厚生労働科学研究費補助金 難治性疾患等克服研究事業(難治性疾患克服研究事業) 分担研究報告書
遺伝性不整脈におけるL型カルシウムチャネルαサブユニットをコードするCACNA1C遺伝子の関与 研究分担者 堀江 稔 滋賀医科大学 内科学講座(循環器・呼吸器) 教授
研究要旨:滋賀医科大学・京都大学に遺伝子解析目的にて登録された約1300家系より、BrS・ERS・
SQTS・IVFと診断された312名の発端者およびLQTSを疑われた278名の発端者を抽出し、CACNA1C 遺伝子を解析した。結果、6名のBrS家系に5種の変異、7名のLQTS家系に5種の変異を同定した。
BrS家系に同定された5変異のうち1変異(CACNA1C-R632R)は塩基置換部位がCACNA1C-exon14の1 塩基目に存在しており、スプライシングエラーを生じるものと考えられた。CACNA1C-R632R保有者 家系の mRNA定量解析を行い、NMDにより mRNA 発現量の低下を引き起こし、機能低下を呈する ことを確認した。LQTS家系に同定された5変異については、パッチクランプ法を用いた機能解析を 行い、2 変異については明らかな機能亢進が示され、Timothy 症候群の心臓外兆候を呈さない変異保 有者が存在することを発見した。本研究は、遺伝性不整脈を有する日本人コホートにおけるCACNA1C 変異の関与を明らかにしたものである。
A.研究目的
遺伝性不整脈は、心臓の興奮・伝導・収縮に関 わる蛋白群をコードする遺伝子の異常により、蛋 白の機能が障害され、結果として多様な不整脈を 起こす病気である。当初より調べられたのはQT延 長症候群である。QT 延長症候群は、心電図上 QT 間隔の延長、意識消失発作、torsade de pointes、心 臓突然死を特徴とする遺伝性不整脈である。原因 遺伝子は現在のところ16種類報告されている。さ らに、特異な胸部誘導V1-3におけるST上昇と心 室細動を特徴とする Brugada 症候群やカテコラミ ン感受性多型性心室頻拍など、10 近い不整脈の原 因遺伝子が同定されている。われわれは、1996 年 から、家族性不整脈症候群に注目し、表1(次ペー ジ)のような疾患について詳しい病像とゲノムを 集積している。また、そのうち現時点で、遺伝子 診断されたコホートについて、内訳を右の図1に 示す。
L 型心筋カルシウムチャネル(L-type Calcium Channel: LTCC) α サ ブ ユ ニ ッ ト を コ ー ド す る
CACNA1C 遺伝子変異が引き起こす遺伝性不整脈
疾患の代表
図1:遺伝子診断例の内訳
的なものとして、チャネル機能亢進による Timothy 症 候群(8 型 QT延長症候群:LQT8)1とチャネル機能低 下による3 型 Brugada 症候群(BrS3)2が挙げられる。
近年では、QT 短縮症候群(SQTS)3や早期再分極症 候群(early repolarization syndrome: ERS)4、特発性心 室細動との関与も指摘されている。LTCC は 4 つのサ ブユニットから構成される巨大タンパクであり5、その遺 伝子解析及び遺伝子変異から生じる異常タンパクの 解析は非常に困難なため、他のイオンチャネル異常と 比較して、報告されている遺伝子異常も少ない。いず れも欧米からの報告であり、日本人を始めとするアジ ア人種での報告はなかった。
今回、我々は遺伝性不整脈を 有 す る 日 本 人 コ ホ ー ト に お け る CACNA1C 変異を同定し、変異保 持者の表現型、及び遺伝子変異 によるチャネルの機能変化につい て比較検討した。
B.研究方法
原因遺伝子の検索目的にて滋 賀医科大学循環器内科および京 都大学大学院循環器内科学講座 に登録された約1800家系(表1) より、BrS・ERS・SQTS・IVF と診断された312名の発端者(家 系)、および臨床的にQT延長症 候群が疑われ、かつ他の原因遺 伝子が同定されていない278名 の発端者をそれぞれ抽出し、
CACNA1C 遺伝子を解析した。
LTCCは遺伝子量が多いため、検索ではまず高解像 度融解(High Resolution Melting: HRM)曲線分析で スクリーニングを実施し、遺伝子変異の可能性が 指摘される発端者を抽出したのち、DNAポリメラ ーゼ連鎖反応(PCR)法で遺伝子配列を読んで詳細 を確認した。
同定された変異のうち、スプライシング変異に ついては、発端者および家族より新たに新鮮末梢 血を採取し、mRNA を抽出した。RT-PCR により mRNAからcDNA合成を行い、direct sequence法に よ る 配 列 確 認 お よ び LightCycler®を 用 い て 、 advanced relative quantification法による定量解析を 行った。
