アラノンジー静注用250mg
に関する資料
本資料に記載された情報に係る権利及び内容の責任はグラクソ・スミ
スクライン株式会社に帰属するものであり、当該情報を適正使用以外
の営利目的に利用することはできません。
グラクソ・スミスクライン株式会社
第
1 部及び第 2 部の略号等一覧
化学名及び構造式 名称 化学名 構造式 ネララビン/Nelarabine(JAN) (治験成分記号:506U78) 2-アミノ-9-β-D-アラビノフラ ノシル-6-メトキシ-9H-プリン 2-Amino-9-β-D -arabinofuranosyl-6-methoxy-9H-purine 略号及び略称 略語(略称) 内容 ADA アデノシンデアミナーゼ AE 有害事象 A/G 比 アルブミン/グロブリン比 ALL 急性リンパ芽球性白血病 ALT アラニンアミノトランスフェラーゼ AML 急性骨髄性白血病 ANC 好中球数 ara-G 9-β-D-アラビノフラノシルグアニン ara-GMP 9-β-D-アラビノフラノシルグアニン一リン酸 ara-GTP 9-β-D-アラビノフラノシルグアニン三リン酸 AST アスパラギン酸アミノトランスフェラーゼ ATS 治療を受けた全患者 AUC 血漿中濃度-時間曲線下面積 (ara-GTP の場合は、最高細胞内濃度-時間曲線下面積) BDC 胆管カニュレーション処置 BFM Berlin-Frankfurt-Munich(欧州の癌研究グループ名) BMT 骨髄移植 BUN 血中尿素窒素 CEM T リンパ芽球系株化細胞 CI 信頼区間 CIV 静脈内持続注入 H N N N N O O H H O H O H H H O C H 3 N H2略語(略称) 内容 Cmax 最高血漿中濃度 (ara-GTP の場合は、最高細胞内濃度) CL クリアランス CL/F 見かけのクリアランス CLcr クレアチニン クリアランス
CLL chronic lymphocytic leukemia, 慢性リンパ性白血病
CML chronic myelogenous leukemia, 慢性骨髄性白血病
CNS 中枢神経系 CR 完全寛解 CR* 造血の回復を伴わない完全寛解を含めた完全寛解 CRF 症例報告書 CRh* 再治療可能なレベルまでの造血の回復を伴う完全寛解 CLp 血漿クリアランス CSF 脳脊髄液 CT 化学療法 CTCL 皮膚T 細胞リンパ腫 CTCv2 共通毒性規準 第2 版 CTEP 癌治療評価プログラム CV 変動係数 CVA 脳血管発作 CYP チトクロームP450 dCF デオキシコホルマイシン dCK デオキシシチジンキナーゼ dGK デオキシグアノシンキナーゼ DFS 無病生存期間 dGTP デオキシグアノシン5'‐三リン酸 dGuo デオキシグアノシン DLT dose-limiting toxicity, 用量制限毒性 DNA デオキシリボ核酸 EHNA erythro-9-(2-hydroxy-3-nonyl)adenine FDA 米国食品医薬品局 FR frank relapse, 明らかな再発(有効性評価に関する略号) GSK グラクソ・スミスクライン HIV ヒト免疫不全ウイルス HLGT 高位グループ用語(ICH 国際医薬用語集)
略語(略称) 内容
IC50 50%阻害濃度
IMP hematological improvement, 血液学的改善(有効性評価に関する略号)
IND 新薬治験申請
IT 髄腔内の
IV 静脈内の
KPS Karnofsky による一般状態
LBL リンパ芽球性リンパ腫
LC-MS/MS Liquid chromatography - Tandem Mass Spectrometry
LDH lactate dehydrogenase, 乳酸脱水素酵素 LPD lymphoproliferative disorder, リンパ増殖症候群 max 最大 mCR 骨髄の完全寛解 MDCKⅡ-MDR1 MDR1 を発現させた MDCKⅡ MDR-1 Multidrug resistance 1 MedDRA ICH 国際医薬用語集 mPR 骨髄の部分寛解
MRD minimal residual disease, 微小残存病変
MRI (核)磁気共鳴映像法
MSC 精神状態変化
MST 生存期間中央値
MTD maximum tolerated dose, 最大耐量
NA Not applicable (該当なし)
NCI National Cancer Institute, 米国立がん研究所
NHL non-Hodgkin's lymphoma, 非ホジキンリンパ腫
NR no response, 反応なし(有効性評価に関する略号)
ODAC Oncologic Drugs Advisory Committee
OS 全生存期間
PCR Polymerase Chain Reaction
PD 薬力学 PD progressive disease, 進行(有効性評価に関する略号) Pgp P-糖蛋白質 PK 薬物動態 PL 前リンパ球性白血病 PLL prolymphocytic leukemia PNP プリンヌクレオシドホスホリラーゼ PNS 末梢神経系 PR 部分寛解
略語(略称) 内容 PTCL 末梢性T 細胞リンパ腫 RNA リボ核酸 SAE 重篤な有害事象 SCT 幹細胞移植 SD 標準偏差 SD stable disease, 安定(有効性評価に関する略号) SGOT 血清グルタミン酸-オキサロ酢酸トランスアミナーゼ (アスパラギン酸・アミノ酸基転移酵素) SGPT 血清グルタミン酸-ピルビン酸トランスアミナーゼ (アラニン・アミノ酸基転移酵素) SOC 器官別大分類 SWOG 米国南西部臨床試験グループ t1/2 消失半減期 tmax 最高血漿中濃度到達時間 (ara-GTP の場合は、最高細胞内濃度到達時間) TK トキシコキネティクス T-ALL T 細胞急性リンパ芽球性白血病 T-CLL T 細胞慢性リンパ性白血病 T-LBL T 細胞リンパ芽球性リンパ腫
T-LPD T-cell lymphoproliferative disorder
T-NHL T-cell non-Hodgkin's lymphoma, T 細胞非ホジキンリンパ腫
Vss 定常状態における分布容積
Vss/F 定常状態における見かけの分布容積
WBC 白血球数
WHO World Health Organization, 世界保健機関
1.4. 特許状況
特
