Molecular Pathological Study on Interstrain
Differences in the Skeletal Muscle Lesions
between Dysferlin-deficient SJL and A/J Mice
著者
Kobayashi Kinji
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(獣医学), 学位記番
号: 論獣第141号, 学位授与年月日: 2010-03-31,
指導教員: 山手丈至.
大阪府立大学博士(獣医学)学位論文
Molecular Pathological Study on Interstrain Differences
in the Skeletal Muscle Lesions
between Dysferlin-deficient SJL and A/J Mice
(Dysferlin を欠損する SJL および A/J マウスにおける
骨格筋病変の系統間差に関する分子病理学的研究)
小林 欣滋
Contents
Preface
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・1
Chapter 1
Pathomorphological Analyses in the Skeletal Muscles of
Dysferlin-deficient SJL and A/J Mice
Introduction
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・5
Materials
and
Methods
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・7
Results
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・10
Discussion
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・13
Summary
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・16
Tables
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・17
Figure
Legends
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・19
Plate
I
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・21
Chapter 2
Association between Endoplasmic Reticulum Stress and
Skeletal Muscle Lesions in SJL and A/J Mice
Introduction
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・25
Materials
and
Methods
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・26
Results
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・29
Discussion
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・30
Summary
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・31
Figure
Legends
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・32
Plate
II
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・34
Chapter 3
Gene Expression Analyses in the Skeletal Muscles of SJL
and A/J Mice
Introduction
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・38
Materials
and
Methods
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・39
Results
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・42
Discussion
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・46
Summary
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・52
Tables
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・53
Figure
Legends
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・58
Plate
III
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・61
Conclusions
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・71
References
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・73
Acknowledgements
・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・84
Preface
Muscular dystrophy is a generic term used to refer to as hereditary muscle disorder characterized clinically in progressive muscle weakness and muscle atrophy, and histopathologically in degeneration/necrosis and regeneration of skeletal muscle fibers. The progression of disease confines patients to a wheelchair or ventilator, and detracts from quality of life (QOL). Although various approaches to therapy of muscular dystrophy, for example, gene therapy including exon skipping or cell based therapy (Arnett et al., 2009; Farini et al., 2009; Wang et al., 2009), are just sought, the standard therapies remain to be established. This group of inherited muscle disorders includes Duchenne muscular dystrophy, Becker muscular dystrophy, Emery-Dreifuss muscular dystrophy, limb girdle muscular dystrophies, distal muscular dystrophies (Miyoshi myopathy, tibial muscular dystrophy, myofibrillary/desmin-related myopathy, Welander myopathy et al.), Fukuyama/non-Fukuyama congenital muscular dystrophies, Nonakel myopathy (also known as distal myopathy with rimmed vacuoles), facioscapulohumeral muscular dystrophy, myotonic dystrophy et al. (Cohn and Campbell, 2000). Out of these, limb girdle muscular dystrophy (LGMD) is a muscle disorder which weakness and muscle atrophy progress slowly and symmetrically in the proximal muscles of limb girdles. Because LGMD included many unclassified muscular dystrophies and were etiologically heterogeneous, a locus-based classification was proposed by a consortium meeting under the auspices of the European Neuromuscular Centre, with the dominant LGMD loci designated LGMD1A, B, C, etc. and the recessive froms as LGMD2A, B, C, etc. in the order of their identification (Bushby, 1999).
LGMD2B and Miyoshi myopathy (MM) are both caused by recessively inherited mutations in the dysferlin gene (Cohn and Campbell, 2000). LGMD2B is characterized by the progressive wasting and weakness of proximal lower limb-girdle muscles. Meanwhile, the distal muscle groups of limb girdle are mostly affected in MM. Both disorders have been considered to be due to a loss of dysferlin protein at the plasma membrane in muscle fibers, which leads to abnormalities in vesicle traffic and membrane repair (Han et al., 2007; Glover and Brown, 2007), and are collectively called ‘dysferlinopathy’.
Two naturally-occurring animal models for LGMD2B, SJL/J (SJL) and A/J mice, have been identified to have mutations in the dysferlin gene associated with phenotypical features of progressive muscular dystrophy (Bittner et al., 1999; Ho et al., 2004). However, the type of dysferlin gene mutation differs between SJL and A/J mice. SJL mice have a splice site mutation that removes a part of the highly conserved C2E domain; the domain is known to bind to calcium, phospholipids or proteins to trigger signaling events and membrane trafficking (Bittner et al., 1999; Vafiadaki et al., 2001; Rizo and Sudhof, 1998). On the other hand, A/J mice bear a unique ETn retrotransposon insertion near the 5’ end (intron 4) of the dysferlin gene (Ho et al., 2004). Interestingly, the two strains show phenotypic divergences differing from each other; A/J mice display a later age of onset and a slower progression of the muscle disease compared with SJL mice (Ho et al., 2004). These findings support the hypothesis that additional enhancers or modulators may be involved in the pathogenesis of skeletal muscle lesions in dysferlinopathy.
To understand the pathogenesis of skeletal muscle lesions in both model mice, in Chapter 1, the distribution of skeletal muscle lesions with age and their characteristics were examined by histopathological and enzyme-/immuno-histochemical examinations, and further the differences were compared between SJL and A/J mice. In Chapter 2, because SJL mice was estimated to produce incomplete dysferlin protein by splicing mutation in coding region, whether or not the endoplasmic reticulum stress induced by aggregation of incomplete proteins contributes to the development of skeletal muscle lesions was investigated by semi-quantitative reverse transcription polymerase chain reaction. In Chapter 3, because there were interstrain differences in the distribution and onset timing of skeletal muscle lesions between SJL and A/J mice in Chapter 1, the gene expression at the age of 10 and 30 weeks were investigated by quantitative real-time polymerase chain reaction using TaqMan® Gene expression assays to identify factors affecting the progression of skeletal muscle lesions in SJL and A/J mice.
This study confirms that there are some interstrain differences in the pathogenesis process of skeletal muscle lesions between A/J and SJL mice, and supports the hypothesis that additional enhancers or modulators may be involved in the pathogenesis of skeletal muscle lesions in animal models of LGMD2B. In addtion, the result convinced that these animal models are useful to understand the pathogenesis in LGMD2B.
Chapter 1
Pathomorphological Analyses in the Skeletal Muscles of
Dysferlin-deficient SJL and A/J Mice
Introduction
The term muscular dystrophy covers a diverse group of inherited disorders characterized by progressive muscle weakness and wasting, in which the primary defect is believed to be in skeletal muscle (Table 1). Limb-girdle muscular dystrophy (LGMD) includes a number of disorders with heterogeneous etiologies. It is still used as a generic term to describe those patients with muscular dystrophy of girdle distribution. A European Neuromuscular Centre meeting in 1995 defined LGMD as a muscular dystrophy with predominantly proximal distribution of weakness which, early in the course of the disease, spares distal muscles as well as facial and extraocular muscles (Bushby, 1999). Limb girdle muscular dystrophy type 2B (LGMD2B) is caused by a deficiency of dysferlin protein due to the mutation of dysferlin gene (Cohn and Campbell, 2000). SJL/J (SJL) and A/J mice were known as animal models for LGMD2B. These animals have been identified to have mutations in the dysferlin gene associated with phenotypical characteristics of progressive muscular degeneration/necrosis with regeneration (Bittner et al., 1999; Ho et al., 2004). However, the onset timing of skeletal muscle lesions varies between SJL and A/J mice, even though both mice have the abnormality of the same gene; A/J mice display a later age of onset and a slower progression of the muscle disease compared with SJL mice (Ho et al., 2004). This difference is estimated to show the association of additional enhancers or modulators with the pathogenesis of skeletal muscle lesions in SJL or A/J mice. Meanwhile, the dysferlinopathies in humans are also a clinically heterogeneous group of disorders (Nguyen et al., 2007). If the detection of additional enhancers or modulators associated with the pathogenesis of dysferlinopathy may help with the
development of more effective therapy to the arrest of progression of disease.
In this Chapter, first of all, the histopathological examination was performed to reveal the difference in distribution of skeletal muscle lesions between SJL and A/J mice. Secondly, the immunohistochemical staining was conducted to identify mononuclear cells infiltrating in the skeletal muscle lesions. Thirdly, the enzyme-histochemical method was applied to confirm the muscle fiber-specific distribution of skeletal muscle lesions.
