症状を呈した患者 5. 過去に適応椎間に外科的治療がされていないこと、又は術後に適応椎間や 隣接椎間で外科的治療(段階的なものを含む)が予定されていない患者 6. 手術時に 18 歳以上である患者
7. 術前の NDI (Neck Disability Index(以下「NDI」という。))スコアが 30 以上である患者
8. 術前の Neck and Arm Pain Questionnaire(頚部痛及び上肢痛に関する質問 表)での頚部痛のスコアが 20 以上である患者 9. 妊娠可能な女性については手術時に妊娠していない患者 10. 治験実施計画書に従う意思があり、同意文書に署名をする意思のある患者 除外基準 以下の除外基準のいずれかに抵触する患者 1. 適応椎間で外科的治療を要する症候性頚椎椎間板疾患以外の脊椎疾患のあ る患者 2. X 線撮影(伸展、屈曲)により頚椎不安定性と診断又は過去に診断された患 者 a) 矢状面で 3.5 mm を超えるトランスレーション b) 矢状面で 20°を超える椎間可動域 3. 外科的治療を要する椎間が 2 椎間以上の患者 4. 適応椎間の隣接椎間に固定術が施されている患者 5. 適応椎間の後方の椎間関節に重度の病変がある患者 6. 過去に適応椎間に外科的治療が施されたことがある患者 7. 過去に骨軟化症又は骨減少症と診断されたことがある患者 8. 骨粗鬆症の疑いのある以下の条件に当てはまる患者(下記のいずれかのリス ク要因に合致する患者は、治験への適格性を検証するため DEXA 法による 骨密度測定を実施すること) a) 黒人以外の閉経後の 60 歳より上の女性で体重が 140 ポンド(63.5 kg) 以下の患者 b) 閉経後の女性で非外傷性の股関節、脊椎、又は手関節を骨折している 患者 c) 70 歳より上の男性 d) 60 歳より上の男性で、非外傷性の股関節又は脊椎を骨折している患者 BMD の T スコアが-3.5 以下(‐3.6、-3.7 など)、又は-2.5 以下(-2.6、 -2.7 など)で脊椎圧迫骨折のある患者は治験から除外する 9. 脊椎に転移性腫瘍のある患者 10. 顕在性又は活動性の、局所又は全身性の細菌性感染症の患者 11. 重度のインスリン依存性糖尿病の患者 12. 慢性又は急性の腎不全、又は腎臓疾患の既往歴のある患者 13. 手術時に口腔内体温が 101℉(38.3℃)より高い患者
16 14. ステンレススチール、チタン、又はチタン合金にアレルギーがあると診断さ れたことのある患者 15. 精神的無能力の患者(疑いのある場合は、精神専門医の診察を受けること) 16. 囚人 17. 妊娠している患者 18. アルコール依存又は薬物乱用の治療を受けている患者 19. 手術予定日の 2 週間以内に、骨代謝の阻害が懸念される薬剤(ステロイド、 メトトレキサートなど)の投与を受ける患者。ただし、周術期に通常使用す る抗炎症剤を除く 20. 骨形成への影響が知られている内分泌又は代謝疾患の既往歴(パジェット 病、腎性骨ジストロフィー、エーラスダンロス症候群、骨形成不全症など) のある患者 21. ステロイドなど、術後に機器の安定性を妨げる薬剤の投与が必要な患者。た だし、血栓予防を目的とした低用量アスピリン、周術期に通常使用する抗炎 症剤を除く 22. 手術前の 28 日以内に他の試験的治療に参加する患者、又は被験機器の埋植 後 16 週間以内に他の治験への参加が予定されている患者 目標とす る被験者 数とその 設定根拠 目標症例数は 275±5 例とした。 [目標とする被験者数設定根拠] 目標とする被験者数は Overall Success の非劣性仮説に基づいて設定した。 過去の椎体間固定機器の治験経験から、本品群と ACDF 群の Overall Success rate を 75%と仮定した。非劣性マージン(Δ)を 10%と設定し、Blackwelder の式か ら両群の非劣性を証明するために必要な症例数は各群 232 例と計算された。しか し、追跡調査が実施できない症例が全体の 10~15%と仮定して、目標とする被験 者数を 275 ± 5 例と設定した。 同意を取得した 299 例のうち、使用機器の使用拒否等により手術が実施されなかった 19 例を除外した 280 例が本治験に登録され、本品を埋植された 280 例が主要解析対象集団 (Primary Dataset)とされ、主要な治験実施計画書からの逸脱が判明した 4 例を除外した 276 例が治験実施計画書に適合した解析対象集団(Per-Protocol Dataset)とされた。また、ACDF 群については、同意を取得した 313 例のうち、同様に手術が実施されなかった 48 例を除外 した 265 例が本治験に登録され、Atlantis を埋植された 265 例が主要解析対象集団(Primary Dataset)とされ、主要な治験実施計画書からの逸脱が判明した 16 例を除外した 249 例が治 験実施計画書に適合した解析対象集団(Per-Protocol Dataset)とされた。Per-Protocol Dataset による解析は、主要評価項目(Overall Success)及びその構成要素のみとし、その統計学的 結果は、副次的解析結果とされた。