1.5. 起原又は発見の経緯及び開発の経緯 1.5.1. 起原又は発見の経緯 ネララビンは、造血器悪性腫瘍の治療薬として開発中のデオキシグアノシン(dGuo)誘 導体で、9-β-D-アラビノフラノシルグアニン(ara-G)のプロドラッグである。dGuo は、細 胞内でデオキシシチジンキナーゼ(dCK)及びデオキシグアノシンキナーゼ(dGK)により リン酸化されてデオキシグアノシン5’-三リン酸(dGTP)となり、DNA 生合成を阻害して 細胞死をもたらす。また、dGuo のリン酸化反応は B 細胞に比べて T 細胞で比較的高いこと
から[Carson, 1977]、dGuo は T 細胞系の腫瘍に対する殺細胞作用が期待された[Carson, 1979; Cohen, 1978]。しかしながら、天然の dGuo はプリンヌクレオシドホスホリラーゼ(PNP)に より異化されることから、PNP による分解に対し抵抗性を有する dGuo 誘導体の開発を行い、 ara-G を見出した。更に、ara-G は水溶性が低いことから、水溶性の高いプロドラッグの創出 をめざし、ネララビンを見出した。 ネララビンは末梢血中でアデノシンデアミナーゼ(ADA)によって速やかに脱メチル化さ れara-G となり、更に細胞内で dCK 及び dGK を介して三リン酸化体(9-β-D-アラビノフラ ノシルグアニン三リン酸、ara-GTP)に変換される。dCK はリンパ系組織に高濃度存在し、 また、その中でも未分化のT 細胞である T リンパ芽球系細胞で高い[Arner, 1995]。dGuo と同 様に、T 細胞における ara-G のリン酸化反応性は B 細胞に比べて高く、ara-GTP の分解は逆 にB 細胞より遅いことから、T 細胞において細胞内 ara-GTP 濃度が高くなる。ネララビンの 主たる作用機序は、活性化されたara-GTP が増殖細胞の DNA に取り込まれ、チェーンター ミネーターとして作用してDNA 合成を停止させることである。 1.5.2. T 細胞急性リンパ芽球性白血病(T-ALL)/T 細胞リンパ芽球性リンパ腫(T-LBL)の臨床的/病態生理学的側面 急性リンパ芽球性白血病(ALL)及びリンパ芽球性リンパ腫(LBL)は、B 細胞系又は T 細胞系の未熟な段階のリンパ芽球の腫瘍である。ALL では腫瘍細胞は骨髄、末梢血を占め、 LBL ではリンパ節又は節外に腫瘤を形成する。病変が腫瘤性で骨髄や末梢血に明らかな腫瘍 細胞の浸潤を認めない場合はLBL と診断される。一方、骨髄や末梢血における腫瘍細胞の 広範な浸潤がみられる場合には通常ALL と診断される。腫瘤性病変があり、骨髄での腫瘍 細胞の浸潤が25%以下の場合は LBL の名称が使われることが多い[陣内, 2004]。 本邦における白血病の年間新規発症患者数は、8,916 人(2005 年)で、このうち小児は 515 人、成人は 8,401 人(2005 年)と推計される[がんの統計編集委員会, 2003; 国立社会保 障・人口問題研究所, 2002]。小児では急性白血病が 95%、慢性白血病が 5%で、急性白血病 のうちリンパ性白血病は80%を占め[林, 2003]、更にその 15%が T 細胞系に分類されるので [陣内, 2004]、小児 T-ALL の年間新規発症患者数(2005 年)は 60 人程度であると推計される。 成人では、急性リンパ性白血病は成人白血病全体の20%を占め、その 20%が T 細胞系と考 えられているので[直江, 2003]、成人 T-ALL の年間新規発症患者数(2005 年)は 300 人程度 であると推計される。 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 1
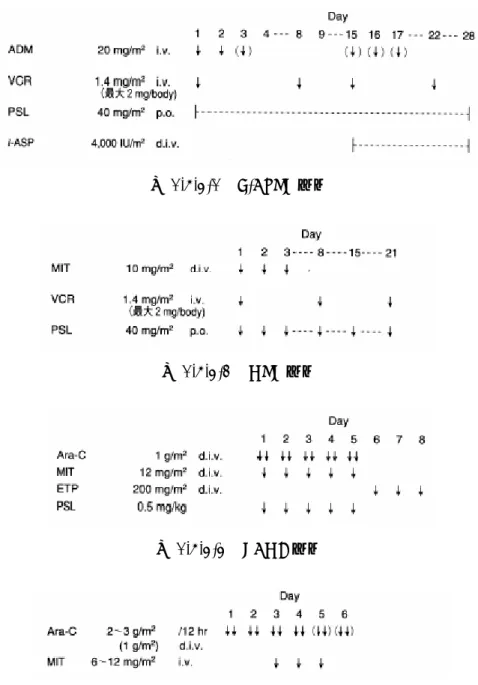
本邦における悪性リンパ腫の年間新規発症患者数は、13,303 人(2005 年)で、このうち小 児は143 人(2005 年)、成人は 13,160 人(2005 年)と推計される[がんの統計編集委員会, 2003; 国立社会保障・人口問題研究所, 2002]。小児では悪性リンパ腫のうち非ホジキンリン パ腫が90%を占め、更に非ホジキンリンパ腫の 30%はリンパ芽球性リンパ腫で、その多くが T 細胞性と考えられているので、小児 T-LBL の年間新規発症患者数(2005 年)は 40 人程度 である。成人では、悪性リンパ腫の1.72%が T-LBL と考えられているので[直江, 2003]、成 人T-LBL の年間新規発症患者数(2005 年)は 200 人程度であると推定される。 1.5.3. T-ALL/T-LBL の治療に対する現状と問題点 ALL の治療の目標は疾患の治癒である。ALL の治療法には、化学療法、放射線療法、造 血幹細胞移植があるが、治療の基本は、ビンクリスチン、副腎皮質ステロイド、ドキソルビ シン(慣用名:アドリアマイシン)、L-アスパラギナーゼ、シクロホスファミド、メトトレ キサートを中心とする複数の抗がん剤を用いた化学療法である。造血幹細胞移植は化学療法 では治癒が困難と考えられる場合に行われる。放射線療法は、白血病細胞の中枢神経への浸 潤予防・治療、移植の前処置に用いられる。 化学療法は、まず完全寛解(CR)を目的とした寛解導入療法に続き、寛解後療法(地固 め療法、強化療法)を行うのが一般的である。近年確立された標準的治療により、小児 ALL の 65%以上[Horibe, 2001]、成人 ALL の 30~40%[Takeuchi, 2002; Kantarjian, 2000]が長期 生存・治癒するようになってきた。しかしながら、急性白血病の寛解導入療法は多種の薬剤 を使用し、白血病細胞の根絶を目指して再発させないことを目標として行われているため、 再発白血病は前治療薬に対して薬剤耐性を獲得していることが多く、治療抵抗性を示す場合 が多い。再発した白血病又は難治性白血病の標準的な治療法は確立されておらず、本邦並び に海外において確立された診療ガイドラインやアルゴリズムは存在しない。寛解導入療法に 反応しない難治例や再発例の予後は極めて不良であり、その長期生存率は10%未満であると 考えられている[第7回 未承認薬使用問題検討会議, 2006]。 米国においてALL の適応で承認されている薬剤は、ビンクリスチン、シタラビン、ドキ ソルビシン、ダウノルビシン、L-アスパラギナーゼ、メトトレキサート、メルカプトプリン、 シクロホスファミド、テニポシド(teniposide、国内未発売)などである。再発・難治性の ALL に対する治療はこれらの中から交叉耐性の少ないと考えられる薬剤の組み合わせや大量 療法が一般的に用いられ、また、小児の再発・難治性ALL に対しては、クロファラビン (clofarabine、国内未発売)が適応を取得している。再発・難治性 ALL の治療として使用す る薬剤は、耐性を獲得している可能性の低い薬剤を選択することが基本とされるが、前治療 で使用した薬剤を再度用いても効果のある場合もあるため、薬剤選択は経験的になされるこ とが多く、確立した標準的な治療法はない。 本邦における再寛解導入療法においても、寛解導入療法に使用する薬剤もしくは大量のシ タラビン(国内販売名:キロサイドN 注)を併用投与することが多い。再発・難治性の ALL に対する治療プロトコールの例を図 1.5.3-1~図 1.5.3-4 に示す[大野, 2001]。 1.5. 起原又は発見の経緯及び開発の経緯