また、LQTS患者に同定されたCACNA1C変異に ついては、whole-cell patch-clamp法を用いて、同定 した遺伝子変異がチャネルに及ぼす機能変化を確 認した。
(表1:症例コホートの内訳)
(倫理面への配慮)
本研究は、ヘルシンキ宣言(世界医師会)ヒトゲ ノム・遺伝子解析研究に関する倫理指針(平成 16 年文部科学省・厚生労働省・経済通産省告示第 1 号)に準拠して実施する。また本研究は、所属施
設の倫理委員会の承認を得ている。
倫理委員会での承認状況:
滋賀医科大学:
家族性不整脈症候群における遺伝子解析
(H21年9月29日更新)承認番号: 21-50)
C.研究結果
Brugada症候群におけるCACNA1C変異
我々のコホートにおいては、BrS の6家系(2.2%) か ら 5 種 の CACNA1C 変 異(N547S, R632Rspl, R1780H, C1855Y and R1910Q(2家系))が確認された。
うち 2 例は、有症候性であった。注目したのは,
安静時心電図では異常を認めなかったが失神を繰 り返し、ピルシカイニド投与下で著明なcoved型の ST上昇が確認された女性患者と、無症状であった が安静時にsaddleback型のST上昇を認め、ピルシ カイニド投与下でcoved型のST上昇が認められた 男性患者であった。図2に 2名の心電図を示す。
彼らは同じR1910Q変異を保持していた。これは、
Brugada 症候群は圧倒的に男性患者が多いとされ
ているが,遺伝子変異の保持率は男女で変わらな いことを裏付ける結果であった。6
LQTS 773
ATS 39
Secondary 16 (hypo-K, hypo-Mg, anorexia, SAH, etc…) Drug-induced LQT 34
BrS 298
PCCD 113 (Juvenile PMI, familial PMI, A-V block)
IVF/VT 84
CPVT 69
ARVC 67
Juvenile/familial AF 64
DCM/HCM 58 (HCM;16)
Syncope 31
ERS 16
SQTS 10
Other 93 MuPVCs, family history of Sudden death, Fabry
図2
5 変異のうち 1 変異(CACNA1C-R632R)は塩基置換 部位がCACNA1C-exon14の1塩基目に存在してい るスプライシング変異であった。CACNA1C-R632R 保有者の臨床経過としては27歳時に心室細動を来 たし、電気生理学検査でも心室細動が再現された ため植え込み型除細動器(ICD)の植え込みが為され ていた。家系内の遺伝子検索では、発端者の長女 に同変異が同定された。発端者の第 2 子である長 男には変異は認めなかった。変異保持者である発 端者及び長女から得たcDNAをdirect sequenceにて 解析したところ、変異アレルが消失していた(図 3 左)。
これは、変異アレルがナンセンス変異依存mRNA 分解機構(nonsense-mediated mRNA decay, NMD)に よって排除されたことを示唆する結果であった。
NMD による mRNA 発現量低下を証明するために 行った定量解析では、同家系内の変異非保持者(長 男)と比較して、変異保持者(発端者及び長女)の CACNA1C-mRNA発現量がそれぞれ約44%(発端者)、
約30%(長女)減少していた(図3右)。7 図3
QT症候群におけるCACNA1C変異
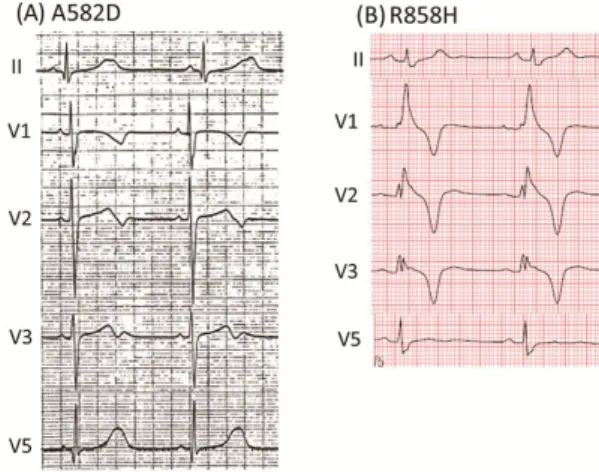
遺伝子スクリーニングにおいて、5 つの新たな CACNA1C変異(P381S, M456I, A582D, R858H(3家 系), G1783C)を7家系に同定した(2.5%) (表2)。図4 に示すように、変異保有者の 12 誘導心電図には、
QT時間の延長に加えて不明瞭なT波終末やノッチ を呈するT波を認めるものがあった。
図4
機能解析では、R858H変異ではチャネル活性化時 のカルシウム電流が正常チャネルに比べて有意に 増加していた。一方、A582D 変異では活性化時の カルシウム電流量は変化がなかったが、不活性化 が有意に遅延していた(図5)。
図5
D.考案
今回の研究で、日本人コホートにおける遺伝性 不整脈患者の CACNA1C 変異頻度を示すことがで きた。下の図6 に、今回同定できた CACNA1C 全 10変異のTopologyを示す。
図 6
本研究では、BrSにおけるCACNA1C変異の頻度は