17 表 4. 実施症例の人口統計学データ 項目 PRESTIGE LP 群 (N=280) ACDF 群 (N=265) p 値* PRESTIGE LP 群 対 ACDF 群 年齢(歳) N 280 265 0.369 平均値 44.5 43.9 SD 8.8 8.8 最小値 23.0 22.0 中央値 44.0 44.0 最大値 78.0 73.0 身長(cm) N 280 265 0.622 平均値 172.0 171.5 SD 10.4 10.7 最小値 152.4 147.3 中央値 170.2 170.2 最大値 195.6 203.2 体重(kg) N 280 264** 0.567 平均値 84.8 83.8 SD 20.4 18.8 最小値 45.4 44.5 中央値 81.6 82.1 最大値 154.2 148.8 性別(n(%)) 男性 129(46.1) 122(46.0) 1.000 女性 151(53.9) 143(54.0) 人種(n(%)) 白人 271(96.8) 243(91.7) 0.043 黒人 7(2.5) 13(4.9) アジア人 0(0.0) 2(0.8) ヒスパニック 1(0.4) 6(2.3) その他 1(0.4) 1(0.4) 喫煙(n(%)) あり 74(26.4) 92(34.7) 0.041 なし 206(73.6) 173(65.3) *連続変数は ANOVA、カテゴリカル変数はフィッシャーの正確確率検定により算出 ** 1 例欠測 によ る 表 5. 術前の医学的状態及び服薬状況 項目 PRESTIGE LP 群 n(%) (N=280) ACDF 群 n(%) (N=265) p 値* PRESTIGE LP 群 対 ACDF 群 症状が出てか ら手術の計画 に至るまでの 時間 6 週未満 22(7.9) 15(5.7) 0.494 6 週から 6 か月 85(30.4) 89(33.6) 6 か月より長い 173(61.8) 161(60.8) 過去に受けた 頚部手術の数 0 277(98.9) 263(99.2) 0.489 1 3(1.1) 1(0.4) 2 0(0.0) 1(0.4) 3 0(0.0) 0(0.0) 4 0(0.0) 0(0.0) 5 以上 0(0.0) 0(0.0)
18 非麻薬製剤** あり 208(74.3) 187(71.1) 0.441 なし 72(25.7) 76(28.9) 弱麻薬製剤** あり 133(47.7) 127(48.3) 0.931 なし 146(52.3) 136(51.7) 強麻薬製剤** あり 62(22.2) 58(22.0) 1.000 なし 217(77.8) 206(78.0) 筋弛緩剤** あり 100(35.8) 114(43.2) 0.095 なし 179(64.2) 150(56.8) *フィッシャーの正確確率検定 、** 1 例又は 2 例 欠測あり 表 6. 被験機器のサイズ及び適応椎間 高さ×奥行き PRESTIGE LP 群 C3/C4 C4/C5 C5/C6 C6/C7 合計 6 mm x 12 mm 1 4 15 11 31(11.1%) 6 mm x 14 mm 1 10 65 37 113(40.4%) 6 mm x 16 mm 2 1 35 23 61(21.8%) 6 mm x 18 mm 0 0 0 0 0(0.0%) 7 mm x 12 mm 0 0 1 1 2(0.7%) 7 mm x 14 mm 0 2 5 8 15(5.4%) 7 mm x 16 mm 0 4 16 9 29(10.4%) 7 mm x 18 mm 0 0 9 11 20(7.1%) 8 mm x 14 mm 0 0 0 3 3(1.1%) 8 mm x 16 mm 0 0 1 4 5(1.8%) 8 mm x 18 mm 0 0 0 1 1(0.4%) 合計 (%) 4(1.4%) 21 (7.5%) 147(52.5%) 108 (38.6%) 280(100.0%) 1)有効性評価 有効性の評価項目として、術後 24 か月時の Overall Success(以下の①~⑤の 5 項目を組 み合わせた評価項目)が主要評価項目とされ、以下の条件を全て満たした症例が「Success 症 例」と定義された。 ① 術後の NDI スコア(表 7)が術前値から 15 ポイント以上改善 ② 術前と比較し術後の神経学的所見(運動、知覚、反射)の維持又は改善
③ Functional Spinal Unit(FSU)Heightの維持(術後 3 か月時以降の FSU Height が術後
6 週時と比較して-2mm 以上高い) ④ 「機器と関連性あり」又は「機器及び手術手技との関連性あり」と判定された重篤 な有害事象の発現がない ⑤ Failure として分類される外科的再処置(リビジョン、抜去(被験者の希望を除く)、 補助的固定)を実施していない なお、主要評価項目のうち、②神経学的所見、④「機器と関連性あり」又は「機器及び手
Functional Spinal Unit(FSU)Height:隣接する 2 個の椎骨とそれらを連結する椎間板等からなるモーシ ョンセグメント
19 術手技との関連性あり」と判定された重篤な有害事象及び⑤Failure として分類される外科 的再処置については、2)安全性評価において後述する。 本治験の評価は非劣性マージンを 10%と設定し、ベイズ流解析を用いて、ACDF 群に対 する非劣性が評価され、事後確率 P(p0-p1<d(delta) | Data;p0:ACDF 群の 95%最高事 後密度区間、p1:本品群の 95%最高事後密度区間)が 0.95 を上回れば非劣性があるものと 評価された。また、同様に ACDF 群に対する優越性が評価され、事後確率 P(p0-p1<0 | Data) が 0.95 を上回れば優越性があるものと設定された。 表 7. NDI スコア
Section 1 - Pain Intensity
0 I have no pain at the moment. 1 The pain is very mild at the moment. 2 The pain is moderate at the moment. 3 The pain is fairly severe at the moment. 4 The pain is very severe at the moment.
