2020 年 9 月改訂(第 18 版)
日本標準商品分類番号
872699医薬品インタビューフォーム
日本病院薬剤師会の IF 記載要領 2018(2019 年更新版)に準拠して作成 アトピー性皮膚炎治療剤(免疫抑制外用剤) タクロリムス水和物 軟膏プロトピック
Ⓡ
軟膏 0.03%小児用
Protopic
Ⓡ
Ointment for Pediatric
剤 形 軟膏剤 製 剤 の 規 制 区 分 劇薬、処方箋医薬品 注) (注意-医師等の処方箋により使用すること) 規 格 ・ 含 量 1g 中 日局 タクロリムス水和物・・・・・・・・・・・・・・・・0.31mg (タクロリムスとして 0.3mg) 一 般 名 和名:タクロリムス水和物(JAN) 洋名:Tacrolimus Hydrate(JAN) 製 造 販 売 承 認 年 月 日 薬 価 基 準 収 載 ・ 販 売 開 始 年 月 日 製造販売承認年月日:2003 年 7 月 17 日 薬価基準収載年月日:2003 年 12 月 12 日 販 売 開 始 年 月 日:2003 年 12 月 12 日 製 造 販 売 ( 輸 入 ) ・ 提 携 ・ 販 売 会 社 名 製 造 販 売:マ ル ホ 株 式 会 社 医薬情報担当者の連絡先 問 い 合 わ せ 窓 口 マルホ株式会社 製品情報センター TEL:0120-12-2834 受付時間:9 時 30 分~17 時 30 分 (土、日、休日および当社休業日を除く) 医療関係者向けホームページ https://www.maruho.co.jp/medical/index.html 本 IF は 2020 年 9 月改訂の添付文書の記載に基づき改訂した。 最新の情報は、独立行政法人 医薬品医療機器総合機構の医薬品情報検索ページで 確認してください。
医薬品インタビューフォーム利用の手引きの概要
―日本病院薬剤師会―
(2020年4月改訂) 1.医薬品インタビューフォーム作成の経緯 医療用医薬品の基本的な要約情報として、医療用医薬品添付文書(以下、添付文書)がある。 医療現場で医師・薬剤師等の医療従事者が日常業務に必要な医薬品の適正使用情報を活用する 際には、添付文書に記載された情報を裏付ける更に詳細な情報が必要な場合があり、製薬企業 の医薬情報担当者(以下、MR)等への情報の追加請求や質疑により情報を補完してきている。 この際に必要な情報を網羅的に入手するための項目リストとして医薬品インタビューフォー ム(以下、IFと略す)が誕生した。 1988年に日本病院薬剤師会(以下、日病薬)学術第2小委員会がIFの位置付け、IF記載様式、 IF記載要領を策定し、その後1998年に日病薬学術第3小委員会が、2008年、2013年に日病薬医薬 情報委員会がIF記載要領の改訂を行ってきた。 IF記載要領2008以降、IFはPDF等の電子的データとして提供することが原則となった。これ により、添付文書の主要な改訂があった場合に改訂の根拠データを追加したIFが速やかに提供 されることとなった。最新版のIFは、医薬品医療機器総合機構(以下、PMDA)の医療用医薬 品情報検索のページ(https://www.pmda.go.jp/PmdaSearch/iyakuSearch/)にて公開されている。日 病薬では、2009年より新医薬品のIFの情報を検討する組織として「インタビューフォーム検討 会」を設置し、個々のIFが添付文書を補完する適正使用情報として適切か審査・検討している。 2019年の添付文書記載要領の変更に合わせ、「IF記載要領2018」が公表され、今般「医療用医 薬品の販売情報提供活動に関するガイドライン」に関連する情報整備のため、その更新版を策 定した。 2.IFとは IFは「添付文書等の情報を補完し、医師・薬剤師等の医療従事者にとって日常業務に必要な、 医薬品の品質管理のための情報、処方設計のための情報、調剤のための情報、医薬品の適正使 用のための情報、薬学的な患者ケアのための情報等が集約された総合的な個別の医薬品解説書 として、日病薬が記載要領を策定し、薬剤師等のために当該医薬品の製造販売又は販売に携わ る企業に作成及び提供を依頼している学術資料」と位置付けられる。 IFに記載する項目配列は日病薬が策定したIF記載要領に準拠し、一部の例外を除き承認の範 囲内の情報が記載される。ただし、製薬企業の機密等に関わるもの及び利用者自らが評価・判 断・提供すべき事項等はIFの記載事項とはならない。言い換えると、製薬企業から提供された IFは、利用者自らが評価・判断・臨床適用するとともに、必要な補完をするものという認識を 持つことを前提としている。 IFの提供は電子データを基本とし、製薬企業での製本は必須ではない。3.IFの利用にあたって 電子媒体のIFは、PMDAの医療用医薬品情報検索のページに掲載場所が設定されている。 製薬企業は「医薬品インタビューフォーム作成の手引き」に従ってIFを作成・提供するが、 IFの原点を踏まえ、医療現場に不足している情報やIF作成時に記載し難い情報等については製 薬企業のMR等へのインタビューにより利用者自らが内容を充実させ、IFの利用性を高める必 要がある。また、随時改訂される使用上の注意等に関する事項に関しては、IFが改訂されるま での間は、製薬企業が提供する改訂内容を明らかにした文書等、あるいは各種の医薬品情報提 供サービス等により薬剤師等自らが整備するとともに、IFの使用にあたっては、最新の添付文 書をPMDAの医薬品医療機器情報検索のページで確認する必要がある。 なお、適正使用や安全性の確保の点から記載されている「Ⅴ.5.臨床成績」や「XII.参考資 料」、「XIII.備考」に関する項目等は承認を受けていない情報が含まれることがあり、その取 り扱いには十分留意すべきである。 4.利用に際しての留意点 IFを日常業務において欠かすことができない医薬品情報源として活用していただきたい。IF は日病薬の要請を受けて、当該医薬品の製造販売又は販売に携わる企業が作成・提供する、医 薬品適正使用のための学術資料であるとの位置づけだが、記載・表現には医薬品、医療機器等 の品質、有効性及び安全性の確保等に関する法律の広告規則や販売情報提供活動ガイドライ ン、製薬協コード・オブ・プラクティス等の制約を一定程度受けざるを得ない。販売情報提供 活動ガイドラインでは、未承認薬や承認外の用法等に関する情報提供について、製薬企業が医 療従事者からの求めに応じて行うことは差し支えないとされており、MR等へのインタビュー や自らの文献調査などにより、利用者自らがIFの内容を充実させるべきものであることを認識 しておかなければならない。製薬企業から得られる情報の科学的根拠を確認し、その客観性を 見抜き、医療現場における適正使用を確保することは薬剤師の本務であり、IFを利用して日常 業務を更に価値あるものにしていただきたい。
目 次
Ⅰ.概要に関する項目 ... 1 1. 開発の経緯 ... 1 2. 製品の治療学的特性 ... 1 3. 製品の製剤学的特性 ... 2 4. 適正使用に関して周知すべき特性 ... 2 5. 承認条件及び流通・使用上の制限事項 ... 2 6. RMP の概要 ... 2 Ⅱ.名称に関する項目 ... 3 1. 販売名 ... 3 2. 一般名 ... 3 3. 構造式又は示性式 ... 3 4. 分子式及び分子量 ... 3 5. 化学名(命名法)又は本質 ... 3 6. 慣用名、別名、略号、記号番号 ... 3 Ⅲ.有効成分に関する項目 ... 4 1. 物理化学的性質 ... 4 2. 有効成分の各種条件下における安定性 ... 4 3. 有効成分の確認試験法、定量法 ... 4 Ⅳ.製剤に関する項目 ... 5 1. 剤形 ... 5 2. 製剤の組成 ... 5 3. 添付溶解液の組成及び容量 ... 5 4. 力価 ... 5 5. 混入する可能性のある夾雑物 ... 6 6. 製剤の各種条件下における安定性 ... 6 7. 調製法及び溶解後の安定性 ... 6 8. 他剤との配合変化(物理化学的変化) ... 6 9. 溶出性 ... 7 10.容器・包装 ... 7 11.別途提供される資材類 ... 7 12.その他 ... 7 Ⅴ.治療に関する項目 ... 8 1. 効能又は効果 ... 8 2. 効能又は効果に関連する注意 ... 8 3. 用法及び用量 ... 8 4. 用法及び用量に関連する注意 ... 8 5. 臨床成績 ... 9 Ⅵ.薬効薬理に関する項目 ... 12 1. 薬理学的に関連ある化合物又は 化合物群 ... 12 2. 薬理作用 ... 12 Ⅶ.薬物動態に関する項目 ... 15 1. 血中濃度の推移 ... 15 2. 薬物速度論的パラメータ ... 