ミリプラチン 製造販売承認申請
CTD 第 2 部
2.7 臨床概要
2.7.3 臨床的有効性の概要
大日本住友製薬株式会社
ミリプラチン 2.7.3 臨床的有効性の概要 Page 1目次
2.7.3 臨床的有効性の概要 2.7.3.1 背景及び概観...11 2.7.3.2 個々の試験結果の要約...22 2.7.3.3 全試験を通しての結果の比較と解析...26 2.7.3.3.1 試験対象集団...26 2.7.3.3.2 全有効性試験の結果の比較検討...38 2.7.3.3.3 部分集団における結果の比較...48 2.7.3.4 推奨用法・用量に関する臨床情報の解析...57 2.7.3.5 効果の持続、耐薬性...61 2.7.3.6 付録...62 ミリプラチン 2.7.3 臨床的有効性の概要 Page 2【本項における用語の説明】 用語 定義、読み替えなど ミリプラチン 化学名: (SP-4-2)-[(1R,2R)-Cyclohexane-1,2-diamine-N,N’]bis(tetradecanoato-O)platinum 化学式(分子量): C34H68N2O4Pt(763.99) 構造式: 懸濁用液 一般名: ヨード化ケシ油脂肪酸エチルエステル ミリプラチン 懸濁液 ミリプラチンをヨード化ケシ油脂肪酸エチルエステルに分散した懸濁液 ジノスタチン スチマラマー 化学名: (4S,6R,11R,12R)-11-[α-D-2,6-Dideoxy-2-(methylamino)galactopyranosyloxy]- 4-[(4R)-2-oxo-1,3-dioxolan-4-yl]-5-oxatricyclo[8.3.0.04,6]trideca-1(13),9-diene- 2,7-diyn-12-yl 2-hydroxy-7-methoxy-5-metylnaphthalene-1-carboxylate 平均分子量: 約15000 構造式: ジノスタチン スチマラマー 懸濁液 ジノスタチン スチマラマーをヨード化ケシ油脂肪酸エチルエステルに分散 した懸濁液 H2 N Pt N H2 H H O O O O H3C H3C ミリプラチン 2.7.3 臨床的有効性の概要 Page 3
用語 定義、読み替えなど シスプラチン 化学名: (SP-4-2)-Diamminedichloroplatinum 化学式(分子量): Cl2H6N2Pt(300.05) 構造式: NH3 Pt NH3 Cl Cl ミリプラチン 2.7.3 臨床的有効性の概要 Page 4
用語 定義、読み替えなど
ECOG の一般状態の基準(Performance Status, PS) PS 0:なんら制限を受けることなく、発病前と同等に社会生活が行える。 1:軽度の症状があり、肉体労働は制限を受けるが、歩行、軽労働や坐業は できる。たとえば軽い家事、事務など。 2:歩行や身の廻りのことはできるが、時に少し介助がいることもある。軽 労働はできないが、日中の50%以上は起居している。 3:身の廻りのある程度のことはできるが、しばしば介助がいり、日中の 50%以上は就床している。 4:身の廻りのこともできず、つねに介助がいり、終日就床を必要としてい る。 この基準は一般状態の指標であり、局所症状で活動性が制限されている場 合は臨床的に判断する。
Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649-55.より抜粋 進行度(Stage)は、各項目別にその患者の進行度値を求め、そのうちの最 も高い数値をあてる。 進行度を次の4 つの Stage に分類する。 T 因子 N 因子 M 因子 Stage I T1 N0 M0 Stage II T2 N0 M0 Stage III T3 N0 M0 T4 N0 M0 Stage IV-A T1、T2、T3、T4 N1 M0 Stage IV-B T1、T2、T3、T4 N0 N1 M1 Stage【第 4 版】 ・T 因子 癌腫の「個数」、「大きさ」、「脈管侵襲」の3 項目によって規定される。複 数の癌腫は多中心性癌腫であっても肝内転移癌腫であってもよい。肝細胞 癌破裂S3はT4として取扱う。 1:腫瘍個数 単発 2:腫瘍径 2 cm 以下 3:脈管侵襲 なし T1 1、2、3 すべて合致 T2 2 項目合致 T3 1 項目合致 T4 すべて合致せず ・N 因子 N0 リンパ節転移を認めない N1 リンパ節転移を認める ・M 因子 M0 遠隔転移を認めない M1 遠隔転移を認める 原発性肝癌取扱い規約 2000 年 11 月【第 4 版】より抜粋 ミリプラチン 2.7.3 臨床的有効性の概要 Page 5
用語 定義、読み替えなど 肉眼的進行度(Stage)は、各項目別にその患者の進行度値を求め、そのう ちの最も高い数値をあてる。 肉眼的進行度を次の4 つの Stage に分類する。 T 因子 N 因子 M 因子 Stage I T1 N0 M0 Stage II T2 N0 M0 T3 N0 M0 Stage III T1-3 N1 M0 Stage IV-A T4 N0-1 M0 Stage IV-B T1-4 N0-1 M1 Stage【第 3 版】 ・T 因子 癌腫の「大きさ」、「単発または複数」、「血管侵襲」の3 項目によって規定 される。複数の癌腫は多中心性癌腫であっても肝内転移癌腫であってもよ い。 T1 :単発した直径2 cm 以下の癌腫で血管侵襲を伴わない T2 :単発した直径2 cm 以下の癌腫であるが血管侵襲を伴う :一葉に限局した最大腫瘍の直径が2 cm 以下の多発性癌腫 :単発した直径2 cm をこえる癌腫であるが血管侵襲を伴わない T3 :単発した直径2 cm をこえる癌腫で血管侵襲を伴う :一葉に限局した直径 2 cm をこえる多発性癌腫 T4:一葉以上を占居する多発性癌腫 :門脈または肝静脈の一次分枝の血管侵襲を伴う ・N 因子 N0:第 1 群リンパ節に転移が認められない N1:第 1 群以上のリンパ節に転移を認められる ・M 因子 M0:遠隔転移が認められない M1:遠隔転移が認められる 原発性肝癌取扱い規約 1992 年 2 月【第 3 版】より抜粋 ミリプラチン 2.7.3 臨床的有効性の概要 Page 6
用語 定義、読み替えなど 各項目のポイントを加算しその合計点で分類する。 A : 5~6 点 B : 7~9 点 C : 10~15 点 原発性肝癌取扱い規約 2000 年 11 月【第 4 版】より抜粋 1 点 2 点 3 点 脳症 ない 軽度 ときどき昏睡 腹水 ない 少量 中等量 血清ビリルビン 値(mg/dL) 2.0 未満 2.0~3.0 3.0 超 血清アルブミン 値(g/dL) 3.5 超 2.8~3.5 2.8 未満 Child-Pugh 分類 プロトロンビン 活性値(%) 70 超 40~70 40 未満 肝細胞癌患者の臨床病期は臨床所見、血液生化学所見により3 期に分類す る。各項目別にその患者の状態を判定して進行度を求め、そのうち2 項目 以上が該当したstage をとる。 原発性肝癌取扱い規約1992 年 2 月【第 3 版】より抜粋 *臨床病期は癌の進行度分類(Stage)と混同しやすいことから「原発性肝 癌取扱い規約2000 年 11 月【第 4 版】で肝障害度 A、B、C に改名された。
I(A) II(B) III(C)
腹水 ない 治療効果がある 治療効果が少ない 血清ビリルビン 値(mg/dL) 2.0 未満 2.0~3.0 3.0 超 血清アルブミン 値(g/dL) 3.5 超 3.0~3.5 3.0 未満 ICG R15(%) 15 未満 15~40 40 超 臨床病期 (肝障害度) プロトロンビン 活性値(%) 80 超 50~80 50 未満 JIS(Japan Integrated Staging)スコア
肝予備能(Child-Pugh 分類)、進行度(Stage)の両者を併せた統合 staging system
Stage I Stage II Stage III Stage IV Child-Pugh C 2 3 4 5 Child-Pugh B 1 2 3 4 JIS スコア
Child-Pugh A 0 1 2 3
用語 定義、読み替えなど 占居部位 肝臓は胆嚢窩と肝上部の下大静脈を結ぶ線によりその左側を左葉、右側を 右葉とし、さらにそれぞれを2 区域に分けたのち、尾状葉とあわせて 5 区 域に大別する。 L:外側区域 lateral segment M:内側区域 medial segment A:前区域 anterior segment P:後区域 posterior segment C:尾状葉 caudate lobe
原発性肝癌取扱い規約 2000 年 11 月【第 4 版】より抜粋
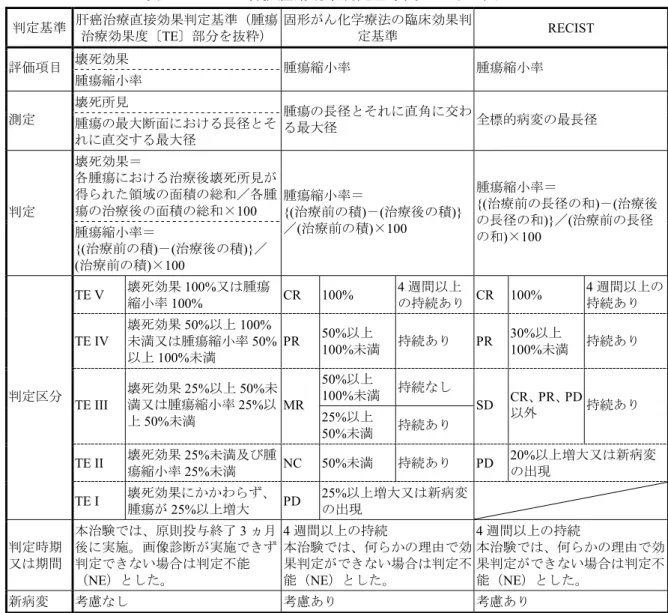