5 The pain is the worst imaginable at the moment.
Section 2 - Personal Care (washing, dressing, etc.)
0 I can look after myself normally without causing extra pain.
1 I can look after myself normally, but it causes extra pain.
2 It is painful to look after myself and I am slow and careful.
3 I need some help, but manage most of my personal care.
4 I need help every day in most aspects of self care.
5 I do not get dressed, wash with difficulty, and stay in bed.
Section 3 - Lifting
0 I can lift heavy weights without extra pain. 1 I can lift heavy weights, but it causes extra pain. 2 Pain prevents me from lifting heavy weights off
the floor, but I can manage if
3 Pain prevents me from lifting heavy weights, but I can manage light to
4 I can lift only very light weights. 5 I cannot lift or carry anything at all.
Section 4 - Reading
0 I can read as much as I want to with no pain in my neck.
1 I can read as much as I want to with slight pain in my neck.
2 I can read as much as I want with moderate pain in my neck.
3 I can't read as much as I want because of moderate pain in my neck.
4 I can hardly read at all because of severe pain in my neck.
Section 6 - Concentration
0 I can concentrate fully when I want to with no difficulty.
1 I can concentrate fully when I want to with slight difficulty.
2 I have a fair degree of difficulty in concentrating when I want to.
3 I have a lot of difficulty in concentrating when I want to.
4 I have a great deal of difficulty in concentrating when I want to. 5 I cannot concentrate at all.
Section 7 - Work
0 I can do as much work as I want to. 1 I can only do my usual work, but no more. 2 I can do most of my usual work, but no
more.
3 I cannot do my usual work. 4 I can hardly do any work at all. 5 I can't do any work at all.
Section 8 - Driving
0 I can drive my car without any neck pain. 1 I can drive my car as long as I want with
slight pain in my neck.
2 I can drive my car as long as I want with moderate pain in my neck.
3 I can't drive my car as long as I want because of moderate pain in my neck. 4 I can hardly drive at all because of severe
pain in my neck.
5 I can't drive my car at all.
Section 9 - Sleeping
0 I have no trouble sleeping.
1 My sleep is slightly disturbed (less than 1 hour sleepless).
2 My sleep is mildly disturbed (1-2 hours sleepless).
3 My sleep is moderately disturbed (2-3 hours sleepless).
4 My sleep is greatly disturbed (3-5 hours sleepless).
20 5 I cannot read at all.
Section 5 - Headaches
0 I have no headaches at all.
1 I have slight headaches which come infrequently.
2 I have moderate headaches which come infrequently.
3 I have moderate headaches which come frequently.
4 I have severe headaches which come frequently. 5 I have headaches almost all the time.
5 My sleep is completely disturbed (5-7 hours sleepless).
Section 10 - Recreation
0 I am able to engage in all my recreation activities with no pain at all.
1 I am able to engage in all my recreation activities, with some pain in my neck. 2 I am able to engage in most, but not all of
my usual recreation activities because of pain in my neck.