16 3. 母集団(ポピュレーション)解析 ... 17 4. 吸収 ... 17 5. 分布 ... 17 6. 代謝 ... 19 7. 排泄 ... 19 8. トランスポーターに関する情報 ... 19 9. 透析等による除去率 ... 19 10.特定の背景を有する患者 ... 19 11.その他 ... 19 Ⅷ.安全性(使用上の注意等)に関する項目 ... 20 1. 警告内容とその理由 ... 20 2. 禁忌内容とその理由 ... 20 3. 効能又は効果に関連する注意と その理由 ... 21 4. 用法及び用量に関連する注意と その理由 ... 21 5. 重要な基本的注意とその理由 ... 21 6. 特定の背景を有する患者に関する注意 ... 22 7. 相互作用 ... 23 8. 副作用 ... 23 9. 臨床検査結果に及ぼす影響 ... 27 10.過量投与 ... 27 11.適用上の注意 ... 28 12.その他の注意 ... 28 Ⅸ.非臨床試験に関する項目 ... 29 1. 薬理試験 ... 29 2. 毒性試験 ... 29 Ⅹ.管理的事項に関する項目 ... 32 1. 規制区分 ... 32 2. 有効期間 ... 32 3. 包装状態での貯法 ... 32 4. 取扱い上の注意 ... 32 5. 患者向け資材 ... 32 6. 同一成分・同効薬 ... 32 7. 国際誕生年月日 ... 32 8. 製造販売承認年月日及び承認番号、 薬価基準収載年月日、販売開始年月日 ... 32 9. 効能又は効果追加、用法及び用量変更 追加等の年月日及びその内容 ... 32 10.再審査結果、再評価結果公表年月日及び その内容 ... 3211.再審査期間 ... 32 12.投薬期間制限に関する情報 ... 33 13.各種コード ... 33 14.保険給付上の注意 ... 33 Ⅺ.文献 ... 34 1. 引用文献 ... 34 2. その他の参考文献 ... 35 Ⅻ.参考資料 ... 36 1. 主な外国での発売状況 ... 36 2. 海外における臨床支援情報 ... 38 ⅩⅢ.備考 ... 40 1. 調剤・服薬支援に際して臨床判断を 行うにあたっての参考情報 ... 40 2. その他の関連資料 ... 40
Ⅰ.概要に関する項目
1. 開発の経緯 1984 年、藤沢薬品(現 アステラス製薬株式会社)は放線菌 Streptomyces tsukubaensis の代謝産物の中から マクロライド系の新規免疫抑制剤タクロリムスを見出した。タクロリムスの臨床開発は移植領域より開 始され、現在、国内では経口剤及び注射剤が、(1)腎移植、肝移植、心移植、肺移植、膵移植及び小腸移植 における拒絶反応の抑制、(2)骨髄移植における拒絶反応及び移植片対宿主病の抑制を効能・効果として 発売され、海外でも 100 ヵ国以上で発売されている。さらに、国内において、重症筋無力症、関節リウマ チ(既存治療で効果不十分な場合に限る)、ループス腎炎(ステロイド剤の投与が効果不十分、又は副作 用により困難な場合)、難治性(ステロイド抵抗性、ステロイド依存性)の活動期潰瘍性大腸炎(中等症 ~重症に限る)及び多発性筋炎・皮膚筋炎に合併する間質性肺炎の効能・効果を経口剤(プログラフのみ) で取得している。 タクロリムスは、リンパ球の一種である T 細胞に作用し免疫抑制作用を発現する。ヘルパーT 細胞は IL-2、IFN-γ 等のサイトカインを産生する Th1 細胞と、IL-4、IL-5 等を産生する Th2 細胞の 2 つのサブセッ トに分類されるが、タクロリムスはこれらのヘルパーT 細胞によるサイトカイン産生をいずれも阻害す る。さらに、炎症性細胞である肥満細胞にも直接作用しヒスタミン遊離を抑制する。 タクロリムスがもつ上記薬理作用から、アトピー性皮膚炎に対する治療効果が期待され、臨床開発を企画 したが、小児における開発は成人における有効性、安全性が確立された後に開始することとし、まず成人 での臨床開発を先行させた。そこで、1989 年 12 月よりタクロリムス外用剤の製剤化研究に着手し、製剤 処方改良を経て、その安全性及び有効性が確認され、1999 年 6 月に成人の「アトピー性皮膚炎」を効能・ 効果としてプロトピック軟膏 0.1%が承認された。 その後、小児を対象とした臨床開発に着手し、0.03%に成人の臨床推奨濃度である 0.1%を加えた 2 濃度を 設定し、有効性、安全性を確認する臨床試験を実施した。その結果、小児アトピー性皮膚炎に対しては 0.03%が臨床推奨濃度であるとの結論を得て、2003 年 7 月にプロトピック軟膏 0.03%小児用が承認され た。 なお、欧米においては本邦に先だって小児アトピー性皮膚炎患者を対象とする臨床開発が成人と並行し て実施され、既に小児に対して欧米ともに 0.03%軟膏が承認されている。 2017 年 10 月に製造販売承認がアステラス製薬株式会社よりマルホ株式会社に承継された。 2. 製品の治療学的特性 (1)非臨床試験成績からみた特徴及び有用性 1)ヒトのアトピー性皮膚炎に類似した病態を形成するラット皮膚炎及び NC マウス自然発症皮膚炎に おける皮膚局所炎症反応、真皮での炎症性細胞の増加を抑制する。(「Ⅵ.2.(2)薬効を裏付ける試験成 績」の項参照) 2)Ⅳ型アレルギー反応(遅延型アレルギー反応)を強く抑制する。(マウス)(「Ⅵ.2.(2)薬効を裏付け る試験成績」の項参照) 3)Ⅰ型アレルギー反応の即時型反応には無効であるが、遅発型反応に対しては軽度の抑制効果を有す る。(マウス)(「Ⅵ.2.(2)薬効を裏付ける試験成績」の項参照) 4)ヒト・ヘルパーT 細胞によるサイトカインの産生をステロイドと同等もしくはより強く抑制する。 (in vitro)(「Ⅵ.2.(2)薬効を裏付ける試験成績」の項参照) 5)ヒト肥満細胞の脱顆粒、好酸球の活性化、ランゲルハンス細胞の抗原提示能をステロイドよりも強 く抑制する。(「Ⅵ.2.(2)薬効を裏付ける試験成績」の項参照) 6)ステロイド外用剤に認められる皮膚萎縮作用を示さない。(「Ⅵ.2.(2)薬効を裏付ける試験成績」の項 参照) (2)臨床試験成績からみた特徴及び有用性 [有効性] 1)小児アトピー性皮膚炎に対して優れた有効性を示す。(「Ⅴ.5.(4)1)有効性検証試験」の項参照) 2)長期使用時においても良好な効果を示し、塗布量の減少にも関わらず、その効果は維持できた。 (「Ⅴ.5.(4)2)安全性試験」の項参照) [安全性] 1)本剤の副作用のうち、最も発現率の高いものは塗布部位にみられる皮膚刺激感(ヒリヒリ感、ほて り感、そう痒感等)であったが、ほとんどが軽度~中等度であった。(「Ⅴ.5.(4).2)安全性試験」の項 参照)また、通常、本剤による皮膚刺激感は治療開始初期に塗布後一過性に発現し、皮膚症状の改 善に伴い発現しなくなる。 2)全身的な副作用の発現頻度は低かった。(「Ⅷ.8.(2)その他の副作用」の項参照)3)承認時までの臨床試験では、小児 356 例中 220 例(61.8%)に、臨床検査値異常を含む副作用が認めら れた。主な副作用は疼痛 130 例(36.5%)、熱感 58 例(16.3%)、毛嚢炎 30 例(8.4%)、そう痒感 28 例 (7.9%)、伝染性膿痂疹 18 例(5.1%)であった。(「Ⅷ.8.(2)その他の副作用」の項参照) (承認時:2003 年 7 月) 3. 製品の製剤学的特性 該当資料なし 4. 適正使用に関して周知すべき特性 該当しない 5. 承認条件及び流通・使用上の制限事項 (1)承認条件 本剤の長期使用例について、免疫抑制作用に伴う有害事象の発現状況を調査すること。 (2)流通・使用上の制限事項 該当しない 6. RMP の概要 該当しない
Ⅱ.名称に関する項目
1. 販売名(1)和名
プロトピック軟膏 0.03%小児用 (2)洋名
Protopic Ointment for Pediatric (3)名称の由来 Prograf(タクロリムス水和物のカプセル剤・顆粒剤及び注射剤の商標)の「Pro」と、Topical 及び Atopic Dermatitis の「topic」を組み合わせたものである 2. 一般名 (1)和名(命名法) タクロリムス水和物(JAN) (2)洋名(命名法)