図 1.5.3-1 L-AdVP 療法
図 1.5.3-2 MVP 療法
図 1.5.3-3 PAME 療法
図 1.5.3-4 Ara-C 大量療法と中等量療法(MIT 併用)
ADM: adriamycin, VCR: vincristine, PSL: prednisone, l-ASP: l-asparaginase, MIT: mitoxantrone, Ara-C: cytarabine, ETP: etoposide
T-LBL は B 細胞 LBL と同様に、悪性リンパ腫の臨床病理分類においてリスクが高いリン パ腫として位置付けられており[Hiddemann, 1996]、T-ALL に準じた治療方針が選択されるが [福原, 2001]、白血化や中枢神経浸潤などを伴うことが多く、初回寛解療法に反応したとして も再発することが多い。再発・難治性のT-LBL は、T-ALL の再発時と同様の治療が行われ ているものの、T-ALL 同様、本邦並びに海外において確立された診療ガイドラインやアルゴ リズムは存在せず、現時点では標準的な治療法はなく予後は不良である。 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 3
再発・難治性のALL/LBL は、再寛解導入療法により再度寛解が得られても一般的に寛解 持続期間は短く長期生存を期待することができない。米国小児悪性腫瘍グループ(COG)が
実施した新規ALL 発症患者を対象とした 2 つの臨床試験(CCG-1952 及び CCG-1961)にお
いて、2 度目の再発を来たした T-ALL 患者において既存治療を行った際の生存率を示す(図 1.5.3-5)。生存率の解析対象は、CCG-1952 及び CCG-1961 試験において寛解導入療法を受
けた後に再発した86 例の ALL 患者(T-ALL:13 例、B-ALL:73 例)であり、これらのすべ
ての被験者における2 度目の再発日を起点とした 1 年後生存率は 9.5%であった。なお、13 例のT-ALL 患者は、2 度目の再発後 183 日以内にすべて死亡した[Sather, 2005]。 0 13 26 39 52 65 78 91 104 117 130 143 Time (Weeks) 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 Pr opo rt io n A li ve 0 13 26 39 52 65 78 91 104 117 130 143 Time (Weeks) 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 Pr opo rt io n A li ve 図 1.5.3-5 T-ALL の 2 度目の再発における既存治療下での生存率(N=13) [Sather, 2005]から一部改変 このように再発・難治性のT-ALL/T-LBL に対する既存の化学療法の効果は限定的である ため、長期生存をめざすには骨髄移植が必要となる[大野, 2004]。移植片対宿主病などの移植 関連合併症の発現が加齢とともに増加するものの、造血幹細胞移植の治療効果は比較的高い。 日本造血細胞移植学会の報告では、ALL の第 2 寛解期における造血幹細胞移植の 5 年後生存 率は小児で59.2%、成人で 32.0%であり、第 3 寛解期における同 5 年後生存率は小児で 31.0%、成人で 20.9%である[平成 13 年度全国調査報告書, 2001]。一方、非寛解期における同 5 年後生存率は小児で 22.6%、成人で 11.3%であり、第 2 寛解期及び第 3 寛解期における造 血幹細胞移植の5 年後生存率と比較して著しく劣る(表 1.5.3-1)。 1.5. 起原又は発見の経緯及び開発の経緯
表 1.5.3-1 1 回目の造血幹細胞移植を受けた ALL の病期別 5 年後生存率 [平成 13 年度全国調査報告書, 2001]を一部改変 小児 成人 例数 5 年後生存率 例数 5 年後生存率 第一寛解期 390 70.3% 819 50.8% 第二寛解期 363 59.2% 275 32.0% 第三寛解期 91 31.0% 68 20.9% 非寛解期 298 22.6% 399 11.3% 以上のとおり、再発・難治性の成人及び小児T-ALL/T-LBL に対する標準的な治療法は現 時点では確立されておらず、有効な治療薬の医療上の必要性は極めて高い。また、これらの 患者において造血幹細胞移植を行う場合であっても、寛解期における移植の長期生存率は非 寛解期のそれと比較して著しく勝るため、再発・難治性の成人及び小児T-ALL/T-LBL に対 する新たな治療薬の早急な開発が望まれている。本剤はこのような再発・難治性の患者にお いても、後述のとおり(1.5.4.1.2 参照)、海外臨床試験において高い奏効率が認められてお り、本剤のような奏効率の高い新たな治療薬が本邦においても開発されることにより、造血 幹細胞移植の成功率を上げるとともに、長期生存をめざして移植を実施する機会を増やすこ とにも貢献できるものと期待される。 1.5.4. 開発の経緯 非臨床試験は1991 年から開始され、薬理、薬物動態及び毒性の評価が行われ、臨床的検 討は海外において1994 年より開始された。 1.5.4.1. 海外における開発の経緯 1.5.4.1.1. 非臨床試験 1991 年からネララビンを候補品として、抗悪性腫瘍用薬としての臨床使用を目的とする 本格的な開発が開始され、薬理試験、薬物動態試験、毒性試験を実施するとともに、合成法、 物理化学的性質が検討された。