3 I am able to engage in a few of my usual recreation activities because of pain in my neck.
4 I can hardly do any recreation activities because of pain in my neck.
5 I can't do any recreation activities at all.
※個々の質問に対する回答は 0~5(各項目の左に記載されているポイント)の 6 段階で判定、合
計の NDI スコアは以下の計算式で算出。
NDI スコア(%)=(個々の回答のスコアの合計) /(個々の質問の最大スコアの合計:全て 回答した場合 50)×100
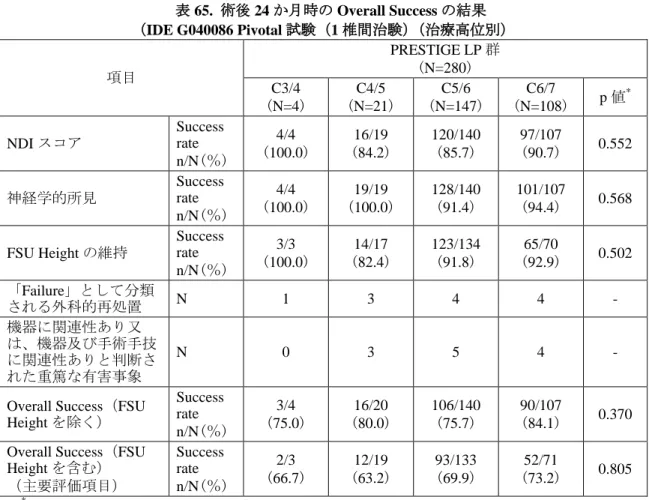
<Overall Success>
本品群の術後 24 か月時の Overall Success(FSU Height を含む)の Success rate は 70.4% (159 例/226 例)、ACDF 群は 63.2%(108 例/171 例)であった(表 8)。また、FSU Height の 欠測率が高く判定不能症例が多くなったことを踏まえ、申請者により、その他の指標による 評価として Overall Success(FSU Height を除く)についての解析も実施された。その結果、 本品群の術後 24 か月時の Overall Success rate は 79.3%(215 例/271 例)、ACDF 群は 66.8% (147 例/220 例)であった。 表 8. 術後 24 か月時の Overall Success の結果 項目 PRESTIGE LP 群 (N=280) ACDF 群 (N=265) NDI スコア Success rate
n/N(%) 237/270 (87.8) 177/219 (80.8) 神経学的所見 Success rate n/N(%) 252/270 (93.3) 184/220 (83.6) FSU Height の維持 Success rate
n/N(%) 205/224 (91.5) 156/164 (95.1) 「Failure」として分類される外科的 再処置 N 12 12 機器に関連性あり又は、機器及び手 術手技に関連性ありと判断された重 篤な有害事象 N 12 10
Overall Success(FSU Height を除く) Success rate n/N(%)
215/271 (79.3)
147/220 (66.8) Overall Success(FSU Height を含む)
(主要評価項目) Success rate n/N(%) 159/226 (70.4) 108/171 (63.2)
21
また、両群とも、いずれの Overall Success rate も術後 24 か月までの術後観察期間 中を通じ安定していたことが示された(表 9 及び表 10)。
表 9. Overall Success rate(FSU Height を含む)の推移
治療群 術 後 (n/N(%)) 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 187/238 (78.6) 189/233 (81.1) 187/234 (79.9) 159/226 (70.4) ACDF 群 (N=265) 113/181 (62.4) 119/174 (68.4) 110/173 (63.6) 108/171 (63.2)
表 10. Overall Success rate(FSU Height を除く)の推移
治療群 術 後 (n/N(%)) 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 223/277 (80.5) 224/271 (82.7) 227/274 (82.8) 215/271 (79.3) ACDF 群 (N=265) 154/239 (64.4) 158/224 (70.5) 150/223 (67.3) 147/220 (66.8)
ベイズ流解析を実施した結果、Overall Success(FSU Height を含む)は、FSU Height の欠 測により判定可能な症例が減少したものの、術後 24 か月時における本品群の ACDF 群に対 する非劣性の事後確率は 99.5%であったことから、本品群の ACDF 群に対する非劣性が認 められた。また、Overall Success(FSU Height を除く)は、術後 24 か月時における本品群の ACDF 群に対する非劣性の事後確率はほぼ 100%であったことから、本品群の ACDF 群に対 する非劣性が認められ、優越性の事後確率は 99.5%であったことから、本品群の ACDF 群 に対する優越性も認められた(表 11)。
表 11. 術後 24 か月時の Overall Success rate のベイズ流統計解析結果
主要評価 項目 術後 24 か月時の Success rate (n/N(%)) 術後 24 か月時の 非劣性の事後確率 (Δ=0.10) 術後 24 か月時の 95%最高事後密度区間 PRESTIGE LP 群 ACDF 群 非劣性 優越性 PRESTIGE LP 群 ACDF 群 Overall Success (FSU Height を 除く) 215/271 (79.3) 147/220 (66.8) ~ 100.0% 99.5% 78.9% (74.1%-84.0%) 67.8% (61.2%-74.0%) Overall Success (FSU Height を 含む) 159/226 (70.4) 108/171 (63.2) 99.5% 73.6% 68.9% (62.7%-75.1%) 65.7% (58.3%-73.4%)
22
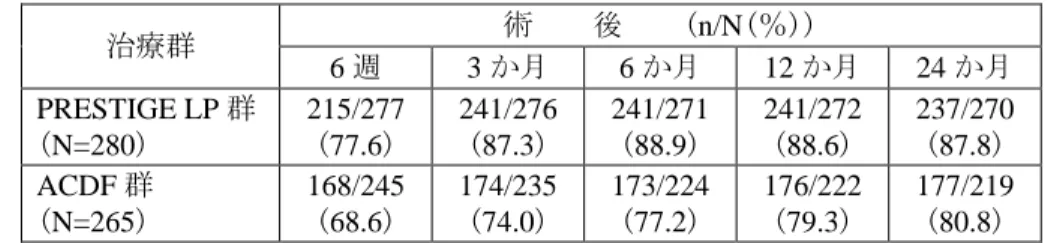