2%台であり、欧米の 8-10%という報告と比較する
と低かった。しかし、有症候性の女性患者が含ま れていたことや、CACNA1Cスプライシング変異に 対する NMD の関与を証明できたことは新たな発 見であった。
遺伝性不整脈分野におけるスプライシング変異の 代表的なものとして LQTS type1 で同定された KCNQ1-A344A変異があるが、この変異はNMDを 生じない8。一方でNMDについては、LQTS type2
において KCNH2 遺伝子のナンセンス変異から
NMD が惹起されるという報告がある9。また、こ れまでの BrS におけるLTCC 変異の報告は、アミ ノ酸変異を伴うチャネル機能異常によるものであ った。本研究は、BrS患者において、NMDを呈す る CACNA1C スプライシング変異について初めて 報告したものである。
一方で CACNA1C 変異による機能亢進異常は、
Timothy 症候群(TS)の原因として報告されており、
本症は多臓器にわたる特徴的かつ重篤な身体症状 を引き起こすとされてきた。また頻度は非常に稀 であり、CACNA1C-G402S・G406Rの二種類のみの 報告であった。2013年にBoczekらは、whole-exome
sequencing を用いたスクリーニングにて 4 種の
CACNA1C variantsを同定し、TSの心臓外兆候を呈 さないLQT8が存在する可能性を示した10。本研究 では、古典的なSanger法を用いて、他の遺伝子異 常が同定できない LQTS 患者において 5 種の CACNA1C変異を同定し、その頻度はこれまで報告 されてきたLQT1-3以外のLQTSで同定される遺伝 子頻度よりも高かった。また変異保持者の表現型、
及び遺伝子変異によるチャネルの機能変化につい ても明らかにした。とりわけ CACNA1C-A582D 変 異においては、Domain II 2-3細胞内linkerに存在す る変異は BrS 等を含めても報告がなく、機能解析 では興味深い結果を示した。不活性化の遅延する タイプの機能亢進であり、TSにおけるG406R変異 の機能障害と近似しているが、A582D 患者は QTc の著明な延長以外は症状を有していなかった。
E.結論
BrSにおいては、日本人コホートでは欧米に比し てCACNA1C変異の頻度は低かったが、CACNA1C のスプライシング変異および NMD による mRNA 発現量の減少がLTCC機能低下を惹起し、BrSを呈
するという新たなメカニズムの存在が示された。
またLQTSについては、CACNA1C変異はTimothy 症候群にみられるような重篤な身体症状を合併し ない LQTS 患者においても同定され、その頻度は これまで報告されてきたLQT1-3以外のLQTSで同 定される遺伝子頻度よりも高かった。他に原因遺 伝 子 が 同 定 さ れ な い LQTS 患 者 に お い て 、
CACNA1C 変異の検索は臨床的に重要であると考
えられた。
関連文献
1. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(v)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell.
2004;119:19-31
2 Berne P, Brugada J. Brugada syndrome 2012. Circ J.
2012;76:1563-1571
3. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C.
Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442-449 4. Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M, Häissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laiño R, Marieb M, Nademanee K, Nam GB, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C, Zimmern S, Veltmann C, Antzelevitch C. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872-1882
5.Catterall WA. Structure and function of voltage-sensitive ion channels. Science.