一連の非臨床試験成績でネララビンはara-G と同程度の細胞 障害活性を示し、毒性試験において認められた神経毒性は臨床使用において十分注意すべき であるものの、リスク・ベネフィットの観点から造血器悪性腫瘍の治療薬としては許容の範 囲内であり、ヒトへの投与を阻むものではないと考えられ、1994 年より臨床試験を開始し た。本剤は細胞障害性の抗悪性腫瘍剤であり、適用対象疾患は難治性・再発性の進行性がん であることなどを踏まえ、安全性薬理試験及び一部の毒性試験は実施されていない。 1.5.4.1.2. 臨床試験 海外においては、成人及び小児の再発・難治性T-ALL/T-LBL をはじめとする難治性の造 血器悪性腫瘍を対象としてGlaxoSmithKline 社(GSK 社)並びに米国立がん研究所(NCI) がネララビンの臨床試験を実施した。海外で実施した臨床試験及びcompassionate use 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 5
(special exceptions)において 20 年 月末現在までに900 人以上に対してネララビンが投 与されている。 第Ⅰ相臨床試験 第Ⅰ相臨床試験(臨床薬理試験)は1994 年から 4 試験を実施した。 1994~1997 年に実施した最初の第Ⅰ相臨床試験(PGAA1001 試験)では、成人及び小児の 難治性造血器悪性腫瘍の再発例を対象に、5 日間連日投与のスケジュールにより、ネララビ ンを5~75mg/kg の範囲で漸増的に投与した。本試験では、用量制限毒性と判断されたグレ ード3 及び 4 の神経系障害が、成人及び小児で各々40 及び 60mg/kg 以上で発現したため、ネ ララビンの第Ⅱ相臨床試験における用量は、成人及び小児とも 1200mg/m2(成人約 30mg/kg、 小児約39mg/kg)の 5 日間連日投与が妥当であると考えられた。1997 年からは、用法・用量 を更に検討するため、3 日間連日投与による第Ⅰ相臨床試験(PGAA1002 試験)並びに 1、3、 5 日目の隔日投与による第Ⅰ相臨床試験(PGAA1003 試験)を並行して実施した。これらの 試験で成人及び小児における安全性及び有効性を検討した結果、成人では2200mg/m2の1、 3、5 日目の隔日投与、小児では 900mg/m2又はそれ以上の5 日間連日投与が推奨用量と考え られた。更に、ネララビン1200mg/m2とフルダラビン30mg/m2を併用して隔日投与する相互 作用試験(PGAA1005 試験)も実施した。 第I 相臨床試験の 4 試験で合計 181 例の患者(成人 141 例、小児 40 例)が組み入れられた。 それぞれの試験の各用量群の患者数が比較的少なかったことから、ネララビン、ara-G 及び 細胞内ara-GTP の薬物動態/薬力学(PK/PD)特性を明らかにするために、試験を通した探索 的な解析を実施した(2.5.3 参照)。 ネララビン、ara-G 及び細胞内 ara-GTP の薬物動態には大きな個人差が認められた。血漿 中のネララビン及びara-G の Cmaxは通常、静脈内投与終了時に得られ、半減期はいずれも短 く(ネララビン:<30 分、ara-G:約 2~3 時間)、連日投与又は隔日投与しても蓄積は生じ なかった。一方、白血病芽球内のara-GTP は概して消失が遅く、1、3 及び 5 日目に隔日投与
するレジメンで蓄積が認められた。また、細胞内ara-GTP の AUC 及び Cmaxは、奏効例(CR
又は部分寛解(PR))の患者では非奏効例の患者よりも高値であった。ネララビン及び ara-G の曝露量には臨床反応との関連性がみられなかった。細胞内 ara-ara-GTP の AUC0-tは、女性成 人患者の方が男性成人患者よりも高値であった。その他の変数(年齢、人種、体表面積、疾 患分類、投与開始前のCLcr概算値、試験)による細胞内ara-GTP の曝露量への明らかな影響 はなかった。(2.7.2 参照) 第Ⅰ相臨床試験を通した探索的なPK/PD の検討の結果、試験中の時期を問わない神経系 障害の指標の発現はネララビンの1日用量及び患者の年齢により良く予測しうることが示唆 された。ネララビンの1 日用量の増加と年齢の上昇、特に患者の年齢が 65 歳以上であるこ とは、概してすべての神経系障害の指標の発現率の上昇との関連性が認められた。1 サイク ル中の累積投与量と神経系障害との関連を検討すると、同じ累積投与量を1 サイクル中に 5 1.5. 起原又は発見の経緯及び開発の経緯
日間かけて投与した場合よりも3 日間で投与した方が神経系障害の発現が多かった。(すな わち、1 日用量の高い方が神経系障害の発現率が高かった。)1 日用量カテゴリー別の投与 日数と神経系障害との関連を検討すると、1 サイクル当たりの投与日数(3 日又は 5 日)に は関係なく、1 日用量が同様であれば、神経系障害の発現頻度は同様であった。曝露量の薬 物動態学的指標(Cmax、AUC)と神経系障害の発現との関連は、一部の例外を除いてみられ なかった。 血液毒性(好中球減少症、血小板減少症、貧血)は成人患者及び小児患者とも多く認めら れたが、ネララビンの血液に及ぼす影響は既知であり、治験対象患者の基礎疾患でもあるた め、用量制限毒性とはみなされなかった(2.7.2 参照)。 第Ⅱ相臨床試験 第Ⅱ相臨床試験は、1997 年から GSK 社と NCI により 3 試験が実施されている。 1. 小児を対象とした臨床試験(PGAA2001 試験) 再発・難治性の小児T-ALL/T-NHL 患者を対象として行われた PGAA2001 試験においては、 ネララビン400、650、900 又は 1200mg/m2を1~5 日目に静脈内投与し、その後 16 日間休薬 する21 日間を 1 サイクルとする用法・用量で実施するよう計画していたが、最初にネララ ビン1200mg/m2を1~5 日目に投与された被験者においてグレード 4 の神経系障害が報告さ れたため、本試験における最高投与量は900mg/m2と設定した。しかし、900mg/m2へ減量し た後にもネララビンと因果関係の否定できない重篤な神経系障害の有害事象が報告されたた め、投与量を更に減量し650mg/m2とした。 650mg/m2投与例におけるCR 率は、本試験組入れ前の寛解導入療法の回数が 1 回 (Stratum 1)の被験者では 42%(13/31)であり、2 回以上(Stratum 2)では 13%(5/39)で あった。