Materials and Methods
Animals
Male SJL and A/J mice at the age of 8 or 30 weeks were obtained from Charles River Laboratories Japan Inc. (Kanagawa, Japan) and Japan SLC, Inc. (Shizuoka, Japan), respectively; six mice at each of 10 and 35 weeks age were examined. Three male BALB/c mice at 10 or 35 weeks of age, obtained from Japan SLC, Inc., served as controls without mutation in the dysferlin gene. These animals were individually housed in plastic cages in an animal room kept under controlled conditions (temperature of 23 ± 2°C, humidity of 30 to 70%, ventilation of 12 times or more per hour, lighting for 12 hours between 06:30 to 18:30) in the test facility (Safety Research Laboratory (Kashima), Mitsubishi Tanabe Pharma Corporation, Osaka, Japan). The mice were given pelleted feed sterilized by 15 kGy gamma irradiation (CRF-1, Oriental Yeast Co., Ltd., Tokyo, Japan) and industrial water ad libitum via an automatic water feeder.
All experimental procedures were approved by the Animal Ethics Committee of Mitsubishi Tanabe Pharma Corporation and were conducted in accordance with the Ethical Guidelines for the USA of Experimental Animals.
Histopathology
Mice were euthanized by exsanguination from the abdominal aorta under ether anesthesia at 10 and 35 weeks of age. The femoral, crural, brachial, forearm, abdominal, pectoral, masseter, lumbar and bulbocavernosus muscles, the diaphragms and tongues were fixed in 10% neutral buffered formalin; skeletal muscles including
bones were decalcified using 50% K-CX (FALMA Co., Ltd., Tokyo, Japan) solution for decalcification, processed and embedded in paraffin. Tissue paraffin blocks were thin-sectioned with a microtome, and each thin section was stained with hematoxylin and eosin (HE). The severity of skeletal muscle lesions such as degeneration/necrosis, central nuclei and atrophy, cellular infiltration, and fatty infiltration were evaluated histopathologically as minimal (±, localized lesions), mild (+, scattered lesions) and moderate (++, multifocal lesions).
Immunohistochemistry
The formalin-fixed paraffin sections of the lumbar muscles at the age of 35 weeks old were examined using rat anti-mouse F4/80 antigen monoclonal antibody (AbD Serotec, Oxford, UK) for macrophages and anti-alpha-smooth muscle actin (α-SMA; Neomarkers, Fremont, CA, USA) antibody for myofibroblasts, to identify types of reactive cells in and around degenerative and/or necrotic muscle fibers. The sections immunostained with the rat anti-mouse F4/80 antigen monoclonal antibody were pretreated with proteinase K (10 µg/mL) for 10 min at room temperature (RT). The sections for these antibodies were immersed in 3% H2O2 solution to inactive
endogenous peroxidase for 10 min at RT, blocked with 5% skim milk for 30 min at RT. The primary antibody for mouse F4/80 antigen diluted at 1:200 or the ready-to-use primary antibody for α-SMA was applied to the sections overnight at 4°C. Immunoperoxidase staining was then performed with the Nichirei-Histofine® Simple Stain Mouse MAX-PO Kit (Nichirei Biosciences Inc., Tokyo, Japan), and the positive staining was visualized with diaminobenzidine. They were then counter-stained
Enzyme-histochemistry
To determine the specificity of muscular dystrophic lesions for the type of muscle fibers (slow-twitch type 1 and fast-twitch type 2 muscle fibers), the enzyme-histochemical stainings for nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) (Novikoff et al., 1961) and succinate dehydrogenase (SDH) (Nachlas et al., 1957) were carried out with modified methods on 6-µm unfixed cryosections of the lumbar and femoral muscles at the age of 35 weeks old; these sections were incubatedat 37°C in NADH staining solution (0.05 M Tris-HCl [pH 7.4], 1 mg/mL nitroblue tetrazolium chloride (NBT), 0.4 mg/mL ß-NADH) for 30min, or in SDH staining solution (0.1 M Tris-HCl [pH 7.4], 0.1 M sodium succinate, 0.1 M MgCl2, 1.5 mg/mL NBT) for2 hours. In both NADH-TR and SDH histochmeistry,
slow-twitch type 1 muscle fibers were stained dark blue, and fast-twitch type 2 muscle fibers were pale blue in color.
Results
Histopathology
The histopathological findings in BALB/c, SJL and A/J mice are shown in Table 2. Fig. 1 indicates typical histopathological findings common to SJL and A/J mice.
10 weeks of age:
No significant changes were observed in the skeletal muscle fibers of BALB/c mice. The femoral, branchial, abdominal, masseter and lumbar muscle fibers in 5 or 6 out of 6 SJL mice, and the crural, forearm, pectoral, diaphragmatic and lingual muscle fibers in 1 to 3 out of 6 SJL mice showed minimal (±) degeneration and/or necrosis. Degenerative and/or necrotic muscle fibers were uniformly stained with eosin. Some of these muscle fibers were associated with infiltration of mononuclear cells. Centrally nucleated fibers were minimally (±) detected in the femoral, crural and abdominal muscles. In contrast, A/J mice did not exhibit histopathological changes in any skeletal muscles other than the masseter muscles with mild centrally nucleated fibers (±) in two cases.
35 weeks of age:
Each minimal (±) degeneration and/or necrosis in the lumbar muscles and centrally nucleated fibers were in 1 out of 3 BALB/c mice; these findings were considered to be incidental, because of very mild lesions. The histopathological lesions of skeletal muscles other than the diaphragm in SJL mice were progressive in
degeneration and/or necrosis in the lumbar muscles and the incidence of centrally nucleated fibers in the femoral, branchial, abdominal and lumbar muscles were moderately-increased (++) as shown in Fig. 1. Moreover, minimal (±) to mild (+) fatty infiltration, variation in muscle fiber size and atrophy in muscle fibers in the femoral (rectus femoris) and lumbar (longissimus and sublumbar) muscles were newly confirmed at 35 weeks of age. The cross-sectional area of lateral longissimus muscles in SJL mice appeared smaller in contrast to those in BALB/c and A/J mice (Fig. 2).
In contrast, the skeletal muscle lesions in A/J mice showed a slow progression with age. Three to 4 out of 6 A/J mice showed minimal (±) to mild (+) degeneration and/or necrosis and variation in muscle fiber size in the femoral muscles, but other muscles (crural, brachial, abdominal, masseter and lumbar muscles (Fig. 2)) showed minimal (±) to mild (+) degeneration and/or necrosis or centrally nucleated fibers in only 1 out of 6 mice, and any mice did not have histopathological lesions in the forearm, pectoral and bulbocavernosus muscles, and diaphragm.
Immunohistochemistry
Mononuclear cells were seen mainly in and around degenerative and/or necrotic muscle fibers in the lumbar muscles of SJL and A/J mice at the age of 35 weeks old. Immunohistochemically, these cells were positive with the rat anti-mouse F4/80 antigen monoclonal antibody (Fig. 3A). On the other hand, α-SMA-positive myofibroblasts were not observed in any skeletal muscle lesion (Fig. 3B).
Enzyme-histochemistry
The NADH-TR histochemistry revealed that most degenerative muscle fibers seen in the rectus femoris and lateral longissimus muscles of SJL mice were fast-twitch type as shown in Fig. 2A; intermyofibrillar networks were indistinct in these muscle fibers. Additionally, the slow-twitch muscle fibers in the same muscles of SJL mice underwent occasionally degeneration as shown in Fig. 4. Although the medial vastus and iliocostalis muscles adjacent to the rectus femoris and lateral longissimus muscles constituted a large majority of the fast-twitch muscle fibers, their muscles had little degeneration/necrosis of muscle fibers in SJL mice. Meanwhile, no significant changes were observed on cryosections in BALB/c and A/J mice.
Discussion
This study showed that there were differences in the progress and prevalent site of skeletal muscle lesions between SJL and A/J mice. In particular, the sensitivity to muscular dystrophic lesions between SJL and A/J mice was most apparent in the lumbar muscles; the extent of histopathological lesions in the lumbar (longissimus and sublumbar) muscles was comparable with those in the femoral (rectus femoris) muscles in SJL mice at 10 and 35 weeks of age, whereas in A/J mice at 35 weeks of age the lesions of lumbar muscles were less severe than those of the femoral muscles. Histopathological changes seen in the lumbar muscles have not been reported in both dysferlinopathy mouse models, and human LGMD2B and MM cases. To the author’s knowledge, it was only reported that fatty degeneration of all erector spinae muscles in the lower thoracic and lumbar region was found out by magnetic resonance imaginUgU in
a 52-year-old woman with LGMD2B (Seror et al., 2008). This may be attributable partly to the fact that the lumbar muscles have not been examined histopathologically or pathophysiologically in LGMD2B and MM. The lumbar muscles should be examined in patients who are suspected to have dysferlinopathy.