【有効性の判定基準】 肝癌治療直接効果判定基準(抜粋) 【治療効果判定】 治療効果判定は、以下に定めるように、腫瘍治療効果度(TE:therapeutic effect)、随伴病巣治療効 果度、新病巣の有無について行う。 腫瘍治療効果度(TE)は、画像診断所見から判定された腫瘍の壊死効果(TN:tumor necrosis)と 腫瘍縮小率(TR:tumor regression)を主体として判定する。 ○壊死効果(TN) 壊死効果の判定は、以下の表に示すように、I・II・III・IV・V の 5 段階で行う。 壊死効果(TN) 腫瘍の壊死効果 TN V TN IV TN III TN II TN I 壊死効果 100% 壊死効果 50%以上、100%未満 壊死効果 25%以上、50%未満 壊死効果 25%未満 壊死効果 なし ○腫瘍縮小率(TR) 腫瘍縮小率の判定は、以下の表に示すように、I・II・III・IV・V の 5 段階で行う。 腫瘍縮小率(TR) 腫瘍縮小率 TR V TR IV TR III TR II TR I 縮小率 100% 縮小率 50%以上、100%未満 縮小率 25%以上、50%未満 縮小率 25%未満 縮小率 25%以上の増大 ○腫瘍治療効果度(TE) 腫瘍治療効果度(TE)の判定は、治療終了後 3 ヵ月以内の TN または TR の最大効果をもって、以 下の表に示すようにI・II・III・IV・V の 5 段階で行う。また、同一症例で、壊死効果(TN)また は腫瘍縮小率(TR)から判定した腫瘍治療効果度(TE)が一致しない場合は、大きい腫瘍治療効 果度(TE)を採用する。 治療効果度 (TE) TE V TE IV TE III TE II TE I 壊死効果100% または腫瘍縮小率 100% 壊死効果50%以上 100%未満 または腫瘍縮小率 50%以上 100%未満 壊死効果25%以上 50%未満 または腫瘍縮小率 25%以上 50%未満 壊死効果25%未満および腫瘍縮小率 25%未満 壊死効果にかかわらず、腫瘍が25%以上増大 ○治療効果の総合評価 治療効果の総合評価は、腫瘍治療効果度(TE)、随伴病巣治療効果、新病巣の有無から以下の表に 定めるようにCR、PR、MR、NC、PD の 5 段階で判定する。 総合評価 腫瘍治療効果度 随伴病巣治療効果 新病巣 CR(著効):complete response TE V D(消失) PR(有効):partial response TE IV
MR(やや有効):minor response TE III R(退縮)
NC(不変):no change TE II N(不変)
無し PD(進行):progressive disease TE I A(進展) 有り
【肝細胞癌治療法についての略号一覧】 略号 省略しない表現 日本語又は説明 CL Chemolipiodolization 肝動脈塞栓療法のうち、抗がん剤と ヨード化ケシ油脂肪酸エチルエステ ルを肝動脈内投与する治療法。 CE Chemoembolization 肝動脈塞栓療法のうち、CL 後、ゼラ チンスポンジ等の塞栓物質を肝動脈 内投与する治療法。
TACE Transcatheter Arterial Chemoembolization 肝動脈塞栓療法
(CL と CE の療法を合わせた総称) TAI Transcatheter Arterial Infusion 肝動注化学療法
RFA Radiofrequency Ablation Therapy ラジオ波焼灼療法 PEIT Percutananeuos Transhepatic Ethanol Injection
Therapy 経皮的エタノール注入療法
PMCT Percutananeuos Microwave Coagulation Therapy 経皮的マイクロウエーブ凝固療法
【臨床検査に関する一般的略号】
略号 省略しない表現 日本語
ALT(GPT) Alanine aminotransferase アラニン・アミノトランスフェラーゼ AST(GOT) Aspartate aminotransferase アスパラギン酸アミノトランスフェラーゼ CRP C-reactive protein C-反応性蛋白 γ-GTP γ-Glutamyl transpeptidase γ-グルタミルトランスペプチダーゼ Hb Hemoglobin ヘモグロビン IgE Immunoglobulin E 免疫グロブリンE LDH Lactate dehydrogenase 乳酸脱水素酵素 NAG N-Acetyl-β-D-Glucosaminidase N-アセチル-β-D-グルコサミニダーゼ PLT Platelet 血小板
WBC White blood cell 白血球
【その他の一般的略号】
略号 省略しない表現 日本語
CT Computerized Tomography コンピューター断層撮影法 ECOG Eastern Cooperative Oncology Group 米国腫瘍学共同研究グループ HBs Hepatitis B virus surface B 型肝炎ウイルス外皮の表面 HCV Hepatitis C virus C 型肝炎ウイルス
2.7.3.1 背景及び概観 ミリプラチンの有効性は第I 相臨床試験、前期第 II 相臨床試験、後期第 II 相臨床試験及 び継続投与試験の成績に基づいて評価した。上記試験の要約を表 2.7.3.1-1 に示した。 (1) 第 I 相臨床試験 本剤の肝動脈内投与における最大許容量の推定と肝細胞癌に対する有効性、薬物動態学 的検討を目的とした。 対象は他の治療法では無効又は適応外の肝細胞癌患者とした。 試験デザインは無対照、非盲検試験(Fibonacci の変法による漸増法)とした。 投与量は腫瘍の大きさに依存して投与液量で規定されるため、イヌ肝動注単回投与毒性 試験(2.6.6.2 参照)より初回濃度を 6 mg/mL とし、12 mg/mL、20 mg/mL と投与液濃度を 増量し、最大6 mL を肝動脈内に投与する Fibonacci の変法による漸増法で実施した。また、 投与回数は原則1 回とした。 目標被験者数は各々の用量レベルに少なくとも3 例登録し、実質臓器(心、肝、腎、血 液、肺など)において、日本癌治療学会の『副作用の記載様式』におけるグレード2 以上 の副作用又は1 グレード以上の動きを伴う副作用が発現した場合、その用量レベルに更に 3 例を加えて検討を行った。 最大許容量は実質臓器(心、肝、腎、血液、肺など)において、「日本癌治療学会『副作 用の記載様式』」におけるグレード2 以上の副作用を 2/3 以上の被験者が経験する最低の投 与量と定義し判定した。 有効性評価は「肝癌治療直接効果判定基準」に準じて抗腫瘍効果の判定を行い、効果・ 安全性評価委員会において効果判定を確定した。転帰(再発、生存・死亡)については可 能な限り調査した。 安全性評価は日本癌治療学会の『副作用の記載様式』に従い判定した。 (2) 前期第 II 相臨床試験 本剤の肝細胞癌に対する有効性、安全性及び体内動態を検討することを目的とした。 対象は肝切除術及びPEIT の適応外の肝細胞癌患者とした。 試験デザインは無対照、非盲検試験とした。 投与液濃度は第I 相臨床試験で得られた推奨用量である 20 mg/ mL とした。また、1 回 目投与4 週経過後できるだけ早期に 2 回目投与を行うこととした。投与間隔は最短で 4 週 間、最長で12 週間とした。ただし、1 回目投与後に CR が得られた場合は 1 回投与とした。 目標被験者数は、ミリプラチンのCR 率を 40%と予測し、95%信頼区間の下限が 15%を 上回るために必要な被験者数として15 例を設定した。 有効性評価は「肝癌治療直接効果判定基準」に準じて抗腫瘍効果の判定を行い、CR の 割合を主要評価項目、CR+PR の割合を副次的評価項目とし、95%信頼区間を算出した。な お、有効性については効果判定委員会の判定を採用した。転帰(再発、生存・死亡)につ ミリプラチン 2.7.3 臨床的有効性の概要 Page 11
いては可能な限り調査した。 安全性評価は「日本癌治療学会薬物有害反応判定基準」に従い判定した。 (3) 後期第 II 相臨床試験 本剤の肝細胞癌に対する有効性、安全性及び薬物動態を検討することを目的とした。 対象は肝切除術及び内科的局所療法の適応外の肝細胞癌患者とした。 試験デザインは対照群を置いた並行群間比較とし、対照薬はヨード化ケシ油脂肪酸エチ ルエステルに懸濁して使用する薬剤として肝細胞癌(肝動脈内投与)の効能を取得した唯 一の薬剤であるジノスタチン スチマラマーを選択した。また、両薬剤の調製方法が異なる ことから非盲検で実施した。 本剤の投与液濃度は20 mg/ mL、ジノスタチン スチマラマーは 1 mg(力価)/mL とし、 いずれも投与液量は6 mL を限度として腫瘍の大きさに従って投与した。1 回目投与後の画 像診断において懸濁液の停滞が不十分でかつ腫瘍濃染像がある場合には、1 回目投与後 4 週間以降12 週以内のできるだけ早期に 2 回目投与を行った。 目標被験者数は実施可能性から120 例とし、ミリプラチンの安全性を評価するためには 可能な限り多くの被験者での使用経験が望ましいことから、ジノスタチン スチマラマー 1 に対して本剤を2 の比で割り付けた。 割付方法は「最大腫瘍径(25 mm 未満と 25 mm 以上)」及び「実施医療機関」を要因と し、確率を用いた動的割付とした。 有効性評価は「肝癌治療直接効果判定基準」に準じて抗腫瘍効果を判定し、TE V の割合 を主要評価項目とした。更に「固形がん化学療法の臨床効果判定基準」及び「RECIST」に よる CR+PR の割合を副次的評価項目とした。有効性については効果判定委員会による効 果判定を採用し、いずれの変数も点推定値及び両側95%信頼区間を算出した。なお、両薬 剤の有効性及び安全性を推定するための情報が不確実であり、実施可能症例数では十分な 検出力が確保できない可能性があることから、投与群間の統計的な比較は行わなかった。 転帰については再発、後治療、生存・死亡などについて調査した。 安全性評価は「日本癌治療学会薬物有害反応判定基準」に従い判定した。 (4) 継続投与試験 本剤の継続投与の希望に対応すること及び継続投与における有効性と安全性を検討する ことを目的とした。 対象は後期第II 相臨床試験に参加し本剤に割り付けられて TE V と判定された後、別部 位に肝細胞癌の再発が認められた患者とした。 試験デザインは無対照、非盲検試験とした。 目標被験者数は最大で80 例とし、有効性及び安全性は後期第 II 相臨床試験と同様に評 価した。また、1 回の投与ごとに登録を行い各々1 例と扱った。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 12