1988;242:50-61
6.Fukuyama M, Ohno S, Wang Q, Kimura H, Makiyama T, Itoh H, Ito M, Horie M. L-type calcium channel mutations in Japanese patients with inherited arrhythmias. Circ J. 2013;77:1799-1806
7. Fukuyama M, Ohno S, Wang Q, Shirayama T, Itoh H, Horie M. Nonsense-mediated mrna decay due to a cacna1c splicing mutation in a patient with brugada syndrome. Heart Rhythm. 2013
8. Murray A, Donger C, Fenske C, Spillman I, Richard P, Dong YB, Neyroud N, Chevalier P, Denjoy I, Carter N, Syrris P, Afzal AR, Patton MA, Guicheney P, Jeffery S. Splicing mutations in KCNQ1: A mutation hot spot at codon 344 that produces in frame transcripts. Circulation. 1999;100:1077-1084 9. Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z.
Nonsense mutations in hERG cause a decrease in mutant mrna transcripts by nonsense-mediated mRNA decay in human long-QT syndrome.
Circulation. 2007;116:17-24
10. Boczek NJ, Best JM, Tester DJ, Giudicessi JR, Middha S, Evans JM, Kamp TJ, Ackerman MJ.
Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ Cardiovasc Genet.
2013;6:279-289
G.研究発表 1. 論文発表
1. Villafane J, Atallah J, Gollob MH, Maury P, Wolpert C, Gebauer R, Watanabe H, Horie M, Anttonen O, Kannankeril P, Faulknier B, Bleiz J, Makiyama T, Hamilton R, Young M-L. Long Term Follow-up of a Pediatric Cohort with Short QT Syndrome. J Am Coll Cardiol. 61(11):
1183-91, 2013.
2. Wang Q, Ohno S, Kato K, Fukuyama M,
Makiyama T, Kimura H, Naiki N, Kawamura M, Hayashi H, Horie M. Genetic Screening of KCNJ8 in Japanese Patients with J-wave Syndromes or Idiopathic Ventricular Fibrillation.
J Arrhythmia 29: 261-264, 2013.
3. Ohno S, Nagaoka I, Fukuyama M, Kimura H, Itoh H, Makiyama T, Shimizu A, Horie M.
Age-dependent clinical and genetic character- ristics in Japanese patients with arrhythmogenic right ventricular cardiomyopathy/ dysplasia. Circ J 77: 1534-1542, 2013.
4. Duchatelet S, Crotti L, Peat R, Denjoy I, Itoh H, Berthet M, Ohno S, Fressart V, Monti C, Crocamo C, Pedrazzini M, Dagradi F, Vicentini
A, Klug D, Brink P, Goosen A, Heikki S, Toivonen L, Lahtinen A, Kontula K, Shimizu W, Horie M, George Jr. AL, Tregouet DA,
Guicheney P, Schwartz PJ. Identification of a KCNQ1 polymorphism acting as a protective modifier against arrhythmic risk in long QT syndrome. Circ Cardiovasc Genet 6: 354-61, 2013.
5. Dochi K, Watanabe H, Kawamura M, Miyamoto A, Ozawa T, Nakazawa Y, Ashihara T, Ohno S, Hayashi H, Ito M, Sakazaki H, Kawata H, Ushinohama H, Kaszynski RH, Minamino T, Sumitomo N, Shimizu W, Horie M. Flecainide reduces ventricular arrhythmias via different actions from β-blockers in catecholaminergic polymorphic ventricular tachycardia. J Arrhythmia 29: 255-260, 2013.
6. Kawamura M, Ohno S, Naiki N, Nagaoka I, Dohchi K, Wang Q, Hasegawa K, Kimura H, Miyamoto A, Mizusawa Y, Itoh H, Makiyama T, Sumitomo N, Ushinohama H, Oyama K,
Murakoshi N, Aonuma K, Horigome H, Honda T, Yoshinaga M, Ito M, Horie M. Genetic
background of catecholaminergic polymorphic ventricular tachycardia in Japan. Circ J 77:
1705-1713, 2013.
7. Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, Rosso R, Bhuiyan ZA, Bikker H, Kannankeril PJ, Horie M, Minamino T, Viskin S, Knollmann BC, Till J, Wilde AA. Effects of flecainide on
exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 10: 542-547, 2013.
8. Horie M, Ohno S. Genetic basis of Brugada syndrome. J Arrhythmia 29: 71–76, 2013.
9. Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, Hattori T, Ohno S, Kita T, Horie M, Yamanaka S, Kimura T. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J. 77(5): 1307-14, 2013.
10. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S,
Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Marec HL, Wilde AA, Probst V, Schott JJ, Dina C, Redon R.
Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 45(9): 1044-9, 2013.
11. Hayashi H, Murakami Y, Horie M. Pitfall of the meta-analysis regarding early repolarization pattern. J Am Coll Cardiol. 62: 86, 2013.