Although there are differences in the age of onset, affected muscles, genetic inheritance and severity among the type of muscular dystrophies, all muscular dystrophies are characterized fundamentally by muscle fiber degeneration, necrosis and inflammatory cell infiltration; subsequently, these lesions may be replaced by fatty and fibrous tissues (Wallace and McNally, 2009; Turk et al., 2006). The muscle fibers may have the regenerative capacity; therefore, centrally nucleated muscle fibers are described as regenerative muscle fibers, and the proportion of thesefibers is used as
a standard method to evaluate the progress of skeletal muscle lesions in dysferlinopathy (Sacco et al., 2005). The similar histopathological characteristics were observed in muscles of the present SJL and A/J mice, although these muscular lesions were exacerbated with age, and particularly more prominent in SJL mice.
Because mononuclear cells seen in or around the degenerative and/or necrotic muscle fibers showed a positive reaction for mouse F4/80 antigen, these cells were identified as macrophages. The F4/80 antibody has been widely used to identify mouse macrophages in lesions (Dixon et al., 1986; Sunderkötter et al., 1993). The previous immunohistochemical analysis using anti-Mac-1 α-chain (also known as CD11b and integrin αM chain) antibody showed that macrophages were a predominant
infiltrating cell type in the muscles of SJL mice (Kostek et al., 2002). Meanwhile, initial infiltration of macrophages and CD4+ lymphocytes was followed by increase in number of CD8+ cells in the experimental allergic myositis in SJL mouse after immunization with muscle fractions (Matsubara et al., 2001). In view of this cellular kinetics, these macrophages seen in the muscle lesions of SJL mice appear to take up cellular debris such as necrotic muscle fibers, denying the autoimmunity mechanism. Necrotic muscle fibers may be replaced by fatty and fibrous tissues in the muscular dystrophies; myogenic cells, such as muscle-derived stem cells that are present at an injured site of muscle, could differentiateinto myofibroblasts under some microenviromnetal conditions (Li and Huard, 2002). The immunostaining for α-SMA, to which antibody has been used to identify myofibroblasts in fibrotic tissues (Brenmoehl et al., 2009), was conducted for the lumbar muscle lesions in SJL and A/J mice, however there were no α-SMA-positive myofibroblasts in the lesions.
differentiation (Brenmoehl et al., 2009). It has been reported that the main source of TGF-β1 is macrophages in fibrotic tissues (Gosselin et al., 2004). Although
F40/80-positive macrophages were seen in dystrophic muscles of the present dysferlinopathy mouse models, the production of TGF-β1 might be insufficient for the
differentiation into myofibroblasts. It is interesting to pursue the relationship between macrophage-producing TGF-β1 and myofibroblast development, in order to understand
the pathogenesis of progressive dysferlinopathy.
In dysferlin deficient patients with the advanced-stage dystrophic pattern, type 1 (slow-twitch) fiber predominance was observed with exceeding 80%, suggesting a selective loss of type 2 (fast-twitch) fibers or a fiber typing conversion process (Fanin and Angelini, 2002). Similarly, the results of NADH-TR and SDH enzyme-histochemical stainings revealed that degeneration of fast-twitch type muscle fibers was predominant in SJL mice. However, the medial vastus and iliocostalis muscles originally constituting a large majority of the fast-twitch muscle fibers had little degeneration/necrosis of muscle fibers. These findings indicated that the sensitivity of the type 2 fibers to injury might be site-specific, as seen in the rectus femoris and lateral longissimus muscles of SJL mice.
Summary
SJL mice showed an earlier age of onset and a faster progression of skeletal muscle lesions as compared with those of A/J mice; there was the difference in distribution of skeletal muscle lesions between SJL and A/J mice; the sensitivity difference to muscular dystrophic lesions between SJL and A/J mice was observed in the lumbar muscles (particularly, lumbar longissimus and sublumbar muscles); the lesions seen mainly in SJL mice at 35 weeks old consisted of degeneration, necrosis, fatty infiltration, variation in muscle fiber size and atrophy in muscle fibers. Immunohistochemically, the most reactive cell type observed in and around degenerative and/or necrotic muscle fibers was macrophages, demonstrable with an anti-F4/80 antibody. However, there were no α-SMA-positive myofibroblasts, which contribute to fibrosis, in the lesions. Enzyme-histochemically, the fast-twitch type muscle fiber was predominant for the degenerative changes seen in the rectus femoris and lateral longissimus muscles of SJL mice.
Table 1. Muscular Dystrophies and Gene Locations*
Disease Mode of
inheritance
Gene locus Gene product
1. X-linked muscular dystrophies
A. Duchenne muscular dystrophy XR Xp21 Dystrophin
B. Becker muscular dystrophy XR Xp21 Dystrophin
C. Emery-Dreifuss muscular dystrophy XR Xq28 Emerin
2. Emery-Dreifuss muscular dystrophy AD 1q11 Lamin A/C 3. Limb-girdle muscular dystrophy (LGMD)
A. LGMD 1A AD 5q22–q34 Myotilin B. LGMD 1B AD 1q11–21 Lamin A/C C. LGMD 1C AD 3p25 Caveolin-3 D. LGMD 1D AD 6q22 (Unknown) E. LGMD 1E AD 7 (Unknown) F. LGMD 2A AR 15q15 Calpain-3 G. LGMD 2B AR 2p13 Dysferlin H. LGMD 2C AR 13q12 γ-Sarcoglycan I. LGMD 2D AR 17q12–q21 α-Sarcoglycan J. LGMD 2E AR 4q12 β-Sarcoglycan K. LGMD 2F AR 5q33–q34 δ-Sarcoglycan L. LGMD 2G AR 17q11–q12 Telethonin M. LGMD 2H AR 9q3–q34 TRIM32 N. LGMD 2I AR 19q13.3 FKRP O. LGMD 2J AR 2q31 Titin P. LGMD 2K AR 9q34 POMT1 Q. LGMD 2L AR 9q31–q33 Fukutin 4. Distal muscular dystrophy
A. Miyoshi myopathy AR 2p13 Dysferlin
B. Tibial muscular dystrophy AD 2q31 Titin
5. Congenital muscular dystrophy (CMD)
A. “Classic” or ”pure” CMD AR 6q22 Laminin α2
B. Fukuyama CMD AR 9q31–q33 Fukutin
C. a7 integrin congenital myopathy AR 12q13 a7 Integrin
D. Rigid spine CMD AR 1p35–36 selenoprotein N
E. Muscle-eye–brain disease AR 1p32–p34 POMT1
6. Other forms of muscular dystrophy
Bethlem myopathy AD 21q22 Collagen VI α1 AD 21q22 Collagen VI α2
AD 2q37 Collagen VI α3
Epidermolysis bullosa and MD AR 8q24–qter Plectin Oculopharyngeal muscular dystrophy AD 14q11.2–q13 PABP2 Facioscapulohumeral muscular dystrophy AD 4q35
Myotonic dystrophy AD 19q13 DMPK AD: autosomal dominant, AR: autosomal recessive, XR: X-linked dominant,
TRIM32: Tripartite motif containing 32, FKRP: Fukutin-related protein, POMT1: Protein O-mannosyl transferase 1, PABP2: Poly A binding protein 2, DMPK: Myotonin-protein kinase
Table 2. Distribution and Incidence of Skeletal Muscle Lesions in BALB/c, SJL and A/J Mice
Strain BALB/c SJL A/J
Age at autopsy (week-old) 10 35 10 35 10 35
Skeletal muscle (femoral muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 6/6 0/6 0/6 3/6
+ 0/3 0/3 0/6 6/6 * 0/6 1/6 ± 0/3 0/3 2/6 0/6 0/6 0/6 Centronuclear muscle fiber
++ 0/3 0/3 0/6 6/6 0/6 0/6
Fatty infiltration (Rectus femoris) ± 0/3 0/3 0/6 5/6 0/6 0/6 Variation in size of muscle fiber 0/3 0/3 0/6 2/6 0/6 3/6
Skeletal muscle (crural muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 1/6 4/6 0/6 1/6