Tacrolimus Hydrate (JAN) tacrolimus (INN) (3)ステム(stem) 免疫抑制剤、ラパマイシン誘導体:-rolimus 3. 構造式又は示性式 4. 分子式及び分子量 分子式:C44H69NO12・H2O 分子量:822.03 5. 化学名(命名法)又は本質 (3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)-5,19-Dihydroxy-3-{(1E)-2-[(1R,3R,4R)-4-hydroxy-3- methoxycyclohexyl]-1-methylethenyl}-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(prop-2-en-1-yl)-15,19-epoxy- 5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone monohydrate (IUPAC)
6. 慣用名、別名、略号、記号番号 治験番号:FR900506、FK506
Ⅲ.有効成分に関する項目
1. 物理化学的性質 (1)外観・性状 白色の結晶又は結晶性の粉末である (2)溶解性 本品はメタノール又はエタノール(99.5)に極めて溶けやすく、N, N-ジメチルホルムアミド又はエタノ ール(95)に溶けやすく、水にほとんど溶けない (3)吸湿性 吸湿性を認めない (4)融点(分解点)、沸点、凝固点 融点:130~133°C (5)酸塩基解離定数 酸塩基解離基を有しない (6)分配係数 1000 以上(1-オクタノール/水系) (7)その他の主な示性値 旋光度〔α〕25 D:-112~-117 主(脱水物に換算したもの 0.2g、N, N-ジメチルホルムアミド、20mL、 100mm) 2. 有効成分の各種条件下における安定性 試験 保存条件 保存形態 保存期間 結果 長 期 保存試験 30°C 二重 ポリエチレン袋 + アイアンドラム 39 カ月 いずれの項目もほとんど変化を認 めず安定 加速試験 40°C/75%RH 6 カ月 いずれの項目もほとんど変化を認 めず安定 苛酷試験 温度 50°C 3 カ月 含量のわずかな低下傾向を認め、 TLC においてわずかに変化を認め た。その他の項目は変化なし。 湿度 30°C/75%RH シャーレ開放 3 カ月 いずれの項目もほとんど変化を認 めず安定 光 室内散光 (1000lx) 50 日 いずれの項目もほとんど変化を認 めず安定 試験項目:性状、確認試験(赤外吸収スペクトル)、旋光度、純度試験(類縁物質)、水分、定量 強制分解による主分解物 (製剤の分解物については「Ⅳ.製剤に関する項目 5.混入する可能性のある夾雑物」の項参照) (1)固体状態における主分解物 光により生成する可能性のある分解物:類縁物質Ⅴ、類縁物質Ⅵ(※)、類縁物質Ⅶ、類縁物質Ⅷ (2)溶液状態における主分解物 熱により生成する可能性のある分解物:類縁物質Ⅹ 光により生成する可能性のある分解物:類縁物質Ⅵ(※)、類縁物質Ⅷ ※類縁物質Ⅴのエピマー 3. 有効成分の確認試験法、定量法 確認試験法 日局「タクロリムス水和物」の確認試験法(呈色反応、赤外吸収スペクトル測定法)による 定量法Ⅳ.製剤に関する項目
1. 剤形 (1)剤形の区別 軟膏剤 (2)製剤の外観及び性状 白色~微黄色の軟膏剤 (3)識別コード 該当しない (4)製剤の物性 適度の固さを有する軟膏剤 稠度(Po):180~290 (5)その他 該当しない 2. 製剤の組成 (1)有効成分(活性成分)の含量及び添加剤 1g 中 日局タクロリムス水和物 0.31mg(タクロリムスとして 0.3mg) 添加剤 炭酸プロピレン、サラシミツロウ、流動パラフィン、パラフィン、白色ワセリン (2)電解質等の濃度 該当しない (3)熱量 該当しない 3. 添付溶解液の組成及び容量 該当しない 4. 力価 該当しない5. 混入する可能性のある夾雑物 熱により生成する可能性のある分解物:類縁物質Ⅸ、ⅩⅠ、ⅩⅤ 光により生成する可能性のある分解物:類縁物質Ⅵ、Ⅷ、ⅩⅠ、ⅩⅤ 類縁物質Ⅵ 類縁物質Ⅷ 類縁物質Ⅸ 類縁物質ⅩⅠ 類縁物質ⅩⅤ 6. 製剤の各種条件下における安定性1) 試験 保存条件 保存形態 保存期間 結果 長 期 保存試験 25°C/60%RH (暗所) アルミチューブ 24 カ月 規格内 苛酷試験 温度 50°C (暗所) アルミチューブ 3 カ月 含量の低下及び類縁物質の 増加を認めたが、規格内であ った。 -20°C⇔30°C注) (暗所) アルミチューブ 8 週間 規格内 湿度 25°C/90%RH (暗所) 無色ガラスビーカー (開放) 3 カ月 類縁物質のわずかな増加を 認めたが、規格内であった。 光 白色蛍光灯下 (1000lx) ガラス板に薄く均一に塗布 7 日 含量の低下及び類縁物質の 増加を認めたが、規格内であ った。 試験項目:性状、稠度、純度試験(類縁物質)、定量 注) -20°C で 2 週間、引き続き 30°C で 2 週間の保存を繰り返した。 7. 調製法及び溶解後の安定性 該当しない 8. 他剤との配合変化(物理化学的変化) 本剤は基剤中に微細な液滴として分散した液滴分散系軟膏である。他剤あるいはワセリンと混合するこ とにより液滴が合一して大きくなるため、混合することは好ましくない。
9. 溶出性 該当しない 10.容器・包装 (1)注意が必要な容器・包装、外観が特殊な容器・包装に関する情報 該当しない (2)包装 チューブ:5g×10 (3)予備容量 該当しない (4)容器の材質 チューブ:アルミニウム キャップ:ポリエチレン 11.別途提供される資材類 該当しない 12.その他 該当しない
Ⅴ.治療に関する項目
1. 効能又は効果 アトピー性皮膚炎 2. 効能又は効果に関連する注意 5.効能・効果に関連する注意 ステロイド外用剤等の既存療法では効果が不十分又は副作用によりこれらの投与ができないなど、 本剤による治療がより適切と考えられる場合に使用する。 (解説) 本剤は 2 年以上の臨床成績がなく、それ以上の長期使用時における安全性は不明である。また、アトピー 性皮膚炎の薬物治療としてはステロイド外用剤が主体と考えられていることから、本剤による治療がよ り適切と考えられる場合に使用すること。 3. 用法及び用量 (1)用法及び用量の解説 通常、小児には 1 日 1~2 回、適量を患部に塗布する。なお、1 回あたりの塗布量は 5g までとするが、 年齢により適宜減量する。 (2)用法及び用量の設定経緯・根拠 (投与方法とその理由) 臨床試験において 1 日 2 回の塗布により十分な効果が認められた。また、使用開始初期は 1 日 2 回塗布の症例が多かったが、使用期間が長期になるに従い、1 日 1~2 回あるいは 1 日 1 回でもコ ントロール可能な症例が増加した。したがって、塗布回数は 1 日 1~2 回とした。なお、副作用発 現頻度に対する塗布回数の影響は認められなかった。 (1 回あたりの塗布量を 5g までとした理由) 臨床試験時の 1 回塗布量の上限を最大 5g とし、その用量の範囲内での有効性及び安全性が確認さ れた。また、アトピー性皮膚炎患者では高い血中濃度が持続する移植患者とは状況が異なるが、移 植領域でみられるような全身副作用の発現を避けるため、市販後においても 1 回塗布量の上限を 5g とした。 4. 用法及び用量に関連する注意 7.用法・用量に関連する注意 7.1 1 回あたりの最大塗布量については、次の表を目安にする。 年齢(体重)区分 1 回塗布量の上限 2 歳~5 歳(20kg 未満) 1g 6 歳~12 歳(20kg 以上 50kg 未満) 2g~4g 13 歳以上(50kg 以上) 5g 参考:臨床試験時の用量[17.1.1 参照] 7.2 皮疹の増悪期には角質層のバリア機能が低下し、血中濃度が高くなる可能性があるので、本剤の使 用にもかかわらず 2 週間以内に皮疹の改善が認められない場合には使用を中止すること。また、皮 疹の悪化をみる場合にも使用を中止すること。 7.3 症状改善により本剤塗布の必要がなくなった場合は、速やかに塗布を中止し、漫然と長期にわたっ て使用しないこと。 7.4 1 日 2 回塗布する場合はおよそ 12 時間間隔で塗布すること。 (解説) 7.1 小児は年齢による体格の違いが大きいことから、本剤の適宜減量時の目安として、国内臨床試験(2 歳 から 15 歳の小児患者を対象)で用いた体重区分(「V.治療に関する項目 5.臨床成績」の項参照)及び 各年齢における標準体重を参考にし、1 回塗布量の上限を年齢(体重)ごとに 3 段階に分けて設定し た。7.2 皮疹の改善が認められない場合に漫然と使用を続けると、高い血中濃度が持続する可能性があるので、 全身性の副作用を避けるために記載した。また、本剤の臨床試験結果にもとづき、症状が改善しない場 合でも 2 週間程度は経過をみることとし、それでも皮疹の改善が認められない場合は使用を中止する ことが妥当であると判断した。 7.3 アトピー性皮膚炎は症状の寛解・増悪を繰り返し、慢性に経過することを特徴とする皮膚疾患であり、 治療の際は症状の改善に応じて塗布量、塗布回数を減らしながら寛解導入の実現を目指すことになる。 