CR の持続期間はばらつきが大きく、寛解導入療法の回数が 1 回の被験者では 0.9~ 260.0 週以上、2 回以上では 4.7~36.4 週であった。寛解導入療法の回数が 1 回の被験者にお いてCR が得られた 13 例のうち 9 例では CR が 4 週間以上持続し、7 例では 130 週間以上持 続した。 造血の回復を伴わない完全寛解を含めた完全寛解(CR*)率は、組入れ前の寛解導入療法 の回数が1 回の被験者では 48%(15/31)、2 回以上では 23%(9/39)であり、CR*持続時間 は、寛解導入療法の回数が1 回の被験者では 0.9~260.0 週以上、2 回以上では 3.3~42.1 週で あった。 1 年後生存率は、寛解導入療法の回数が 1 回の被験者では 33%であり、2 回以上では 14% であった。CR が得られた被験者における生存期間は、寛解導入療法の回数が 1 回の被験者 では7.9~262.6 週間以上であり、2 回以上では 16.6~57.4 週間であった(表 1.5.4-1、2.5.4 参 照)。 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 7
表 1.5.4-1 PGAA2001 試験(小児)における CR/CR*率及び生存期間 表2.5.4-2 を再掲 1 回の寛解導入療法 31 例 2 回以上の寛解導入療法 39 例 CR 及びその期間 CR 例数(%) [95%CI] 13 (42%) [25%~61%] 5 (13%) [4%~27%] CR 持続期間(週) 0.9~260.0 以上 4.7~36.4 CR*及びその期間 CR*例数(%) [95%CI] 15 (48%) [30%~67%] 9 (23%) [11%~39%] CR*持続期間(週) 0.9~260.0 以上 3.3~42.1 生存期間及び1 年後生存率 生存期間 中央値(週) [95%CI] 33.3 [24.1~93.6] 13.1 [8.7~17.4] 1 年後生存率(%) [95%CI] 33% [16%~50%] 14% [3%~26%] なお、ネララビン投与直前の寛解導入療法が無効であった難治例におけるCR 率及び CR* 率は、寛解導入療法が1 回の被験者において、各々44%、56%であり、2 回以上の被験者に おいては各々18%、27%であった。また、1 年後生存率は、ネララビン投与前の寛解導入療 法が1 回の被験者においては 56%、2 回以上では 14%であった(2.5.4 参照)。 本試験においてネララビン650mg/m2が投与された84 例における有害事象発現率は 79% (66/84)であり、高頻度に報告された有害事象は血液障害(ヘモグロビン減少:38%、血小 板数減少:30%、白血球数減少:38%及び好中球数減少:37%)であった。血液障害以外の 有害事象のうち、650mg/m2投与例で認められた主な有害事象(発現率が10%以上)は、頭 痛(17%)、トランスアミナーゼ上昇(12%)、血中カリウム減少(11%)、血中アルブミ ン減少、血中ビリルビン減少及び嘔吐(10%)であった。650mg/m2投与例におけるグレード 3 の有害事象発現率は、64%(54/84)であり、グレード 4 以上の発現率は 48%(40/84)であ った。主なグレード3 の有害事象(発現率が 10%以上)は、ヘモグロビン減少(23%)、白 血球数減少(14%)及び好中球数減少(10%)であり、主なグレード 4 以上の有害事象(発 現率が10%以上)は、好中球数減少(26%)、血小板数減少(19%)及び白血球数減少 (17%)であった。 本試験でのネララビン650mg/m2投与例における神経系障害の有害事象発現率は、38% (32/84)であり、5%以上の神経系障害の有害事象は、頭痛(17%)、傾眠(7%)、感覚減 退、末梢性感覚ニューロパシー及び末梢性ニューロパシー(各6%)であった。650mg/m2投 与例におけるグレード3 の神経系障害の有害事象発現率は 14%(12/84)であり、グレード 4 以上の発現率は8%(7/84)であった。グレード 3 の有害事象は、末梢性感覚ニューロパシ ー(6%)、頭痛及び感覚減退(各 4%)、末梢性ニューロパシー及び末梢性運動ニューロパ シー(各2%)、失調、筋緊張亢進、運動機能障害、錯感覚、傾眠(各 1%)であった。また、 グレード4 の有害事象は、痙攣(4%)、頭痛(2%)、大発作型痙攣、第 3 脳神経麻痺、傾 眠、及び第6 脳神経麻痺(各 1%)であり、グレード 5 の有害事象は、てんかん重積状態 1.5. 起原又は発見の経緯及び開発の経緯
(1%)であった。グレード 4 の痙攣・大発作痙攣を発現した 4 例のうち 3 例は、中枢神経 系の疾患を合併していたか、又は、試験に組み入れられる2 ヵ月以内に中枢神経系の病変が 認められた被験者であった。グレード5 のてんかん重積状態を発現した被験者は、既往歴と して2 回の原因不明の発作があった。 なお、650mg/m2投与例において報告された神経系障害の有害事象80 件のうち、試験終了 時までに回復したものは63%(50/80 件)、未回復(死亡例の 1 件を含む):18%(14/80 件)、不明:20%(16/80 件)であった(2.5.5 参照)。 2. 成人を対象とした臨床試験(PGAA2002 試験、PGAA2003 試験) 再発・難治性の成人 T-ALL/T-LBL 患者を対象として行われた PGAA2002 試験においては、 21 日間を 1 サイクルとしてネララビン 1500mg/m2を1、3、5 日目に静注点滴投与した。本試 験におけるネララビンの最高投与量は2200mg/m2と計画されていたが、並行して実施してい たPGAA2003 試験においてネララビン 2200mg/m2が投与された被験者において重篤な上行 性末梢性ニューロパシーが報告されたため、本試験における投与量を1500mg/m2と変更した。 PGAA2003 試験では、フルダラビンが無効である成人の難治性慢性リンパ性白血病 (CLL)患者を対象として、ネララビンの 1500mg/m2又は2200mg/m2を1、3、5 日目に静注 点滴投与した。なお、PGAA2003 試験の対象は難治性 CLL であり、米国で承認された適応 症や本邦において承認申請する効能・効果とは異なるため、本項においては安全性の成績に ついてのみ述べる。 