Centronuclear muscle fiber ± 0/3 0/3 1/6 0/6 0/6 0/6 (Anterior tibial muscle) + 0/3 0/3 0/6 5/6 0/6 0/6 Skeletal muscle (brachial muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 5/6 5/6 0/6 1/6 Centronuclear muscle fiber + 0/3 0/3 0/6 3/6 0/6 0/6 ++ 0/3 0/3 0/6 2/6 0/6 0/6
Skeletal muscle (forearm muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 1/6 0/6 0/6 0/6
Centronuclear muscle fiber ± 0/3 0/3 0/6 6/6 0/6 0/6
Skeletal muscle (abdominal muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 5/6 0/6 0/6 1/6 + 0/3 0/3 0/6 6/6 * 0/6 0/6 Centronuclear muscle fiber ± 0/3 1/3 3/6 0/6 0/6 0/6
++ 0/3 0/3 0/6 6/6 0/6 0/6 Variation in size of muscle fiber 0/3 0/3 0/6 3/6 0/6 0/6
Skeletal muscle (pectoral muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 3/6 4/6 0/6 0/6 Centronuclear muscle fiber + 0/3 0/3 0/6 6/6 0/6 0/6 Skeletal muscle (masseter muscle)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 5/6 5/6 0/6 0/6 Centronuclear muscle fiber ± 0/3 0/3 0/6 5/6 2/6 1/6 + 0/3 0/3 0/6 1/6 0/6 0/6
Skeletal muscle (lumbar muscle)
Degenerative/Necrotic muscle fiber ± 0/3 1/3 6/6 0/6 0/6 0/6 + 0/3 0/3 0/6 1/6 * 0/6 1/6 *
++ 0/3 0/3 0/6 5/6 * 0/6 0/6
Centronuclear muscle fiber ++ 0/3 0/3 0/6 6/6 0/6 0/6 Fatty infiltration
(Longissimus, Sublumbar) ± 0/3 0/3 0/6 5/6 0/6 0/6 Atrophic muscle fiber ± 0/3 0/3 0/6 5/6 0/6 0/6 Variation in size of muscle fiber 0/3 0/3 0/6 5/6 0/6 0/6
Skeletal muscle (diaphragm)
Degenerative/Necrotic muscle fiber ± 0/3 0/3 1/6 1/6 0/6 0/6
Skeletal muscle (bulbocavernosus muscle)
Centronuclear muscle fiber ± 0/3 0/3 0/6 1/6 0/6 0/6 Lingual muscle
Inflammatory cell infiltration in the ± 0/3 0/3 1/6 0/6 0/6 0/6 interstitium + 0/3 0/3 0/6 0/6 0/6 1/6
Centronuclear muscle fiber ± 0/3 0/3 0/6 2/6 0/6 0/6
±: Minimal, +: Mild, ++: Moderate, ■: Frequent percentage >50%
Figure Legends
Fig. 1. Typical histopathological findings common to SJL and A/J mice. First of all,
degenerative/necrotic muscle fibers showing hyalinization or swelling appear (A). Secondly, mononuclear cells infiltrate around or into the muscle degenerative/necrotic fibers (B). Thirdly, the muscle fibers having central nuclei (arrows) considered as regenerative process are observed (C). With further progression, these muscles are accompanied by fatty infiltration (D). Bar = 30 μm (A, B), 100 μm (C, D)
Fig. 2. Histopathology of the lumbar muscles in BALB/c, SJL and A/J mice. The
cross-sectional area of lateral sides (asterisk) in the lumbar longissimus muscle (area enclosed in solid line) of SJL mice appears smaller in area in contrast to those in BALB/c and A/J mice (upper figures). In particular, fatty infiltration, variation in muscle fiber sizes and minimal atrophy in muscle fibers are observed in SJL mice (lower figures). HE staining; Bar = 1000 μm (Upper); Bar = 100 μm (Lower).
Fig. 3. Immunohistopathology with the antibody against mouse F4/80 antigen (A)
and α-smooth muscle actin (α-SMA) (B) in the lumbar muscles of SJL mouse. Infiltrating mononuclear cells seen in and around a degenerative muscle fiber (asterisk) in the lumbar muscles of SJL mice show a positive for the antibody, indicating macrophages (A). There are no α-SMA-positive myofibroblasts
in the observed specimens (B). An arrow shows the vascular smooth muscles of arteriole as the internal control for α-SMA. Bar = 50 μm (A), 25 μm (B)
Fig. 4. Nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR)
enzyme-histochemistry in the lumbar longissimus muscles of SJL mice. (A) Intermyofibrillar networks are indistinct in a fast-twitch muscle fiber indicated by black arrows. (B) Intermyofibrillar networks are indistinct with mosaic dark blue regions in slow-twitch muscle fibers indicated by white arrows. Bar = 50 μm
Plate I
Fig. 2
Fig. 3
*
Chapter 2
Association between Endoplasmic Reticulum Stress
and Skeletal Muscle Lesions in SJL and A/J Mice
Introduction
Recently, it has been reported that dysferlin is conventionally degraded by endoplasmic reticulum (ER)-associated degradation system composed of ubiquitin and proteasome. However, the novel mutant (L1341P) dysferlin proteins, which are spontaneously aggregated in the ER, are able to induce eukaryotic translation initiation factor 2a (eIF2a) phosphorylation and LC3 conversion; this process is a key step for autophagosome formation, resulting in the ER stress cell death (Fujita et al., 2007). Some novel variants of limb girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy (MM) have shown a patchy sarcolemmal immunostaining for mutant dysferlin or its intracellular aggregates in the muscle fibers of human patients (Ikezoe et al., 2003; Wenzel et al., 2006). However, the possible relationship between ER stress and skeletal muscle lesions has not yet been investigated in both SJL and A/J mice.
As shown in Fig. 1, the mRNA transcript of XBP1 is known to have 26 nucleotides spliced out of its coding sequence post-transcriptionally as a part of the unfolded protein response to ER stress (Yoshida et al., 2001).
In the present study, to confirm whether or not the ER stress contributes to the development of skeletal muscle lesions in SJL/J (SJL) and A/J mice, the gene expression level of spliced XBP1 mRNA as an ER stress marker in the femoral and lumbar muscles of these mice was measured by semi-quantitative reverse transcription polymerase chain reaction (RT-PCR).
Materials and Methods
Animals
Used animals, rearing conditions, feeding, water supply and an approval by the Animal Ethics Committee of Mitsubishi Tanabe Pharma Corporation followed Chapter 1.
Semi-quantitative Reverse Transcription Polymerase Chain Reaction
The femoral and lumbar muscles were removed from mice at 35 weeks of age, frozen immediately in liquid nitrogen and stored in a freezer at –80°C until use. As the positive control, 80% confluent culture of mouse embryo fibroblasts (MEF) were incubated for 18 hours in the growth medium (D-MEM; Invitrogen, Carlsbad, CA, USA) including 10% fetal bovine serum (Invitrogen) and 5 µg/ml tunicamycin (Calbiochem, Bad Soden, Germany) in dimethyl sulfoxide (Hybri-Max™; Sigma-Aldrich, St.Louis, MO, USA) (Yamamoto et al., 2003). The same volume of dimethyl sulfoxide as tunicamycin solution in the positive control was added in 80% confluent culture of MEF as the negative control.
Total RNA was extracted from 100 mg skeletal muscles and cultured MEF using ISOGEN (NIPPON GENE Co., Ltd., Tokyo, Japan), according to the manufacturer’s instruction. Extracted RNA samples were dissolved in DEPC-treated water and stored at –25°C. The concentration of RNA was estimated using the spectrophotometer (GeneQuant, GE Healthcare UK Ltd., Buckinghamshire, UK).
μg of total RNA, 50 ng of random hexamers and 1 mM dNTP mix was prepared as template, incubated at 65°C for 5 min and placed on ice for at least 1 min. Ten μL of cDNA synthesis mix (20 mM Tris-HCl (pH 8.4), 50 mM KCl, 5 mM MgCl2, 10 mM
DTT, 40 U of RNaseOUTTM, 200 U of SuperScriptTM III RT) was added to a tube containing RNA/Primer mix. The mixture was incubated at 50ºC for 50 min, terminated at 85ºC for 5 min, added 2 U of E. coli RNase H for removing RNA, placed at 37ºC for 20 min, and removed 2 μL aliquot for PCR.