従って、症状改善後の必要以上の塗布、あるいは予防的な使用は避けるべきであることから、症状の改 善により本剤塗布の必要がなくなった場合は速やかに塗布を中止すること。 7.4 1 日 2 回の場合、塗布間隔が短いと血中濃度が高くなる可能性があるので、1 日 2 回使用の場合の最 大間隔であるおよそ 12 時間間隔で塗布する旨を記載した。 5. 臨床成績 (1)臨床データパッケージ 試験 目的 対象 塗布部分、用法・用量 第Ⅲ相試験 比較試験 (対照:軟膏基剤) アトピー性皮膚炎患者、 中等度以上 軟膏基剤又はタクロリムス軟膏 0.03%、0.1%を 1 日 2 回単純塗布・3 週間 長期観察試験 アトピー性皮膚炎患者、 中等度以上 タクロリムス軟膏 0.03%、0.1%を 1 日 1~2 回 単純塗布・52 週間 第Ⅲ相試験後、継続試験を実施 (2)臨床薬理試験 小児を対象とした該当資料はないが、参考として健康成人男子を対象とした臨床薬理試験の成績を以 下に記載する2)。 <参考> 健康成人男子 12 例への 0.1~2%タクロリムス軟膏 30mg/2cm2の単回塗布試験で、パッチテスト、光 パッチテストともに刺激性は認められず、健康成人男子 6 例への 2%タクロリムス軟膏 30mg/4×4cm の 1 日 2 回、7 日間反復塗布試験でも皮膚刺激性は認められなかった。両塗布試験で自他覚症状、臨 床検査、理学的検査等に異常所見は認められなかった。なお、タクロリムスは血中において定量限界 付近の濃度が検出されただけで、反復塗布によっても血中濃度の蓄積性はみられなかった。 注)本剤の承認されている用法・用量は「通常、小児には 1 日 1~2 回、適量を患部に塗布する。なお、1 回あた りの塗布量は 5g までとするが、年齢により適宜減量する。」である。 なお、本試験におけるタクロリムス軟膏は製剤処方改良にて添加剤を変更する前のものであるが、現行の製 品に含有される添加剤はすべて含まれている。 (3)用量反応探索試験 該当資料なし (4)検証的試験 1)有効性検証試験 2 歳以上 16 歳未満の中等症以上の小児アトピー性皮膚炎患者を対象に、タクロリムス軟膏 0.1%、 0.03%又は軟膏基剤を 1 日 2 回(1 回量最大 5g)、3 週間単純塗布による二重盲検比較試験を実施した3)。 試験デザイン:多施設共同二重盲検群間比較試験(検証的試験) 対象:中等症~重症の小児アトピー性皮膚炎患者
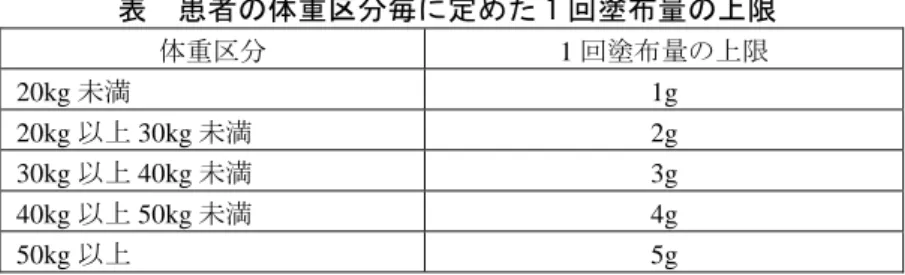
選択基準:中等症~重症(Rajka and Langeland の基準)のアトピー性皮膚炎患者(Hanifinand Rajka の基 準)と診断された患者。患者の体重区分毎に定めた治験薬の 1 回塗布量の上限ですべての皮疹 に塗布可能な患者。躯幹又は四肢に中等度以上(治験薬の有効性を評価できる程度)の皮疹が 存在する患者。年齢:2 歳以上 16 歳未満の患者。 除外基準:治験薬塗布部位にアトピー性皮膚炎以外の皮膚疾患を有する患者。治験薬塗布開始前 4 週 間以内に PUVA 療法を含む光線療法、ステロイド剤及び免疫抑制剤の全身投与を受けた患者。 併用禁止薬及び併用禁止療法を中止できない患者。薬物過敏症を有する患者。悪性腫瘍の合併 あるいは既往のある患者。感染症を合併している患者。高度の腎機能障害、高度の高カリウム 血症を合併する患者。又はそれらに起因する症状を有する患者。明らかな心・肝・腎・膵・血 液疾患を合併する患者。タクロリムスを使用中あるいは使用した経験のある患者。 試験方法:治験薬(タクロリムス軟膏 0.03%、タクロリムス軟膏 0.1%又は軟膏基剤)を被験部位に 1 日 2 回(およそ 12 時間間隔)、3 週間単純塗布する。
表 患者の体重区分毎に定めた 1 回塗布量の上限 体重区分 1 回塗布量の上限 20kg 未満 1g 20kg 以上 30kg 未満 2g 30kg 以上 40kg 未満 3g 40kg 以上 50kg 未満 4g 50kg 以上 5g 主要評価項目:主有効性観察・評価部位における改善度 治験薬塗布開始 3 週後(又は中止時)の開始日(第 1 日)に対する皮膚症状スコアの合計の変 化(変化率)を治験依頼者が計算し、以下の改善度判定基準に従い機械的に改善度を求める。 副次的に治験薬塗布開始 1、2 週後も同様に行う。なお、治験薬塗布開始日の症状スコア 4(高 度)で以降悪化した 4 を◎で囲んだものについては症状スコア 5(高度からの悪化)とする。 改善度判定基準 改善度判定 変化率(%) 1.寛解 100 2.著明改善 67~99 3.中等度改善 34~66 4.軽度改善 1~33 5.不変 0 6.悪化 <0 副次評価項目:主有効性観察・評価部位における皮膚症状スコアの合計、皮膚症状スコアの変化。治験 薬塗布部位(顔面・頸部、躯幹、上肢、下肢別)における皮膚症状スコアの合計、皮膚症状ス コアの変化。治験薬塗布部位(顔面・頸部、躯幹、上肢、下肢別)における皮疹面積の変化。 そう痒の程度の変化。治験薬塗布部位全体における全般改善度 統計手法:有意水準は両側 5%とし、被験者背景因子の不均衡は両側 15%を指標として検討する。 有効性:主有効性観察・評価部位における改善度(「著明改善」以上の改善率)を算出し、Dunnett 型 多重比較を用いて軟膏基剤とタクロリムス軟膏各群(0.03%群、0.1%群)間の比較を行う。副 次的にタクロリムス軟膏 0.03%群と 0.1%群の比較を Fisher の直接確率法を用いて行う。 安全性:副作用について発現率を算出し、Fisher の直接確率法を用いて各群間の対比較を行う。なお、 因果関係にかかわらずすべての有害事象についての解析も行う。 結果: 最終時改善度では 0.03%、0.1%両群ともそれぞれ 66.7%(48/72 例)、75.7%(53/70 例)と高い改 善率(「著明改善」以上)を示し、両濃度とも軟膏基剤群 12.7%(9/71 例)に比べ有意に優れた有 効性を認めたが、両濃度間に有意差はなかった。 同様に観察時期別の改善度についても、各観察時期において 0.03%群及び 0.1%群は基剤群よりも 高い改善率で推移し、いずれの時期においても基剤群と 0.03%群及び 0.1%群の間にはそれぞれ有 意差が認められた。一方、両濃度間ではいずれの時期においても有意差は認められなかった。 副作用発現頻度は、本剤群で 49.3%(36/73 例)であった。主な副作用は塗布部位の刺激感(刺激 感、ほてり感、そう痒感等)45.2%(33/73 例)、塗布部位の感染症(毛包炎(毛嚢炎)、伝染性軟 属腫、伝染性膿痂疹)9.6%(7/73 例)であった。 注)0.1%軟膏は成人にのみ承認されている。また、本剤の承認されている用法・用量は「通常、小児には 1 日 1~2 回、適量を患部に塗布する。なお、1 回あたりの塗布量は 5g までとするが、年齢により適宜減量する。」である。 2)安全性試験 長期観察試験4),5) 小児アトピー性皮膚炎患者に、タクロリムス軟膏 0.03%又は 0.1%を 1 日 1~2 回(1 回量最大 5g)、 単純塗布による長期使用時(1 年間)の安全性及び有効性を検討した。その結果、塗布部位の刺激感 が 0.03%群で 50%(52/104 例)、0.1%群で 62.4%(68/109 例)に認められたが、両濃度群の発現率に 有意差はなかった。また、刺激感の大半は軽度~中等度で、多くは塗布開始後 1 週間以内に発現した が、皮疹の改善とともに軽快した。副作用発現頻度は 66.3%(69/104 例)であった。主な副作用は塗 布部位の刺激感(ヒリヒリ感、そう痒感、ほてり感等)50.0%(52/104 例)、塗布部位の感染症(毛包 炎(毛嚢炎)、伝染性膿痂疹、単純疱疹等)33.7%(35/104 例)であった。なお、本剤との因果関係を