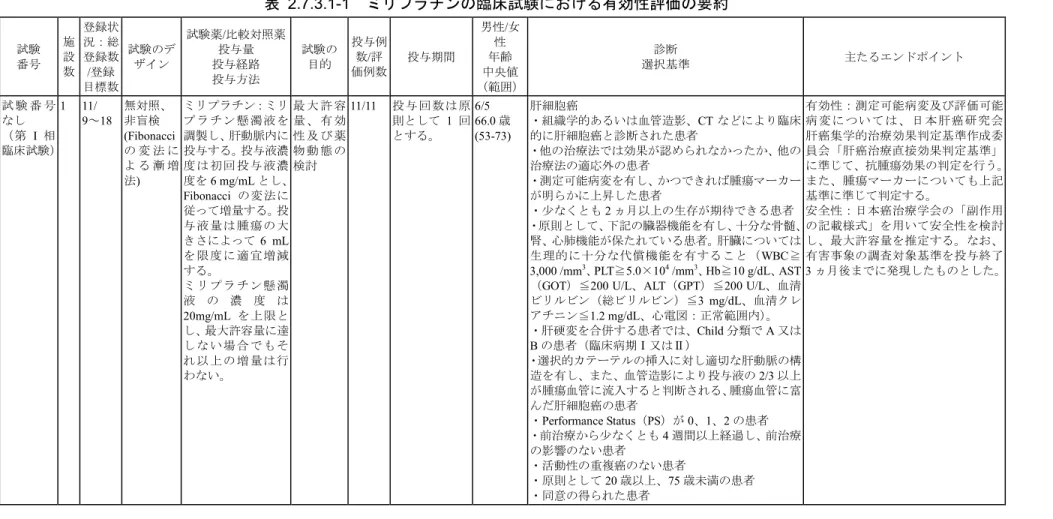
表 2.7.3.1-1 ミリプラチンの臨床試験における有効性評価の要約 試験 番号 施 設 数 登録状 況:総 登録数 /登録 目標数 試験のデ ザイン 試験薬/比較対照薬 投与量 投与経路 投与方法 試験の 目的 投与例 数/評 価例数 投与期間 男性/女 性 年齢 中央値 (範囲) 診断 選択基準 主たるエンドポイント 試 験 番 号 なし (第 I 相 臨床試験) 1 11/ 9~18 無対照、 非盲検 (Fibonacci の 変 法 に よ る 漸 増 法) ミリプラチン:ミリ プ ラ チ ン 懸 濁 液 を 調製し、肝動脈内に 投与する。投与液濃 度 は 初 回 投 与 液 濃 度を6 mg/mL とし、 Fibonacci の変法に 従って増量する。投 与 液 量 は 腫 瘍 の 大 きさによって6 mL を 限 度 に 適 宜 増 減 する。 ミ リ プ ラ チ ン 懸 濁 液 の 濃 度 は 20mg/mL を上限と し、最大許容量に達 し な い 場 合 で も そ れ 以 上 の 増 量 は 行 わない。 最 大 許 容 量 、 有 効 性 及 び 薬 物 動 態 の 検討 11/11 投与回数は原 則として 1 回 とする。 6/5 66.0 歳 (53-73) 肝細胞癌 ・組織学的あるいは血管造影、CT などにより臨床 的に肝細胞癌と診断された患者 ・他の治療法では効果が認められなかったか、他の 治療法の適応外の患者 ・測定可能病変を有し、かつできれば腫瘍マーカー が明らかに上昇した患者 ・少なくとも2 ヵ月以上の生存が期待できる患者 ・原則として、下記の臓器機能を有し、十分な骨髄、 腎、心肺機能が保たれている患者。肝臓については 生理的に十分な代償機能を有すること(WBC≧ 3,000 /mm3、PLT≧5.0×104 /mm3、Hb≧10 g/dL、AST
(GOT)≦200 U/L、ALT(GPT)≦200 U/L、血清 ビリルビン(総ビリルビン)≦3 mg/dL、血清クレ アチニン≦1.2 mg/dL、心電図:正常範囲内)。 ・肝硬変を合併する患者では、Child 分類で A 又は B の患者(臨床病期Ⅰ又はⅡ) ・選択的カテーテルの挿入に対し適切な肝動脈の構 造を有し、また、血管造影により投与液の2/3 以上 が腫瘍血管に流入すると判断される、腫瘍血管に富 んだ肝細胞癌の患者 ・Performance Status(PS)が 0、1、2 の患者 ・前治療から少なくとも4 週間以上経過し、前治療 の影響のない患者 ・活動性の重複癌のない患者 ・原則として20 歳以上、75 歳未満の患者 ・同意の得られた患者 有効性:測定可能病変及び評価可能 病 変 に つい て は 、日 本 肝癌 研究 会 肝癌集学的治療効果判定基準作成委 員会「肝癌治療直接効果判定基準」 に準じて、抗腫瘍効果の判定を行う。 また、腫瘍マーカーについても上記 基準に準じて判定する。 安全性:日本癌治療学会の「副作用 の記載様式」を用いて安全性を検討 し、最大許容量を推定する。なお、 有害事象の調査対象基準を投与終了 3 ヵ月後までに発現したものとした。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 13
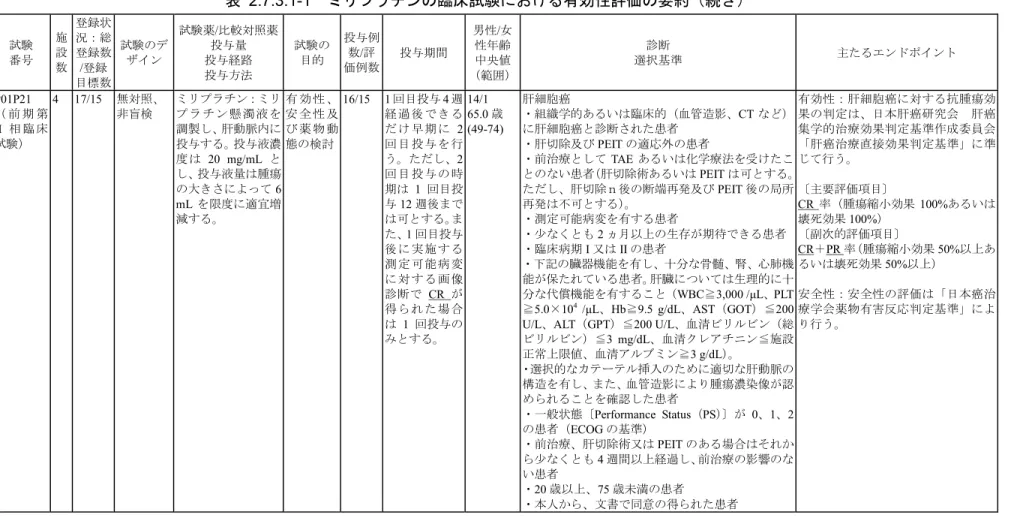
表 2.7.3.1-1 ミリプラチンの臨床試験における有効性評価の要約(続き) 試験 番号 施 設 数 登録状 況:総 登録数 /登録 目標数 試験のデ ザイン 試験薬/比較対照薬 投与量 投与経路 投与方法 試験の 目的 投与例 数/評 価例数 投与期間 男性/女 性年齢 中央値 (範囲) 診断 選択基準 主たるエンドポイント P01P21 ( 前 期 第 II 相 臨 床 試験) 4 17/15 無対照、 非盲検 ミリプラチン:ミリ プ ラ チ ン 懸 濁 液 を 調製し、肝動脈内に 投与する。投与液濃 度は 20 mg/mL と し、投与液量は腫瘍 の大きさによって6 mL を限度に適宜増 減する。 有 効 性 、 安 全 性 及 び 薬 物 動 態の検討 16/15 1 回目投与 4 週 経 過 後 でき る だ け 早 期 に 2 回 目 投 与を 行 う。ただし、2 回 目 投 与の 時 期は 1 回目投 与12 週後まで は可とする。ま た、1 回目投与 後 に 実 施す る 測 定 可 能病 変 に 対 す る画 像 診断で CR が 得 ら れ た場 合 は 1 回投与の みとする。 14/1 65.0 歳 (49-74) 肝細胞癌 ・組織学的あるいは臨床的(血管造影、CT など) に肝細胞癌と診断された患者 ・肝切除及びPEIT の適応外の患者 ・前治療としてTAE あるいは化学療法を受けたこ とのない患者(肝切除術あるいはPEIT は可とする。 ただし、肝切除n後の断端再発及びPEIT 後の局所 再発は不可とする)。 ・測定可能病変を有する患者 ・少なくとも2 ヵ月以上の生存が期待できる患者 ・臨床病期I 又は II の患者 ・下記の臓器機能を有し、十分な骨髄、腎、心肺機 能が保たれている患者。肝臓については生理的に十 分な代償機能を有すること(WBC≧3,000 /µL、PLT ≧5.0×104 /µL、Hb≧9.5 g/dL、AST(GOT)≦200 U/L、ALT(GPT)≦200 U/L、血清ビリルビン(総 ビリルビン)≦3 mg/dL、血清クレアチニン≦施設 正常上限値、血清アルブミン≧3 g/dL)。 ・選択的なカテーテル挿入のために適切な肝動脈の 構造を有し、また、血管造影により腫瘍濃染像が認 められることを確認した患者 ・一般状態〔Performance Status(PS)〕が 0、1、2 の患者(ECOG の基準) ・前治療、肝切除術又はPEIT のある場合はそれか ら少なくとも4 週間以上経過し、前治療の影響のな い患者 ・20 歳以上、75 歳未満の患者 ・本人から、文書で同意の得られた患者 有効性:肝細胞癌に対する抗腫瘍効 果の判定は、日本肝癌研究会 肝癌 集学的治療効果判定基準作成委員会 「肝癌治療直接効果判定基準」に準 じて行う。 〔主要評価項目〕 CR 率(腫瘍縮小効果 100%あるいは 壊死効果100%) 〔副次的評価項目〕 CR+PR 率(腫瘍縮小効果 50%以上あ るいは壊死効果50%以上) 安全性:安全性の評価は「日本癌治 療学会薬物有害反応判定基準」によ り行う。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 14
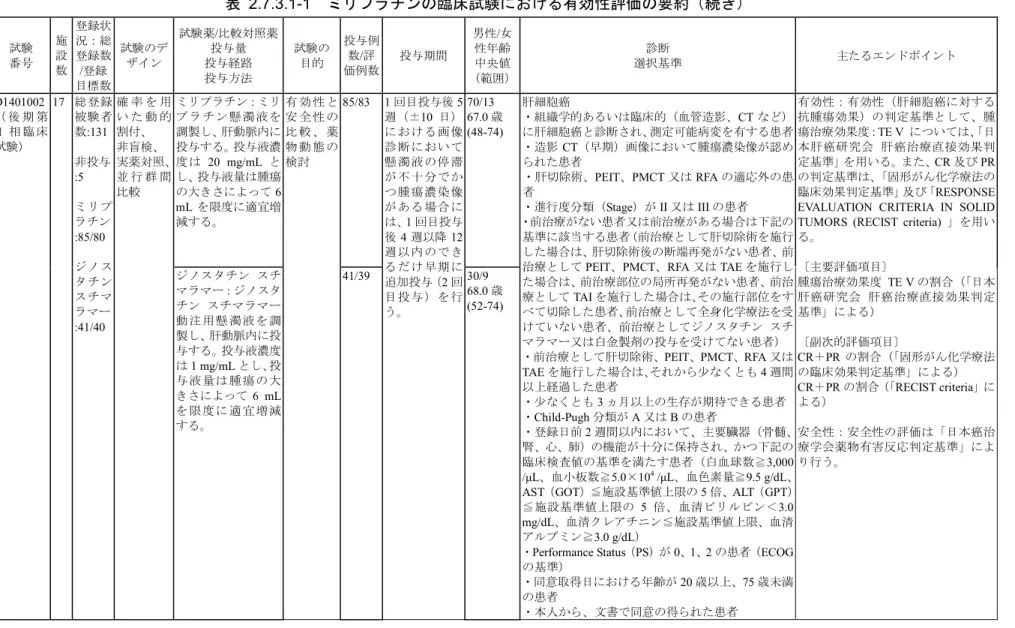
表 2.7.3.1-1 ミリプラチンの臨床試験における有効性評価の要約(続き) 試験 番号 施 設 数 登録状 況:総 登録数 /登録 目標数 試験のデ ザイン 試験薬/比較対照薬 投与量 投与経路 投与方法 試験の 目的 投与例 数/評 価例数 投与期間 男性/女 性年齢 中央値 (範囲) 診断 選択基準 主たるエンドポイント ミリプラチン:ミリ プ ラ チ ン 懸 濁 液 を 調製し、肝動脈内に 投与する。投与液濃 度は 20 mg/mL と し、投与液量は腫瘍 の大きさによって6 mL を限度に適宜増 減する。 85/83 70/13 67.0 歳 (48-74) D1401002 ( 後 期 第 II 相 臨 床 試験) 17 総登録 被験者 数:131 非投与 :5 ミリプ ラチン :85/80 ジノス タチン スチマ ラマー :41/40 確 率 を 用 い た 動 的 割付、 非盲検、 実薬対照、 並 行 群 間 比較 ジノスタチン スチ マラマー:ジノスタ チン スチマラマー 動 注 用 懸 濁 液 を 調 製し、肝動脈内に投 与する。投与液濃度 は1 mg/mL とし、投 与 液 量 は 腫 瘍 の 大 きさによって6 mL を 限 度 に 適 宜 増 減 する。 有 効 性 と 安 全 性 の 比 較 、 薬 物 動 態 の 検討 41/39 1 回目投与後 5 週(±10 日) に お け る画 像 診 断 に おい て 懸 濁 液 の停 滞 が 不 十 分で か つ 腫 瘍 濃染 像 が あ る 場合 に は、1 回目投与 後4 週以降 12 週 以 内 ので き る だ け 早期 に 追加投与(2 回 目 投 与 )を 行 う。 30/9 68.0 歳 (52-74) 肝細胞癌 ・組織学的あるいは臨床的(血管造影、CT など) に肝細胞癌と診断され、測定可能病変を有する患者 ・造影CT(早期)画像において腫瘍濃染像が認め られた患者 ・肝切除術、PEIT、PMCT 又は RFA の適応外の患 者 ・進行度分類(Stage)が II 又は III の患者 ・前治療がない患者又は前治療がある場合は下記の 基準に該当する患者(前治療として肝切除術を施行 した場合は、肝切除術後の断端再発がない患者、前 治療としてPEIT、PMCT、RFA 又は TAE を施行し た場合は、前治療部位の局所再発がない患者、前治 療としてTAI を施行した場合は、その施行部位をす べて切除した患者、前治療として全身化学療法を受 けていない患者、前治療としてジノスタチン スチ マラマー又は白金製剤の投与を受けてない患者) ・前治療として肝切除術、PEIT、PMCT、RFA 又は TAE を施行した場合は、それから少なくとも 4 週間 以上経過した患者 ・少なくとも3 ヵ月以上の生存が期待できる患者 ・Child-Pugh 分類が A 又は B の患者 ・登録日前2 週間以内において、主要臓器(骨髄、 腎、心、肺)の機能が十分に保持され、かつ下記の 臨床検査値の基準を満たす患者(白血球数≧3,000 /µL、血小板数≧5.0×104 /µL、血色素量≧9.5 g/dL、 AST(GOT)≦施設基準値上限の 5 倍、ALT(GPT) ≦施設基準値上限の 5 倍、血清ビリルビン<3.0 mg/dL、血清クレアチニン≦施設基準値上限、血清 アルブミン≧3.0 g/dL)