The expression of spliced XBP1 mRNA was semi-quantitatively measured by a combination of RT-PCR and PstI restriction enzyme treatment as shown in Fig. 2 (Hirota et al., 2006). Primer sequences for semi-quantitative RT-PCR of XBP1 mRNA were designed as follows: 5’-TGAGAACCAGGAGTTAAGAACACGC-3’ for forward primer and 5’-TTCTGGGTAGACCTCTGGGAGTTCC-3’ for reverse primer. The semi-quantitative approach was according to the protocol annexed QuantumRNA™ 18S Internal Standards Kit (Ambion Inc., Montrouge, France). The protocol consisted of several steps, including the determination of linear range and optimum ratio of marker primers to competimers, before undertaking semi-quantitative PCR. The results led to the conclusion that the midpoint cycle of the linear range amplification cycle was 30 cycle, and the 3:7 18S rRNA primer: competimer ratio was the best. PCR was performed in a final volume of 50 μL containing 2 µL cDNA aliquot of template, 0.1 μM of each XBP1-specific primers, 18S primer: competimer mixture (each 0.5 μM), 1X AccuPrime™ PCR Buffer I (60 mM Tris-SO4 (pH 8.9), 18
mM (NH4)2SO4, 2 mM MgSO4, 0.2 mM of each dNTPs, thermostable AccuPrimeTM
protein), and 1 U AccuPrime™ Taq DNA Polymerase High Fidelity (Invitrogen). For measurement of spliced XBP1 mRNA, XBP1 double-stranded cDNA was
synthesized under the following thermal cycling conditions: 1 cycle at 94ºC for 30 sec, 2 cycle at 94ºC for 15 sec, 55ºC for 30 sec and 68ºC for 60 sec, and 1 cycle at 68ºC for 120 sec in a thermal cycler (PTC-200 DNA Engine™, MJ Research, MS, USA). Then, 9 U of restriction enzyme PstI (Takara Bio, Inc., Shiga, Japan) was added to the reaction mixture to digest the double-stranded cDNA of unspliced XBP1 at 37ºC for 60 min. The remaining spliced XBP1 cDNA was amplified by PCR (1 cycle at 94ºC for 30 sec, 28 cycle at 94ºC for 15 sec, 55ºC for 30 sec and 68ºC for 60 sec, and 1 cycle at 68ºC for 120 sec). Total (without PstI treatment; unspliced form + spliced form) XBP1 cDNA was amplified by the following condition: 1 cycle at 94ºC for 30 sec, 30 cycle at 94ºC for 15 sec, 55ºC for 30 sec and 68ºC for 60 sec 30 cycles, and 1 cycle at 68ºC for 120 sec. These PCR products were resolved by electrophoresis on 3% agarose gel. After visualization by ethidium bromide staining, the optical density (OD) values of bands on agarose gel photographed by digital camera (COOLPIX 990, Nikon, Tokyo, Japan) were analyzed using NIH Image J software (http://rsb.info.nih.gov/ij/). The ratio (OD value for band representing the XBP1-specific product divided by that for 18S rRNA co-amplified in the same sample) was recorded as the level of expression of XBP1 gene in the skeletal muscle relative to the expression of 18S rRNA in the same tissue.
DNA sequencing analysis of the amplicons from PCRs obtained from total RNA of the skeletal muscle of a BALB/c mouse was performed by ABI PRISM® 310 Genetic Analyzer (Applied Biosystems, CA, USA), and showed that the products were derived from the unspliced and spliced XBP1mRNAs (GenBank accession no.: NM_013842).
Results
To investigate the relationship between ER stress and the skeletal muscle lesions in SJL and A/J mice, the expression of spliced XBP1 mRNA was semi-quantitatively evaluated by a combination of RT-PCR and PstI restriction enzyme treatment. The quantity of spliced XBP1 mRNA was observed to be lower in SJL and A/J mice than in BALB/c mice, and there was no difference between XBP1 splicing levels in SJL and A/J mice, as shown in Fig. 3. Meanwhile, MEF exposed to tunicamycin for 18 hours as a positive control significantly increased in the expression level of spliced XBP1 mRNA compared with that of MEF treated with DMSO as a negative control.
Discussion
In mammalian, there are three unfolded protein response (UPR) signaling pathway through three UPR transducers (inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (Atf6) and protein kinase-like ER kinase (PERK) (Zhang and Kaufman, 2004). These UPR transducers are controlled by glucose related protein 78 (BiP/Grp78), which is one master regulator and a member of the heat shock protein 70 chaperone family. Under unstress condition, BiP/Grp78 binds to the lumenal domain of IRE1, Atf6 and PERK (Ellgaard and Helenius, 2003; Gething, 1999; Shen et al., 2002). In response to accumulation of unfolded or misfolded proteins in the ER, BiP/Grp78 is released from these transducers. IRE1 dissociated from BiP/Grp78 is dimerized and activated, and acquires RNase activity to initiate XBP1 mRNA splicing. Spliced XBP1 mRNA is translated to a potent transcription factor that binds to the cis-acting ER stress response element and the unfolding protein response element (Yoshida et al., 2001). In other words, the splicing process of XBP1 mRNA is regulated by BiP/Grp78. Therefore, the author thought that it was more important to measure expression levels of spliced XBP1 mRNA as an ER stress marker. In this study, because the expression level of XBP-1 splicing form was lower in SJL and A/J mice than that in BALB/c mice, it was considered that BiP/Grp78-madiated Atf6 and PERK pathways were not up-regulated as well. Accordingly, the author deduced that ER stress did not affect the development of skeletal muscle lesions in SJL mice as the enhancing or modulating factor.
Summary
To confirm whether or not the ER stress contributes to the development of skeletal muscle lesions in SJL and A/J mice, the gene expression level of spliced XBP1 mRNA as an ER stress marker in the femoral and lumbar muscles of these mice was measured by the semi-quantitative RT-PCR.
Because the analyses of spliced XBP1 mRNA did not show the increased expression, it was considered that ER stress did not affect the progression of skeletal muscle lesions in SJL mice with the advanced stage of dysferlinopathy.
Figure Legends
Fig. 1. Three UPR signaling pathway through three UPR transducers (IRE1, Atf6 and
PERK). Under unstress condition, BiP/Grp78 binds to the lumenal domain of IRE1, Atf6 and PERK, but BiP/Grp78 is released from these transducers in response to accumulation of unfolded or misfolded proteins in the ER. IRE1 released from BiP/Grp78 is dimerized and acquires RNase activity to initiate XBP1 mRNA splicing. Spliced XBP1 mRNA by activated IRE1 is translated to a potent transcription factor that binds to the cis-acting ER stress response element and the unfolding protein response element, and induces various ER responses.
Fig. 2. The principle of spliced XBP1-specific RT-PCR. Total RNA was
reverse-transcribed and double-strand cDNA was synthesized by PCR using specific sense and anti-sense primers for the XBP1 gene. The reaction mixture was treated with PstI, then subjected to PCR reaction. The double-strand cDNA derived from unspliced XBP1 mRNA was digested at the
PstI site, and so was not amplified by the PCR reaction. However,
double-strand cDNA derived from spliced XBP1 mRNA was not digested with PstI, because of loss of the PstI site due to the splicing in response to ER stress, was amplified by PCR.
Fig. 3. Semi-quantitative RT-PCR results for total and spliced XBP1 mRNA using
total RNA extracted from the lumbar muscles of BALB/c, SJL and A/J mice. The expression level of spliced XBP1 mRNA is observed to be lower in SJL and A/J mice than in BALB/c mice, and there is not difference between XBP1 splicing levels in SJL and A/J mice. MEF incubated for 18 hours with 5 µg/ml tunicamycin (Tm) as a positive control increases in the expression level of spliced XBP1 mRNA compared with that of MEF treated with DMSO as a negative control. Numerical values under bands of spliced XBP-1 show a ratio (percentage) of optical density of normalized spliced XBP-1 mRNA to optical density of normalized total XBP-1 mRNA.