以上の結果から、いずれの解析方法でも非劣性が示され、Overall Success(FSU Height を 除く)においては優越性も示されたことから、申請者は、本品を用いる治療法は頚椎変性疾 患に対する既存の標準治療である ACDF と同様に安全かつ有効であることが示されたとし ている。 <NDI スコア> 術前後の日常生活における頚部疼痛の影響について、NDI スコアを用いて評価し た。術後の NDI スコアが術前値から 15 ポイント以上改善した症例が Success 例と 定義され、Success Rate は全ての術後観察期で本品群が ACDF 群を上回っていた。
表 12. NDI スコアの評価における Success rate の推移
治療群 術 後 (n/N(%)) 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 215/277 (77.6) 241/276 (87.3) 241/271 (88.9) 241/272 (88.6) 237/270 (87.8) ACDF 群 (N=265) 168/245 (68.6) 174/235 (74.0) 173/224 (77.2) 176/222 (79.3) 177/219 (80.8) 表 13. NDI スコアの推移 治療群 項目 術前 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 280 277 276 271 272 270 平均値 55.5 22.8 17.1 15.8 14.9 15.6 SD 14.7 17.9 17.8 18.3 18.0 18.3 最小値 30.0 0.0 0.0 0.0 0.0 0.0 中央値 54.0 20.0 12.0 10.0 8.0 8.0 最大値 98.0 84.0 86.0 88.0 86.0 82.0 ACDF 群 (N=265) 例数 264 245 236 225 223 220 平均値 56.4 32.1 26.8 24.5 23.4 22.4 SD 15.9 18.4 20.9 20.9 21.3 21.5 最小値 26.0 0.0 0.0 0.0 0.0 0.0 中央値 58.0 32.0 24.0 20.0 18.0 16.0 最大値 100.0 72.0 88.0 93.3 84.0 88.0 また、NDI スコアの平均は本品群、ACDF 群ともに術後の全ての観察期間におい て術前に比べて改善しており、これらの改善は統計学的に有意であった(p<0.001: paired t-test)。ベイズ流統計解析結果では、術後 24 か月時点の ACDF 群に対する 本品群の非劣性の事後確率はほぼ 100%であり、統計学的に非劣性であることが示 された。
23 <FSU Height>
FSU Height の維持又はインプラントの沈み込みの有無を評価するため、FSU Height が 測定され、術後 3 か月時以降の FSU Height が術後 6 週時と比較して-2mm 以上高い症例 が Success 例と定義された。適応椎間と解剖学的な関係で治療高位の画像が撮像できなか った、あるいは画像の悪さが原因で FSU Height が評価できなかった症例があり、術後 24 か月時における評価可能症例数は本品群 224 例/280 例、ACDF 群 164 例/265 例であった。 術後 24 か月時の FSU Height の Success rate は本品群 91.5%(205 例/224 例)、ACDF 群 95.1%(156 例/164 例)であり、ベイズ流統計解析の結果、本品群の ACDF 群に対する非 劣性の事後確率は 99.2%であり、統計学的な非劣性が示された。
表 14. FSU Height の Success rate
治療群 術 後 (n/N(%)) 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 229/235 (97.4) 227/230 (98.7) 225/233 (96.6) 205/224 (91.5) ACDF 群 (N=265) 182/182 (100.0) 174/175 (99.4) 164/172 (95.3) 156/164 (95.1) 有効性の副次評価項目として、X 線画像評価(motion の維持)、全般的健康状態(SF-36)、 疼痛評価(頚部痛、上肢痛)、被験者の満足度、治療結果に対する被験者の評価、歩行評価 (Nurick 分類3)、椎間孔圧迫テストが設定された。 <X 線画像評価(motion の維持)> 適応椎間の motion(可動性)の維持に関し、X 線画像評価を用いて、屈曲伸展の椎間可動 域、骨梁形成、トランスレーション(並進可動域)、左右側屈が評価された。Success の定義 は下記のとおりとされ、評価は二名の独立した放射線科医により測定され、画像所見に相違 があった場合には三番目の放射線科医により判定された。 X 線機能撮影(屈曲、伸展)画像を用いて椎間可動域を測定した結果 、本品群の平 均椎間可動域は術前 5.67°、術後 24 か月時 7.51°であり、術後の全観察期間を通じて 約 7°から 8°の範囲で維持されていた(表 15)。術後 24か月時の Success rate は 67.8% であった(表 16)。 Success の定義 (PRESTIGE LP 群) a. 4°< 屈曲伸展の椎間可動域 < 20° b. 上下椎体間で繋がった骨梁が形成されていないこと (ACDF 群) a. 屈曲伸展の椎間可動域≦ 4° b. 繋がった骨梁が形成されていること。少なくとも側方又は前後方及び/又 はインプラントを取り囲む自家骨周りで、上下椎体の連続した骨の癒 合が認められること c. 上位又は下位椎体表面の 50%以上を覆う透過性がないこと