・Performance Status(PS)が 0、1、2 の患者(ECOG の基準) ・同意取得日における年齢が20 歳以上、75 歳未満 の患者 ・本人から、文書で同意の得られた患者 有効性:有効性(肝細胞癌に対する 抗腫瘍効果)の判定基準として、腫 瘍治療効果度:TE V については、「日 本肝癌研究会 肝癌治療直接効果判 定基準」を用いる。また、CR 及び PR の判定基準は、「固形がん化学療法の 臨床効果判定基準」及び「RESPONSE EVALUATION CRITERIA IN SOLID TUMORS (RECIST criteria) 」を用い る。 〔主要評価項目〕 腫瘍治療効果度 TE V の割合(「日本 肝癌研究会 肝癌治療直接効果判定 基準」による) 〔副次的評価項目〕 CR+PR の割合(「固形がん化学療法 の臨床効果判定基準」による) CR+PR の割合(「RECIST criteria」に よる) 安全性:安全性の評価は「日本癌治 療学会薬物有害反応判定基準」によ り行う。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 15
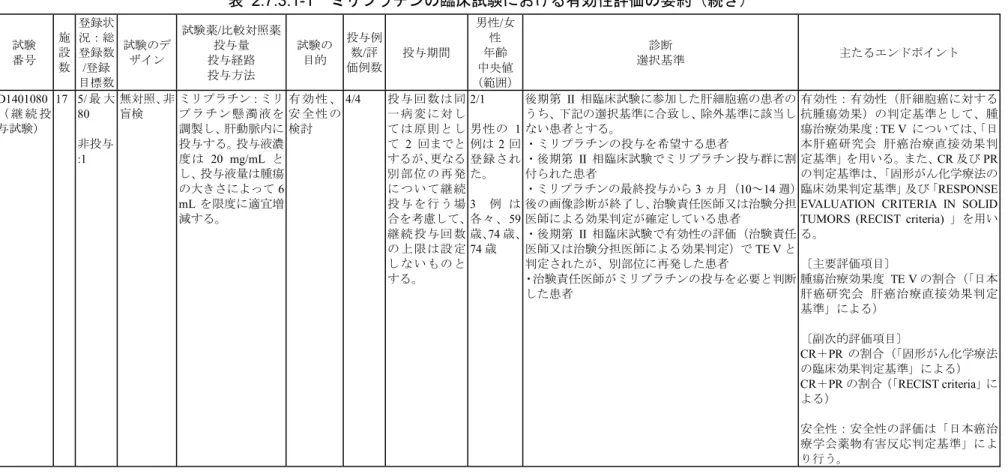
表 2.7.3.1-1 ミリプラチンの臨床試験における有効性評価の要約(続き) 試験 番号 施 設 数 登録状 況:総 登録数 /登録 目標数 試験のデ ザイン 試験薬/比較対照薬 投与量 投与経路 投与方法 試験の 目的 投与例 数/評 価例数 投与期間 男性/女 性 年齢 中央値 (範囲) 診断 選択基準 主たるエンドポイント D1401080 ( 継 続 投 与試験) 17 5/ 最 大 80 非投与 :1 無対照、非 盲検 ミリプラチン:ミリ プ ラ チ ン 懸 濁 液 を 調製し、肝動脈内に 投与する。投与液濃 度は 20 mg/mL と し、投与液量は腫瘍 の大きさによって6 mL を限度に適宜増 減する。 有 効 性 、 安 全 性 の 検討 4/4 投 与 回 数は 同 一 病 変 に対 し て は 原 則と し て 2 回までと するが、更なる 別 部 位 の再 発 に つ い て継 続 投 与 を 行う 場 合を考慮して、 継 続 投 与回 数 の 上 限 は設 定 し な い もの と する。 2/1 男性の 1 例は2 回 登録され た。 3 例 は 各々、59 歳、74 歳、 74 歳 後期第 II 相臨床試験に参加した肝細胞癌の患者の うち、下記の選択基準に合致し、除外基準に該当し ない患者とする。 ・ミリプラチンの投与を希望する患者 ・後期第 II 相臨床試験でミリプラチン投与群に割 付られた患者 ・ミリプラチンの最終投与から3 ヵ月(10~14 週) 後の画像診断が終了し、治験責任医師又は治験分担 医師による効果判定が確定している患者 ・後期第 II 相臨床試験で有効性の評価(治験責任 医師又は治験分担医師による効果判定)でTE V と 判定されたが、別部位に再発した患者 ・治験責任医師がミリプラチンの投与を必要と判断 した患者 有効性:有効性(肝細胞癌に対する 抗腫瘍効果)の判定基準として、腫 瘍治療効果度:TE V については、「日 本肝癌研究会 肝癌治療直接効果判 定基準」を用いる。また、CR 及び PR の判定基準は、「固形がん化学療法の 臨床効果判定基準」及び「RESPONSE EVALUATION CRITERIA IN SOLID TUMORS (RECIST criteria) 」を用い る。 〔主要評価項目〕 腫瘍治療効果度 TE V の割合(「日本 肝癌研究会 肝癌治療直接効果判定 基準」による) 〔副次的評価項目〕 CR+PR の割合(「固形がん化学療法 の臨床効果判定基準」による) CR+PR の割合(「RECIST criteria」に よる) 安全性:安全性の評価は「日本癌治 療学会薬物有害反応判定基準」によ り行う。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 16
上記4 試験における有効性評価に関わる主なデザインの相違点を表 2.7.3.1-2 に示した。 表 2.7.3.1-2 有効性評価に関わる主なデザインの相違点 選択基準(抜粋) 試験名 試験 デザイン 用法・用量 (ミリプラチン) 除外基準(抜粋) 対照薬 主なエンドポイント (有効性) 後期第II 相臨床 試験 確率を用い た動的割 付、 非盲検、 実薬対照、 並行群間比 較 投与液濃度は20 mg/mL とし、投与液量 は腫瘍の大きさによ って6 mL を限度に適 宜増減する。 投与回数は最大2 回。 投与間隔は4~12 週。 ・肝切除術、PEIT、PMCT 又はRFA の適応外の患者 ・進行度分類(Stage)が II 又はIII の患者 ・前治療がない患者又は前治 療がある場合は下記の基準 に該当する患者(肝切除術後 の断端再発がない患者、前治 療としてPEIT、PMCT、RFA 又はTAE を施行した場合は 前治療部位の局所再発がな い患者、前治療としてTAI を施行した場合は、その施行 部位をすべて切除した患者 等) ・Child-Pugh 分類が A 又は B の患者 ジノスタ チン ス チマラマ ー [主要評価] TE V の割合 ・肝癌治療直接効果判定基 準 [副次的評価] CR+PR の 割合 ・固形がん化学療法の臨床 効果判定基準 ・RECIST ・肝切除及びPEIT の適応外 の症例 ・前治療としてTAE あるい は化学療法を受けたことの ない症例 ・臨床病期I 又は II の症例 前期第II 相臨床 試験 無対照、 非盲検 投与液濃度は20 mg/mL とし、投与液量 は腫瘍の大きさによ って6 mL を限度に適 宜増減する。 投与回数は最大2 回。 投与間隔は4~12 週。・肝細胞癌の遠隔転移を有す る症例 なし 肝癌治療直接効果判定基 準 [主要評価]CR の割合 [副次的評価] CR+PR の 割合 ・他の治療法では効果が認め られなかったか、他の治療法 の適応外の症例 ・肝硬変を合併する症例で は、臨床病期I 又は II 第I 相臨 床試験 無対照、 非盲検 (Fibonacci の変法によ る漸増法) 初回濃度を6 mg/mL とし、12 mg/mL、20 mg/mL と増量し、投与 液量は腫瘍の大きさ によって6 mL を限度 に適宜増減する。投与 回数は原則として1 回。 ・肝細胞癌の広範な遠隔転移 を有する症例 なし 肝癌治療直接効果判定基 準に準じて抗腫瘍効果判 定を行う。 継続投 与試験 無対照、 非盲検 投与液濃度は20 mg/mL とし、投与液量 は腫瘍の大きさによ って6 mL を限度に適 宜増減する。 投与回数は同一病変 に対しては原則2 回 まで。ただし継続投与 回数の上限はなし。 ・ミリプラチンの投与を希望 する患者 ・後期第II 相臨床試験で有 効性の評価をTE V と判定さ れたが、別部位に再発した患 者 なし [主要評価] TE V の割合 ・肝癌治療直接効果判定基 準 [副次的評価] CR+PR の 割合 ・固形がん化学療法の臨床 効果判定基準 ・RECIST 【用法・用量】 第I 相臨床試験では、体表面積当たりの投与量を順次増量する方法をとることが多いが、 本剤の対象は肝細胞癌であり、通常の静脈内投与あるいは水溶性薬剤の肝動脈内投与とは 異なり、油性懸濁液を肝動脈より腫瘍局所に向けて投与し、腫瘍血管に懸濁液が充満した 時点で終了するため、投与液量は腫瘍の大きさ、血行動態、カテーテルの位置、肝障害の ミリプラチン 2.7.3 臨床的有効性の概要 Page 17
程度を勘案して個別に決定される。従って、投与液量は上限のみ設定し、用量の設定とし ては投与液濃度を増量する方法を採用することとした。初回濃度を動物モデルによる毒性 試験成績から6 mg/mL と設定し、12 mg/mL、20 mg/mL とヨード化ケシ油脂肪酸エチルエ ステルへの最大懸濁濃度まで増量し、腫瘍の大きさに従って最大6 mL まで投与可能とし た。投与回数は原則として1 回とした。投与液量の上限設定にあたっては、投与液が標的 外部位へ流入した場合の危険性を鑑み、CL として安全に投与可能な最大量はジノスタチ ン スチマラマーについて承認された「1 回 6 mL」であると考え、投与液量の上限を 6 mL と設定した。 前期第II 相臨床試験において投与液濃度は、第 I 相臨床試験の結果より推奨用量とされ た20 mg/mL とした。前期第 II 相臨床試験及び後期第 II 相臨床試験共に、投与液量は腫瘍 の大きさに従って最大6 mL まで投与可能とし、投与回数は最大 2 回、投与間隔は 4 週か ら12 週以内とした。 後期第II 相臨床試験は、前期第 II 相臨床試験及び第 I 相臨床試験の結果より投与液濃度 を20 mg/mL とした。 継続投与試験では後期第II 相臨床試験と同用法・用量とし、投与回数は同一病変に対し ては原則2 回までとしたが、更なる別部位の再発があった場合を考慮し投与回数の上限は 設定しなかった。 【対象疾患】 いずれの試験も対象疾患は肝細胞癌としたが、詳細な組み入れ基準は各試験で異なった。 第I 相臨床試験は他の治療で効果が認められなかったか、他の治療法の適応外の患者を 対象とした。Stage について治験実施時に直接的な規定はしていないが、除外基準で「肝細 胞癌の広範な遠隔転移を有する被験者」と定めているため、原発性肝癌取扱い規約 1992 年2 月【第 3 版】における Stage IV-B を除く I、II、III、IV-A の患者を対象とした。