PGAA2002 試験において、ネララビン 1500mg/m2が投与された被験者におけるCR 率は、 本試験組入れ前の寛解導入療法の回数が1 回の被験者では 18%(2/11)であり、2 回以上に おいても18%(5/28)であった。CR の持続期間は寛解導入療法の回数が 1 回の被験者 (CR:2 例)では 51.0 週及び 212.0 週、2 回以上では 15.1~195.4 週以上であった。 CR*率は、組入れ前の寛解導入療法の回数が 1 回の被験者では 27%(3/11)、2 回以上で は21%(6/28)であり、CR*持続時間は、寛解導入療法の回数が 1 回の被験者では 4.7~ 215.0 週であり、2 回以上では 4.0~195.4 週以上であった。 また、1 年後生存率は、寛解導入療法の回数が 1 回の被験者では 36%、2 回以上では 29% であり、これらをあわせた全体では31%であった(表 1.5.4-2、2.5.4 参照)。 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 9
表 1.5.4-2 PGAA2002 試験(成人)における CR/CR*率及び生存期間 表2.5.4-3 を再掲 1 回の寛解導入療法 11 例 2 回以上の寛解導入療法 28 例 合計 39 例 CR 及びその期間 CR 例数(%) [95%CI] 2 (18%) [2%~52%] 5 (18%) [6%~37%] 7 (18%) [8%~34%] CR 持続期間(週) 51.0 及び 212.0 15.1~195.4 以上 15.1~212.0 CR*及びその期間 CR*例数(%) [95%CI] 3 (27%) [6%~61%] 6 (21%) [8%~41%] 9 (23%) [11%~39%] CR*持続期間(週) 4.7~215.0 4.0~195.4 以上 4.0~215.0 生存期間及び1 年後生存率 生存期間 中央値(週) [95%CI] 20.1 [12.0~220] 20.6 [10.4~36.4] 20.4 [12.9~36.4] 1 年後生存率 [95%CI] 36% [8%~65%] 29% [12%~45%] 31% [16%~45%] なお、ネララビン投与直前の寛解導入療法が無効であった難治例におけるCR 率は 18%で あり、この3 例の寛解持続期間は 15.1 週間、29.4 週間及び 156.3 週間以上であった(2.5.4 参 照)。 成人を対象とした第Ⅱ相臨床試験(PGAA2002 試験及び PGAA2003 試験)においてネララ ビン1500mg/m2投与時の有害事象発現率は96%(99/103)であり、主な有害事象(発現率が 20%以上)は、疲労(50%)、悪心(41%)、ヘモグロビン減少(30%)、血小板数減少 (26%)、咳嗽(25%)、好中球数減少及び傾眠(各 23%)、下痢及び嘔吐(各 22%)、便 秘及び浮動性めまい(各21%)、呼吸困難(20%)であった。グレード 3 の有害事象発現率 は59%(61/103)、グレード 4 の有害事象発現率は 26%(27/103)であった。主なグレード 3 の有害事象(発現率が 5%以上)は、疲労(10%)、発熱性好中球減少症(9%)、ヘモグ ロビン減少及び血小板数減少(各7%)、胸水、発熱及び筋力低下(各 5%)であった。また、 主なグレード4 の有害事象(発現率が 2%以上)は、好中球数減少(12%)、血小板数減少 (10%)、疲労、ヘモグロビン減少及び呼吸困難(各 2%)であった。 また、神経障害系の有害事象の発現率は72%(74/103)であった。主な神経系障害の有害 事象(発現率が10%以上)は、傾眠(23%)、浮動性めまい(21%)、感覚減退(17%)、 錯感覚及び頭痛(各15%)及び末梢性感覚ニューロパシー(13%)であった。これらの神経 系障害の有害事象の多くはグレード1 又はグレード 2 であり、グレード 3 の神経系障害の有 害事象発現率は10%(10/103)、グレード 4 は 3%(3/103)であった。2 例以上に発現した グレード3 又はグレード 4 の神経系障害の有害事象は、失調(グレード 3、2 例)、感覚減 退(グレード3、2 例)であり、3 例以上に発現したグレード 3 又はグレード 4 の神経系障害 の有害事象はなかった。 PGAA2003 試験において報告された神経系障害の有害事象は、156 件であった。これらの うち、試験終了時までに回復したものは66%(103/156 件)、未回復(死亡例の 3 件を含 1.5. 起原又は発見の経緯及び開発の経緯
む):34%(53/156 件)であった。PGAA2002 試験においては、61 件の神経系障害の有害事 象が報告されたが、これらの有害事象の転帰に関する情報は入手していない(2.5.5 参照)。 米国及び欧州における申請・承認状況及び外国規制当局による審査 米国においては、前述の成績に基づき、2003 年 10 月にファーストトラックの指定を、 2004 年 8 月には希少疾病用医薬品(オーファンドラッグ)の指定を受け、表 1.5.4-3 に示す 効能・効果で2005 年 4 月に承認申請を行った。本剤は 2005 年 9 月の Oncologic Drugs Advisory Committee(ODAC)に諮られ、成人の適応については満場一致で、小児の適応に ついては11 対 1 で承認が勧告され、2005 年 10 月に申請どおりの効能・効果で承認を取得し た。 表 1.5.4-3 米国における効能・効果 米国における効能・効果 INDICATIONS AND USAGE
ARRANON is indicated for the treatment of patients with cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma whose disease has not responded to or has relapsed following treatment with at least two chemotherapy regimens. This use is based on the induction of complete responses. Randomized trials demonstrating increased survival or other clinical benefit have not been conducted.