24 表 15. 椎間可動域(屈曲・進展)の推移 治療群 項目 術前 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 260 265 264 258 266 264 平均値 5.67° 6.88° 7.51° 7.47° 7.85° 7.51° SD 3.69° 3.79° 4.05° 4.46° 4.32° 4.87° 最小値 0.27° 0.45° 0.10° 0.23° 0.34° 0.19° 中央値 4.79° 6.44° 7.44° 7.18° 7.27° 7.27° 最大値 18.10° 21.27° 19.45° 21.03° 19.75° 26.43° ACDF 群 (N=265) 例数 220 162 174 164 161 161 平均値 7.87° 0.53° 0.33° 0.31° 0.33° 0.35° SD 4.32° 1.94° 0.34° 0.23° 0.30° 0.33° 最小値 0.74° 0.04° 0.01° 0.01° 0.02° 0.02° 中央値 7.29° 0.29° 0.28° 0.27° 0.28° 0.29° 最大値 21.34° 23.10° 2.71° 2.36° 3.09° 3.42°
表 16. X 線画像評価における Success rate の推移(PRESTIGE LP 群)
項目 Success 症例数及び Success rate (%)
6 週 3 か月 6 か月 12 か月 24 か月 4°<可動域<20° 197 201 189 204 181 骨梁なし 278 274 268 269 253 Success/Failure 197/67 201/56 189/67 203/61 179/85 Success rate 74.6% 78.2% 73.8% 76.9% 67.8% 椎間のトランスレーション(並進可動域) は、側面伸屈の X 線画像により評価さ れ、本品群の全ての術後評価時点において平均値は 0.90 から 1.03 mm の間であった (表 17)。 表 17. トランスレーション(並進可動域)の推移 治療群 項目 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 257 253 250 260 255 平均値 0.90 mm 0.99 mm 0.96 mm 0.97 mm 1.03 mm SD 0.58 mm 0.59 mm 0.59 mm 0.67 mm 0.70 mm 最小値 0.00 mm 0.00 mm 0.00 mm 0.00 mm 0.00 mm 中央値 0.80 mm 0.87 mm 0.89 mm 0.80 mm 0.85 mm 最大値 2.98 mm 2.91 mm 3.15 mm 3.65 mm 3.71 mm ACDF 群 (N=265) 例数 198 200 201 209 193 平均値 0.16 mm 0.15 mm 0.20 mm 0.15 mm 0.15 mm SD 0.12 mm 0.13 mm 0.62 mm 0.12 mm 0.13 mm 最小値 0.00 mm 0.00 mm 0.00 mm 0.00 mm 0.00 mm 中央値 0.14 mm 0.12 mm 0.13 mm 0.13 mm 0.13 mm 最大値 0.66 mm 0.84 mm 8.69 mm 0.61 mm 0.60 mm
25 左右側屈は、左右側屈位の X 線画像により評価され、本品群の全ての術後評価時 点において、平均値は 6.15°から 6.78°の範囲内で一定であった(表 18)。 表 18. 左右側屈の推移 治療群 項目 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 274 274 268 270 264 平均値 6.25° 6.60° 6.78° 6.58° 6.15° SD 3.06° 3.34° 3.58° 3.75° 3.81° 最小値 0.35° 0.27° 0.33° 0.36° 0.04° 中央値 6.03° 6.19° 6.55° 5.82° 5.63° 最大値 16.55° 15.83° 16.60° 17.52° 18.17° <全般的健康状態(SF-36)>
全般的な健康状態は SF-36(Medical Outcomes Study 36-Item Short Form Health Survey)を 用いて評価された。身体的側面の QOL(Quality of Life)サマリースコア(PCS:Physical Component Summary)及び精神的側面の QOL サマリースコア(MCS:Mental Component Summary)に要約され、スコアが高いほどよい健康状態を示す。Success の定義は、それぞ れ術後の PCS-術前の PCS≧0、術後の MCS-術前の MCS≧0 と定義された。PCS 及び MCS は、本品群、ACDF 群ともに術前より高い数値を示しており、QOL が改善していることが 示された。 PCS 及び MCS のベイズ流統計解析の結果、術後 24 か月時の ACDF 群に対する本品群の 非劣性の事後確率は、PCS では 93.6%と統計学的な非劣性は示されなかったが、MCS では ほぼ 100%であり、非劣性が示されるとともに、優越性の事後確率は 99.2%であったことか ら、本品群の ACDF 群に対する優越性も確認された。 表 19. SF-36(PCS)の推移 治療群 項目 術前 術後 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 279 267 270 265 平均値 32.2 47.3 47.6 46.6 SD 7.4 10.9 10.4 11.4 中央値 32.4 50.2 51.7 51.0 ACDF 群 (N=265) 例数 263 222 222 218 平均値 32.0 42.9 43.4 44.4 SD 7.5 11.7 11.7 12.0 中央値 31.5 43.2 46.4 47.9