前期第II 相臨床試験では、肝切除及び PEIT の適応外で、前治療として TACE あるいは 化学療法を受けたことのない患者、かつ、Stage については、第 I 相臨床試験と同様原発性 肝癌取扱い規約 1992 年 2 月【第 3 版】における Stage IV-B を除く I、II、III、IV-A の患者 を対象とした。 後期第 II 相臨床試験では、肝切除術、PEIT、PMCT 又は RFA の適応外で、前治療がな い又は前治療があるとしても局所治療で治療し前治療部位に局所再発がないか、TAI を施 行した場合はその施行部位をすべて切除した患者で、かつ、原発性肝癌取扱い規約 2000 年11 月【第 4 版】における Stage II、III の患者を対象とした。 継続投与試験の目的は、後期第II 相臨床試験に参加し本剤投与により TE V と判定され た後で、別部位に再発した患者の継続投与の希望に対応すること、及び継続投与における 有効性と安全性を検討することであり、特にStage 等の条件を厳密に規定しなかった。 後期第II 相臨床試験では前期第 II 相臨床試験との相違点として RFA 又は PMCT の適応 ミリプラチン 2.7.3 臨床的有効性の概要 Page 18
患者を除いているが、これは治療技術の進歩により後期第II 相臨床試験実施時期には上記 の内科的局所療法が普及していたためである。また、後期第 II 相臨床試験では前期第 II 相臨床試験に比べてStage I 及び IV-A の患者を除いているが、これは本剤の代表的な適応 患者を明確にした上で評価するために規定した。すなわち、Stage I は単発の 2 cm 以下の 腫瘍であり、肝切除術、PEIT、PMCT 又は RFA によって確実な効果の見込まれる場合が多 く、これらの治療法がTACE よりも優先されるため後期第 II 相臨床試験の対象から除いた。 Stage IV-A は 2000 年 11 月に改訂された原発性肝癌取扱い規約【第 4 版】で「脈管侵襲あ り」に該当することになり、TACE による治療では効果が得られ難いと考えられるため後 期第II 相臨床試験の対象から除いた。なお、実際の治療現場においては背景肝の状況や年 齢によって肝切除術が適切でない場合や、超音波で描出が不十分である腫瘍や占居部位に 問題がある腫瘍等の内科的局所療法が適切でない場合などは、腫瘍の大きさや数や Stage には限定されずにTACE が実施されている。 【有効性評価項目】 抗がん剤の抗腫瘍効果判定基準は従来から存在する「固形がん化学療法の臨床効果判定 基準」や国際的な基準である「RECIST」の他に、肝細胞癌治療に特化した基準として「肝 癌治療直接効果判定基準」がある。各抗腫瘍効果判定基準間の主な差異を表 2.7.3.1-3 に示 した。 抗腫瘍効果の判定はいずれの試験も「肝癌治療直接効果判定基準」を採用した。「肝癌治 療直接効果判定基準」は、腫瘍の縮小に加え、造影X 線 CT による腫瘍の壊死を加味して 抗腫瘍効果を判定する基準であり、肝細胞癌において、TACE や RFA、PEIT、PMCT など 腫瘍壊死を来す治療では、腫瘍が完全壊死となっても明らかな腫瘍縮小や消失を示さない 事例が見られることから、日本肝癌研究会により設定された。従って腫瘍縮小率のみで判 定する「固形がん化学療法の臨床効果判定基準」や「RECIST」を用いるよりは「肝癌治療 直接効果判定基準」を判定基準として採用することが妥当と考えた。また、「固形がん化学 療法の臨床効果判定基準」や「RECIST」では 4 週間以上の効果の持続を見ているが、「肝 癌治療直接効果判定基準」においても投与終了3 ヵ月後の時点で判定するため、実質的に 効果の持続を含めて判定していると考えられる。 各試験の主要評価項目について下記に示した。 第I 相臨床試験では、造影 X 線 CT による腫瘍壊死を従来の腫瘍縮小効果と合わせて効 果判定の基準とした「肝癌治療直接効果判定基準」を用いて抗腫瘍効果を判定したが、主 要評価項目は特定しなかった。 前期第II 相臨床試験においては「肝癌治療直接効果判定基準」に準じ、壊死効果と腫瘍 縮小率から判定する TE、新病巣の有無、随伴病巣治療効果を併せて判定する総合評価の CR の割合を主要評価項目とした。本治療法が肝動脈内投与という局所療法であることを 考慮すると、より高い水準の有効性が求められると考えたため、CR+PR の割合ではなく ミリプラチン 2.7.3 臨床的有効性の概要 Page 19
CR の割合を主要評価項目とした。なお、CTD における解析では、CR を判定する上で TE も評価しているため、前期第II 相臨床試験の TE も提示した。 後期第II 相臨床試験及び継続投与試験では、「肝癌治療直接効果判定基準」によるTE を 採用した。これは肝内再発あるいは多中心性発癌を含め治療後再発率の高い肝細胞癌にお いては局所を継続的に制御していくことが極めて重要であり、かつ、本療法は標的とする 病変のみへの効果を期待する治療法であることから、新病巣の有無、随伴病巣治療効果を 併せて評価する総合評価よりも、治療部位のみの効果を判定する TE が適切と考えたから である。また、より治癒や延命につながると考えられるTE V の割合(壊死効果 100%又は 腫瘍縮小率100%)を主要評価項目とした。なお、 臨床試験開始後の2004 年に 改訂された「肝癌治療直接効果判定基準」では治療効果判定は局所治療効果すなわち「狙 った病巣に対する効果」で評価することが明記された。更に後期第II 相臨床試験において は、従来からの抗がん剤の判定基準として用いられてきた腫瘍の縮小と新病変の有無を加 味した「固形がん化学療法の臨床効果判定基準」及び腫瘍の縮小と新病変の有無を加味し た基準で国際的に知られた基準である「RECIST」による CR+PR の割合を副次的評価項目 とした。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 20
表 2.7.3.1-3 各抗腫瘍効果判定基準間の主な差異 判定基準 肝癌治療直接効果判定基準(腫瘍治療効果度〔TE〕部分を抜粋) 固形がん化学療法の臨床効果判定基準 RECIST 壊死効果 評価項目 腫瘍縮小率 腫瘍縮小率 腫瘍縮小率 壊死所見 測定 腫瘍の最大断面における長径とそ れに直交する最大径 腫瘍の長径とそれに直角に交わ る最大径 全標的病変の最長径 壊死効果= 各腫瘍における治療後壊死所見が 得られた領域の面積の総和/各腫 瘍の治療後の面積の総和×100 判定 腫瘍縮小率= {(治療前の積)-(治療後の積)}/ (治療前の積)×100 腫瘍縮小率= {(治療前の積)-(治療後の積)} /(治療前の積)×100 腫瘍縮小率= {(治療前の長径の和)-(治療後 の長径の和)}/(治療前の長径 の和)×100 TE V 壊死効果縮小率 100%又は腫瘍 100% CR 100% 4 週間以上 の持続あり CR 100% 4 週間以上の 持続あり TE IV 壊死効果50%以上 100% 未満又は腫瘍縮小率50% 以上100%未満 PR 50%以上100%未満 持続あり PR 30%以上100%未満 持続あり 50%以上 100%未満 持続なし TE III 壊死効果25%以上 50%未 満又は腫瘍縮小率25%以 上50%未満 MR 25%以上 50%未満 持続あり SD CR、PR、PD以外 持続あり TE II 壊死効果瘍縮小率25%未満及び腫25%未満 NC 50%未満 持続あり PD 20%以上増大又は新病変の出現 判定区分 TE I 壊死効果にかかわらず、腫瘍が25%以上増大 PD 25%以上増大又は新病変の出現 判定時期 又は期間 本治験では、原則投与終了3 ヵ月 後に実施。画像診断が実施できず 判定できない場合は判定不能 (NE)とした。 4 週間以上の持続 本治験では、何らかの理由で効 果判定ができない場合は判定不 能(NE)とした。 4 週間以上の持続 本治験では、何らかの理由で効 果判定ができない場合は判定不 能(NE)とした。 新病変 考慮なし 考慮あり 考慮あり ミリプラチン 2.7.3 臨床的有効性の概要 Page 21