Plate II
Bip: immunoglobulin heavy-chain binding protein
(Grp78: 78 kDa glucose-regulated protein)
PERK: protein kinase-like endoplasmic reticulum kinase eIF2: eukaryotic translation-initiation factor 2α
ATF4 (Atf4): activating transcription factor 4 AARE: amino acid response element
CHOP: CCAAT/enhancer binding protein (C/EBP) homologous protein ATF6 (Atf6): activating transcription factor 6
S1P: site-1 protease S2P: site-2 protease
ERSE: ER-stress response elements XBP1: X-box binding protein 1
PDI: protein disulfide isomerase IRE1: inositol-requiring enzyme 1
TRAF2: tumor necrosis factor receptor-associated factor 2 ASK1: apoptosis signalregulating kinase 1
JNK: Jun N-terminal kinase
Fig.2
First strand cDNA
Chapter 3
Gene Expression Analyses
Introduction
As part of discovering additional enhancers or modifiers associated with phenotypic divergences between SJL/J (SJL) and A/J mice, the author investigated whether or not endoplasmic reticulum (ER) stress contributes to the development of skeletal muscle lesions in SJL and A/J mice in Chapter 2. Although spliced XBP1 mRNA, which is known as an ER stress marker, was measured by semi-quantitative reverse transcription polymerase chain reaction (RT-PCR), there was no difference between XBP1 splicing levels in SJL and A/J mice. Accordingly, it was concluded that ER stress was less likely to be an additional enhancer or modifier in the skeletal muscle lesions of SJL and A/J mice.
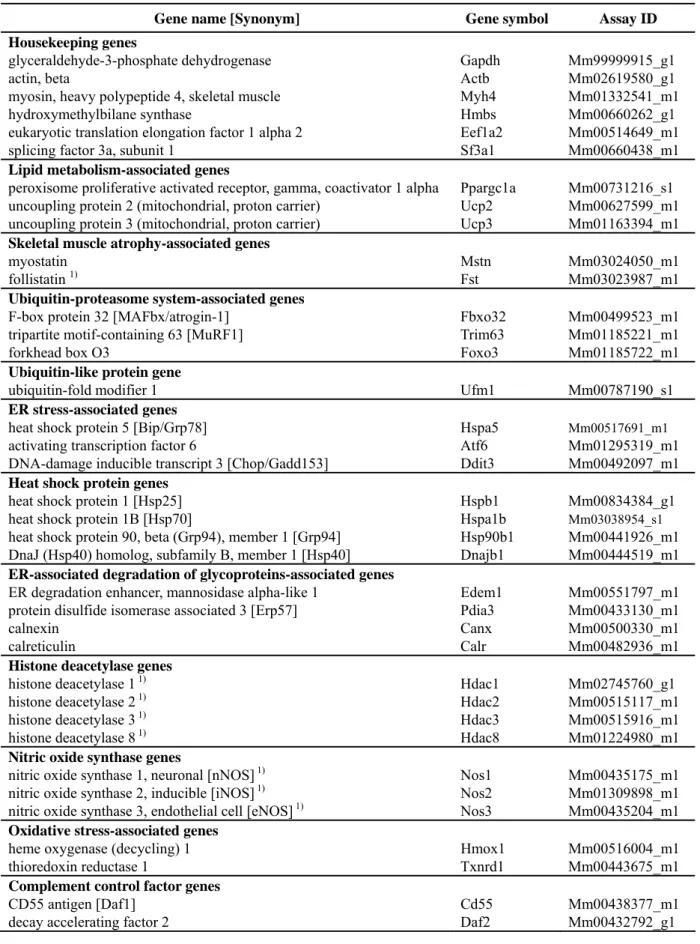
In this Chapter, the expression of 37 genes including ER stress-associated genes other than XBP-1 at the age of 10 and 30 weeks was investigated by quantitative real-time polymerase chain reaction (RT-PCR) using TaqMan® Gene expression assays to identify factors affecting the progression of skeletal muscle lesions in SJL and A/J mice. The linkages or signal transduction pathways of principal genes are schematically shown in Figs. 1 to 8.
Materials and Methods
Animals
Male BALB/c, SJL and A/J mice at the age of 7 or 8 weeks were obtained from Charles River Laboratories Japan Inc. (Kanagawa, Japan) and Japan SLC, Inc. (Shizuoka, Japan), respectively; three mice at each of 10 and 30 weeks age were examined. The rearing conditions, feeding, water supply and an approval by the Animal Ethics Committee of Mitsubishi Tanabe Pharma Corporation followed Chapter 1.
Quantitative Real-time Polymerase Chain Reaction analysis
The rectus femoris and longissimus lumborum were removed from mice at 10 and 30 weeks of age, quickly cut it into slices less than 0.5 cm thick and incubated overnight in an RNAlater RNA Stabilization Reagent (Qiagen, Valencia, CA, USA) at 2–8ºC. The tissue samples in reagents were transferred to a freezer at –20°C and stored until use.
For quantitative RT-PCR analysis, total RNA wasisolated from each isolated muscle with an RNeasy® Fibrous Tissue Mini Kit (Qiagen) according to the instructions of the manufacturer. Total RNA samples were quantitated using a Quant-iTTM RNA assay kit (Invitrogen) and a QubitTM Fluorometer (Invitrogen). Equal quantities of total RNA obtained from three mice in each strain were mixed and pooled together in a freezer at –80°C.
For each sample, 2 µg of pooled total RNA was reverse transcribedin 20 µL of the reaction mixture using a High CapacityRNA-to-cDNA Kit (Applied Biosystems,
Foster City, CA, USA). Afterthe initial step at room temperature for 10 min, the reaction mixture was incubated at 37ºC for 60 min, terminated at 95ºC for 5 min and held at 4ºC. The gene-specific primers and probes used for quantitative RT-PCR analysis were available as TaqMan® Gene expression assays (Applied Biosystems) (Table 1). Quantitative RT-PCR reactions for each gene were performed with doubling-diluted cDNA samples. The quantitative RT-PCR reaction was conducted on a7500 Fast Real-Time PCR System (Applied Biosystems) in 20 µLof the reaction mixture containing 1x TaqMan Fast Universal PCR MasterMix; No AmpErase® UNG (Applied Biosystems), 1x Gene Expression Assay mix, and5 µL of diluted cDNA sample as a template. MicroAmp® Fast 96-Well Reaction Plate, covered by optical adhesive covers (Applied Biosystems), were used. Amplification was conducted according to the following thermal profile: 1 cycle at 95 °C for 20 sec, and 40 cycles at 95 °C for 3 sec and 60 °C for 30 sec. Initial raw data analysis was performedusing the Sequence Detection Software version 1.3.1. Relative mRNA levels were calculated by the comparative threshold cycle (Ct) method (Pfaffl, 2001; Pfaffl et al., 2002), as describedin Applied Biosystems User Bulletin Number 2 (P/N 4303859).
The program calculates ΔCt and ΔΔCt with the formulas: ΔΔCt =ΔCt sample [Ct Gapdh (from BALB/c mice at 30 weeks of age, SJL or A/J mice) – Ct target gene (from BALB/c mice at 30 weeks of age, SJL or A/J mice)] – ΔCt control [Ct Gapdh (from BALB/c mice at 10 weeks of age) – Ct target gene (from BALB/c mice at 10 weeks of age)]. The relative gene expression was calculated by the expression 2–(ΔΔCt).
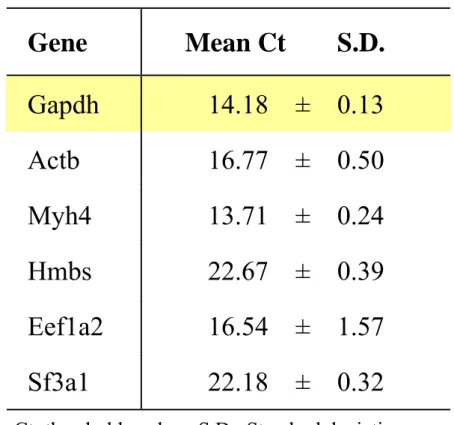
expression profile of six candidate reference genes (i.e. Actb (β-actin), MYH4 (myosin, heavy polypeptide 4, skeletal muscle), Hmbs (hydroxymethylbilane synthase 9), Eef1a2 (eukaryotic translation elongation factor 1α2), Sf3a1 (splicing factor 3a, subunit 1) and Gapdh) in randomly selected different samples (cDNA from the rectus femoris and longissimus lumborum of BALB/c mice at 10 and 30 weeks old). The rationale was to select the stable gene that exhibited relatively less volatility from 10 to 30 weeks old. Gapdh exhibited the least variability among the samples as determined by analysis of the standard deviation of the Ct values (Table 2).