12. Smith JL, Reloj AR, Nataraj PS, Bartos DC, Schroder EA, Moss AJ, Ohno S, Horie M, Anderson CL, January CT, Delisle BP.
Pharmacological Correction of Long QT-linked Mutations in KCNH2 (hERG) Increases the Trafficking of Kv11.1 Channels Stored in the Transitional ER. Am J Physiol -Cell Physiology 305: C919-30, 2013.
13. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C.
HRS/EHRA/APHRS Expert Consensus
Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm.
10: 1932-63, 2013.
14. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C;
Document Reviewers, Ackerman M, Belhassen B, Estes NA 3rd, Fatkin D, Kalman J, Kaufman E, Kirchhof P, Schulze-Bahr E, Wolpert C, Vohra J, Refaat M, Etheridge SP, Campbell RM, Martin ET, Quek SC. Executive summary:
HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes.
Europace. 15(10): 1389-406, 2013.
15. Hayashi H, Shibukawa T, Horie M. Restoration of aberrant conduction induced by premature ventricular contractions. Intern Med. 52:1425, 2013.
16. Hasegawa K, Ohno S, Itoh H, Makiyama T, Aiba T, Nakano Y, Shimizu W, Matsuura H, Horie M.
A rare KCNE1 polymorphism, D85N, as a genetic modifier of long QT syndrome. J Arrhythmia 2014 (in press)
2. 学会発表
1. Ohno S, Hasegawa K, Makiyama T, Doi S, Horie M. Different regulation of IKS channels by two KCNE 1 C- terminus variants predicts the QTc response to the exercise stress. The Heart Rhythm Society's 34th Annual Scientific Sessions (2013.05.08-11, Denver, CO, U.S.A.) 2. Itoh H, Berthet M, Fressart V, Denjoy I ,
Maugenre S, Klug D, Mizusawa Y, Makiyama T, Hofman N, Husemann A, Shimizu W, Wilde AA , Schulze-Bahr E, Horie M, du Montcel ST, Guicheney P. Asymmetry of parental origin in Long QT syndrome. European Human Genetics Conference (2013.06.08-11, Paris, France) 3. Ohno S, Omura M, Kawamura M, Kimura H,
Itoh H, Makiyama T, Ushinohama H, Makita N, Horie M. Exon-3 deletion of RyR2 encoding cardiac ryanodine receptor related to left
ventricular non-compaction (LVNC) with ventricular arrhythmia and bradycardia. EHRA EUROPACE 2013 (2013.06.23- 26, Athens, Greece)
4. Fukuyama M, Wang Q, Kato K, Ohno S, Kimura H, Makiyama T, Itoh H, Ito M, Matsuura H, Horie M. Novel CACNA1C mutations in Long QT syndrome patients- The subtype of Long QT syndrome type 8. Denis Escande Symposium 2013(2013.8.30-31, Amsterdam, The Nether- lands)
5. Ohno S, Fukuyama M, Itoh H, Makiyama T, Horie M. Copy number variation in KCNQ1 gene were frequently identified in the pediatric patients of long QT syndrome and caused exercise related QT prolongation. ESC
CONGRESS 2013(2013.8.31-9.4, Amsterdam, The Netherlands)
6. Horie M: Genetic and Molecular Basis of ARVC.
VT Workshop 2-ARVC Session. 6th APHRS &
CardioRhythm 2013.(2013.10.03-06, Hong Kong, China)
7. Horie M: Genetic Testing in ARVC. Genetic and Inherited Syndrome 1-Update on Clinical Applications of Genetic Testing. 6th APHRS &
CardioRhythm 2013.(2013.10.03-06, Hong Kong, China)
8. Ohno S, Wang Q, Hasegwas K, Itoh H, Makiyama T, Horie M. Phenotypic
characterization of three patients with lethal arrhythmia related to KCNH2-R148W missense mutation. 6th APHRS & CardioRhythm
2013.(2013.10.03-06, Hong Kong, China) 9. Hasegawa K, Watanabe H, Ohno S, Itoh H,
Makiyama T, Ashihara T, Hayashi H, Horie M.
The High Prevalence of Early Repolarization in Genotyped Long QT Syndrome. AHA Scientific Sessions 2013.(2013.11.16-20, Dallas, Texas, U.S.A.)
H.知的財産権の出願・登録状況(予定を含む。)
1. 特許取得 なし
2. 実用新案登録 なし
3. その他 (研究協力者)
大野聖子 (滋賀医科大学 呼吸循環器内科)