臨床効果は、両濃度とも 1 年間の観察期間中、良好に維持された。塗布部位全体での「著明改善」以 上の改善率は、0.1%群(76.3%)が 0.03%群(62.2%)に比べて高かった。中等度改善率は 0.03%群でも塗 布開始後経時的に上昇し、高い有効性を示した。また、塗布回数については、使用開始初期は 1 日 2 回塗布を必要とする症例が多かったが、その後は 1 日 1 回塗布でも 1 日 2 回塗布と同様の効果が得ら れた。 注)0.1%軟膏は成人にのみ承認されている。また、本剤の承認されている用法・用量は「通常、小児には 1 日 1~2 回、適量を患部に塗布する。なお、1 回あたりの塗布量は 5g までとするが、年齢により適宜減量する。」である。 (5)患者・病態別試験 該当資料なし (6)治療的使用 1)使用成績調査(一般使用成績調査、特定使用成績調査、使用成績比較調査)、製造販売後データベー ス調査、製造販売後臨床試験の内容 該当資料なし 2)承認条件として実施予定の内容又は実施した調査・試験の概要 本剤のがん原性に関する試験結果6) 0.1%プロトピック軟膏をマウスの皮膚に 2 年間塗布投与すると悪性リンパ腫の発生が増加すること を 0.1%プロトピック軟膏の審査過程において既に報告7)しているが、本試験でも同様の成績が得られた。 しかし、本試験では 52 週間塗布の時期に関しては、マウス生涯の前半あるいは後半の塗布時期によ る発生頻度の差は認められなかった。マウスの寿命の 1/4 である 26 週間の塗布では、塗布時期に関 わらず本剤は悪性リンパ腫発生頻度に影響を与えなかった。また、無処置の 2 群、sham 群と軟膏基 剤塗布群の計 4 群の対照群間で悪性リンパ腫の発生頻度に統計学的な有意差が認められなかった。軟 膏塗布により増加した悪性リンパ腫は、その組織型や免疫組織型、超微形態的特徴や遺伝解析におい て、各対照群に自然発生した悪性リンパ腫と同等であった。その他、血液学検査、脾臓及びリンパ節 の経時的なリンパ球サブセット解析を実施したが、いずれも有意な変動はなかった。 本試験結果から、0.1%プロトピック軟膏塗布で増加する悪性リンパ腫の発生は、B6c3F1 マウスに自 然発生する悪性リンパ腫の発生が増強されたものと考えられ、これはマウスでは薬物血中濃度が持続 するため全身性免疫低下によることが示唆された。(塗布がん原性試験については「Ⅸ.非臨床試験に 関する項目 2.毒性試験 (4)がん原性試験」の項参照) (7)その他 該当資料なし
Ⅵ.薬効薬理に関する項目
1. 薬理学的に関連ある化合物又は化合物群 ステロイド等の免疫抑制剤 注意:関連のある化合物の効能・効果等は、最新の添付文書を参照すること。 2. 薬理作用 (1)作用部位・作用機序 タクロリムスは T 細胞、肥満細胞、好酸球、ランゲルハンス細胞等の炎症性細胞の働き、中でも T 細 胞からのサイトカインの産生を強く抑制し、これらの炎症性細胞の相互作用により誘発されるアトピ ー性皮膚炎に対して抑制作用を示すと考えられる。 1)サイトカイン産生抑制作用8) ヒト・ヘルパーT 細胞による IL-2、IL-3、IL-4、IL-5、インターフェロン-γ、GM-CSF 等のサイトカ インの産生をステロイドと同等もしくはより強く抑制する(in vitro)。 2)肥満細胞脱顆粒抑制作用9),10)抗 IgE 抗体刺激によるヒト肥満細胞からのヒスタミン遊離をステロイドより強く抑制する(in vitro)。 3)好酸球脱顆粒抑制作用11) カルシウムイオノフォア刺激によるヒト好酸球からの塩基性蛋白(ECP)の遊離をステロイドより 強く抑制する(in vitro)。 4)抗原提示能抑制作用12) ヒト皮膚ランゲルハンス細胞をタクロリムスで前処理することにより、ランゲルハンス細胞を抗原 提示細胞とする混合リンパ球反応を抑制する(in vitro)。 (2)薬効を裏付ける試験成績 1)動物皮膚炎モデルに対する作用 ①ラット抗原連続塗布皮膚炎モデルに対する作用13) Brown Norway 系雄性ラット耳介部でのジニトロクロロベンゼン連続塗布皮膚炎モデルを用いて、 0.1%及び 0.3%タクロリムス軟膏(各 n=9)、0.12%ベタメタゾン吉草酸エステル軟膏(n=8-9)及 び 0.1%アルクロメタゾンプロピオン酸エステル軟膏(n=8)の作用を検討した。統計学的な検定 は一元配置分散分析及び Tukey-Kramer の多重比較を用いて行った。 無塗布群(n=9)では耳浮腫及び炎症部位における肥満細胞及び好酸球の増加というアトピー性 皮膚炎に類似の所見が認められた。 タクロリムス軟膏群ではいずれの濃度においてもほぼ同等に明らかな耳浮腫の抑制効果を示し、 軟膏基剤群(n=9)との間に有意な差が認められ、その作用はベタメタゾン吉草酸エステル軟膏 より強く、アルクロメタゾンプロピオン酸エステル軟膏より弱かった。また、無塗布群及び軟膏 基剤群でみられた真皮における肥満細胞及び好酸球の増加は、いずれの軟膏群においても抑制も しくは抑制される傾向を示した。 ②NC マウス自然発症皮膚炎に対する作用14) 雌雄の NC/Nga(NC)マウスでの自然発症皮膚炎モデルを用いてタクロリムス軟膏の皮膚炎に対す る作用を検討した。 皮膚炎未発症 NC マウス(5~8 週齢)の頸部、頭部及び顔面に 0.1%、0.3%、0.5%及び 1%タクロ リムス軟膏(各 n=11)、0.12%ベタメタゾン吉草酸エステル軟膏(n=15~16)及び 0.1%アルクロ メタゾンプロピオン酸エステル軟膏(n=17)を塗布(100mg/匹、週 2 回、約 9 週間)したところ、 無塗布群(n=11)では皮膚炎が発症し、真皮での炎症性細胞の増加及び IL-4、IL-5、IgE の上昇 が認められたが、タクロリムス軟膏群では濃度依存性はみられないものの、軟膏基剤群(n=12) と比べて明らかな皮膚炎発症抑制がみられ、真皮での炎症性細胞の増加抑制、IL-4、IL-5 の低下 及び濃度に依存した IgE 値の低下作用が認められた。両ステロイド軟膏群では、明らかな皮膚炎 抑制作用はみられなかったが、真皮での炎症性細胞の増加抑制及び IL-4、IgE 値の低下作用がみ られた。 また、既に皮膚炎を発症した 11~15 週齢の NC マウスに 0.1~0.5%タクロリムス軟膏を塗布 (100mg/匹、週 2 回、約 9 週間)する治療的投与においても、皮膚炎の進展が抑制された。
③Ⅰ型(即時型及び遅発型)皮膚アレルギー反応に対する作用 i)マウス抗原誘発即時型及び遅発型皮膚アレルギー反応 BALB/c 系雌性マウスでのアスカリス抽出物溶液腹腔内投与による能動感作モデル(n=8)を用い て、0.1~1%タクロリムス軟膏、0.12%ベタメタゾン吉草酸エステル軟膏及び 0.1%アルクロメタ ゾンプロピオン酸エステル軟膏を反応誘発前後に塗布し、2 相性(即時型及び遅発型)反応に対 する作用を検討した。耳の厚さの測定及び漏出色素量の測定結果の統計解析は、一元配置分散分 析及び Tukey-Kramer の多重比較で行い、各々p<0.05 の時、有意差ありとした。 無塗布群では誘発 1 時間後及び 24 時間後を極大反応とする即時型、遅発型浮腫反応が観察され、 タクロリムス軟膏は即時型の反応には作用を示さなかったが、遅発型の反応には濃度依存的に抑 制作用を示し、両ステロイド軟膏は両反応に対して明らかな抑制作用を示した15)。 なお、タクロリムスは受動感作したマウスで抗原により誘発した即時型の反応に無効であるが、 遅発型の反応を抑制することが報告されている16)。 ii)マウス受身皮膚アナフィラキシー(PCA)反応15) C3H 系雄性マウスでの耳介内側への同種抗 dinitrophenyl(DNP)IgE モノクローナル抗体皮内投与 による受動感作モデル(n=9~12)を用いて、0.01~1%タクロリムス軟膏、0.12%ベタメタゾン吉 草酸エステル軟膏及び 0.1%アルクロメタゾンプロピオン酸エステル軟膏を反応誘発前に塗布し、 PCA 反応に対する作用を検討した。 タクロリムス軟膏は本反応に対し抑制作用を示さず、両ステロイド軟膏は明らかな抑制作用を示 した。 ④Ⅳ型(遅延型)皮膚アレルギー反応に対する作用 i)マウス接触性皮膚遅延型反応 BDF1系雌性マウスでの腹部皮膚へのオキサゾロン塗布による感作モデル(n=10)を用いて、0.1 ~1%タクロリムス軟膏、0.12%ベタメタゾン吉草酸エステル軟膏及び 0.1%アルクロメタゾンプ ロピオン酸エステル軟膏を反応誘発前後に塗布し、オキサゾロン接触性遅延型反応に対する作用 を検討した。無塗布群では誘発 24 時間後に浮腫反応(耳の厚さの増加)が観察され、タクロリ ムス軟膏群では濃度依存的に本反応を抑制し、その抑制率は約 53~75%を示した。両ステロイド 軟膏は本反応に対して完全な抑制作用を示し、タクロリムス軟膏より明らかに強かったが、両ス テロイドは正常マウスの耳の厚さを減少させ、遅延型反応抑制作用に皮膚萎縮作用も関与するも のと考えられた。タクロリムス軟膏は正常マウスの耳の厚さに影響を与えなかった15)。 なお、局所投与したタクロリムスはジニトロフルオロベンゼンにより誘発したブタ皮膚遅延型反 応を抑制することが報告されている17)。 ii)マウス・ツベルクリン反応15)