Quadruplicate measurements per gene were conducted and represented as the mean and standard deviation. Changes in gene expression were reported as fold changes relative to controls (the Ct values in BALB/c mice at the age of 10 weeks). We considered that the gene expression was up-regulated or down-regulated if the relative gene expression was more than twice or less than one-half that of the control. Data were not statistically analyzed, because quantitative RT-PCR analysis was performed using pooled samples of equal quantities of total RNA obtained from three mice in each strain.
Results
Tables 3 and 4 show the results of quantitative RT-PCR analysis. Figs. 9 and 10 indicate the changes of principal genes in the longissimus lumborum of BALB/c, SJL and A/J mice.
U
Lipid metabolism-associated genesU:
Uncoupling protein 2 (Ucp2) was up-regulated in the rectus femoris and longissimus lumborum of SJL mice and down-regulated in these muscles of A/J mice. Uncoupling protein 3 (Ucp3) was down-regulated in the longissimus lumborum of SJL and A/J mice and the rectus femoris of A/J mice at 30 weeks of age. There was no difference in the gene expression level of peroxisome proliferative activated receptor-γ coactivator 1α (PGC-1α) in SJL and A/J mice at 10 and 30 weeks of age compared with that of the control.
U
Skeletal muscle atrophy-associated genesU:
Expression of myostatin mRNA was decreased in the longissimus lumborum of SJL mice at 30 weeks of age. There was no difference in the gene expression level of follistatin in the rectus femoris of SJL and A/J mice at 10 and 30 weeks of age compared with that of the control.
U
Ubiquitin-proteasome system-associated genesU:
The gene expression level of atrogin-1/muscle atrophy F-box (MAFbx) in the rectus femoris of BALB/c mice at 30 weeks of age was higher than that of the control. On the other hand, the gene expression level of atrogin-1/MAFbx in the rectus femoris and longissimus lumborum of SJL mice at 10 weeks of age was lower than that of the
lumborum of SJL mice at 30 weeks of age was down-regulated compared with that of the control.
U
Ubiquitin-like protein geneU:
There was no difference in the gene expression level of ubiquitin-fold modifier 1 (Ufm1) in the rectus femoris and longissimus lumborum of BALB/c mice at 30 weeks of age, SJL and A/J mice at 10 and 30 weeks of age compared with that of the control.
U
ER stress-associated genesU:
There were no changes of ER stress-associated genes (immunoglobulin heavy-chain binding protein (Bip)/78 kDa glucose-regulated protein (Grp78), Atf6, C/EBP homologous protein (Chop)/Gadd153) in the rectus femoris and longissimus lumborum of BALB/c mice at 30 weeks of age, SJL and A/J mice at 10 and 30 weeks of age compared with that of the control.
U
Heat shock protein genesU:
The gene expression level of heat shock protein 70 (Hsp70) in the rectus femoris and longissimus lumborum of BALB/c mice at 30 weeks of age was more than 30 times higher than that of the control. The gene expression level of heat shock protein 40 (Hsp40), which is known to be a co-chaperone regulating Hsp70, in the rectus femoris and longissimus lumborum of BALB/c mice at 30 weeks of age was also more than four times higher than that of the control. Hsp70 was up-regulated in the rectus femoris and longissimus lumborum of SJL mice at 10 and 30 weeks of age more than 10 times higher than that of the control. Hsp40 was up-regulated in the rectus femoris and longissimus lumborum of SJL mice at 10 weeks of age more than three times higher than that of the control. However, the gene expression levels of Hsp70 and Hsp40 in SJL mice were lower than those of BALB/c mice at 30 weeks of age.
Hsp70 and Hsp40 were down-regulated in the longissimus lumborum or rectus femoris of A/J mice at 30 weeks of age compared with those of the control. 94 kDa glucose-regulated protein (Grp94) was up-regulated in the longissimus lumborum of SJL mice at 30 weeks of age compared with that of the control.
U
ER-associated degradation of glycoproteins-associated genesU:
ER degradation enhancer, mannosidase alpha-like 1 (Edem1) mRNA in the rectus femoris and longissimus lumborum, and 57 kDa ER protein (Erp57) mRNA in the longissimus lumborum showed high expression levels in SJL mice at 30 weeks of age compared with those in the control.
U
Histone deacetylase genesU:
There was no difference in the gene expression level of histone deacetylase (HDAC) 1, 2, 3 and 8 in the rectus femoris of BALB/c mice at 30 weeks of age, or SJL and A/J mice at 10 and 30 weeks of age compared with that of the control (data not shown).
U
Nitric oxide synthase genesU:
The gene expression level of neuronal nitric oxide synthase (nNOS) in the rectus femoris of SJL mice at 10 weeks of age was lower than that of the control. The inducible nitric oxide synthase (iNOS) gene in the rectus femoris of SJL and A/J mice at 30 weeks of age was expressed at a lower level than that of the control.
U
Oxidative stress-associated genesU:
The expression level of heme oxygenase 1 (Hmox1) mRNA in the rectus femoris and longissimus lumborum of SJL mice at 30 weeks of age was more than two times higher than that of the control. On the other hand, thioredoxin reductase 1 (Txnrd1)
level than that of the control.
U
Complement control factor genesU:
The gene expression level of CD55 antigen (decay-accelerating factor: Daf1) in the rectus femoris and longissimus lumborum of SJL mice at 10 and 30 weeks of age was lower than that of the control. Because the Ct values were over 30, the data of decay accelerating factor 2 (Daf2) were treated as informal data. However, the gene expression level of Daf2 in the rectus femoris and longissimus lumborum of most SJL mice at 10 and 30 weeks of age fell below the detectable limit, while the threshold cycle was recorded as mathematical values in quantitative RT-PCR analysis of BALB/c and A/J mice.
Discussion
This study showed that there were differences in the gene expression profiling of skeletal muscles between SJL and A/J mice.
SJL mice have a mutation in the Tbc1d1 gene that results in a truncated protein lacking the TBC Rab–GTPase-activating protein domain (Chadt et al., 2008). Recombinant congenic mice lacking TBC1D1 showed reduced body weight, decreased respiratory quotient, increased fatty acid uptake/oxidation and reduced glucose uptake in isolated skeletal muscle. Meanwhile, transgenic mice, in which skeletal muscle-specific overexpression of PGC-1α was induced, showed reduced body weight, increased fatty acid oxidation, reduced glucose transporter 4 (GLUT4) mRNA expression and atrophy of skeletal muscle, especially type 2B fiber-rich muscles, with increasing mitochondrial number and replacement of atrophic muscle fibers with adipocytes (Miura et al., 2003 and 2006). It is estimated that ATP deprivation associated with increased uncoupling of oxidative phosphorylation in the skeletal muscles causes atrophy of type 2B fiber-rich muscles in PGC-1α transgenic mice. Because there are some similarities between SJL and PGC-1α transgenic mice, we analyzed the expression of lipid metabolism associated genes (PGC-1α, Ucp2 and Ucp3) in the skeletal muscles of SJL and A/J mice. As a result, expression of Ucp2 increased more than twice, and that of Ucp3 mRNA reduced slightly in the rectus femoris and longissimus lumborum of SJL mice. The reduced ATP production may affect the process of skeletal muscle damage in SJL mice.
muscle mass (Joulia-Ekaza and Cabello, 2006) in the longissimus lumborum of SJL mice at 30 weeks of age. It is known that myostatin mRNA dramatically declines during muscle regeneration following an injury (Armand et al., 2003; Shibata et al., 2006). Centrally nucleated fibers as a regenerative change were observed in the skeletal muscles of SJL mice at this age. Therefore, the down-regulation of myostatin in SJL mice appears in the repair process of skeletal muscles at 30 weeks of age. Fibrosis was scarcely apparent and alpha-smooth muscle actin-positive myofibroblasts contributing to fibrosis were not observed in any skeletal muscle lesions of SJL mice as identified in Chapter 1. Because myostatin directly regulates skeletal muscle fibrosis (Li et al., 2008), it is estimated that these histopathological findings are associated with the down-regulation of myostatin in SJL mice.
Atrogin-1/MAFbx up-regulated in the rectus femoris of BALB/c mice at 30 weeks of age was ubiquitin-ligase E3 related to muscle atrophy (Kandarian and Jackman, 2006). However, the gene expression of other ubiquitin ligase E3 (MuRF1) and its transcription-regulated factor (Foxo3) was not altered and there was no atrophic change of skeletal muscles in BALB/c mice at this age. Therefore, the cause of up-regulation of atrogin-1/MAFbx is not known.