2.7.3.2 個々の試験結果の要約 第I 相臨床試験、前期第 II 相臨床試験、後期第 II 相臨床試験及び継続投与試験の結果に ついて以下に要約した。 (1) 第 I 相臨床試験 本剤の肝動脈内投与における最大許容量の推定と肝細胞癌に対する有効性、薬物動態学 的検討を目的として、他の治療法では無効か適応外の肝細胞癌患者11 例を対象として無対 照、非盲検試験(Fibonacci の変法による漸増法)を実施した。 抗がん剤の第I 相臨床試験では、体表面積当たりの投与量を順次変更する方法を選択す ることが多い。本剤は通常の静脈内投与あるいは水溶性薬剤の肝動脈内投与ではなく、油 性懸濁液を肝動脈より局所投与し、腫瘍血管に懸濁液が充満した時点で終了するため、投 与液量は腫瘍の大きさ、血行動態、カテーテルの位置、肝障害の程度を勘案して個別に決 定される。従って投与液濃度を変更する方法を選択した。初回濃度を動物モデルによる毒 性試験より6 mg/mL と設定し、Fibonacci の変法に従って副作用の程度を見ながら 12 mg/mL、 20 mg/mL に増量した。また、投与液量は上限を 6 mL とし、本剤が腫瘍血管に充満した時 点で投与終了とした。 6 mg/mL 及び 12 mg/mL では各 3 例の被験者に投与され、安全性上特に問題となる副作 用はなく20 mg/mL に増量した。20 mg/mL では 3 例の投与が終了した段階で、1 例に 1 グ レードの変動を伴うグレード3 の血小板数減少が認められ、3 例の追加が必要となった。 追加で2 例に投与された時点で、グレード 2 以上の副作用を経験した被験者は 5 例中 3 例 となった。あと1 例追加して 6 例とする場合、追加する 1 例がグレード 2 以上の副作用を 発現する場合には、最大許容量は20 mg/mL(投与液量上限 6 mL)と推定できるが、本剤 の懸濁可能な濃度は最大20 mg/mL であることを勘案すると、6 例目の投与結果に関わらず 第II 相臨床試験の推奨用量は 20 mg/mL(投与液量上限 6 mL)となるため、6 例目の投与 は行わず、最大許容量は20 mg/mL 以上(投与液量上限 6 mL)と推定した。 有効性については12 mg/mL で CR 1 例、20 mg/mL で PR 1 例が得られ、有効率(CR+PR の割合)は18.2%(2/11)であった。 以上より本剤の最大許容量は20 mg/mL 以上(投与液量上限 6 mL)と推定されたが、懸 濁可能な最大濃度が20 mg/mL であることを踏まえ、前期第 II 相臨床試験の推奨用量を 20 mg/mL(投与液量上限 6 mL)とした。また本治験は第 I 相段階であり、低い用量レベルで の被験者が含まれていること、進行した被験者が多く全例再発例でシスプラチンによる前 治療無効例が11 例中 7 例含まれていることを考慮すると、本剤は肝細胞癌に対して有用で ある可能性が示唆された。 (2) 前期第 II 相臨床試験 肝細胞癌に対する本剤の第 I 相臨床試験で得られた推奨用量における有効性、安全性及 ミリプラチン 2.7.3 臨床的有効性の概要 Page 22
び体内動態を検討する目的で、肝細胞癌患者17 例を対象として無対照、非盲検試験を実施 した。有効性の主要評価項目はCR の割合(肝癌治療直接効果判定基準)、副次的評価項目 はCR+PR の割合(肝癌治療直接効果判定基準)とし、投与回数は最大 2 回、投与液量は 1 回当たり6 mL を上限として腫瘍の大きさに従って投与した。 有効性及び安全性の評価対象は15 例で、主要評価項目である CR の割合は 60.0%(9/15)、 PR と判定された被験者がいなかったため副次的評価項目である CR+PR の割合も 60.0% (9/15)であった。 安全性については、グレード2 以下で発現頻度の高い有害事象として発熱や疼痛が認め られたが肝動脈内投与時によく見られる投与直後の一過性の事象であった。グレード3 以 上の有害事象として肝機能に関連する臨床検査値異常が認められたが、いずれも慢性肝炎 あるいは肝硬変を合併しており、肝動脈内投与されることを考慮すると必ずしも臨床的に 重篤な事象ではないと考えた。また、抗がん剤に特有の骨髄障害や白金製剤に顕著な重度 の腎機能障害に類する有害事象は認められなかった。 本剤に特徴的な有害事象として全例に好酸球増多が発現したが1 回目投与より 2 回目投 与で程度が軽減しており、すべての被験者が投与4 週後には回復していること、IgE 上昇 が見られなかったことを考慮するとアレルギーやアナフィラキシーなどの免疫学的な変化 に結びつくものではないと判断した。 以上より本剤は肝細胞癌に対する抗腫瘍効果が期待され、安全性についてもグレード 4 の重篤な事象は認められずほとんどが投与4~6 週後には回復していることから、コントロ ール可能で忍容可能であると考えられた。 (3) 後期第 II 相臨床試験 肝細胞癌に対する本剤の有効性、安全性、薬物動態を検討することを目的に、肝細胞癌 患者 131 例を対象としてジノスタチン スチマラマーを対照薬とした並行群間比較試験を 実施した。有効性の主要評価項目はTE V の割合(肝癌治療直接効果判定基準)、副次的評 価項目はCR+PR の割合(固形がん化学療法の臨床効果判定基準及び RECIST)、安全性は 「日本癌治療学会薬物有害反応判定基準」に従い判定し、いずれの薬剤も投与回数は最大 2 回、投与液量は 1 回当たり 6 mL を上限として腫瘍の大きさに従って投与した。 有効性及び安全性の評価対象はミリプラチン投与群が83 例、ジノスタチン スチマラマ ー投与群が 39 例で、主要評価項目である TE V の割合はミリプラチン投与群が 26.5% (22/83)、ジノスタチン スチマラマー投与群が 17.9%(7/39)であり、両群で同程度であ った。副次的評価項目は CR+PR の割合(固形がん化学療法の臨床効果判定基準)はミリ プラチン投与群が 20.5%(17/83)、ジノスタチン スチマラマー投与群が 23.1%(9/39)、 CR+PR の割合(RECIST)はミリプラチン投与群が 24.1%(20/83)、ジノスタチン スチマ ラマー投与群が25.6%(10/39)であり、両群で同程度であった。 ミリプラチン投与群は、1 回目投与後に比較して 2 回目投与後に有害事象の発現割合が ミリプラチン 2.7.3 臨床的有効性の概要 Page 23
減少する傾向があったが、ジノスタチン スチマラマー投与群では 2 回目投与後に有害事 象の発現割合が増加する傾向があった。死亡、重篤、投与の中止に至る有害事象の発現リ スクはミリプラチンとジノスタチン スチマラマーで大きくは異ならないと考えられた。本 剤特有の有害事象として好酸球数の増加があった。好酸球の増加は発熱を伴うものの2 回 目投与後の発現割合は低くなっており特に処置なく回復し、アナフィラキシー反応に結び つく可能性は低いと考えられた。また、腎障害の発現はなかったものの腎機能に関連する 臨床検査値異常変動はミリプラチン投与群に多かった。一方、ジノスタチン スチマラマー 投与群は投与部位における血管障害が高頻度に発現すること、及び不可逆的な肝臓に関連 する有害事象発現リスクが高いことなど、再発を繰り返す肝細胞癌において後治療への支 障や予後に悪影響を与えるリスクが高いことが示唆され、総合すると、本剤の方が安全で 使いやすい薬剤であると考えられた。 以上より本剤は、ヨード化ケシ油脂肪酸エチルエステルに懸濁し肝動脈内に投与するCL として唯一の既承認医薬品であるジノスタチン スチマラマーに比較して同程度の抗腫瘍 効果を示し、安全で使いやすく、有用な薬剤であると考えられた。 20 年 月 日に実施した 相談(# 、1.13-01 参照)における「 。」との助言を踏まえ、後期第 II 相臨床試験に組み 入れられた被験者に関して、各実施医療機関の治験責任医師に「登録時点でCE が優先さ れる被験者」であったか否かを調査した。後期第II 相臨床試験の有効性及び安全性の解析 対象集団122 例(ミリプラチン投与群 83 例、ジノスタチン スチマラマー投与群 39 例) 全例について調査結果が得られ、「CE が優先される被験者(以下、CE の対象であった被 験者)」以外の被験者(以下、CL の対象であった被験者)は 39 例(ミリプラチン投与群 27 例、ジノスタチン スチマラマー投与群 12 例)であった。CL の対象であった被験者に 関しては、CL を選択した理由を調査した。CL を選択した理由として、「肝機能へのダメ ージが大きいと予測されるため」が92.3%(36/39)、「選択的に塞栓できず、胆癌領域に比 べ塞栓範囲が大きくなると思われるため」が17.9%(7/39)、「そのほか」が7.7%(3/39)(重 複回答)であった(表2.7.6.3-33 参照)。追加調査結果は、参考資料として第 5 部に添付し た(5.3.5.1-参 01 参照)。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 24
(4) 継続投与試験 本剤の継続投与の希望に対応すること及び継続投与における有効性と安全性を検討する ことを目的として、後期第II 相臨床試験に参加し本剤投与群に割り付けられて TE V と判 定された後、別部位に肝細胞癌の再発が認められた患者を対象として無対照、非盲検試験 を実施し、のべ4 例(うち 1 例は 2 回投与され、2 例と扱った)に投与された。有効性の 主要評価項目はTE V の割合(肝癌治療直接効果判定基準)、副次的評価項目は CR+PR の 割合(固形がん化学療法の臨床効果判定基準及びRECIST)、安全性は「日本癌治療学会薬 物有害反応判定基準」に従い判定し、投与液量は1 回当たり 6 mL を上限として腫瘍の大 きさに従って投与した。投与回数については新たな再発がある場合を考慮し、継続投与が 可能な投与回数の上限は設定しないが、原則として1 つの部位当たりの投与回数は 2 回ま でとした。 主要評価項目であるTE V は 4 例中 1 例であった。副次的評価項目の CR+PR(固形がん 化学療法の臨床効果判定基準及びRECIST)はいずれも 4 例中 1 例であった。 安全性については後期第 II 相臨床試験の安全性成績と比較して新たな有害事象は各々1 例に発現した「血中アルブミン増加」、「点滴部位紅斑」のみであり、肝細胞癌の治療にお いて特に問題となる事象は認められなかった。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 25
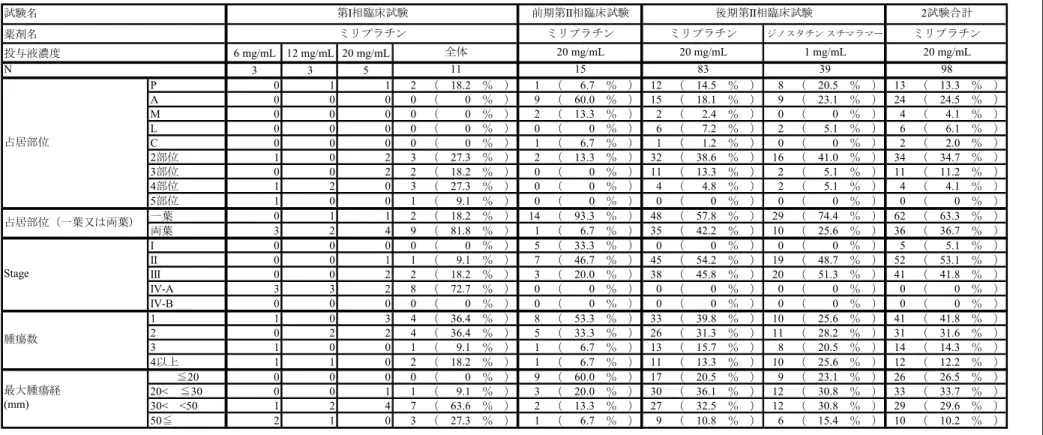
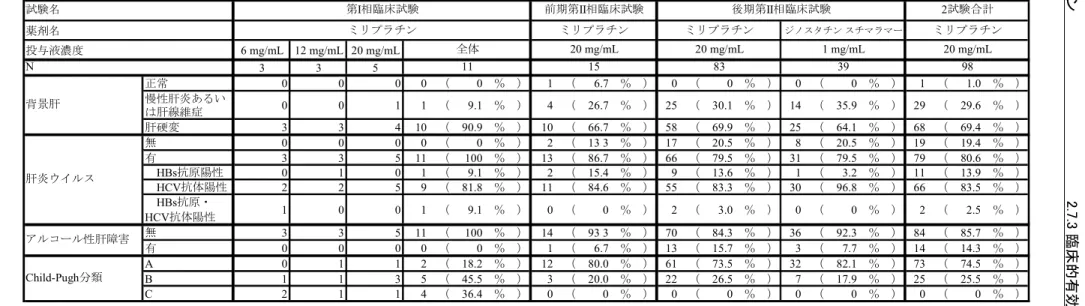
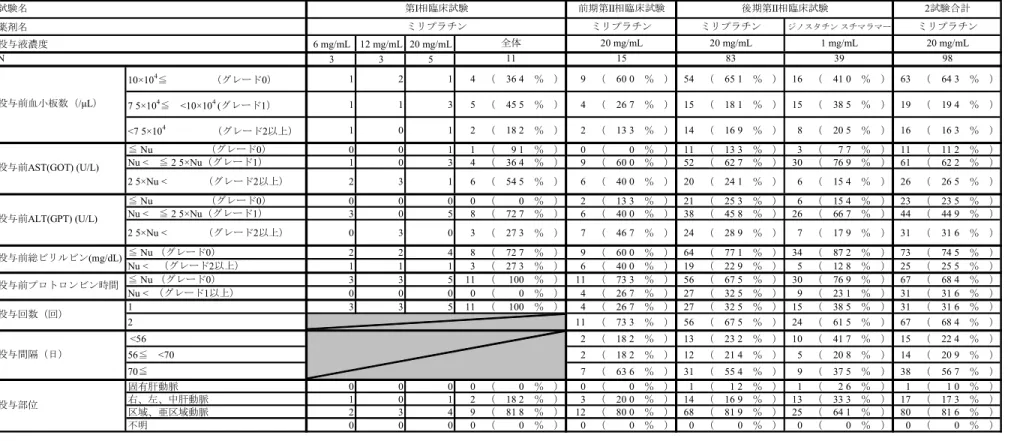