ARRANON:米国における本剤の販売名 米国における効能・効果は、少なくとも2 回以上の寛解導入療法に対して再発・難治性の T-ALL/T-LBL であり、1 回の寛解導入療法後の再発・難治例に使用される、いわゆる 2nd line としての効能・効果は取得していないが、これは、19 年 月のFDA との協議におけ る合意に基づき設定されたものである。本邦と同様に米国においてもT-ALL/LBL の 2nd line としての標準的な治療法は、確立しておらず、1st line の薬剤の中から耐性を獲得している可 能性の低い薬剤を経験的に選択することが多い。しかしながら、2nd line として実施されて いる多剤併用化学療法によりある程度の治療効果が期待できることから、2nd line として適 応を取得するのであれば無作為化試験を実施すべきであるとの見解がFDA より示された。 一方、2 回以上の寛解導入療法に対して再発・難治性の T-ALL/T-LBL は、1 回の寛解導入療
法後の再発・難治例よりも更に予後不良であり、このunmet medical needs に対して本剤の有
用性が期待できると判断され、FDA より無作為化、非盲検・非対照試験の成績を受け入れ る姿勢が示された。このように、米国においては、いわゆる2nd line としての効能・効果は 取得していないが、これは本剤の有効性が認められないと判断されたためではない。 米国においては、第Ⅲ相臨床試験を実施することが迅速承認の条件とされ、現在実施中で ある。本試験は30 歳未満の新規 T-ALL 発症者を対象とし、68 週間の治療コース中に本剤 650mg/m2を地固め療法・維持療法として計4 サイクル投与し、標準療法を対照として 4 年
間の無病生存(event free survival)率を比較・検討することを目的とし、GlaxoSmithKline 社
が承認後に実施予定の試験として提案したもので、米国食品医薬品局(FDA)に提案どおり
認められたものである。なお、米国においてFDA より実施を指示されている試験は、次の
一試験のみである。
1.5. 起原又は発見の経緯及び開発の経緯
• 第Ⅲ相 AALL0434 試験 新たにT-ALL と診断された 30 歳未満の患者における高用量メトトレキサート、ネララ ビン及び半増量BMF 療法(無作為化、非盲検・非対照試験、目標 640 例) 本剤は、欧州においても2005 年 6 月に希少疾病用医薬品の指定を受けており、2006 年 5 月に承認申請を行った。 1.5.4.2. 本邦における開発の経緯 前述のとおり、海外において実施された再発・難治性のT-ALL/T-LBL を対象とした第Ⅱ 相臨床試験において高いCR 率及び 1 年後生存率が得られており、また、リスク/ベネフィ ットも許容の範囲内にあるもとのと考えられ、標準的な治療法の確立されていない再発・難 治性のT-ALL/T-LBL に対し、臨床的に有意義な利益を提供できることが期待される。本邦 における患者数は非常に少ないため、通常規模の臨床試験の実施は極めて困難である状況下、 本剤をより早く臨床使用に供するため、海外臨床試験成績に基づき承認申請を行うことを考 慮し、その妥当性について、平成 年 月 日に独立行政法人医薬品医療機器総合機構 (機構)に対して医薬品申請前相談を行った。 その結果、 とされた。あわせて、本剤の特性に ついての分析及び考察について機構の意見が示され、「 」との助言が得られた(5.4.1 参照)。 また、ネララビンは「平成17 年 10 月~12 月に欧米 4 ヵ国のいずれかの国で新たに承認さ れた医薬品(類型Ⅰ)」及び「学会・患者団体から追加で検討要望があった医薬品」として、 第7 回未承認薬使用問題検討会議(平成 18 年 1 月 19 日開催)に諮られた。同検討会議にお いて、ワーキンググループより「対象疾患患者数は決して多くはないものの、本剤の医療上 の必要度は高いと判断される。本剤の臨床開発は、これまで本邦では全く実施されておらず、 FDA 承認後もこのままの状態を放置すると、本邦の当該疾患患者に不利益が及ぶことが懸 念されることから、我が国での治験が早期に開始されるよう検討すべきである。(中略)日 本人患者に対する薬物動態学的検討を含むfeasibility study を全く実施せずに本剤を承認する ことは、投与対象患者の安全性確保の観点より適切ではない。しかし、上記に述べた様に、 対象患者数が著しく少ないことを考慮すると、日本人患者少数例を対象とするphaseⅠ/Ⅱ study を実施しつつ、海外データをできるだけ利用した臨床開発を行うことが現実的な方向 性と考えられる。」との検討結果が報告され[第7回 未承認薬使用問題検討会議, 2006]、本 報告どおり本邦において早期に治験が開始されるよう検討すべきと結論された。 1.5. 起原又は発見の経緯及び開発の経緯
以上の相談結果に基づき、また、未承認薬使用問題検討会議における議論も考慮に入れ、 本邦において成人及び小児の再発・難治性T-ALL/T-LBL を対象とした第Ⅰ相臨床試験を実 施し、可能な限りの症例を収集して、海外で得られた安全性並びに薬物動態の成績が日本人 T-ALL/T-LBL の成績と大きな違いがないことを確認することとした。同時に、相談時に得 られた機構の意見及び助言を踏まえて資料を整備し、以下の効能・効果、用法・用量にて承 認申請を行うこととした。 【効能・効果】 再発又は難治性の下記疾患: ・T 細胞急性リンパ性白血病 ・T 細胞リンパ芽球性リンパ腫 【用法・用量】 通常、成人には、ネララビンとして1500mg/m2(体表面積)を1 日 1 回 2 時間以上かけて点 滴静注する。これを1、3、5 日目に投与し、その後 16 日間休薬する。21 日間を 1 クールと して、繰り返す。 通常、小児には、ネララビンとして650mg/m2(体表面積)を1 日 1 回 1 時間以上かけて点 滴静注する。これを 5 日間連日投与し、その後 16 日間休薬する。21 日間を 1 クールとして、 繰り返す。 なお、1.5.2 に述べたとおり、本剤の適応対象患者数はきわめて少なく、また、本剤の適 応対象となる疾患は予後不良の重篤な疾患であり、かつ、代替する適切な医薬品等又は治療 法がないことから、平成5 年 8 月 25 日付薬発第 725 号厚生省薬務局長通知「薬事法及び医 薬品副作用被害救済・研究振興基金法の一部を改正する法律の施行について」記第2 の 1 の (1)に示される指定の基準を満たすものと考え、平成 18 年 3 月 16 日に希少疾病用医薬品等指 定申請を行い、平成18 年 6 月に指定を受けた。 以上の開発の経緯を図 1.5.4-1 に示す。 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 13
試験項目 品質に関する試験 薬理 ADME 毒性 第Ⅰ相 試験/ 臨床薬 理試験 臨床 試験 * 2007 年 8 月時点において実施中 図 1.5.4-1 開発の経緯図 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 14
1.5.5. 特徴及び有用性 海外において実施された再発・難治性の成人及び小児T-ALL/T-LBL を対象とした臨床試 験成績から得られているネララビンの特徴は以下のとおりである。 1. 標準的な治療法が現時点では確立されていない再発・難治性の T-ALL/T-LBL に対 し、ネララビンの単独投与は既存の多剤併用療法と比較して同程度以上のCR 率であ った(2.5.6.2 参照)。 2. ネララビン単独投与により、1 回の寛解導入療法に対する再発・難治性の T-ALL/T-LBL のみならず、2 回以上の再発・難治例に対して高い CR 率を示した(2.5.4.3 参 照)。 3. 既存療法と比較して高い 1 年後生存率であった(2.5.4.3 参照)。 4. 多剤併用による寛解導入療法が無効であった難治性の T-ALL/T-LBL に対し、ネララ ビンの単独投与によりCR が得られた被験者が報告されている(2.5.4.3 参照)。 5. ネララビン投与時の安全性は、既存の多剤併用療法と比較して許容しうる範囲内であ った(2.5.6.3 参照)。 以上の成績から、本剤は、標準的治療法の確立されていない成人及び小児における再発・ 難治性のT-ALL/T-LBL 患者に対し、臨床的に有意義な利益を提供できることが期待され、 また、本剤のリスク/ベネフィットも許容の範囲内にあるものと考えられる。 1.5. 起原又は発見の経緯及び開発の経緯 1.5 - p. 15
1.5.6. 参考文献
Arner ESJ, Eriksson S. Mammalian deoxyribonucleoside kinases. Pharmacol Ther. 1995;14:155-86. Carson DA, Kaye J, Matsumoto S, et al. Biochemical basis for the enhanced toxicity of
deoxyribonucleosides towards malignant human T cell lines. Proc Natl Acad Sci USA. 1979;76:2430-3.