There was no difference in the gene expression level of ER stress-associated genes (Bip/Grp78, Atf6 and Chop/Gadd153). This result was consistent with the author’s previous conclusion through analyses of spliced XBP1 mRNA, in that ER stress did not affect the progression of skeletal muscle lesions in SJL mice, as indicated in Chapter 2.
Although the gene expression of Hsp70 and Hsp40 was up-regulated in the rectus femoris and longissimus lumborum of BALB/c mice at 30 weeks of age, there were no
causative lesions in these muscles. Atrogin-1/MAFbx was also up-regulated in BALB/c mice at this age. Hsp70 suppresses the gene expression of atrogin-1/MAFbx and MuRF1 through inhibition of forkhead box O3 (Foxo3) and nuclear factor-kappa B (NF-κB) transcriptional activities (Senf et al., 2008). The up-regulation of Hsp70 and Hsp40 genes may show an adaptive response for atrogin-1/MAFbx-induced factor. In SJL mice, Hsp70 was sustainably up-regulated in the rectus femoris and longissimus lumborum at 10 and 30 weeks of age, and Hsp40 showed a high level of gene expression in the same muscles at only 10 weeks of age. Skeletal muscle lesions in SJL mice began to be observed from 10 weeks of age. As the author discusses later, because Hmox1 mRNA as an oxidative stress marker was expressed at a high level, it is estimated that induction of Hsp70 and Hsp40 as well as induction of Hmox1 reflects a protective mechanism for skeletal muscle damage. On the other hand, the gene expression of Hsp70 and Hsp40 in the rectus femoris and longissimus lumborum of A/J mice was down-regulated. The loss of fer-1, dysferlin homolog, in C. elegans causes down-regulation of hsp-70 (Krajacic et al., 2009). It is possible that the down-regulation of Hsp70 gene expression in the skeletal muscles of A/J mice is caused by the functional loss of dysferlin. Hsp70 in SJL mice may be induced by a different pathway from the dysferlin-mediated pathway
In addition, Grp94 was up-regulated in the longissimus lumborum of SJL mice at 30 weeks of age. Grp94 is localized in the ER, and its overexpression is accompanied by accelerated myotube formation; thus, cell-surface expression of Grp94is necessary for maintenance of fusion competence (Gorza and Vitadello, 2000). As previously mentioned, because regenerative change was observed in the skeletal muscles of SJL
the myotube formation as part of a repair process of skeletal muscles at 30 weeks of age.
Edem1 and Erp57 mRNA in the rectus femoris and/or longissimus lumborum showed high expression levels in SJL mice at 30 weeks of age. Edem1 accelerates ER-associated degradation (ERAD) by stimulating de-mannosylation of terminally misfolded glycoproteins (Olivari et al., 2006). Erp57, a member of the protein disulfide isomerase family, is also an important component involved in the calnexin/calreticulin system for folding of newly synthesized proteins and glycoproteins (Michalak et al., 2009). The up-regulation of these genes was not accompanied by the up-regulation of genes associated with ERAD, and therefore, the significance of these gene changes is unknown.
Histone acetylation plays a critical role in both transcription initiation and elongation of Hsp70 gene (Zhao et al., 2006). In addition, nitric oxide (NO) is a key regulator of multiple HDAC functions in mammalian neurons (Watson and Riccio, 2009). Consequently, the author quantitated the gene expression levels of NOS (nNOS, iNOS, eNOS) and HDAC (HDAC 1, 2, 3, 8). However, there was no remarkable change in the gene expression of HDAC, and SJL and/or A/J mice showed low expression levels of nNOS and iNOS genes. Accordingly, it was concluded that HDAC and NO were not related to the induction of Hsp70 and Hsp40 in BALB/c or SJL mice.
Hmox1 provides the first line of defense against oxidative stress because it rapidly responds to oxidants (Islam et al., 2008). Txnrd1 regulates the antioxidant functions of thioredoxin in cytoplasm (Hama et al., 2009; Nordberg and Arnér, 2001). It is considered that up-regulation of Hmox1 in SJL mice at 30 weeks of age, as well as
up-regulation of Hsp70 and Hsp40, indicates a protective process for skeletal muscle damage.
Hmox1 induction in the skeletal muscles follows a fiber type-specific pattern, and is expressed predominantly in the soleus muscle, which contains a high percentage of red fibers (type I and IIA predominant), whereas Hmox1 mRNA in the extensor digitorum longus (EDL) muscle (type IIB predominant) was only slightly affected (Vesely et al., 1999). In addition, the levels of inducible Hsp70 continuously accumulate in the soleus muscle following exercise, while they rise only transiently in the EDL muscle (Hernando and Manso, 1997). Notably, numerous studies have shown that the soleus muscle and, in general, muscles rich in type I fibers are more resistant to oxidative injury mediated by ischemia-reperfusion than either EDL or other muscles composed primarily of type IIB fibers (Woitaske and McCarter, 1998; Bushell et al., 1995; Fridén et al., 1994). Otherwise, the results of enzyme-histochemical stainings revealed that degeneration of fast-twitch type (type II) fibers was predominant in SJL mice as shown in Chapter 1. There is a possibility that the up-regulation of Hmox1 and Hsp70 protects the slow-twitch type (type I) fibers from any injurious factors in SJL mice.
It was reported that the gene expression of Daf1/CD55 as a complement inhibitor was down-regulated in the skeletal muscles of LGMD2B patients or SJL mice (Wenzel et al., 2005). Moreover, the serum concentration of the fifth component of complement (C5) in SJL mice is known to be significantly greater than that of other strains (Lynch and Kay, 1995). When checking the gene expression of Daf1/CD55 in A/J mice, there was no predominant difference in the gene expression levels of
difference in sensitivity to complement dependent cytotoxicity causes the difference in phenotype between the two dysferlin-deficient mice.
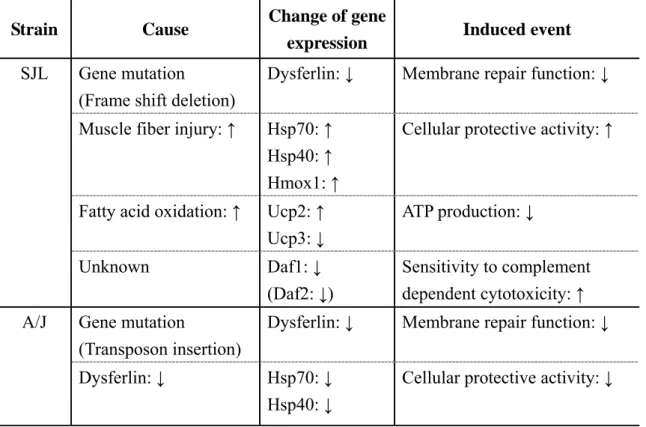
This result confirms that there are some interstrain differences in the pathogenesis process of skeletal muscle lesions between A/J and SJL mice, and supports the hypothesis that additional enhancers or modulators may be involved in the pathogenesis of skeletal muscle lesions in dysferlinopathy. Meanwhile, the dysferlinopathies in humans are a clinically heterogeneous group of disorders ranging from asymptomatism to severe functional disability (Nguyen et al., 2007). If the above hypothesis is verified, the author may be able to indicate and explain the cause of phenotype diversity in human dysferlinopathies. Although the effect of heat shock proteins on the protective process, alteration of ATP production associated with uncoupling proteins, oxidative stress, and the difference in sensitivity to complement dependent cytotoxicity throughout complement inhibitors are considered as candidate causes of interstrain differences in the pathogenesis process between two strains (Table 5), the verification of these hypotheses requires further investigation.
Summary
In this Chapter, the expression of 37 genes including ER stress-associated genes other than XBP-1 (examined in Chapter 2) at the age of 10 and 30 weeks were investigated by quantitative RT-PCR to identify factors affecting the progression of skeletal muscle lesions in SJL and A/J mice.
These results confirmed that there were some interstrain differences in the pathogenesis process of skeletal muscle lesions between A/J and SJL mice, and supported the hypothesis that additional enhancers or modulators may be involved in the pathogenesis of skeletal muscle lesions in dysferlinopathy. The effect of heat shock proteins on the protective process, alteration of ATP production associated with uncoupling proteins, oxidative stress, and the difference in sensitivity to complement dependent cytotoxicity throughout complement inhibitors are considered as candidate causes of interstrain differences in the pathogenesis process between two strains, although the verification of these hypotheses requires further investigation.