2.7.3.3 全試験を通しての結果の比較と解析 2.7.3.3.1 試験対象集団 (1) 人口統計学的特性及びその他の基準値 全試験の有効性評価対象例について、人口統計学的特性及びその他の基準値の特性を表 2.7.3.3-1 に示した。 人口統計学的特性及びその他の基準値のうち、腫瘍数、最大腫瘍径については客観的に かつ統一的に測定された効果判定委員会データを用いた。Stage は測定可能病変以外も含ん だ腫瘍の進行度を分類するという観点から投与前に治験責任医師又は治験分担医師にて判 定されたデータを用いた。なお、第I 相臨床試験及び前期第 II 相臨床試験については原発 性肝癌取扱い規約(2000 年 11 月【第 4 版】)に従って新たに Stage を導出した。また、投 与部位については、複数の投与部位から投与された被験者は最も末梢の動脈に分類した。 【第I 相臨床試験】 有効性評価対象例は11 例であった。内訳は 6 mg/mL が 3 例、12 mg/mL が 3 例、20 mg/mL が5 例であった。 性別は男性6 例、女性 5 例、平均年齢は 67 歳、全例が前治療有であった。 腫瘍数は1 個及び 2 個が各々36.4%(4/11)と多かった。最大腫瘍径は 30 mm 超が 90.9% (10/11)、Stage は Stage IV-A が 72.7%(8/11)と多かった。占居部位は 1 部位では P が 18.2% (2/11)のみであり、複数部位が多くを占めた。 背景肝は肝硬変が90.9%(10/11)、肝炎ウイルス有の被験者のうち HCV 抗体陽性が 81.8% (9/11)と多かった。Child-Pugh 分類は A が 18.2%(2/11)、B が 45.5%(5/11)であり、C も存在した36.4%(4/11)。 投与前臨床検査値では、血小板数はグレード0 が 36.4%(4/11)、AST(GOT)はグレー ド0 が 9.1%(1/11)、ALT(GPT)はグレード 0 が 0%(0/11)、総ビリルビンはグレード 0 が72.7%(8/11)、プロトロンビン時間はグレード 0 が 100%(11/11)であった。 投与回数は原則1 回で、投与部位は区域、亜区域動脈が 81.8%(9/11)と多かった。 【前期第II 相臨床試験】 有効性評価対象例は15 例であった。 性別は男性14 例、女性 1 例、平均年齢は 64 歳、前治療無は 73.3%(11/15)であった。 腫瘍数は1 個が 53.3%(8/15)と多く、次いで 2 個が 33.3%(5/15)であった。最大腫瘍 径は20 mm 以下が 60.0%(9/15)と多かった。Stage は Stage I が 33.3%(5/15)、Stage II が 46.7%(7/15)、Stage III が 20.0%(3/15)であった。占居部位は 1 部位では P、A が各々6.7% (1/15)、60.0%(9/15)で右葉が多く、複数部位は 2 部位が 13.3%(2/15)であった。
背景肝は肝硬変が 66.7%(10/15)、慢性肝炎が 26.7%(4/15)、肝炎ウイルス有の被験者 のうちHBs 抗原陽性が 15.4%(2/15)、HCV 抗体陽性が 84.6%(11/15)、Child-Pugh 分類は A が 80.0%(12/15)、B が 20.0%(3/15)であった。
投与前臨床検査値では、血小板数はグレード0 が 60.0%(9/15)、AST(GOT)はグレー ド0 が 0%(0/15)、ALT(GPT)はグレード 0 が 13.3%(2/15)、総ビリルビンはグレード 0 が60.0%(9/15)、プロトロンビン時間はグレード 0 が 73.3%(11/15)であった。 投与回数は1 回が 26.7%(4/15)、2 回が 73.3%(11/15)、2 回投与例の投与間隔は 70 日 以上が63.6%(7/15)と多く、投与部位は区域、亜区域動脈が 80.0%(12/15)と多かった。 【後期第II 相臨床試験】 有効性評価対象例はミリプラチン投与群 83 例、ジノスタチン スチマラマー投与群 39 例であった。 ミリプラチン投与群83 例のうち、性別は男性 70 例、女性 13 例、平均年齢は 66 歳、前 治療無は69.9%(58/83)であった。前治療有のうち、手術が 48.0%(12/25)と多く、前治 療回数は1 回が 52.0%(13/25)と多かった。 腫瘍数は1 個が 39.8%(33/83)と多く、次いで 2 個が 31.3%(26/83)であった。最大腫 瘍径は「20 mm< ≤30 mm」が 36.1%(30/83)と多かった。Stage は Stage II が 54.2%(45/83)、 Stage III が 45.8%(38/83)であった。占居部位は 1 部位では P、A が各々14.5%(12/83)、 18.1%(15/83)で右葉が多く、複数部位では 2 部位が 38.6%(32/83)と最も多かった。ま た、一葉が57.8%(48/83)、両葉が 42.2%(35/83)であった。 背景肝は慢性肝炎あるいは肝繊維症が30.1%(25/83)、肝硬変が 69.9%(58/83)、肝炎ウ イルス有の被験者のうちHBs 抗原陽性が 13.6%(9/66)、HCV 抗体陽性が 83.3%(55/66)、 Child-Pugh 分類は A が 73.5%(61/83)、B が 26.5%(22/83)であった。 投与前臨床検査値では、血小板数はグレード 0 が 65.1%(54/83)、AST(GOT)はグレ ード0 が 13.3%(11/83)、ALT(GPT)はグレード 0 が 25.3%(21/83)、総ビリルビンはグ レード0 が 77.1%(64/83)、プロトロンビン時間はグレード 0 が 67.5%(56/83)であった。 投与回数は1 回が 32.5%(27/83)、2 回が 67.5%(56/83)、2 回投与例の投与間隔は 70 日 以上が55.4%(31/56)と多かった。投与部位は区域、亜区域動脈が 81.9%(68/83)と多か った。 被験者背景のうちミリプラチン投与群とジノスタチン スチマラマー投与群と比較して 20%以上の差が認められたカテゴリーを含む項目は「投与前血小板数」及び「投与前 ALT (GPT)」であった。 後期第II 相臨床試験で実施した追加調査の結果 CE の対象であった集団及び CL の対象 であった集団の人口統計学的特性及びその他の基準値の特性を表2.7.3.3-2 に示した。 CE の対象であった被験者 83 例(以下、CE)と CL の対象であった被験者 39 例(以下、 CL)について分布に 20%以上の相違が見られた項目は、占居部位〔CE:3 部位 3.6%(3/83)、 CL:3 部位 25.6%(10/39)〕、占居部位(一葉又は両葉)〔CE:一葉 75.9%(63/83)、両葉 24.1%(20/83)、CL:一葉 35.9%(14/39)、両葉 64.1%(25/39)〕、腫瘍数〔CE:1 個 44.6% (37/83)、4 個以上 9.6%(8/83)、CL:1 個 15.4%(6/39)、4 個以上 33.3%(13/39)〕、最大 ミリプラチン 2.7.3 臨床的有効性の概要 Page 27
腫瘍径〔CE:20 mm 以下 12.0%(10/83)、CL:20 mm 以下 41.0%(16/39)〕、Child-Pugh 分 類〔CE:A 84.3%(70/83)、B 15.7%(13/83)、CL:A 59.0%(23/39)、B 41.0%(16/39)〕、 投与部位〔CE:右、左、中肝動脈 15.7%(13/83)、区域、亜区域動脈 83.1%(69/83)、CL: 右、左、中肝動脈35.9%(14/39)、区域、亜区域動脈 61.5%(24/39)〕であった。CL の対 象であった集団は、CE の対象であった集団に比較して腫瘍の占居部位が広い被験者、腫 瘍数の多い被験者、最大腫瘍径が20 mm 以下の被験者、Child-Pugh 分類 B の被験者が多く、 また、投与部位に関しては区域、亜区域動脈から投与した被験者は少なかった。 (2) 人口統計学的特性及びその他の基準値に関する試験間の差異 人口統計学的特性及びその他の基準値のうち、腫瘍因子及び肝機能因子に関して、後期 第II 相臨床試験と前期第 II 相臨床試験における分布の相違を検討した。 【腫瘍因子】 <腫瘍数> 後期第II 相臨床試験では 3 個が 15.7%(13/83)、4 個以上が 13.3%(11/83)、前期第 II 相 臨床試験では3 個、4 個以上のいずれも 6.7%(1/15)であり、後期第 II 相臨床試験におい て腫瘍数が3 個以上の被験者の割合が高かった。 <最大腫瘍径> 後期第II 相臨床試験では「20 mm< ≤30 mm」が 36.1%(30/83)と最も多く、20 mm 以下は20.5%(17/83)であったが、前期第 II 相臨床試験では 20 mm 以下が 60.0%(9/15) と半数以上を占めており、後期第II 相臨床試験において最大腫瘍径が大きい被験者が多か った。 <占居部位> 腫瘍数が多くなれば、腫瘍の占居部位が複数にまたがる可能性が高いと考えられるが、 後期第II 相臨床試験では一葉占居が 57.8%(48/83)、両葉占居が 42.2%(35/83)であった が、前期第 II 相臨床試験では 1 例を除いた 93.3%(14/15)が一葉占居であり、後期第 II 相臨床試験において腫瘍が両葉を占居している被験者が多かった。 <Stage>
後期第II 相臨床試験では Stage II と Stage III が約半数ずつであったが、前期第 II 相臨床 試験ではStage I が 33.3%(5/15)、II が 46.7%(7/15)、III が 20.0%(3/15)であった。