26 表 20. SF-36(MCS)の推移 治療群 項目 術前 術後 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 279 267 270 265 平均値 44.5 51.3 52.0 52.6 SD 11.5 10.5 10.3 9.6 中央値 46.5 55.1 56.2 56.2 ACDF 群 (N=265) 例数 263 222 222 218 平均値 42.7 49.4 48.8 50.2 SD 12.4 10.7 11.4 11.1 中央値 42.1 52.7 52.6 52.9 <疼痛評価(頚部痛及び上肢痛)>
頚部痛及び上肢痛は、Measuring Health4より一部適用した NRS(Numerical Rating Scale)を用いて、痛みの強さ及び痛みの頻度を 0 から 10(スコアが低いほど痛みが 少ない)で評価し、それらを掛け合わせることで 0 から 100 となる疼痛スコアを算出 した。Success は、術前のスコア-術後のスコア≧ 0(スコア= 痛みの強さ×痛みの 頻度)と定義された。 頚部痛及び上肢痛のスコアの平均は NDI スコアと同様、両群ともに、術後の全て の観察時期において、術前に比べて有意に改善していた (表 21 及び表 22)。また、 本品群の術後 24 か月時の頚部痛、上肢痛の Success rate はともに 96.3%(260 例/270 例及び 258 例/268 例)であった。ベイズ流統計解析により、ACDF 群に対する本品群 の非劣性の事後確率は頚部痛、上肢痛ともにほぼ 100%であり、統計学的な非劣性が 示された。 表 21. 頚部痛のスコアの推移 治療群 項目 術前 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 280 277 276 270 274 270 平均値 67.0 13.3 10.4 10.3 11.8 10.6 SD 20.8 19.4 16.8 18.5 20.4 19.2 最小値 20.0 0.0 0.0 0.0 0.0 0.0 中央値 70.0 4.0 2.0 1.5 1.5 1.0 最大値 100.0 81.0 90.0 100.0 100.0 90.0 ACDF 群 (N=265) 例数 264 245 237 223 222 220 平均値 69.3 20.2 17.9 18.5 19.5 16.6 SD 21.5 22.8 23.0 23.3 25.1 24.4 最小値 20.0 0.0 0.0 0.0 0.0 0.0 中央値 72.0 12.0 6.0 7.0 6.0 4.0 最大値 100.0 100.0 100.0 100.0 90.0 90.0
27 表 22. 上肢痛のスコアの推移 治療群 項目 術前 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (N=280) 例数 280 277 276 271 274 268 平均値 59.6 10.8 8.4 8.5 9.4 8.5 SD 26.3 19.3 18.0 19.1 19.7 18.3 最小値 0.0 0.0 0.0 0.0 0.0 0.0 中央値 64.0 1.0 0.0 0.0 0.0 0.0 最大値 100.0 100.0 100.0 100.0 100.0 100.0 ACDF 群 (N=265) 例数 264 245 237 223 221 220 平均値 62.4 13.2 12.7 13.3 16.1 14.2 SD 28.5 23.0 22.6 23.1 25.6 24.3 最小値 0.0 0.0 0.0 0.0 0.0 0.0 中央値 67.0 2.0 1.0 2.0 3.0 1.0 最大値 100.0 100.0 100.0 100.0 100.0 100.0 <被験者の満足度> 割り付けられた治療に対する被験者の満足度が、以下の 3 つの質問を用いて評価さ れた。回答は「確実に真実(Definitely True)」から「確実に誤り(Definitely False)」 までの 5 段階評価とされ、「確実に真実(Definitely True)」又は「ほぼ真実(Mostly True)」と評価された場合に Success と定義された。 質問 1. 手術結果に満足している 質問 2. 期待通りの効果であった 質問 3. 同様の状態になった時、同じ治療を受けようと思う 3 つの質問において、本品群の術後 24 か月時の Success 例はそれぞれ 90.0%(243 例/270 例)、87.4%(236 例/270 例)、90.7%(245 例/270 例)であり、ACDF 群の術 後 24 か月時の Success 例はそれぞれ 87.7%(192 例/219 例)、82.2%(180 例/219 例)、 83.5%(183 例/219 例)であり、2 群間で大きな差は認められなかった 。 <治療結果に対する被験者の評価> 割り付けられた治療に対する治療結果について 、被験者により「完全に回復した (Completely Recovered)」から「非常に悪化した(Vastly Worsened)」までの 7 段階 評価で評価され、「完全に回復した (Completely Recovered)」又は「とても良くな った(Much Improved)」、「少し良くなった (Slightly Improved)」と評価された場 合に Success と定義された。 術後 24 か月時の Success 症例は本品群では 95.5%(258 例/270 例)であり、ACDF 群の 90.8%(199 例/219 例)よりも高いことが示された。