BALB/c 系雌性マウスでの腋下及び鼠径部への結核死菌を含む Freund’s incomplete adjuvant 懸濁 液皮下注射による感作モデル(n=8)を用いて、0.1~1%タクロリムス軟膏、0.12%ベタメタゾン 吉草酸エステル軟膏及び 0.1%アルクロメタゾンプロピオン酸エステル軟膏を反応誘発前後に塗 布し、ツベルクリン遅延型反応に対する作用を検討した。 無塗布群では誘発 24 時間後に浮腫反応が観察されたが、タクロリムス軟膏は明らかな抑制作用 を示し、その抑制率は約 80~90%、ベタメタゾン吉草酸エステル軟膏及びアルクロメタゾンプロ ピオン酸エステル軟膏では各々約 98%、約 83%であった。
2)in vitro試験 試験項目 動物種 試験結果 IC50a)(ng/mL) タクロリムス ベタメタゾン 吉草酸 エステル アルクロメタゾン プロピオン酸 エステル T 細胞からのサイトカイン産生 1)末梢血単核球からのサイトカ イン産生(抗 CD3・抗 CD2 抗体 刺激)8) IL-2 IL-3 IL-4 IL-5 IFN-γ GM-CSF 2)脾臓細胞からのサイトカイン 産生(Con A 刺激)18) IL-2 IL-3 IL-4 IL-5 IFN-γ GM-CSF ヒト マウス 0.02 0.02 0.02 0.07 0.11 0.07 0.04 0.04 0.08 0.46 0.10 0.18 0.27 0.17 0.16 0.08 0.72 0.10 0.26 0.08 0.08 0.07 0.09 0.07 5.54 1.76 2.09 0.89 3.26 0.76 1.89 0.83 0.37 0.59 0.63 0.56 肥満細胞及び好塩基球からのヒスタ ミン遊離及びサイトカイン産生 1)皮膚肥満細胞ヒスタミン遊離 (抗 IgE 抗体刺激)9) 2)末梢血好塩基球ヒスタミン遊離 (抗 IgE 抗体刺激)19) 3)腹腔肥満細胞ヒスタミン遊離 (抗原刺激)20) 4)好塩基球性白血病細胞 ヒスタミン遊離(抗原刺激)21) 5)好塩基球性白血病細胞 TNF-α 産生 (抗 IgE 抗体刺激)21) ヒト ヒト ラット ラット ラット 1.8b) 3.1 1000ng/mL で無効 3.2 12.0 -c) 1000ng/mL で無効 1000ng/mL で無効 1000ng/mL で無効 0.5 - c) 1000ng/mL で無効 1000ng/mL で無効 1000ng/mL で無効 3.5 好酸球の脱顆粒 末梢血好酸球からの ECP 遊離 (calcium ionophore 刺激)11) ヒト 10 ~ 100ng/mL で 約 42~45%の抑制 100ng/mL で無効 100ng/mL で無効 ランゲルハンス細胞の抗原提示能 皮膚ランゲルハンス細胞抗原提 示能(皮膚混合リンパ球反応)12) ヒト 3.0 222.4 - c) a)50% inhibitory concentration
b)IC40値 c)未実施
(3)作用発現時間・持続時間 該当資料なし
Ⅶ.薬物動態に関する項目
1. 血中濃度の推移 (1)治療上有効な血中濃度 該当しない(吸収されて作用を示す薬剤ではない) (2)臨床試験で確認された血中濃度 1)反復塗布(外国人データ)22) 通常用量での該当資料はないが、参考としてタクロリムス軟膏 0.1%を反復塗布したデータを以下に記 載する。 小児アトピー性皮膚炎患者 39 例を塗布面積により 3 群に分け、タクロリムス軟膏 0.1%を 14 日間反復 塗布(初日及び 14 日目は 1 日 1 回、2 日目から 13 日目までは 1 日 2 回塗布)した。その結果、塗布後 の全身移行性は低く、全測定試料中 92%で血中濃度は 1ng/mL 以下であり、17%は定量限界(0.025ng/mL) 以下であった。また、タクロリムスの全身移行性は塗布面積とともに増加する傾向にあったが、塗布前 の全血中濃度(C0)及び塗布後 24 時間の全血中濃度(Cmin)の比較からタクロリムスの蓄積はないと考えら れた。 反復塗布時の薬物動態パラメータ 塗布面積 範囲(cm2) 平均塗布 面積(cm2) 例 数 測定日 (日) 塗布量 (g) Cmax (ng/mL) Tmaxa) (h) C0b) (ng/mL) Cmin (ng/mL) AUC0-24h (ng・h/mL) ≦1500 1001±443 16 1 2.3 ±1.2 0.44 ±0.76 4 - 0.12 ±0.17 5.17 ±8.82 4 - - - 0.29 ±0.28 - - 14 2.1 ±1.0 0.20 ±0.19 6 0.16 ±0.16 0.11 ±0.08 3.34 ±2.50 >1500 ≦3000 2317±466 14 1 3.8 ±1.3 0.99 ±1.37 23 - 0.95 ±1.37 17.48 ±25.74 4 - - - 0.96 ±0.90 - - 14 3.7 ±1.1 0.83 ±1.34 4 0.67 ±1.12 0.59 ±1.08 15.44 ±28.80 >3000 ≦5000 3903±534 9 1 4.8 ±1.1 1.03 ±1.13 4 - 0.78 ±1.18 11.03 ±11.88 4 - - - 0.96 ±1.58 - - 14 4.2 ±1.0 0.98 ±1.03 2.5 0.32 ±0.30 0.41 ±0.40 11.35 ±8.66 a):中央値、b):塗布前血中濃度、-:算出せず (平均値±S.D.) 注)小児で承認された製剤は 0.03%軟膏である。また、本剤の承認されている用法・用量は「通常、小児には 1 日 1~2 回、適量を患部に塗布する。なお、1 回あたりの塗布量は 5g までとするが、年齢により適宜減量する。」である。2)長期使用時4) 小児アトピー性皮膚炎患者 104 例に 0.03%軟膏を 1 回最大 5g、1 日 1~2 回塗布し 52 週後まで血中濃度 を測定したところ下表のとおりであった。 長期使用時の血中濃度 測定時期 測定例数 血中濃度(ng/mL) 平均値b)± S.D. N.D. a) <1a) 1≦~<3a) 3≦~<5 5≦ 最小値~最大値 4 日目 52 0.07±0.26 48(92.3) 3(5.8) 1(1.9) N.D.~1.50 1 週後 104 0.04±0.17 99(95.2) 4(3.8) 1(1.0) N.D.~1.39 2 週後 101 0.03±0.13 97(96.0) 4(4.0) N.D.~0.93 12 週後 98 0.01±0.06 97(99.0) 1(1.0) N.D.~0.59 28 週後 96 0.02±0.12 93(96.9) 3(3.1) N.D.~0.86 52 週後 97 0.01±0.05 96(99.0) 1(1.0) N.D.~0.54 N.D.:定量限界(0.50ng/mL)未満 a)例数(%) b)測定された全例の平均(N.D.を 0ng/mL として計算) (3)中毒域 該当資料なし <参考>移植領域におけるデータ 移植領域での経口剤・注射剤の臨床試験成績の分析では、血中トラフ濃度が 20ng/mL を超える期間が 長い場合、副作用が発現しやすくなることがわかっている。 (4)食事・併用薬の影響 該当資料なし 2. 薬物速度論的パラメータ (1)解析方法 該当資料なし (2)吸収速度定数 該当資料なし (3)消失速度定数 該当資料なし (4)クリアランス 該当資料なし <参考>静脈内投与時のデータ(ラット、ブタ) ラット 23)及びブタ 24)にタクロリムス 1mg/kg を静脈内投与したところ、全身クリアランス(平均値 ±S.E. )は各々1.47±0.03L/h/kg(n=5)、0.26±0.03L/h/kg(n=3)であった。 (5)分布容積 該当資料なし <参考>静脈内投与時のデータ(ラット、ブタ) ラット23)及びブタ24)にタクロリムス 1mg/kg を静脈内投与したところ、定常状態での分布容積(平均 値±S.E. )は各々17.3±1.8L/kg(n=5)、5.27±0.43L/kg(n=3)であった。 (6)その他 該当資料なし