Carson DA, Seegmiller JE. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: possible role of nucleoside kinase(s). Proc Natl Acad Sci USA. 1977;74:5677-5681.
Cohen A, Gudas LJ, Ammann AJ, et al. Deoxyguanosine triphosphate as a possible toxic metaboloite in immunodeficiency associated with purine nucleoside phosphorylase deficiency. J Clin Invest. 1978;61:1405-1409.
Hiddemann W, Longo DL, Coiffier B, Fisher RI, Cabanillas F, Cavalli F, et al. Lymphoma
classification-the gap between biology and clinical management is closing. Blood. 1996;88:4085-9. Horibe K, Tsukimoto I, Ohno R. Clinicopathologic characteristics of leukemia in Japanese children and young adults. Leukemia. 2001;15:1256–1261.
Kantarjian HM, O’Brien S, Smith TL, Cortes J, Giles FJ, et al. Results of Treatment With Hyper-CVAD, a Dose-Intensive Regimen, in Adult Acute Lymphocytic Leukemia. J Clin Oncol. 2000;18:457.
Sather. Available from URL:
http://www.fda.gov/ohrms/dockets/ac/05/slides/2005-4174S2_03_01-GSK-Presentation.ppt. COG (uppublished), USA; 2005.
Takeuchi J, Kyo T, Naito K, Sao H, Takahashi M, Miyawaki S, et al. Induction therapy by frequent administration of doxorubicin with four other drugs, followed by intensive consolidation and maintenance therapy for adult acute lymphoblastic leukemia: the JALSG-ALL93 study. Leukemia. 2002;16:1259-66. がんの統計編集委員会. がんの統計<2003 年版>. 財団法人がん研究振興財団. 2003:40-47. 国立社会保障・人口問題研究所. 日本の将来推計人口 平成 13 (2001)~62 (2050)年 平成 14 年 1 月推計. 財団法人厚生統計協会. 2002:73. 陣内 逸郎. B および T 前駆細胞の腫瘍 新 WHO 分類による白血病・リンパ系腫瘍の病態学. 中外医学社. 2004:101-8. 1.5. 起原又は発見の経緯及び開発の経緯
大野 竜三, 宮脇修一. 白血病の基礎と臨床、成人 ALL とその再発難治例. 医薬ジャーナル社.
2004:401-9.
大野 竜三, 小寺 良尚. 白血病治療マニュアル 改訂第 2 版. 南江堂. 2001:62-66.
第7回 未承認薬使用問題検討会議. Available from URL:
http://www.mhlw.go.jp/shingi/2006/01/dl/s0119-4a4-1.pdf. 厚生労働省, 日本; 2006. ワーキンググ ループ検討結果報告書(ネララビン). 直江 知樹. 白血病と類縁疾患. 日本臨床腫瘍学会 編. 癌と化学療法社. 2003:944-53. 福原 資郎, 藤本正博. エビデンスに基づく標準的治療法、悪性リンパ腫2-b NHL. 造血器腫瘍 白血病・悪性リンパ腫・多発性骨髄腫. 治癒をめざした診断・治療の実践. 93-8 ed. 日本:南江 堂; 2001.
平成13 年度全国調査報告書. Available from URL: http://www.jshct.com/report_2001/index.html.
日本造血細胞移植学会, 日本; 2001.
林 泰秀. 小児の白血病と悪性リンパ腫. 日本臨床腫瘍学会 編. 癌と化学療法社. 2003:933-43.
1.5. 起原又は発見の経緯及び開発の経緯
![表 1.5.3-1 1 回目の造血幹細胞移植を受けた ALL の病期別 5 年後生存率 [平成 13 年度全国調査報告書, 2001]を一部改変 小児 成人 例数 5 年後生存率 例数 5 年後生存率 第一寛解期 390 70.3% 819 50.8% 第二寛解期 363 59.2% 275 32.0% 第三寛解期 91 31.0% 68 20.9% 非寛解期 298 22.6% 399 11.3% 以上のとおり、再発・難治性の成人及び小児 T-ALL/T-LBL に対](https://thumb-ap.123doks.com/thumbv2/123deta/6527001.666320/11.918.137.783.152.265/年度調査報告改変小児成人例数年後生存例数年後生存とおりに対.webp)
![表 1.5.4-1 PGAA2001 試験(小児)における CR/CR*率及び生存期間 表 2.5.4-2 を再掲 1 回の寛解導入療法 31 例 2 回以上の寛解導入療法 39 例 CR 及びその期間 CR 例数(%) [95%CI] 13 (42%) [25%~61%] 5 (13%) [4%~27%] CR 持続期間(週) 0.9~260.0 以上 4.7~36.4 CR*及びその期間 CR*例数(%) [95%CI] 15 (48%) [30%~67%] 9](https://thumb-ap.123doks.com/thumbv2/123deta/6527001.666320/14.918.136.785.148.423/おける再掲寛解導入療法回以上及びその期間CR例数以上CR及びそのCR.webp)
![表 1.5.4-2 PGAA2002 試験(成人)における CR/CR*率及び生存期間 表 2.5.4-3 を再掲 1 回の寛解導入療法 11 例 2 回以上の寛解導入療法28 例 合計 39 例 CR 及びその期間 CR 例数(%) [95%CI] 2 (18%) [2%~52%] 5 (18%) [6%~37%] 7 (18%) [8%~34%] CR 持続期間(週) 51.0 及び 212.0 15.1~195.4 以上 15.1~212.0 CR*及びそ](https://thumb-ap.123doks.com/thumbv2/123deta/6527001.666320/16.918.125.796.148.424/おける再掲寛解導入回以上合計及びその期間CR例数CI期間以上CR及び.webp)