後期第II 相臨床試験では選択基準で Stage II 又は III と規定したため、Stage I は存在しな かった。 【肝機能因子】 <投与前AST(GOT)> 後期第II 相臨床試験ではグレード 2 以上が 24.1%(20/83)であったが、前期第 II 相臨床 試験では40.0%(6/15)であり、前期第 II 相臨床試験においてグレード 2 以上の被験者が ミリプラチン 2.7.3 臨床的有効性の概要 Page 28
多かった。 <投与前ALT(GPT)> 後期第II 相臨床試験ではグレード 2 以上が 28.9%(24/83)であったが、前期第 II 相臨床 試験では46.7%(7/15)であり、前期第 II 相臨床試験においてグレード 2 以上の被験者が 多かった。 <投与前総ビリルビン> 後期第II 相臨床試験ではグレード 2 以上が 22.9%(19/83)であったが、前期第 II 相臨床 試験では40.0%(6/15)であり、前期第 II 相臨床試験においてグレード 2 以上の被験者が 多かった。 ミリプラチン 2.7.3 臨床的有効性の概要 Page 29
表 2.7.3.3-1 人口統計学的特性及びその他の基準値の特性 薬剤名 投与液濃度 6 mg/mL 12 mg/mL 20 mg/mL 3 3 5 男 0 3 3 6 ( 54 5 % ) 14 ( 93 3 % ) 70 ( 84 3 % ) 30 ( 76 9 % ) 84 ( 85 7 % ) 女 3 0 2 5 ( 45 5 % ) 1 ( 6 7 % ) 13 ( 15 7 % ) 9 ( 23 1 % ) 14 ( 14 3 % ) 年齢(歳) 平均値 68 65 67 <65 0 1 2 3 ( 27 3 % ) 7 ( 46 7 % ) 25 ( 30 1 % ) 11 ( 28 2 % ) 32 ( 32 7 % ) 65≦ <70 2 2 0 4 ( 36 4 % ) 5 ( 33 3 % ) 31 ( 37 3 % ) 14 ( 35 9 % ) 36 ( 36 7 % ) 70≦ 1 0 3 4 ( 36 4 % ) 3 ( 20 0 % ) 27 ( 32 5 % ) 14 ( 35 9 % ) 30 ( 30 6 % ) 無 0 0 0 0 ( 0 % ) 11 ( 73 3 % ) 58 ( 69 9 % ) 26 ( 66 7 % ) 69 ( 70 4 % ) 有 3 3 5 11 ( 100 % ) 4 ( 26 7 % ) 25 ( 30 1 % ) 13 ( 33 3 % ) 29 ( 29 6 % ) 手術 0 0 0 0 ( 0 % ) 1 ( 25 0 % ) 12 ( 48 0 % ) 7 ( 53 8 % ) 13 ( 44 8 % ) PEIT 0 0 0 0 ( 0 % ) 1 ( 25 0 % ) 4 ( 16 0 % ) 1 ( 7 7 % ) 5 ( 17 2 % ) PMCT 0 0 0 0 ( 0 % ) 0 ( 0 % ) 1 ( 4 0 % ) 1 ( 7 7 % ) 1 ( 3 4 % ) RFA 0 0 0 0 ( 0 % ) 0 ( 0 % ) 3 ( 12 0 % ) 2 ( 15 4 % ) 3 ( 10 3 % ) TAE 0 1 2 3 ( 27 3 % ) 0 ( 0 % ) 3 ( 12 0 % ) 1 ( 7 7 % ) 3 ( 10 3 % ) 〈Chemolipiodolization or TAE〉 +切除 0 0 0 0 ( 0 % ) 2 ( 50 0 % ) 0 ( 0 % ) 1 ( 7 7 % ) 2 ( 6 9 % ) Chemolipiodolization 3 2 3 8 ( 72 7 % ) 0 ( 0 % ) 1 ( 4 0 % ) 0 ( 0 % ) 1 ( 3 4 % ) 放射線 0 0 0 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) その他 0 0 0 0 ( 0 % ) 0 ( 0 % ) 1 ( 4 0 % ) 0 ( 0 % ) 1 ( 3 4 % ) 1 1 1 1 3 ( 27 3 % ) 1 ( 25 0 % ) 13 ( 52 0 % ) 9 ( 69 2 % ) 14 ( 48 3 % ) 2 1 0 1 2 ( 18 2 % ) 0 ( 0 % ) 6 ( 24 0 % ) 2 ( 15 4 % ) 6 ( 20 7 % ) 3以上 1 2 3 6 ( 54 5 % ) 3 ( 75 0 % ) 6 ( 24 0 % ) 2 ( 15 4 % ) 9 ( 31 0 % ) 年齢分布(歳) 67 前治療回数 (回) 試験名 N 第I相臨床試験 性別 11 全体 ミリプラチン 20 mg/mL 20 mg/mL 20 mg/mL 1 mg/mL 39 2試験合計 ミリプラチン 後期第II相臨床試験 ミリプラチン ミリプラチン 前期第II相臨床試験 ジノスタチン スチマラマー 64 83 66 15 66 66 98 前治療 前治療 (初回治療) 2 試験合計:前期第 II 相臨床試験+後期第 II 相臨床試験 ミリプラチン 2.7.3 臨床的有効性の概要 Page 30
表 2.7.3.3-1 人口統計学的特性及びその他の基準値の特性(続き) 薬剤名 投与液濃度 6 mg/mL 12 mg/mL 20 mg/mL 3 3 5 P 0 1 1 2 ( 18.2 % ) 1 ( 6.7 % ) 12 ( 14.5 % ) 8 ( 20.5 % ) 13 ( 13.3 % ) A 0 0 0 0 ( 0 % ) 9 ( 60.0 % ) 15 ( 18.1 % ) 9 ( 23.1 % ) 24 ( 24.5 % ) M 0 0 0 0 ( 0 % ) 2 ( 13.3 % ) 2 ( 2.4 % ) 0 ( 0 % ) 4 ( 4.1 % ) L 0 0 0 0 ( 0 % ) 0 ( 0 % ) 6 ( 7.2 % ) 2 ( 5.1 % ) 6 ( 6.1 % ) C 0 0 0 0 ( 0 % ) 1 ( 6.7 % ) 1 ( 1.2 % ) 0 ( 0 % ) 2 ( 2.0 % ) 2部位 1 0 2 3 ( 27.3 % ) 2 ( 13.3 % ) 32 ( 38.6 % ) 16 ( 41.0 % ) 34 ( 34.7 % ) 3部位 0 0 2 2 ( 18.2 % ) 0 ( 0 % ) 11 ( 13.3 % ) 2 ( 5.1 % ) 11 ( 11.2 % ) 4部位 1 2 0 3 ( 27.3 % ) 0 ( 0 % ) 4 ( 4.8 % ) 2 ( 5.1 % ) 4 ( 4.1 % ) 5部位 1 0 0 1 ( 9.1 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 一葉 0 1 1 2 ( 18.2 % ) 14 ( 93.3 % ) 48 ( 57.8 % ) 29 ( 74.4 % ) 62 ( 63.3 % ) 両葉 3 2 4 9 ( 81.8 % ) 1 ( 6.7 % ) 35 ( 42.2 % ) 10 ( 25.6 % ) 36 ( 36.7 % ) I 0 0 0 0 ( 0 % ) 5 ( 33.3 % ) 0 ( 0 % ) 0 ( 0 % ) 5 ( 5.1 % ) II 0 0 1 1 ( 9.1 % ) 7 ( 46.7 % ) 45 ( 54.2 % ) 19 ( 48.7 % ) 52 ( 53.1 % ) III 0 0 2 2 ( 18.2 % ) 3 ( 20.0 % ) 38 ( 45.8 % ) 20 ( 51.3 % ) 41 ( 41.8 % ) IV-A 3 3 2 8 ( 72.7 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) IV-B 0 0 0 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 0 ( 0 % ) 1 1 0 3 4 ( 36.4 % ) 8 ( 53.3 % ) 33 ( 39.8 % ) 10 ( 25.6 % ) 41 ( 41.8 % ) 2 0 2 2 4 ( 36.4 % ) 5 ( 33.3 % ) 26 ( 31.3 % ) 11 ( 28.2 % ) 31 ( 31.6 % ) 3 1 0 0 1 ( 9.1 % ) 1 ( 6.7 % ) 13 ( 15.7 % ) 8 ( 20.5 % ) 14 ( 14.3 % ) 4以上 1 1 0 2 ( 18.2 % ) 1 ( 6.7 % ) 11 ( 13.3 % ) 10 ( 25.6 % ) 12 ( 12.2 % ) ≦20 0 0 0 0 ( 0 % ) 9 ( 60.0 % ) 17 ( 20.5 % ) 9 ( 23.1 % ) 26 ( 26.5 % ) 20< ≦30 0 0 1 1 ( 9.1 % ) 3 ( 20.0 % ) 30 ( 36.1 % ) 12 ( 30.8 % ) 33 ( 33.7 % ) 30< <50 1 2 4 7 ( 63.6 % ) 2 ( 13.3 % ) 27 ( 32.5 % ) 12 ( 30.8 % ) 29 ( 29.6 % ) 50≦ 2 1 0 3 ( 27.3 % ) 1 ( 6.7 % ) 9 ( 10.8 % ) 6 ( 15.4 % ) 10 ( 10.2 % ) 98 83 15 39 2試験合計 ミリプラチン 後期第II相臨床試験 ミリプラチン ミリプラチン 前期第II相臨床試験 ジノスタチン スチマラマー 20 mg/mL 20 mg/mL 20 mg/mL 1 mg/mL 第I相臨床試験 11 全体 ミリプラチン 試験名 N Stage 占居部位 腫瘍数 最大腫瘍経 (mm) 占居部位(一葉又は両葉) 2 試験合計:前期第 II 相臨床試験+後期第 II 相臨床試験 ミリプラチン 2.7.3 臨床的有効性の概要 Page 31