3. 母集団(ポピュレーション)解析 (1)解析方法 該当資料なし (2)パラメータ変動要因 該当資料なし 4. 吸収 該当資料なし <参考>バイオアベイラビリティ(ラット、ブタ) ラットの健常及び角質層を除去した損傷皮膚(n=5~7)に 0.5%タクロリムス軟膏 100mg/10cm2を密封法で 単回塗布したところ、AUC から算出した経皮投与によるタクロリムスのシステミックアベイラビリティ は健常皮膚で 4.7%、損傷皮膚で 62.4%であった23)。また、ブタの健常皮膚(n=3)に 0.1%タクロリムス 軟膏 100mg/10cm2/kg を密封法で単回塗布したところ、システミックアベイラビリティは 0.94%であった24)。 <参考>吸収率(ラット)25) ラットの健常及び角質層を除去した損傷皮膚(n=3)に 0.5%14C-タクロリムス軟膏 320mg/kg を密封法で 単回塗布したときの尿及び糞中への排泄率の合計から、健常及び損傷皮膚からの吸収率は各々4.6%、 56.0%と推定された。なお、ラット健常皮膚(n=3)への単純塗布では 5.6%と推定された。 5. 分布 (1)血液-脳関門通過性 該当資料なし <参考>脳内移行性(ラット)25) 角質層を除去したラット損傷皮膚に 0.5%14C-タクロリムス軟膏 320mg/kg を密封法で単回塗布したと きの組織内放射能濃度は以下のとおりである。 組織 放射能濃度(ng eq./g) 塗布後 30 分 2 時間 8 時間 24 時間 72 時間 大脳 8±2 9±1 8±0 10±1 10±1 小脳 N.D. 7±3 N.D. 7±1 N.D. 平均値±S.E.(n=3) N.D.:検出限界以下 (2)血液-胎盤関門通過性 該当資料なし <参考> 移植領域におけるデータ(外国人データ)26) タクロリムスを経口投与された妊婦 8 名(外国人移植患者:腎臓 4 例、腎臓/膵臓 1 例、腎臓/心臓 1 例、肝臓 2 例)の分娩後速やかに採取された母体血と臍帯静脈血の平均濃度は、それぞれ 9.0±3.4ng/mL (8 例)、6.6±1.8ng/mL(7 例)であった。また、平均血漿中濃度は、それぞれ 0.40±0.20ng/mL(7 例)、 0.09±0.04ng/mL(6 例)であった。 移植領域におけるデータ(外国人データ)27) タクロリムスを投与された妊婦の分娩 3 日後までの平均血漿中濃度*は 1.46ng/mL(15 例)、臍帯血 濃度は 0.71ng/mL(13 例)、羊水中濃度は 0.2ng/mL 未満(2 例)で、このとき新生児の平均血漿中濃 度*は 0.54ng/mL(7 例)、髄液中濃度(1 例)は測定限界以下であった(外国人肝移植患者)。 *血漿中濃度:国内では通常、全血(whole blood)にて血中濃度測定が行われている。ヘマトクリット値等の条 件により多少異なるが、血漿中濃度は全血濃度の約 1/10 である。 (3)乳汁への移行性 該当資料なし <参考>移植領域におけるデータ(外国人データ)27) タクロリムスを投与された妊婦の分娩 3 日後までの平均血漿中濃度*は 1.46ng/mL(15 例)で、この とき平均初乳中濃度は 0.79ng/mL(6 例)と母体血漿中濃度*のほぼ半分であった(外国人肝移植患 者)。
*血漿中濃度:国内では通常、全血(whole blood)にて血中濃度測定が行われている。ヘマトクリット値等の条 件により多少異なるが、血漿中濃度は全血濃度の約 1/10 である。 (4)髄液への移行性 該当資料なし <参考>移植領域におけるデータ(外国人データ)28) 外国人患者 1 例で、髄液中に本剤は検出されず(<0.1ng/mL)、このときの血漿中濃度*は 3.3ng/mL であった。 *血漿中濃度:国内では通常、全血(whole blood)にて血中濃度測定が行われている。ヘマトクリット値等の条 件により多少異なるが、血漿中濃度は全血濃度の約 1/10 である。 (5)その他の組織への移行性 該当資料なし <参考>組織への分布(ラット)25) 角質層を除去したラット損傷皮膚に 0.5%14C-タクロリムス軟膏 320mg/kg を密封法で単回 塗布したときの臓器・組織内放射能濃度は以下のとおりである。 組 織 塗布後 30 分 放射能濃度(ng eq./mL 又は g) 2 時間 8 時間 24 時間 72 時間 血漿 全血 大脳 小脳 下垂体 眼球 ハーダー腺 耳下腺 舌下腺 下顎腺 甲状腺 胸腺 心臓 肺 肝臓 腎臓 副腎 脾臓 膵臓 筋肉 白色脂肪 褐色脂肪 骨髄 皮膚 睾丸 前立腺 リンパ節 胃 小腸 大腸 膀胱 筋肉(塗布部) 皮膚(塗布部) 26±5 44±5 8±2 N.D. 135±53 22±8 53±12 73±26 102±28 147±49 495±138 48±16 499±147 1469±97 361±142 446±113 815±289 312±111 249±91 54±22 60±27 534±151 110±44 47±15 6±2 61±21 95±38 160±48 123±57 57±19 34±15 71±27 146556±53208 13±2 28±4 9±1 7±3 272±90 30±7 91±22 199±48 211±70 281±78 548±78 92±26 556±90 1134±137 528±122 501±86 1036±203 575±137 355±88 55±19 90±27 490±82 171±42 92±21 9±2 101±22 214±50 288±79 209±82 140±50 73±25 100±14 16052±6110 N.D. N.D. 8±0 N.D. 271±38 23±1 88±10 115±7 112±13 168±13 155±4 116±10 199±16 310±24 199±18 170±7 282±30 232±21 207±25 83±11 55±6 184±12 63±14 72±1 11±1 91±9 183±27 131±13 121±8 93±7 89±18 73±3 1311±131 N.D. N.D. 10±1 7±1 127±42 14±4 33±11 39±12 32±7 55±14 N.D. 91±36 83±19 81±21 76±30 65±15 66±25 61±18 87±25 28±10 11±4 70±3 N.D. 41±15 12±6 25±7 58±19 81±24 78±32 87±31 106±36 33±12 729±135 N.D. N.D. 10±1 N.D. N.D. 9±1 12±2 10±1 N.D. 17±1 N.D. 23±3 31±2 25±2 51±3 34±4 N.D. 16±2 25±2 7±2 N.D. 23±4 N.D. 12±3 11±1 8±0 15±3 42±1 26±2 58±4 100±18 10±1 313±78 平均値±S.E.(n=3) N.D.:検出限界以下 (6)血漿蛋白結合率 ヒト血漿蛋白との結合率は以下のとおりである29)。 血漿蛋白結合率 測定法 化合物 濃度(ng/mL) 蛋白結合率(%) 平衡透析法(in vitro) タクロリムス 1.0 10 >98.5 99.0±0.2 平衡透析法(in vitro) 3H-dihydro-タクロリムス 0.5
5.0
99.2±0.5 99.0±0.2
6. 代謝 (1)代謝部位及び代謝経路 該当資料なし <参考>(ラット、外国人データ) ラットの皮膚ミクロゾームと14C-タクロリムスを NADPH 存在下で好気的に反応させると、13 位 O-脱メチル体の生成が認められたが、その代謝活性は、肝ミクロゾームに比べ約 1/2700 と非常に弱か った。また、ラットに 0.5%14C-タクロリムス軟膏を 14 日間反復塗布したときの最終塗布後 1 及び 7 日の皮膚(塗布部)中の未変化体濃度の測定においても、放射能の 90%以上が未変化体であった30)。 したがって、タクロリムスはラットの皮膚中でほとんど代謝を受けないことが考えられた。 なお、外国人肝移植患者での血中、尿中代謝物は主として脱メチル体であったが、胆汁中代謝物は主 として水酸化体であった31)。 (2)代謝に関与する酵素(CYP 等)の分子種、寄与率 主として CYP3A4 で代謝される32)(in vitro)。
(3)初回通過効果の有無及びその割合 該当しない (4)代謝物の活性の有無及び活性比、存在比率 該当資料なし <参考>(in vitro)33) ラット肝ミクロゾームによる in vitro 反応系を用いて同定された 8 種類の代謝物のうち、主代謝物で ある 13 位の O-脱メチル体の薬理活性はタクロリムスに比べて非常に弱かった。 7. 排泄 該当資料なし <参考>排泄率(ラット)25) ラットの健常皮膚及び角質層を除去した損傷皮膚(n=3)に 0.5%14C-タクロリムス軟膏 320mg/kg を密封 法で単回塗布したときの 168 時間までの尿及び糞中への放射能排泄率は、健常皮膚で各々0.4%、4.2%、 損傷皮膚で各々2.4%、53.6%であった。また、ラット健常皮膚(n=3)への単純塗布法では各々0.5%、5.1% であった。 8. トランスポーターに関する情報 In vitro 試験の結果、タクロリムスは P-糖蛋白質の基質であると考えられる34)。 9. 透析等による除去率 該当資料なし <参考>移植領域におけるデータ(外国人データ)28) 透析液中の濃度は検出限界以下であり、透析により除去されない。 10.特定の背景を有する患者 該当資料なし 11.その他 該当資料なし