28 <歩行評価> 歩行評価は、術前及び術後に Nurick 分類を使用して評価された。Nurick 分類は、0 ~5 までの Grade で評価し、スコアが高いほど神経学的所見に起因する重い機能障害 が あ る こ と を 示 す 指 標 で あ る 。 神 経 根 症 又 は 脊 髄 症 状 が な い 歩 行 が 正 常 な 症 例 は Normal に分類された。Success の定義は、術前のスコア-術後のスコア ≧ 0 とされ た。 Nurick 分類 Grade 0 Grade 1 Grade 2 Grade 3 Grade 4 Grade 5 :根障害はあるが、脊髄障害はない。 :脊髄障害の兆候はあるが、歩行障害はない。 :軽微な歩行障害はあるが、全時間就労を妨げるものではない。 :歩行障害のため、全時間就労できず、また家事の全ては行えない。 しかし他人の援助は要さない。 :歩行に他人の補助か歩行器が必要である。 :車椅子生活又は寝たきり。 術前の歩行評価は本品群の 93.6%(262 例/280 例)、ACDF 群の 76.9%(203 例/264 例)が「Normal」であり、術後 24 か月時では PRESTIGE LP 群の 99.3%(268 例/270 例)、ACDF 群の 96.4%(212 例/220 例)が「Normal」となった。術後 24 か月時の Success rate は本品群 99.3%、ACDF 群 99.5%であった。
<椎間孔圧迫テスト> 神経根の圧迫の有無が椎間孔圧迫テストにより評価され、術前は両群ともに 40% 以上の症例が「Positive(陽性)」であったが、術後 24 か月時では両群とも「Negative (陰性)」の症例が 94%を超え、同様であった(表 23)。 表 23. 椎間孔圧迫テストによる評価の推移 治療群 項目 術前 術後 6 週 3 か月 6 か月 12 か月 24 か月 PRESTIGE LP 群 (n(%)) (N=280) Negative 160(57.1) 271(97.5) 273(98.9) 269(99.3) 271(99.3) 265(98.1) Positive 120(42.9) 7(2.5) 3(1.1) 2(0.7) 2(0.7) 5(1.9) ACDF 群 (n(%)) (N=265) Negative 121(46.0) 242(96.0) 228(95.0) 220(96.5) 211(94.2) 208(94.5) Positive 142(54.0) 10(4.0) 12(5.0) 8(3.5) 13(5.8) 12(5.5) また、有効性に関するその他の評価項目として、隣接椎間の評価、就労状況及び治 療に対する医師の評価が設定された。
29 <隣接椎間の評価> 本治療の隣接椎間への有効性への影響の有無を評価するために、適応椎間の上下椎 の可動域及びトランスレーション(並進可動域)について評価した。 隣接椎間の可動域は全ての観察期間において ACDF 群の可動域が、本品群に比し て大きく、本品群では、術後 24 か月時点において、それぞれの可動域の平均値は、 適応椎間 7.51°(表 15)、上位隣接椎間 10.40°、下位隣接椎間 6.77°であった(表 24)。 表 24. 隣接椎間の可動域(屈曲・進展)の推移(平均±SD) PRESTIGE LP 群 (N=280) ACDF 群 (N=265) 症例数 可動域 症例数 可動域 上位隣接椎間 術前 274 8.51° ± 4.13° 230 10.77° ± 4.71° 術後 6 週 271 7.83° ± 3.82° 207 9.66° ± 3.75° 術後 3 か月 269 8.82° ± 3.95° 218 11.03° ± 4.11° 術後 6 か月 268 9.35° ± 4.31° 216 11.33° ± 4.49° 術後 12 か月 272 9.79° ± 4.43° 212 12.05° ± 4.78° 術後 24 か月 267 10.40° ± 4.26° 211 11.88° ± 4.56° 下位隣接椎間 術前 187 6.09° ± 4.02° 128 7.77° ± 4.17° 術後 6 週 199 5.68° ± 3.76° 113 8.22° ± 4.51° 術後 3 か月 199 6.25° ± 3.99° 114 9.24° ± 4.64° 術後 6 か月 187 6.63° ± 4.31° 121 8.71° ± 4.73° 術後 12 か月 195 6.95° ± 4.33° 120 9.53° ± 4.79° 術後 24 か月 188 6.77° ± 4.38° 120 9.10° ± 4.82° 適応椎間の隣接椎間のトランスレーション(並進可動域)は、術後観察期間を通じ て安定していたことが示された(表 25)。 表 25. 隣接椎間のトランスレーション(並進可動域)の推移 (平均±SD) PRESTIGE LP 群 (N=280) ACDF 群 (N=265) 症例数 可動域 症例数 可動域 上位隣接椎間 術後 12 か月 272 1.82 mm ± 0.91 mm 212 1.47 mm ± 0.80 mm 術後 24 か月 267 1.94 mm ± 0.96 mm 211 1.46 mm ± 0.79 mm 下位隣接椎間 術後 12 か月 180 1.10 mm ± 0.80 mm 120 0.85 mm ± 0.61 mm 術後 24 か月 177 1.12 mm ± 0.82 mm 120 0.89 mm ± 0.66 mm <就労状況> 就労状況について、術前に仕事に従事していた症例は本品群で 67.1%(188 例/280 例)、 ACDF 群で 62.6%(166 例/265 例)、術後 24 か月時では本品群で 73.4%(199 例/271 例)、 ACDF 群で 75.9%(167 例/220 例)であり、両群とも同様であった。術後仕事に復帰するま での日数を Kaplan-Meier 法で解析した結果、日数の中央値は本品群で 40 日、ACDF 群で 60 日であり、統計学的な有意差が認められた(p=0.0019;Log-Rank 検定、p=0.0001;Wilcoxon 検定)。