Ⅷ.安全性(使用上の注意等)に関する項目
1. 警告内容とその理由 1.警告 1.1 本剤の使用は、小児のアトピー性皮膚炎の治療法に精通している医師のもとで行うこと。 1.2 マウス塗布がん原性試験において、高い血中濃度の持続に基づくリンパ腫の増加が認められている。 また、本剤使用例において関連性は明らかではないが、リンパ腫、皮膚がんの発現が報告されてい る。本剤の使用にあたっては、これらの情報を患者又は代諾者に対して説明し、理解したことを確 認した上で使用すること。[15.2.2 参照] 1.3 潰瘍、明らかに局面を形成しているびらんに使用する場合には、血中濃度が高くなり、腎障害等の 副作用が発現する可能性があるので、あらかじめ処置を行い、潰瘍、明らかに局面を形成している びらんの改善を確認した後、本剤の使用を開始すること。[2.1 参照] (解説) 1.1 小児のアトピー性皮膚炎患者に本剤をより適切に使用するために、小児のアトピー性皮膚炎の治療法 に精通している医師のもとで本剤を使用するよう、「警告」の項に記載し、注意を喚起することとし た。 1.2 マウスの全体表面積の 40%相当部位にタクロリムス軟膏 0.03%、0.1%を 2 年間塗布した実験におい て、0.1%群で高い血中濃度の持続に基づく内臓のリンパ腫の増加が認められている(「IX.非臨床試験 に関する項目 2.毒性試験 (4)がん原性試験」の項参照)。本剤を適正に使用した場合には全身性の免 疫抑制作用が発現するほど高い血中濃度が持続する可能性はほとんどないと考えられるが、本剤は長 期使用(例えば 5~10 年程度)の成績はなく、長期使用時におけるリンパ腫、皮膚がんの発現に及ぼ す影響については不明である。また、自然発症の可能性が考えられるあるいは情報不足等、本剤との 因果関係は明らかではないが、タクロリムス軟膏を使用したアトピー性皮膚炎患者でリンパ腫、皮膚 がんが報告されている。したがって、本剤の使用にあたっては、これらの情報を患者に対して説明し、 理解したことを確認した上で使用することとした。 1.3 本剤の臨床試験において、特に高いタクロリムス血中濃度は検出されていない。しかし、本剤の経皮 吸収が高まる可能性がある皮膚の損傷が激しい部分に本剤を使用した場合は、タクロリムス血中濃度 が持続し、タクロリムス経口剤・注射剤を投与した移植患者で認められている腎障害等の副作用が発 現する可能性を否定できない。したがって、潰瘍、明らかに局面を形成しているびらんでは皮膚の損 傷が激しいことから、タクロリムス血中濃度の上昇による全身性副作用の発現を避けるため、これら の部位への使用を禁忌とし、十分な注意喚起を行うために「警告」の項にも記載した。潰瘍、明らか に局面を形成しているびらんへの使用にあたっては、本剤使用前にあらかじめ亜鉛華軟膏等で処置を 行い、改善を確認した後に使用を開始すること。 2. 禁忌内容とその理由 2.禁忌(次の患者には投与しないこと) 2.1 患部に潰瘍、明らかに局面を形成しているびらんのある患者[1.3 参照] 2.2 高度の腎障害、高度の高カリウム血症の患者[9.1.1、9.2.1 参照] 2.3 魚鱗癬様紅皮症を呈する疾患(Netherton 症候群等)の患者〔経皮吸収が高く、本剤の血中濃度が高 くなり、腎障害等の副作用が発現する可能性がある。〕[9.1.2 参照] 2.4 低出生体重児、新生児、乳児又は 2 歳未満の幼児[9.7 参照] 2.5 本剤の成分に対し過敏症の既往歴のある患者 2.6 PUVA 療法等の紫外線療法を実施中の患者[10.1、15.2.1 参照] (解説) 2.1 「1.警告内容とその理由 1.3」の項参照 2.2 移植領域での経口剤・注射剤の投与において腎障害、高カリウム血症が高頻度にみられていることか ら、高度の腎障害、高度の高カリウム血症のある患者は「禁忌」とし、腎障害、高カリウム血症のあ る患者は「特定の背景を有する患者に関する注意」に設定した。 2.3 Netherton 症候群の患者で本剤使用後にタクロリムス血中濃度が上昇し、腎不全を認めた症例が報告さ れたことから、先天的に角質バリア機能が高度に障害されている Netherton 症候群のような患者には 何らかの使用制限が必要と考えられた。また、Netherton 症候群患者の皮膚の状態として「魚鱗癬様紅 皮症」を呈することが知られているので、皮膚の臨床像から早期に判断できるよう「魚鱗癬様紅皮症2.4 本剤の臨床試験は国内外ともに 2 歳以上の小児を対象としており、低出生体重児、新生児、乳児又は 2 歳未満の幼児では使用経験がなく、安全性は確立していないので使用しないこと。 2.5 一般に、ある薬剤の成分により過敏症を生じた患者に、同一成分を含有する薬剤が再投与された場合、 アレルギー症状を呈する可能性が高く、ショック等の重篤な副作用を生じるおそれがある。 2.6 アルビノ無毛マウスに 40 週間にわたり UVA 及び UVB を照射し、その後 12 週間無処置期間を設け て観察すると試験動物のすべてに皮膚腫瘍が発生するが、この試験系において紫外線照射と並行して 本剤を塗布すると皮膚腫瘍の発生時期が早まることが示されている(「Ⅸ.非臨床試験に関する項目 2. 毒性試験 (7)その他の特殊毒性 3)」の項参照)。 3. 効能又は効果に関連する注意とその理由 「Ⅴ.2.効能又は効果に関連する注意」を参照すること。 4. 用法及び用量に関連する注意とその理由 「Ⅴ.4.用法及び用量に関連する注意」を参照すること。 5. 重要な基本的注意とその理由 8.重要な基本的注意 8.1 重度の皮疹もしくは塗布面積が広範囲にわたる場合は、血中濃度が高くなる可能性があるので、本 剤使用開始の 2~4 週間後に 1 回、その後は必要に応じて適宜腎機能検査を行い、異常が認められ た場合には、直ちに使用を中止し、適切な処置を行うこと。 8.2 密封法及び重層法での臨床使用経験はないので、密封法及び重層法は行わないこと。 8.3 本剤使用時は日光への曝露を最小限にとどめること。また、日焼けランプ/紫外線ランプの使用を避 けること。[15.2.1 参照] 8.4 2 年以上の長期使用時の局所免疫抑制作用(結果として、感染症を増加させたり、皮膚がんの誘因 となる可能性がある)については、臨床試験成績がなく不明である。 8.5 皮膚感染症を伴うアトピー性皮膚炎患者には使用しないことを原則とするが、やむを得ず使用する 場合には、感染部位を避けて使用するか、又はあらかじめ適切な抗菌剤、抗ウイルス剤、抗真菌剤 による治療を行う、もしくはこれらとの併用を考慮すること。[9.1.3 参照] 8.6 使用後、一過性に皮膚刺激感(灼熱感、ほてり感、疼痛、そう痒感等)が高頻度に認められるが、通 常、皮疹の改善とともに発現しなくなるので、皮膚刺激感があることについて患者に十分説明するこ と。 (解説) 8.1 タクロリムス経口剤・注射剤を投与した移植患者では腎障害、高カリウム血症が高頻度にみられてお り、これらはタクロリムスの血中濃度上昇が大きな要因である。重度の皮疹もしくは塗布面積が広範 囲にわたる場合はタクロリムスの血中濃度が高くなる可能性があることから、皮膚の状態が悪くタク ロリムスの血中移行が高いと考えられる使用開始 2~4 週後に 1 回、その後は必要に応じて適宜腎機 能検査を行うこと。 8.2 本剤での密封法及び重層法による臨床使用経験はなく、安全性は確立していない。 8.3 アルビノ無毛マウスに 40 週間にわたり UVA 及び UVB を照射し、その後 12 週間無処置期間を設け て観察すると試験動物のすべてに皮膚腫瘍が発生するが、この試験系において紫外線照射と並行して 本剤を塗布すると皮膚腫瘍の発生時期が早まることが示されている。また、過度の紫外線曝露は皮膚 腫瘍のリスクファクターであることから、日光への曝露を最小限にとどめ、日焼けランプ/紫外線ラン プの使用は避けること(「Ⅸ.非臨床試験に関する項目 2.毒性試験 (7)その他の特殊毒性 3)」の項参照)。 8.4 本剤の使用について、外国では 2 年の臨床試験成績があるが、それ以上の期間の使用経験はなく、長 期使用によるリスクについては不明であるため、その旨を記載した。 8.5 本剤は免疫抑制作用を有しており、皮膚感染症を伴う患者では皮膚感染症が増悪するおそれがある。 8.6 本剤使用後における皮膚刺激感の発現頻度はかなり高いこと、多くは皮疹の改善に伴い発現しなくな る4)ことから、患者の不安を緩和するため皮膚刺激感があることをあらかじめ十分説明すること。ま た、刺激感は入浴時に増強することがある。 なお、使用期間中に高度の刺激感が持続する場合には休薬もしくは中止する。